
Tetrachlorocobaltate-Catalyzed Methane Oxidation to Methyl Trifluoroacetate

Abstract
:
1. Introduction
2. Results and Discussion
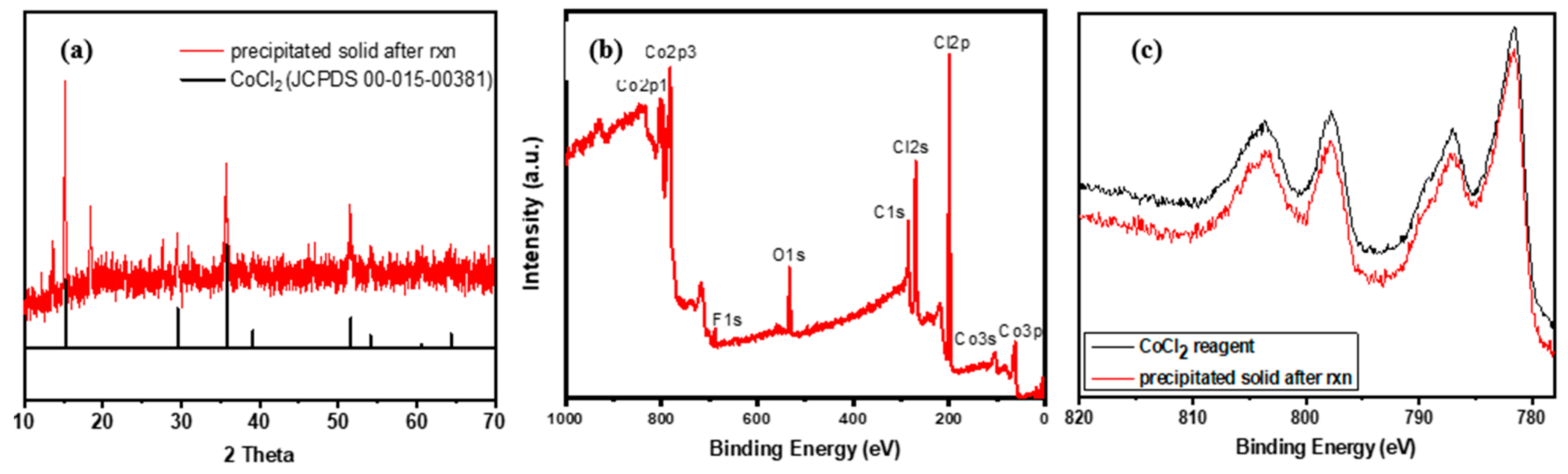
2.1. [Me4N]2CoCl4-Catalyzed Methane Oxidation
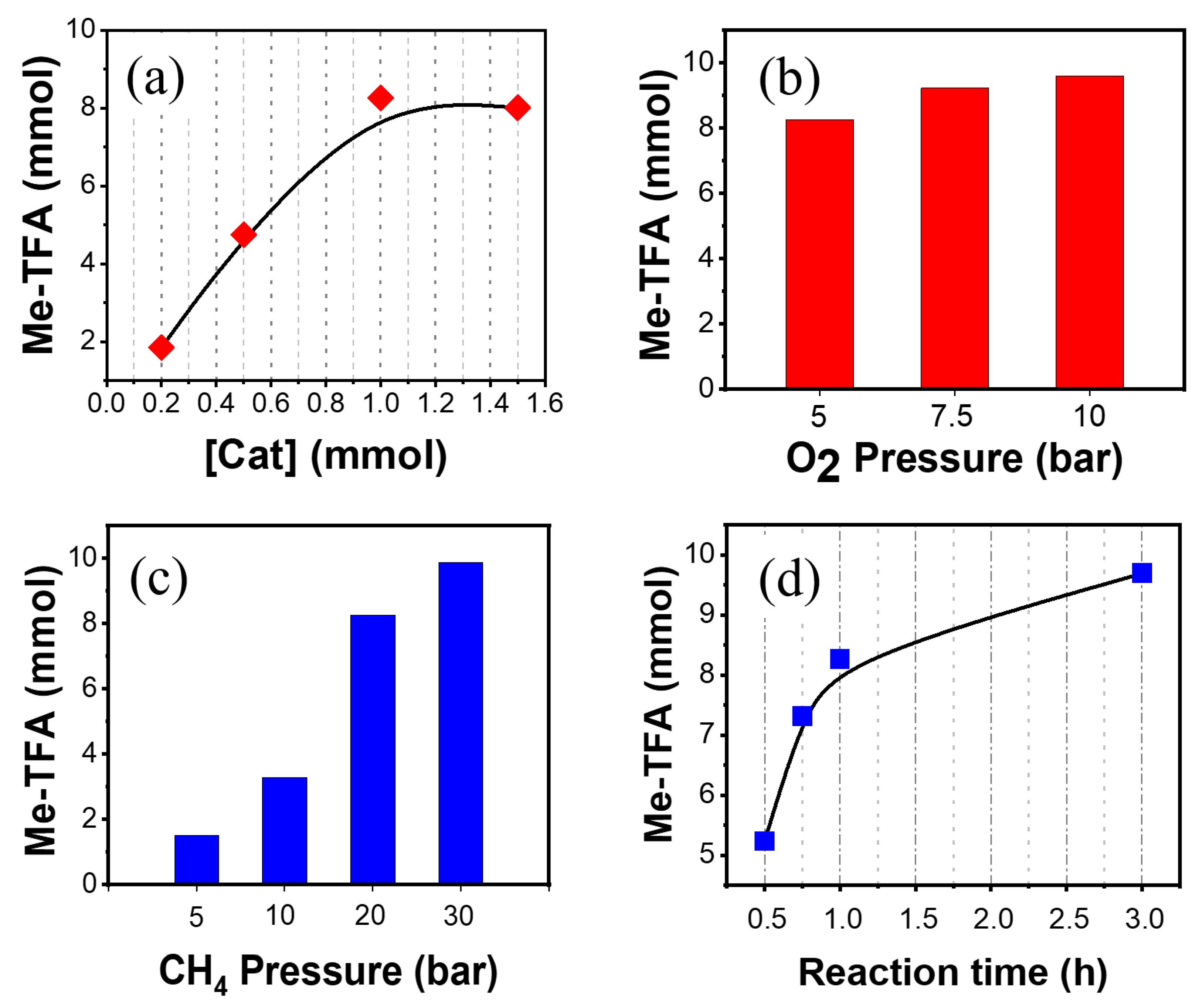
2.2. Effects of Reaction Conditions
3. Experimental Section
3.1. General Information
3.2. Catalyst Synthesis
3.3. Methane Oxidation Reaction
3.4. Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Olah, G.A. Beyond oil and gas: The methanol economy. Angew. Chem. Int. Ed. 2005, 44, 2636–2639. [Google Scholar] [CrossRef] [PubMed]
- Simon Araya, S.; Liso, V.; Cui, X.; Li, N.; Zhu, J.; Sahlin, S.L.; Jensen, S.H.; Nielsen, M.P.; Kær, S.K. A Review of The Methanol Economy: The Fuel Cell Route. Energies 2020, 13, 596. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Galvan, M.C.; Mota, N.; Ojeda, M.; Rojas, S.; Navarro, R.M.; Fierro, J.L.G. Direct methane conversion routes to chemicals and fuels. Catal. Today 2011, 171, 15–23. [Google Scholar] [CrossRef]
- Meng, X.; Cui, X.; Rajan, N.P.; Yu, L.; Deng, D.; Bao, X. Direct Methane Conversion under Mild Condition by Thermo-, Electro-, or Photocatalysis. Chem 2019, 5, 2296–2325. [Google Scholar] [CrossRef]
- Gesser, H.D.; Hunter, N.R. The Direct Conversion of Methane to Methanol by Controlled Oxidation. Chem. Rev. 1985, 85, 235–244. [Google Scholar] [CrossRef]
- Stahl, S.S.; Labinger, J.A.; Bercaw, J.E. Homogeneous oxidation of alkanes by electrophilic late transition metals. Angew. Chem. Int. Ed. 1998, 37, 2181–2192. [Google Scholar] [CrossRef]
