
Pt-Mo/C, Pt-Fe/C and Pt-Mo-Sn/C Nanocatalysts Derived from Cluster Compounds for Proton Exchange Membrane Fuel Cells


 , ,
, , 
Abstract
:1. Introduction
2. Results and Discussion
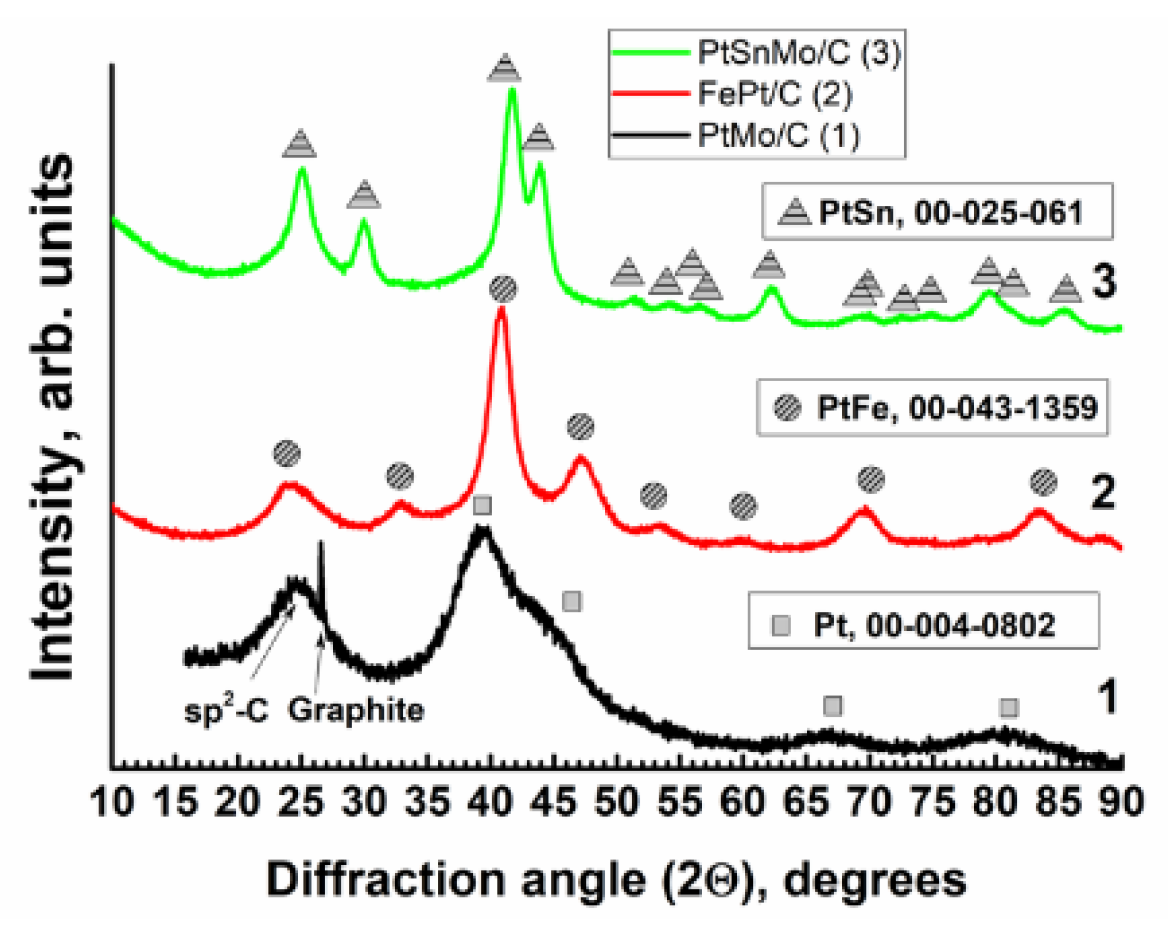
