

CYP153A71 from Alcanivorax dieselolei: Oxidation beyond Monoterminal Hydroxylation of n-Alkanes

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Heterologous Expression of CYP153A71 and Associated Redox-Partner Proteins
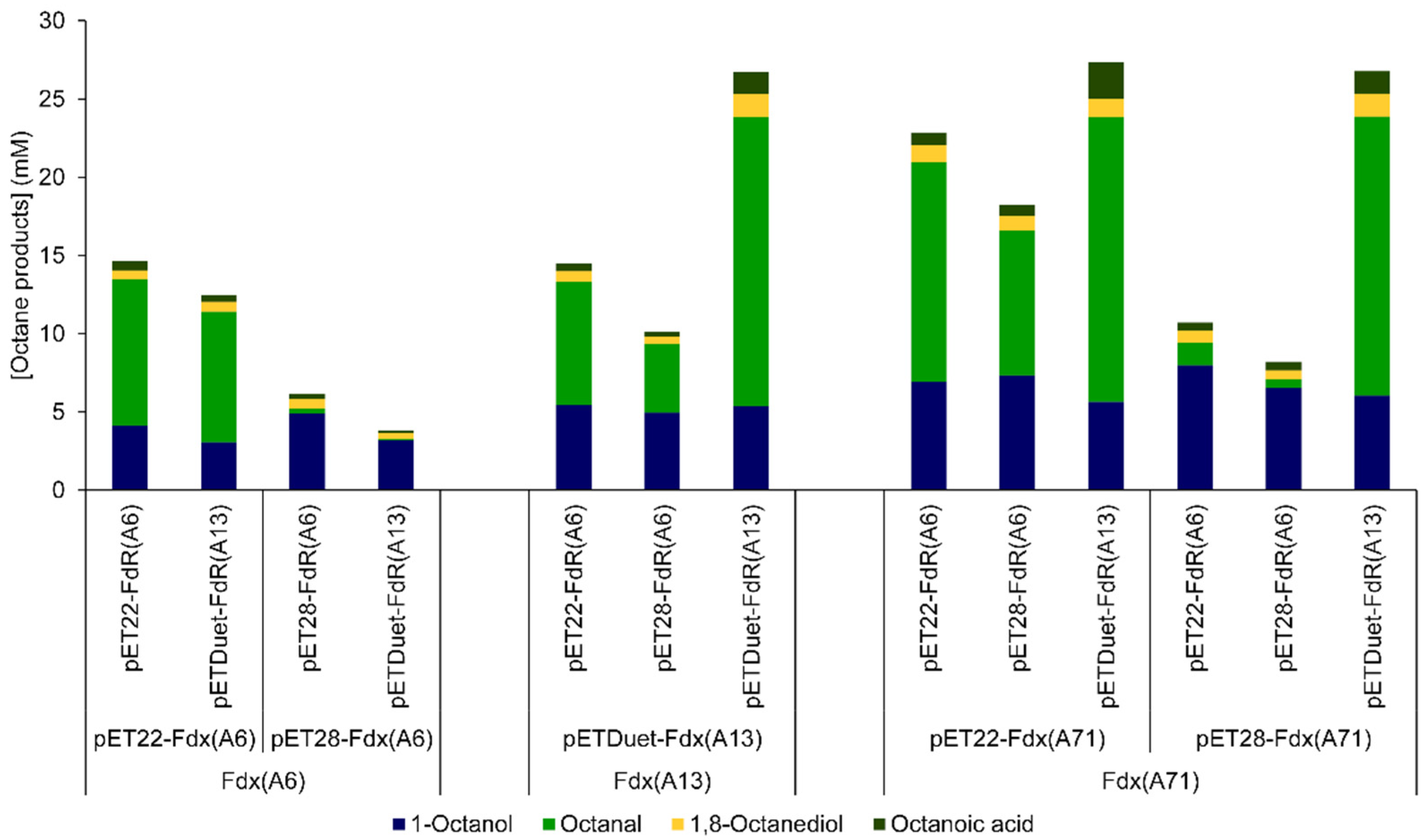
2.2. Redox-Partner Coupling
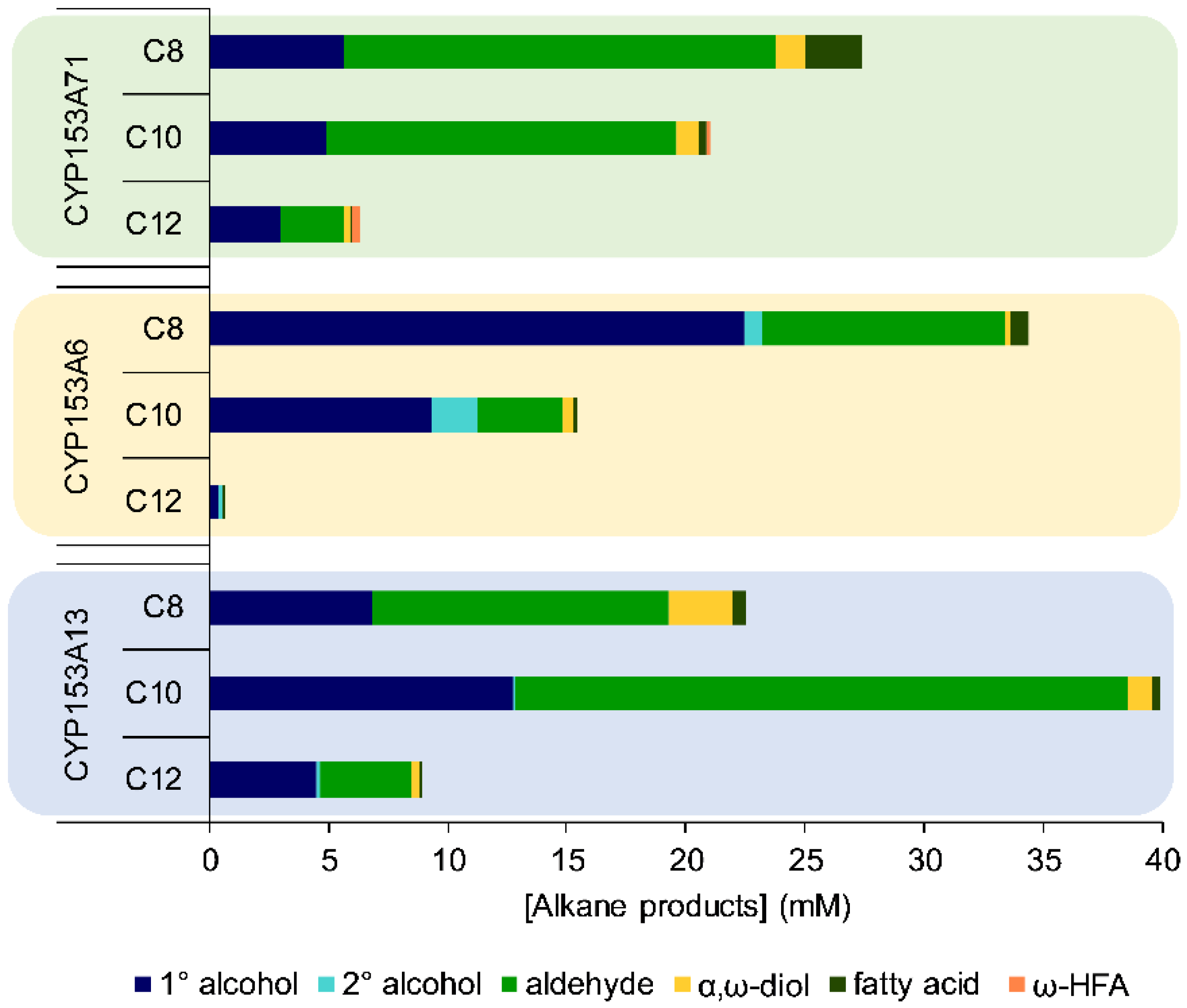
2.3. Substrate Scope
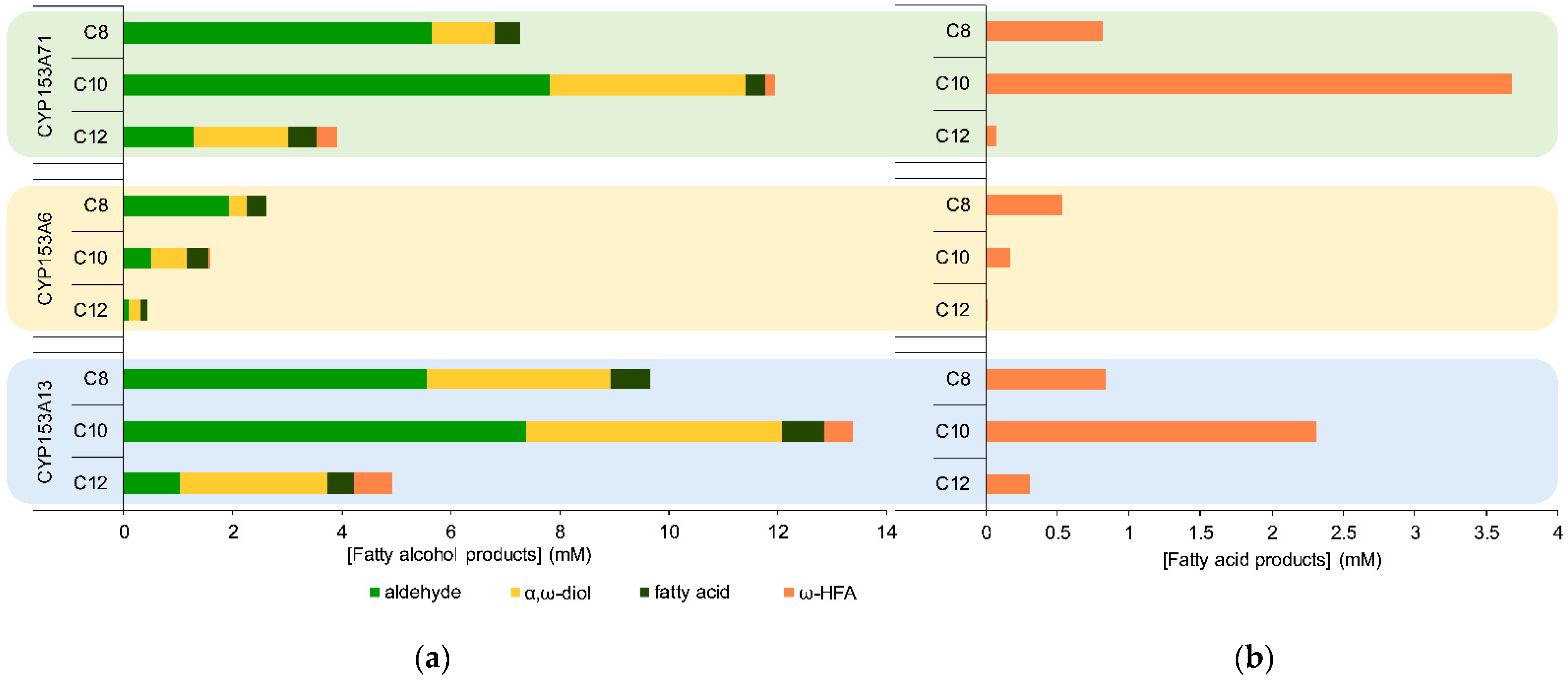
2.4. Factors Affecting Over-Hydroxylation
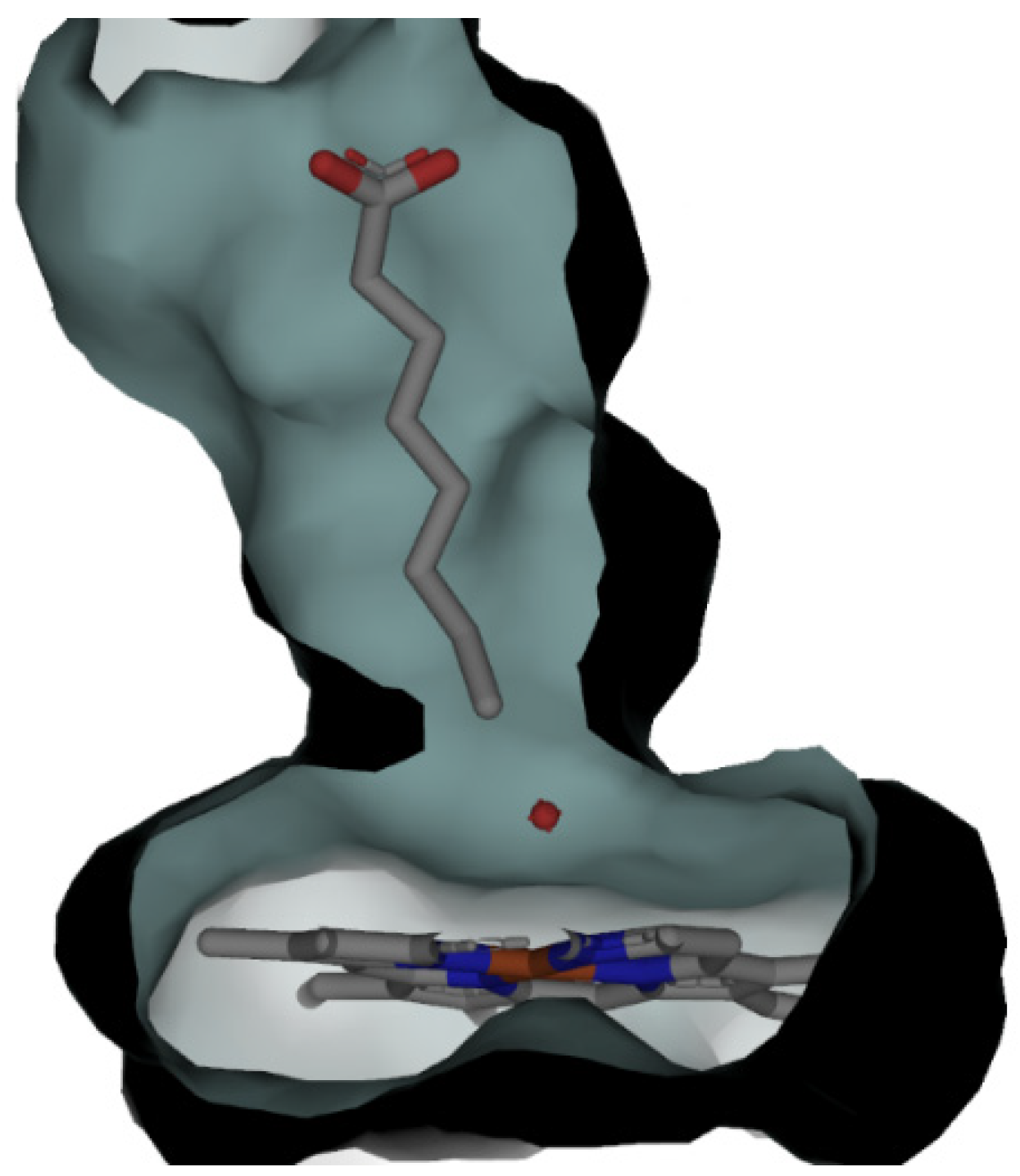
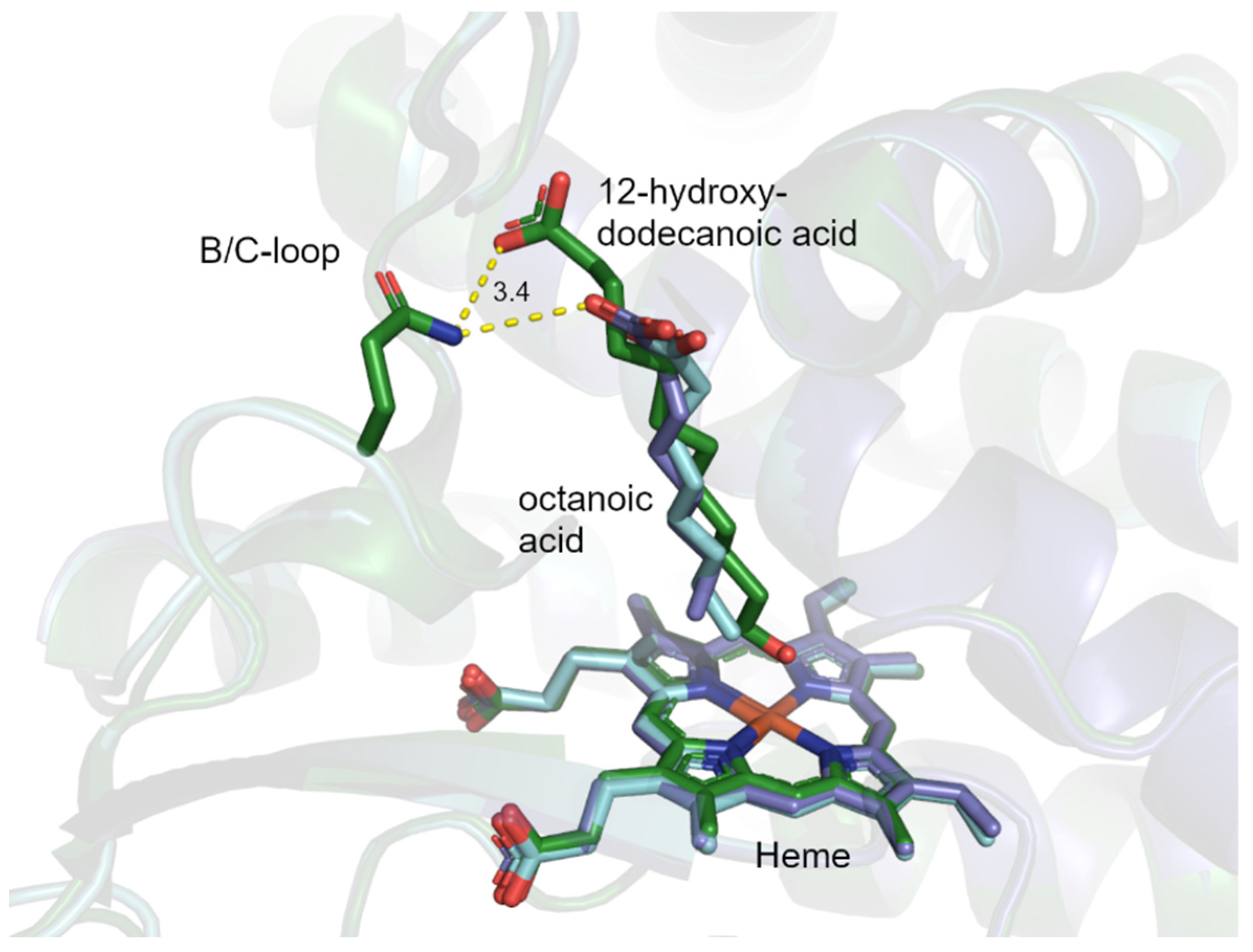
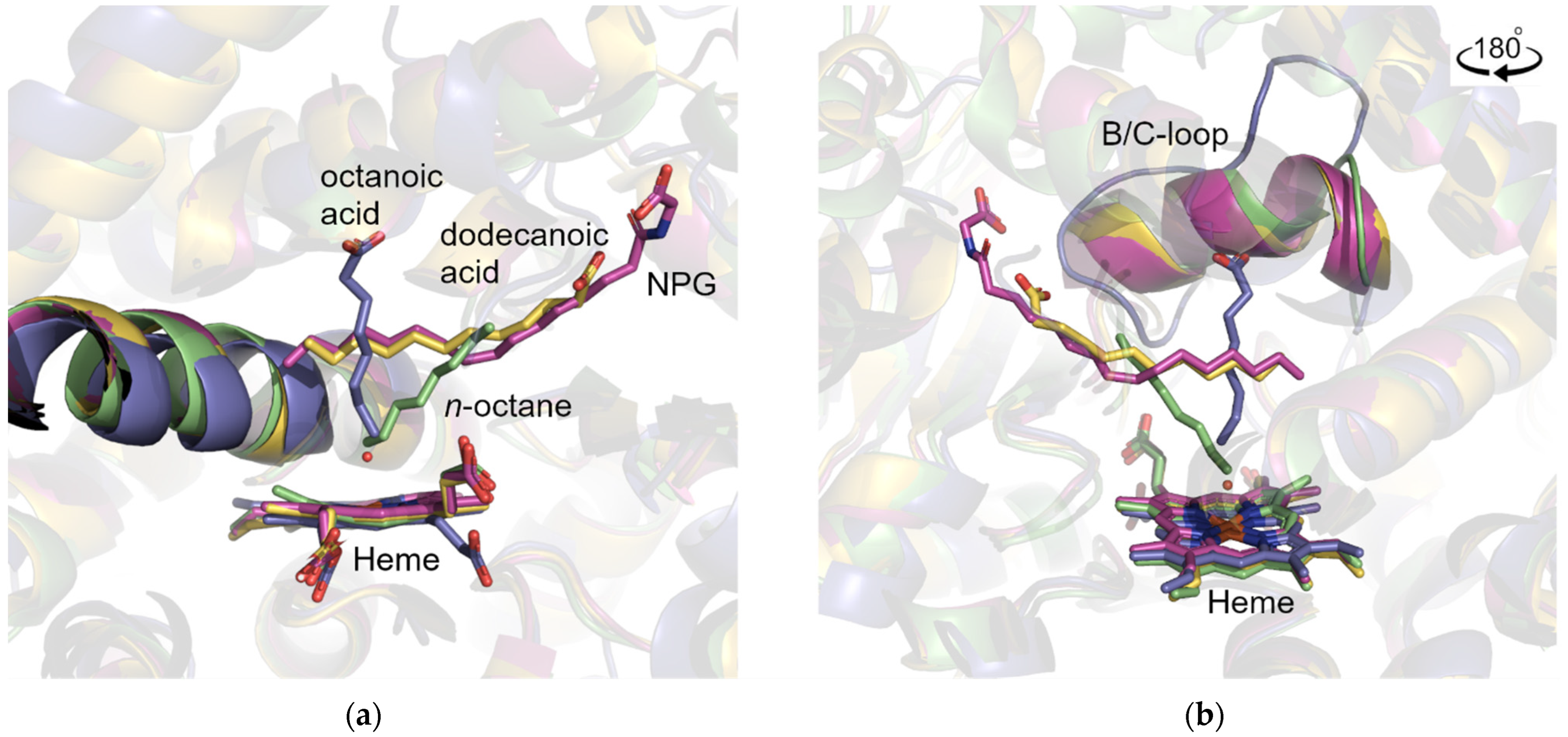
2.5. CYP153A71 Structure Determination
3. Materials and Methods
3.1. Expression Constructs
3.2. Heterologous Expression
3.3. Biotransformations Using Cell-Free Extracts (CFEs)
3.4. Protein Purificaton
3.5. Biotransformations Using Purified Enzyme
3.6. Crystallization and Structure Determination of CYP153A71
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sterckx, H.; Morel, B.; Maes, B.U.W. Catalytic Aerobic Oxidation of C(Sp3)−H Bonds. Angew. Chem. Int. Ed. 2019, 58, 7946–7970. [Google Scholar] [CrossRef]
- Münch, J.; Püllmann, P.; Zhang, W.; Weissenborn, M.J. Enzymatic Hydroxylations of Sp3-Carbons. ACS Catal. 2021, 11, 9168–9203. [Google Scholar] [CrossRef]
- Guengerich, F.P. Mechanisms of Cytochrome P450 Substrate Oxidation: MiniReview. J. Biochem. Mol. Toxicol. 2007, 21, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Hammerer, L.; Winkler, C.K.; Kroutil, W. Regioselective Biocatalytic Hydroxylation of Fatty Acids by Cytochrome P450s. Catal. Lett. 2018, 148, 787–812. [Google Scholar] [CrossRef] [Green Version]