- Ravi, M.; Ranocchiari, M.; van Bokhoven, J.A. The Direct Catalytic Oxidation of Methane to Methanol—A Critical Assessment. Angew. Chem. Int. Ed. Engl. 2017, 56, 16464–16483. [Google Scholar] [CrossRef]
- Periana, R.A.; Taube, D.J.; Evitt, E.R.; Löffler, D.G.; Wentrcek, P.R.; Voss, G.; Masuda, T. A Mercury-Catalyzed, High-Yield System for the Oxidation of Methane to Methanol. Science 1993, 259, 340–343. [Google Scholar] [CrossRef]
- Periana, R.A.; Taube, D.J.; Gamble, S.; Taube, H.; Satoh, T.; Fujii, H. Platinum catalysts for the high-yield oxidation of methane to a methanol derivative. Science 1998, 280, 560–564. [Google Scholar] [CrossRef]
- Zimmermann, T.; Soorholtz, M.; Bilke, M.; Schüth, F. Selective Methane Oxidation Catalyzed by Platinum Salts in Oleum at Turnover Frequencies of Large-Scale Industrial Processes. J. Am. Chem. Soc. 2016, 138, 12395–12400. [Google Scholar] [CrossRef]
- Dang, H.T.; Lee, H.W.; Lee, J.; Choo, H.; Hong, S.H.; Cheong, M.; Lee, H. Enhanced Catalytic Activity of (DMSO)2PtCl2 for the Methane Oxidation in the SO3–H2SO4 System. ACS Catal. 2018, 8, 11854–11862. [Google Scholar] [CrossRef]
- Lee, H.W.; Dang, H.T.; Kim, H.; Lee, U.; Ha, J.-M.; Jae, J.; Cheong, M.; Lee, H. Pt black catalyzed methane oxidation to methyl bisulfate in H2SO4-SO3. J. Catal. 2019, 374, 230–236. [Google Scholar] [CrossRef]
- Meyer, D.; Strassner, T. CH-activation of methane—Synthesis of an intermediate? J. Organomet. Chem. 2015, 784, 84–87. [Google Scholar] [CrossRef]