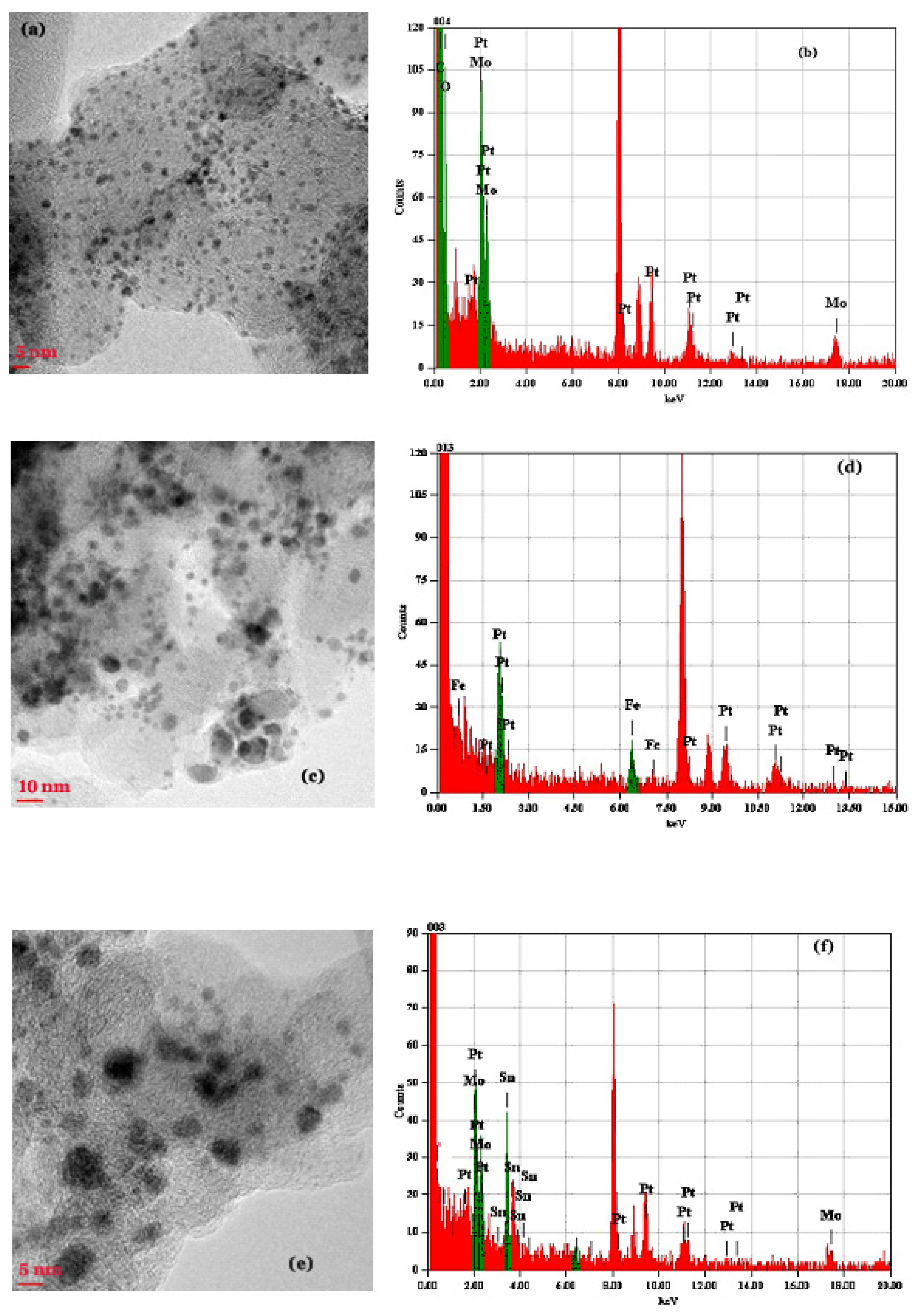
2.1. XRD, TEM and X-ray Fluorescence Studies
2.1.1. The PtMo/C Catalyst
2.1.2. The PtFe/C Catalyst