- Hannemann, F.; Bichet, A.; Ewen, K.M.; Bernhardt, R. Cytochrome P450 Systems-Biological Variations of Electron Transport Chains. Biochim. Biophys. Acta 2007, 1770, 330–344. [Google Scholar] [CrossRef]
- Van Beilen, J.B.; Funhoff, E.G.; van Loon, A.; Just, A.; Kaysser, L.; Bouza, M.; Holtackers, R.; Röthlisberger, M.; Li, Z.; Witholt, B. Cytochrome P450 Alkane Hydroxylases of the CYP153 Family Are Common in Alkane-Degrading Eubacteria Lacking Integral Membrane Alkane Hydroxylases. Appl. Environ. Microbiol. 2006, 72, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Beilen, J.B.; Holtackers, R.; Lüscher, D.; Bauer, U.; Witholt, B.; Duetz, W.A. Biocatalytic Production of Perillyl Alcohol from Limonene by Using a Novel Mycobacterium sp. Cytochrome P450 Alkane Hydroxylase Expressed in Pseudomonas Putida. Appl. Environ. Microbiol. 2005, 71, 1737–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudiminchi, R.K.; Randall, C.; Opperman, D.J.; Olaofe, O.A.; Harrison, S.T.L.; Albertyn, J.; Smit, M.S. Whole-Cell Hydroxylation of n-Octane by Escherichia coli Strains Expressing the CYP153A6 Operon. Appl. Microbiol. Biotechnol. 2012, 96, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Funhoff, E.G.; Salzmann, J.; Bauer, U.; Witholt, B.; van Beilen, J.B. Hydroxylation and Epoxidation Reactions Catalyzed by CYP153 Enzymes. Enzym. Microb. Technol. 2007, 40, 806–812. [Google Scholar] [CrossRef]
- Li, A.; Liu, J.; Pham, S.Q.; Li, Z. Engineered P450pyr Monooxygenase for Asymmetric Epoxidation of Alkenes with Unique and High Enantioselectivity. Chem. Commun. 2013, 49, 11572. [Google Scholar] [CrossRef] [PubMed]