- Ravi, M.; van Bokhoven, J.A. Homogeneous Copper-Catalyzed Conversion of Methane to Methyl Trifluoroacetate in High Yield at Low Pressure. ChemCatChem 2018, 10, 2383–2386. [Google Scholar] [CrossRef]
- Muehlhofer, M.; Strassner, T.; Herrmann, W.A. New Catalyst Systems for the Catalytic Conversion of Methane into Methanol. Angew. Chem. Int. Ed. 2002, 41, 1745–1747. [Google Scholar] [CrossRef]
- Campbell, A.N.; Stahl, S.S. Overcoming the Oxidant problem Strategies to use O2 as the oxidant in organometallic CH oxidation reactions catalyzed by Pd and Cu. Acc. Chem. Res. 2012, 45, 851–863. [Google Scholar] [CrossRef]
- Kalman, S.E.; Munz, D.; Fortman, G.C.; Boaz, N.C.; Groves, J.T.; Gunnoe, T.B. Partial oxidation of light alkanes by periodate and chloride salts. Dalton Trans. 2015, 44, 5294–5298. [Google Scholar] [CrossRef]
- Fortman, G.C.; Boaz, N.C.; Munz, D.; Konnick, M.M.; Periana, R.A.; Groves, J.T.; Gunnoe, T.B. Selective monooxidation of light alkanes using chloride and iodate. J. Am. Chem. Soc. 2014, 136, 8393–8401. [Google Scholar] [CrossRef]
- Liebov, N.S.; Goldberg, J.M.; Boaz, N.C.; Coutard, N.; Kalman, S.E.; Zhuang, T.; Groves, J.T.; Gunnoe, T.B. Selective Photo-Oxygenation of Light Alkanes Using Iodine Oxides and Chloride. ChemCatChem 2019, 11, 5045–5054. [Google Scholar] [CrossRef]
- Schwartz, N.A.; Boaz, N.C.; Kalman, S.E.; Zhuang, T.; Goldberg, J.M.; Fu, R.; Nielsen, R.J.; Goddard, W.A.; Groves, J.T.; Gunnoe, T.B. Mechanism of Hydrocarbon Functionalization by an Iodate/Chloride System: The Role of Ester Protection. ACS Catal. 2018, 8, 3138–3149. [Google Scholar] [CrossRef]
- Cheong, S.-H.; Kim, D.; Dang, H.T.; Kim, D.; Seo, B.; Cheong, M.; Hong, S.H.; Lee, H. Methane oxidation to methyl trifluoroacetate by simple anionic palladium catalyst: Comprehensive understanding of K2S2O8-based methane oxidation in CF3CO2H. J. Catal. 2022, 413, 803–811. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, M.; Han, Z.; Huang, S.; Yuan, D.; Su, W. Atmosphere-Pressure Methane Oxidation to Methyl Trifluoroacetate Enabled by a Porous Organic Polymer-Supported Single-Site Palladium Catalyst. ACS Catal. 2021, 11, 1008–1013. [Google Scholar] [CrossRef]
- Munz, D.; Strassner, T. Alkane C-H functionalization and oxidation with molecular oxygen. Inorg. Chem. 2015, 54, 5043–5052. [Google Scholar] [CrossRef] [PubMed]
- Strassner, T.; Ahrens, S.; Muehlhofer, M.; Munz, D.; Zeller, A. Cobalt-Catalyzed Oxidation of Methane to Methyl Trifluoroacetate by Dioxygen. Eur. J. Inorg. Chem. 2013, 3659–3663. [Google Scholar] [CrossRef]
- An, Z.; Pan, X.; Liu, X.; Han, X.; Bao, X. Combined redox couples for catalytic oxidation of methane by dioxygen at low temperature. J. Am. Chem. Soc. 2006, 128, 16028–16029. [Google Scholar] [CrossRef]
- Yuan, J.; Wang, L.; Wang, Y. Direct Oxidation of Methane to a Methanol Derivative Using Molecular Oxygen. Ind. Eng. Chem. Res. 2011, 50, 6513–6516. [Google Scholar] [CrossRef]
- Yuan, J.; Liu, L.; Wang, L.; Hao, C. Partial Oxidation of Methane with the Catalysis of Palladium(II) and Molybdovanadophosphoric Acid Using Molecular Oxygen as the Oxidant. Catal. Lett. 2012, 143, 126–129. [Google Scholar] [CrossRef]