- Kubota, M.; Nodate, M.; Yasumoto-Hirose, M.; Uchiyama, T.; Kagami, O.; Shizuri, Y.; Misawa, N. Isolation and Functional Analysis of Cytochrome P450 CYP153A Genes from Various Environments. Biosci. Biotechnol. Biochem. 2005, 69, 2421–2430. [Google Scholar] [CrossRef] [PubMed]
- Malca, S.H.; Scheps, D.; Kühnel, L.; Venegas-Venegas, E.; Seifert, A.; Nestl, B.M.; Hauer, B. Bacterial CYP153A Monooxygenases for the Synthesis of Omega-Hydroxylated Fatty Acids. Chem. Commun. 2012, 48, 5115. [Google Scholar] [CrossRef] [PubMed]
- Funhoff, E.G.; Bauer, U.; García-Rubio, I.; Witholt, B.; van Beilen, J.B. CYP153A6, a Soluble P450 Oxygenase Catalyzing Terminal-Alkane Hydroxylation. J. Bacteriol. 2006, 188, 5220–5227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, T.; Narikawa, T.; Sumisa, F.; Arisawa, A.; Takeda, K.; Kato, J. Production of α,ω-Alkanediols Using Escherichia coli Expressing a Cytochrome P450 from Acinetobacter sp. OC4. Biosci. Biotechnol. Biochem. 2006, 70, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Scheps, D.; Malca, S.H.; Hoffmann, H.; Nestl, B.M.; Hauer, B.; Malca, S.H.; Hoffmann, H.; Nestl, B.M.; Hauer, B. Regioselective ω-Hydroxylation of Medium-Chain n-Alkanes and Primary Alcohols by CYP153 Enzymes from Mycobacterium marinum and Polaromonas sp. strain JS666. Org. Biomol. Chem. 2011, 9, 6727–6733. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, J.; Li, Z. Engineering of P450pyr Hydroxylase for the Highly Regio- and Enantioselective Subterminal Hydroxylation of Alkanes. Angew. Chem. Int. Ed. 2014, 53, 3120–3124. [Google Scholar] [CrossRef]