- Blankenship, A.N.; Ravi, M.; Newton, M.A.; van Bokhoven, J.A. Heterogeneously Catalyzed Aerobic Oxidation of Methane to a Methyl Derivative. Angew. Chem. Int. Ed. 2021, 60, 18138–18143. [Google Scholar] [CrossRef]
- Coutard, N.; Goldberg, J.M.; Valle, H.U.; Cao, Y.; Jia, X.; Jeffrey, P.D.; Gunnoe, T.B.; Groves, J.T. Aerobic Partial Oxidation of Alkanes Using Photodriven Iron Catalysis. Inorg. Chem. 2022, 61, 759–766. [Google Scholar] [CrossRef]
- Coutard, N.; Musgrave, C.B.; Moon, J.; Liebov, N.S.; Nielsen, R.M.; Goldberg, J.M.; Li, M.; Jia, X.; Lee, S.; Dickie, D.A.; et al. Manganese Catalyzed Partial Oxidation of Light Alkanes. ACS Catal. 2022, 12, 5356–5370. [Google Scholar] [CrossRef]
- Tang, R.; Kochi, J.K. Cobalt (III) Trifluoroacetate: An Electron Transfer Oxidant. J. Inorg. Nucl. Chem. 1973, 35, 3845–3856. [Google Scholar] [CrossRef]
- Vargaftik, M.N.; Stolarov, I.P.; Moiseev, I.I. Highly Selective Parital Oxidation of Methane to Methyl Trifluoroacetate. Chem. Commun. 1990, 15, 1049–1050. [Google Scholar] [CrossRef]
- Wiesner, J.R.; Srivastava, R.C.; Kennard, C.H.L.; Divaira, M.; Lingafelter, E.C. The Crystal Structures of Tetramethylammonium Tetrachloro-cobaltate (II), -nickelate (II) and -zincate (II). Acta Crystallogr. 1966, 23, 565–574. [Google Scholar] [CrossRef]
- Stolarov, I.P.; Vargaftik, M.N.; Shishkin, D.I.; Moiseev, I.I. Oxidation of Ethane and Propane with Cobalt (II) Catalyst: Unexpected Formation of 1,2-Diol Esters and C-C Bond Cleavage. J. Chem. Soc. Chem. Commun. 1991, 14, 938–939. [Google Scholar] [CrossRef]
- Puri, M.; Verma, R.D. Bis(tetramethylammonium) tetrakis (trifluoroacetato) metallates (II). Indian J. Chem. 1983, 22, 418–419. [Google Scholar]
- Bergman Jr, J.G.; Cotton, F.A. The Preparation, Properties, and Struture of Tetraphenylarsonium Tetrakis (trifluoroacetato) cobaltate (II). Inorg. Chem. 1966, 5, 1420–1424. [Google Scholar] [CrossRef]
- Baillie, M.J.; Brown, D.H.; Moss, K.C.; Sharp, D.W.A. Anhydrous Metal Trifluoroacetates. J. Chem. Soc. A 1968, 3110–3114. [Google Scholar] [CrossRef]
- Agambar, C.A.; Orrell, K.A. Trifuoroacetate Complexes of Cobalt(II), Nickel(II), and Copper(II) with pyridine-type ligands. J. Chem. Soc. 1974, 8, 897–904. [Google Scholar]
- Clifford, A.A.; Waters, W.A. Oxidations of organic compounds by cobaltic salts. Part VI. Kinetic and product studies of the oxidation of some carboxylic acids. J. Chem. Soc. 1965, 2796–2804. [Google Scholar] [CrossRef]
- Lande, S.S.; Kochi, J.K. Formation and Oxidation of Alkyl Radicals by Cobalt (III) Complexes. J. Am. Chem. Soc. 1968, 90, 5196–5207. [Google Scholar] [CrossRef]
- Zargari, N.; Winter, P.; Liang, Y.; Lee, J.H.; Cooksy, A.; Houk, K.N.; Jung, K.W. Unexpected, Latent Radical Reaction of Methane Propagated by Trifluoromethyl Radicals. J. Org. Chem. 2016, 81, 9820–9825. [Google Scholar] [CrossRef] [PubMed]