- Nebel, B.A.; Scheps, D.; Malca, S.H.; Nestl, B.M.; Breuer, M.; Wagner, H.G.; Breitscheidel, B.; Kratz, D.; Hauer, B. Biooxidation of N-Butane to 1-Butanol by Engineered P450 Monooxygenase under Increased Pressure. J. Biotechnol. 2014, 191, 86–92. [Google Scholar] [CrossRef]
- Yang, Y.; Chi, Y.T.; Toh, H.H.; Li, Z. Evolving P450pyr Monooxygenase for Highly Regioselective Terminal Hydroxylation of N-Butanol to 1,4-Butanediol. Chem. Commun. 2015, 51, 914–917. [Google Scholar] [CrossRef]
- Hoffmann, S.M.; Danesh-Azari, H.R.; Spandolf, C.; Weissenborn, M.J.; Grogan, G.; Hauer, B. Structure-Guided Redesign of CYP153AM.Aq for the Improved Terminal Hydroxylation of Fatty Acids. ChemCatChem 2016, 8, 3178. [Google Scholar] [CrossRef] [Green Version]
- Notonier, S.; Gricman, Ł.; Pleiss, J.; Hauer, B. Semirational Protein Engineering of CYP153AM.Aq.-CPRBM3 for Efficient Terminal Hydroxylation of Short- to Long-Chain Fatty Acids. ChemBioChem 2016, 17, 1550–1557. [Google Scholar] [CrossRef]
- Duan, Y.; Ba, L.; Gao, J.; Gao, X.; Zhu, D.; de Jong, R.M.; Mink, D.; Kaluzna, I.; Lin, Z. Semi-Rational Engineering of Cytochrome CYP153A from Marinobacter aquaeolei for Improved ω-Hydroxylation Activity towards Oleic Acid. Appl. Microbiol. Biotechnol. 2016, 100, 8779–8788. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.; Park, B.G.; Yoo, H.W.; Kim, J.; Choi, K.Y.; Kim, B.G. Semi-Rational Engineering of CYP153A35 to Enhance ω-Hydroxylation Activity toward Palmitic Acid. Appl. Microbiol. Biotechnol. 2018, 102, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Rapp, L.R.; Marques, S.M.; Zukic, E.; Rowlinson, B.; Sharma, M.; Grogan, G.; Damborsky, J.; Hauer, B. Substrate Anchoring and Flexibility Reduction in CYP153A M.Aq Leads to Highly Improved Efficiency toward Octanoic Acid. ACS Catal. 2021, 11, 3182–3189. [Google Scholar] [CrossRef]