- Hori, H.; Tanako, Y.; Koike, K.; Takeuchi, K.; Einaga, H. Decomposition of Environmentally Persistent Trifluoroacetic Acid to Fluoride Ions by Homogeneous Photocatalyst in Water. Environ. Sci. Technol. 2003, 37, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Puri, J.K.; Chhoker, R. Synthesis and Characterization of Metal Tribromoacetates and Their Alkali Metal Derivatives. J. Chem. 2013, 2013, 392310. [Google Scholar] [CrossRef] [Green Version]
- Piao, D.-g.; Inoue, K.; Shibasaki, H.; Taniguchi, Y.; Kitamura, T.; Fujiwara, Y. An efficient partial oxidation of methane in trifluoroacetic acid using vanadium-containing heteroplolyacid catalysts. J. Organomet. Chem. 1999, 574, 116–120. [Google Scholar] [CrossRef]
- Yamanaka, I.; Soma, M.; Otsuka, K. Oxidation of methane to methanol with oxygen catalysed by Europium trichloride at room temperature. Chem. Commun. 1995, 2235–2236. [Google Scholar] [CrossRef]
- Chen, W.; Kocal, J.A.; Brandvold, T.A.; Bricker, M.L.; Bare, S.R.; Broach, R.W.; Greenlay, N.; Popp, K.; Walenga, J.T.; Yang, S.S.; et al. Manganese oxide catalyzed methane partial oxidation in trifluoroacetic acid: Catalysis and kinetic analysis. Catal. Today 2009, 140, 157–161. [Google Scholar] [CrossRef]
- Ingrosso, G.; Midollini, N. Palladium(II)- or copper(II)-catalysed solution-phase oxyfunctionalisation of methane and other light alkanes by hydrogen peroxide in trifluoroacetic anhydride. J. Mol. Catal. A Chem 2003, 204–205, 425–431. [Google Scholar] [CrossRef]
- Seki, Y.; Min, J.S.; Misono, M.; Mizuno, N. Reaction Mechanism of Oxidation of Methane with Hydrogen Peroxide Catalyzed by 11-Molybdo-1-vanadophosphoric Acid Catalyst Precursor. J. Phys. Chem. B 2000, 104, 5940–5944. [Google Scholar] [CrossRef]