- Dong, Y.; Chong, G.; Li, C.; Chen, Q.; Pan, J.; Li, A.; Xu, J. Carving the Active Site of CYP153A7 Monooxygenase for Improving Terminal Hydroxylation of Medium-Chain Fatty Acids. ChemBioChem 2022, 23, e202200063. [Google Scholar] [CrossRef]
- Liu, C.; Wang, W.; Wu, Y.; Zhou, Z.; Lai, Q.; Shao, Z. Multiple Alkane Hydroxylase Systems in a Marine Alkane Degrader, Alcanivorax dieselolei B-5. Environ. Microbiol. 2011, 13, 1168–1178. [Google Scholar] [CrossRef]
- Wang, W.; Shao, Z. The Long-Chain Alkane Metabolism Network of Alcanivorax Dieselolei. Nat. Commun. 2014, 5, 5755. [Google Scholar] [CrossRef] [Green Version]
- Otomatsu, T.; Bai, L.; Fujita, N.; Shindo, K.; Shimizu, K.; Misawa, N. Bioconversion of Aromatic Compounds by Escherichia coli That Expresses Cytochrome P450 CYP153A13a Gene Isolated from an Alkane-Assimilating Marine Bacterium Alcanivorax borkumensis. J. Mol. Catal. B Enzym. 2010, 66, 234–240. [Google Scholar] [CrossRef]
- Park, H.; Bak, D.; Jeon, W.; Jang, M.; Ahn, J.; Choi, K. Engineering of CYP153A33 with Enhanced Ratio of Hydroxylation to Overoxidation Activity in Whole-Cell Biotransformation of Medium-Chain 1-Alkanols. Front. Bioeng. Biotechnol. 2022, 9, 817455. [Google Scholar] [CrossRef]
- Guengerich, F.P.; Sohl, C.D.; Chowdhury, G. Multi-Step Oxidations Catalyzed by Cytochrome P450 Enzymes: Processive vs. Distributive Kinetics and the Issue of Carbonyl Oxidation in Chemical Mechanisms. Arch. Biochem. Biophys. 2011, 507, 126–134. [Google Scholar] [CrossRef] [Green Version]
- Jung, E.; Park, B.G.; Ahsan, M.M.; Kim, J.; Yun, H.; Choi, K.-Y.; Kim, B.-G. Production of ω-Hydroxy Palmitic Acid Using CYP153A35 and Comparison of Cytochrome P450 Electron Transfer System in Vivo. Appl. Microbiol. Biotechnol. 2016, 100, 10375–10384. [Google Scholar] [CrossRef]
- Joo, S.Y.; Yoo, H.W.; Sarak, S.; Kim, B.G.; Yun, H. Enzymatic Synthesis of ω-Hydroxydodecanoic Acid by Employing a Cytochrome P450 from Limnobacter sp. 105 MED. Catalysts 2019, 9, 54. [Google Scholar] [CrossRef] [Green Version]
- Kochius, S.; van Marwijk, J.; Ebrecht, A.C.; Opperman, D.J.; Smit, M.S. Deconstruction of the CYP153A6 Alkane Hydroxylase System: Limitations and Optimization of In Vitro Alkane Hydroxylation. Catalysts 2018, 8, 531. [Google Scholar] [CrossRef] [Green Version]
- Fiorentini, F.; Hatzl, A.M.; Schmidt, S.; Savino, S.; Glieder, A.; Mattevi, A. The Extreme Structural Plasticity in the CYP153 Subfamily of P450s Directs Development of Designer Hydroxylases. Biochemistry 2018, 57, 6701–6714. [Google Scholar] [CrossRef]
- Amaya, J.A.; Batabyal, D.; Poulos, T.L. Proton Relay Network in the Bacterial P450s: CYP101A1 and CYP101D1. Biochemistry 2020, 59, 2896–2902. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.H.; Baer, B.R.; Rettie, A.E.; Johnson, E.F. The Crystal Structure of Cytochrome P450 4B1 (CYP4B1) Monooxygenase Complexed with Octane Discloses Several Structural Adaptations for ω-Hydroxylation. J. Biol. Chem. 2017, 292, 5610–5621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitehouse, C.J.C.; Bell, S.G.; Wong, L.L. P450 BM3 (CYP102A1): Connecting the Dots. Chem. Soc. Rev. 2012, 41, 1218–1260. [Google Scholar] [CrossRef] [PubMed]