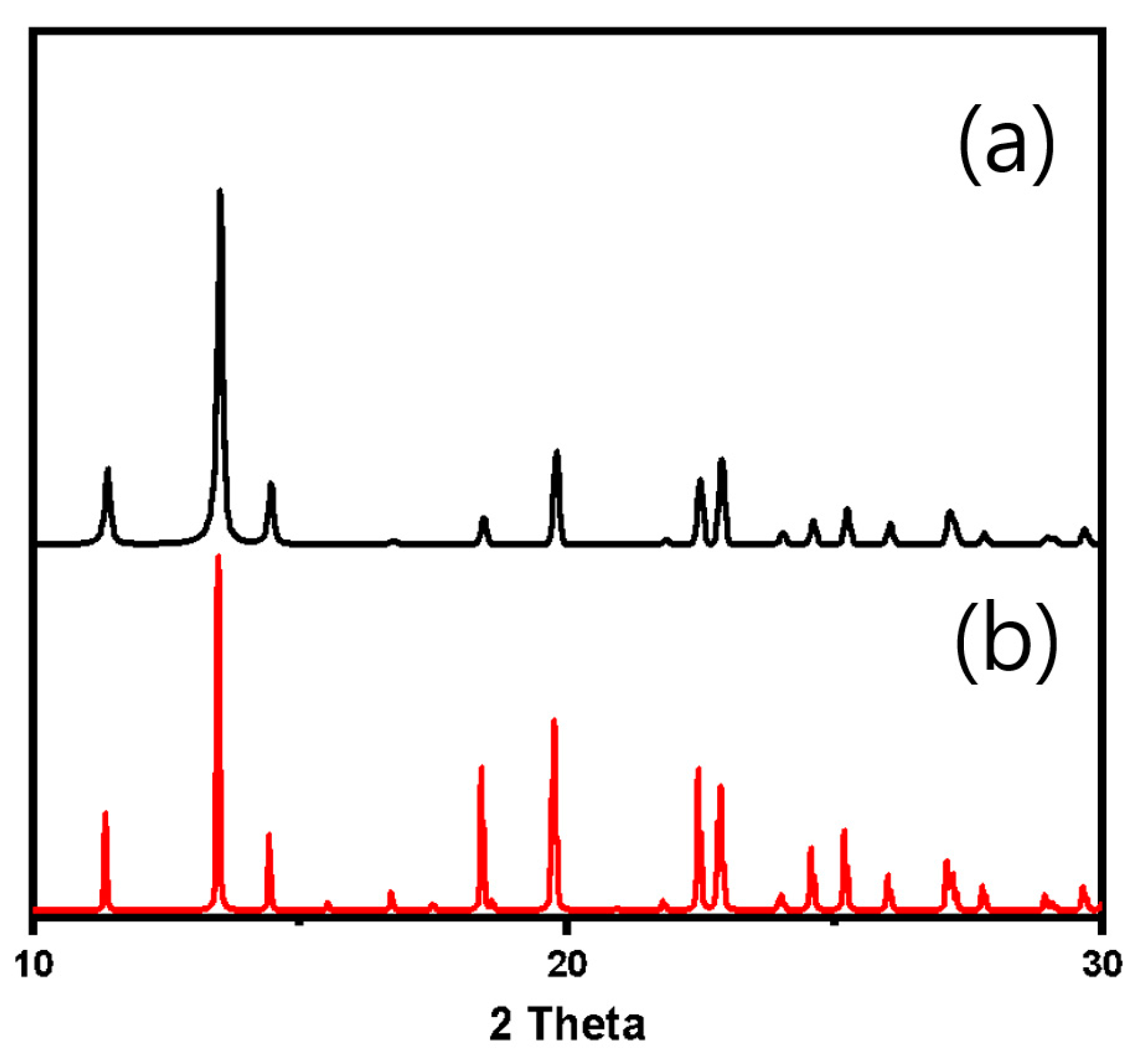


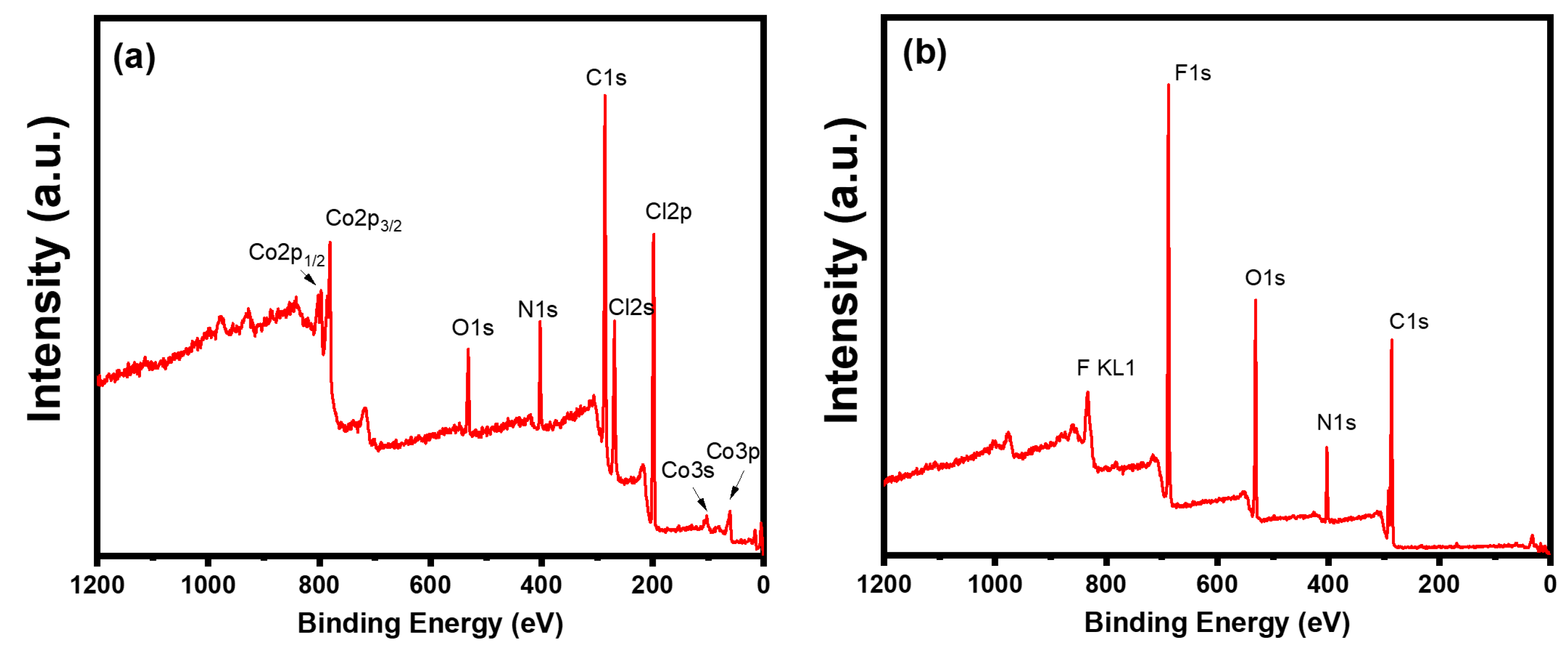
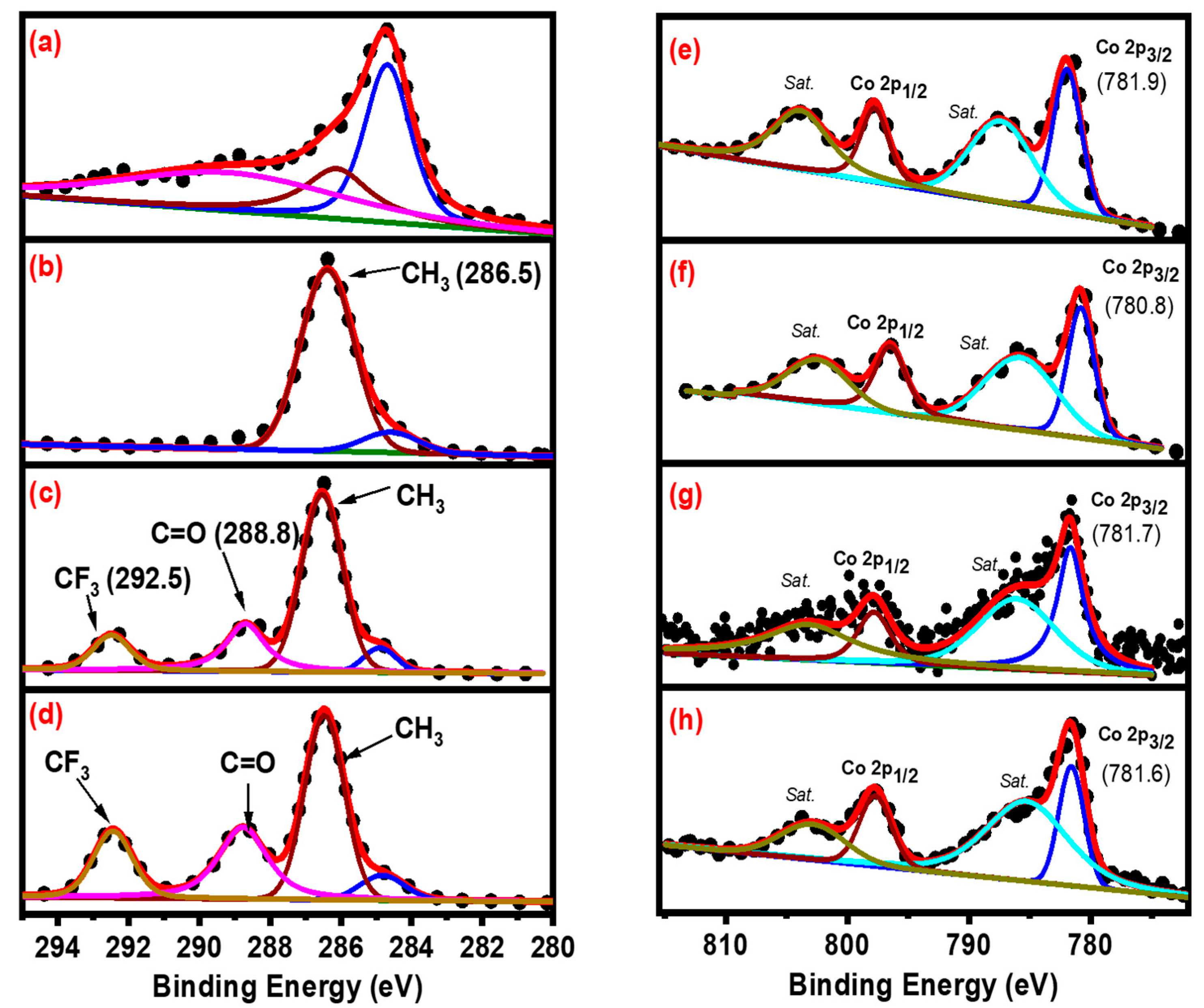
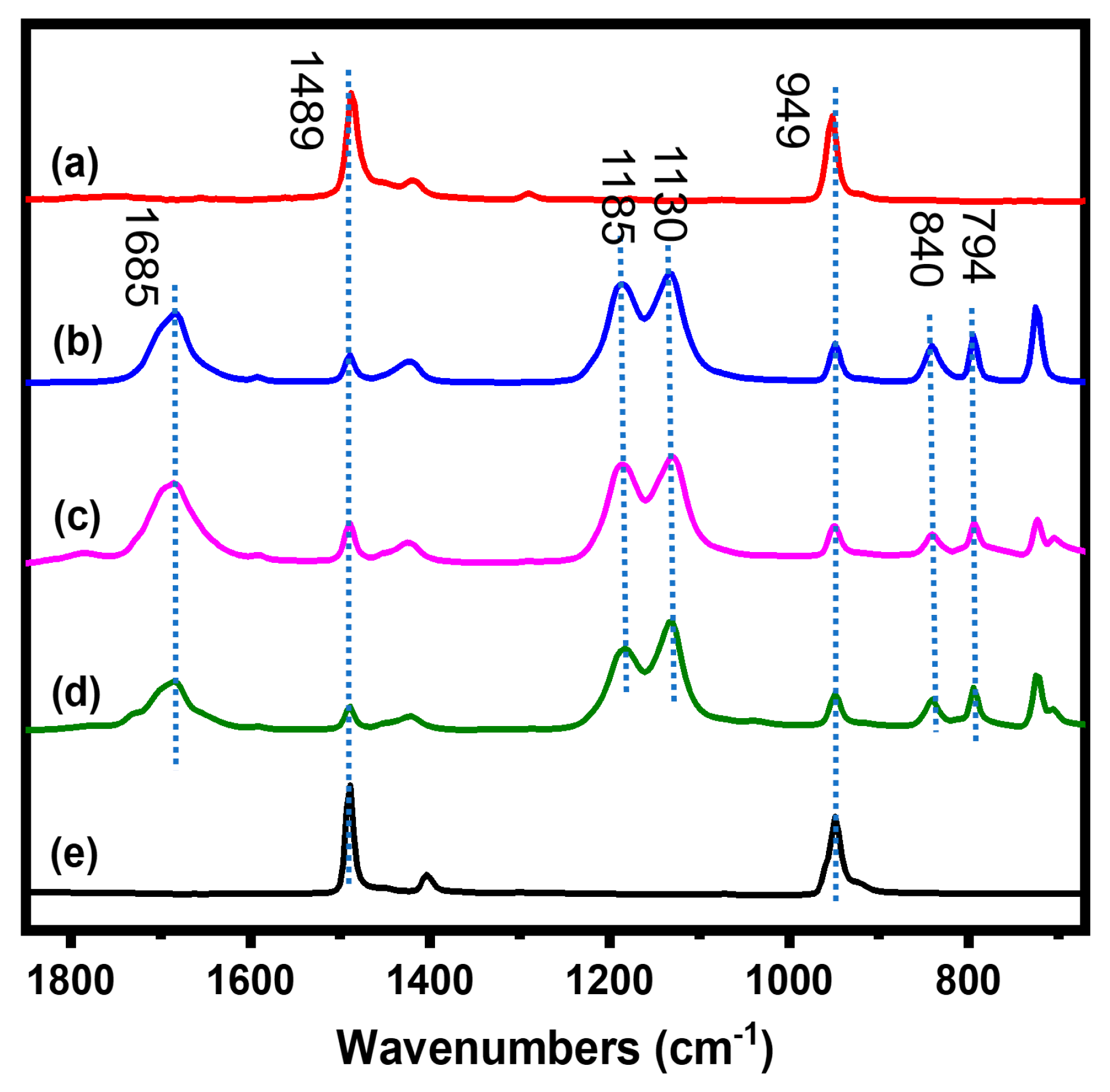

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst System | MeTFA (mmol, molester molmetal−1h−1) | CO2 (mmol) |
|---|---|---|---|
| 1 | [Me4N]2CoCl4 | 8.26 | 5.00 |
| 2 b | [Me4N]2CoCl4 | 0 | 10.59 |
| 3 c | [Me4N]2CoCl4 | 9.73 | 9.62 |
| 4 | CoCl2 | 0.33 | 1.50 |
| 5 | CoCl2 + 2Me4NCl | 5.52 | 3.82 |
| 6 | Co(NO3)2.6H2O | 4.38 | 9.18 |
| 7 | Co(TFA)2 | 4.74 | 9.86 |
| 8 | CoBr2 | 0.12 | 2.49 |
| 9 | CoSO4.7H2O | 0.10 | 1.93 |
| 10 | CoTFA4(Me4N)2 | 5.40 | 7.56 |
| 11 | CoTFA4(Me4N)2 + 2Me4NCl | 5.75 | 3.76 |
| 12 | - | - | 0.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dang, H.T.; Cheong, S.; Kim, J.; Tran, N.T.; Kim, H.; Lee, H. Tetrachlorocobaltate-Catalyzed Methane Oxidation to Methyl Trifluoroacetate. Catalysts 2022, 12, 1419. https://doi.org/10.3390/catal12111419
Dang HT, Cheong S, Kim J, Tran NT, Kim H, Lee H. Tetrachlorocobaltate-Catalyzed Methane Oxidation to Methyl Trifluoroacetate. Catalysts. 2022; 12(11):1419. https://doi.org/10.3390/catal12111419
Chicago/Turabian StyleDang, Huyen Tran, Seokhyeon Cheong, Jiyun Kim, Ngoc Tuan Tran, Honggon Kim, and Hyunjoo Lee. 2022. "Tetrachlorocobaltate-Catalyzed Methane Oxidation to Methyl Trifluoroacetate" Catalysts 12, no. 11: 1419. https://doi.org/10.3390/catal12111419