- Aschenbrenner, J.C.; Ebrecht, A.C.; Tolmie, C.; Smit, M.S.; Opperman, D.J. Structure of the Fungal Hydroxylase, CYP505A30, and Rational Transfer of Mutation Data from CYP102A1 to Alter Regioselectivity. Catal. Sci. Technol. 2021, 11, 7359–7367. [Google Scholar] [CrossRef]
- Fasan, R.; Meharenna, Y.T.; Snow, C.D.; Poulos, T.L.; Arnold, F.H. Evolutionary History of a Specialized P450 Propane Monooxygenase. J. Mol. Biol. 2008, 383, 1069–1080. [Google Scholar] [CrossRef] [Green Version]
- Urban, P.; Lautier, T.; Pompon, D.; Truan, G. Ligand Access Channels in Cytochrome P450 Enzymes: A Review. Int. J. Mol. Sci. 2018, 19, 1617. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Poulos, T.L. The Structure of the Cytochrome P450BM-3 Haem Domain Complexed with the Fatty Acid Substrate, Palmitoleic Acid. Nat. Struct. Biol. 1997, 4, 140–146. [Google Scholar] [CrossRef]
- Pennec, A.; Jacobs, C.L.; Opperman, D.J.; Smit, M.S. Revisiting Cytochrome P450-Mediated Oxyfunctionalization of Linear and Cyclic Alkanes. Adv. Synth. Catal. 2015, 357, 118–130. [Google Scholar] [CrossRef]
- Studier, F.W. Protein Production by Auto-Induction in High Density Shaking Cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P.; Martin, M.V.; Sohl, C.D.; Cheng, Q. Measurement of Cytochrome P450 and NADPH-Cytochrome P450 Reductase. Nat. Protoc. 2009, 4, 1245–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonrhein, C.; Flensburg, C.; Keller, P.; Sharff, A.; Smart, O.; Paciorek, W.; Womack, T.; Bricogne, G. Data Processing and Analysis with the AutoPROC Toolbox. Acta Cryst. 2011, D67, 293–302. [Google Scholar]
- Evans, P. Scaling and Assessment of Data Quality. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.R.; Murshudov, G.N. How Good Are My Data and What Is the Resolution? Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, D69, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser Crystallographic Software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC 5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP 4 Suite and Current Developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. PyMOL: An Open-Source Molecular Graphics Tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacobs, C.L.; do Aido-Machado, R.; Tolmie, C.; Smit, M.S.; Opperman, D.J. CYP153A71 from Alcanivorax dieselolei: Oxidation beyond Monoterminal Hydroxylation of n-Alkanes. Catalysts 2022, 12, 1213. https://doi.org/10.3390/catal12101213
Jacobs CL, do Aido-Machado R, Tolmie C, Smit MS, Opperman DJ. CYP153A71 from Alcanivorax dieselolei: Oxidation beyond Monoterminal Hydroxylation of n-Alkanes. Catalysts. 2022; 12(10):1213. https://doi.org/10.3390/catal12101213
Chicago/Turabian StyleJacobs, Cheri Louise, Rodolpho do Aido-Machado, Carmien Tolmie, Martha Sophia Smit, and Diederik Johannes Opperman. 2022. "CYP153A71 from Alcanivorax dieselolei: Oxidation beyond Monoterminal Hydroxylation of n-Alkanes" Catalysts 12, no. 10: 1213. https://doi.org/10.3390/catal12101213
APA StyleJacobs, C. L., do Aido-Machado, R., Tolmie, C., Smit, M. S., & Opperman, D. J. (2022). CYP153A71 from Alcanivorax dieselolei: Oxidation beyond Monoterminal Hydroxylation of n-Alkanes. Catalysts, 12(10), 1213. https://doi.org/10.3390/catal12101213