2.1.3. The PtMoSn/C Catalyst
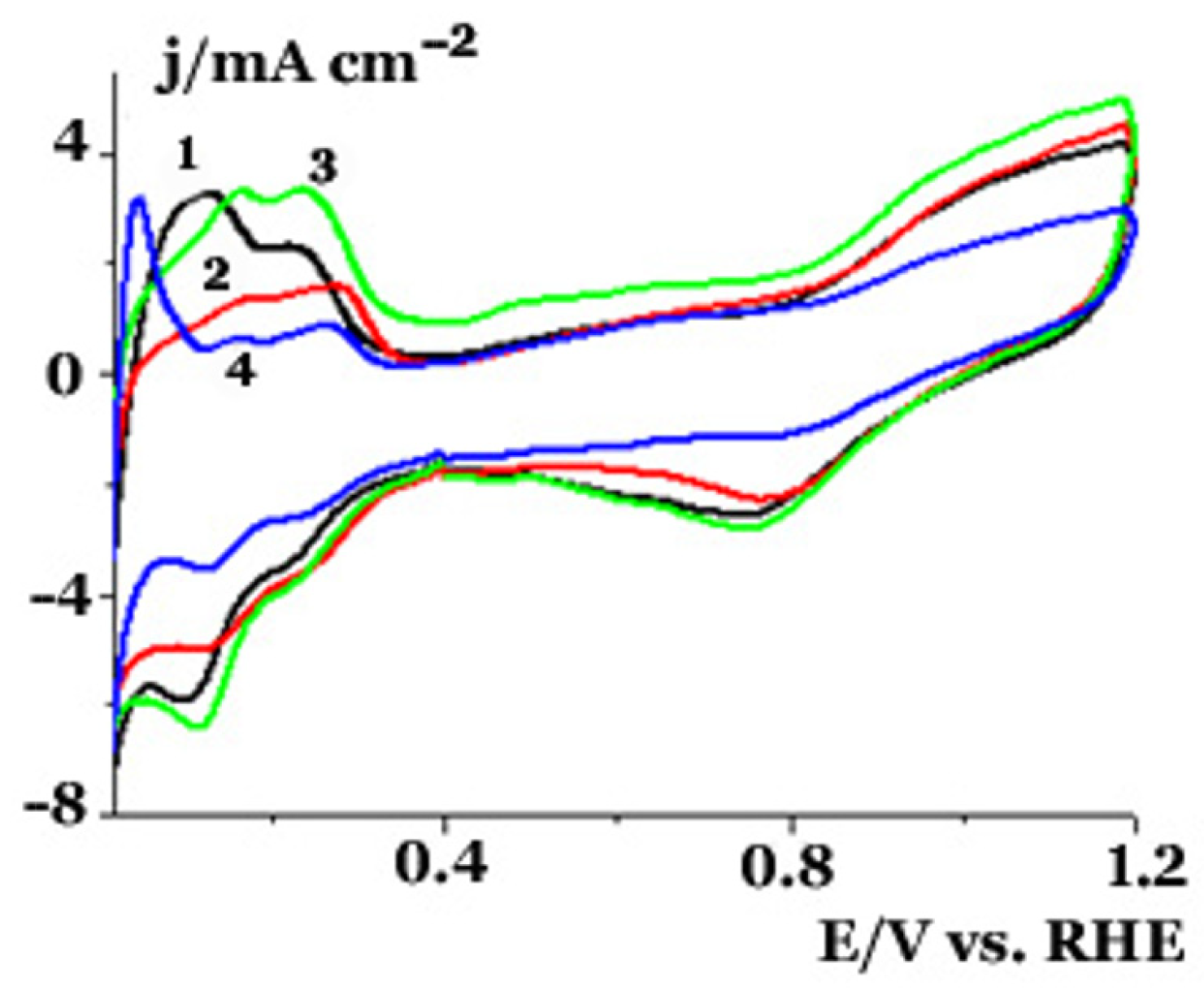
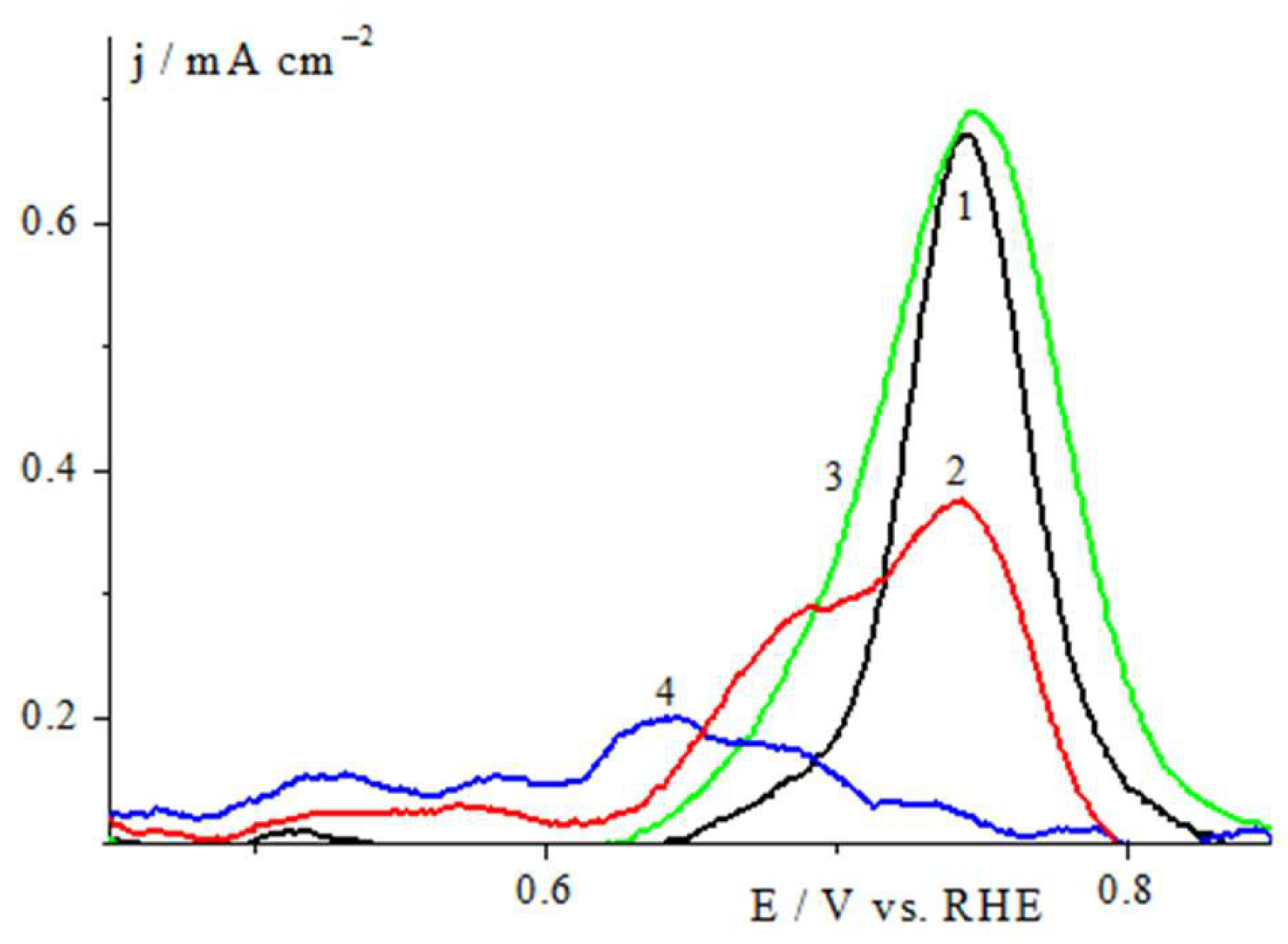
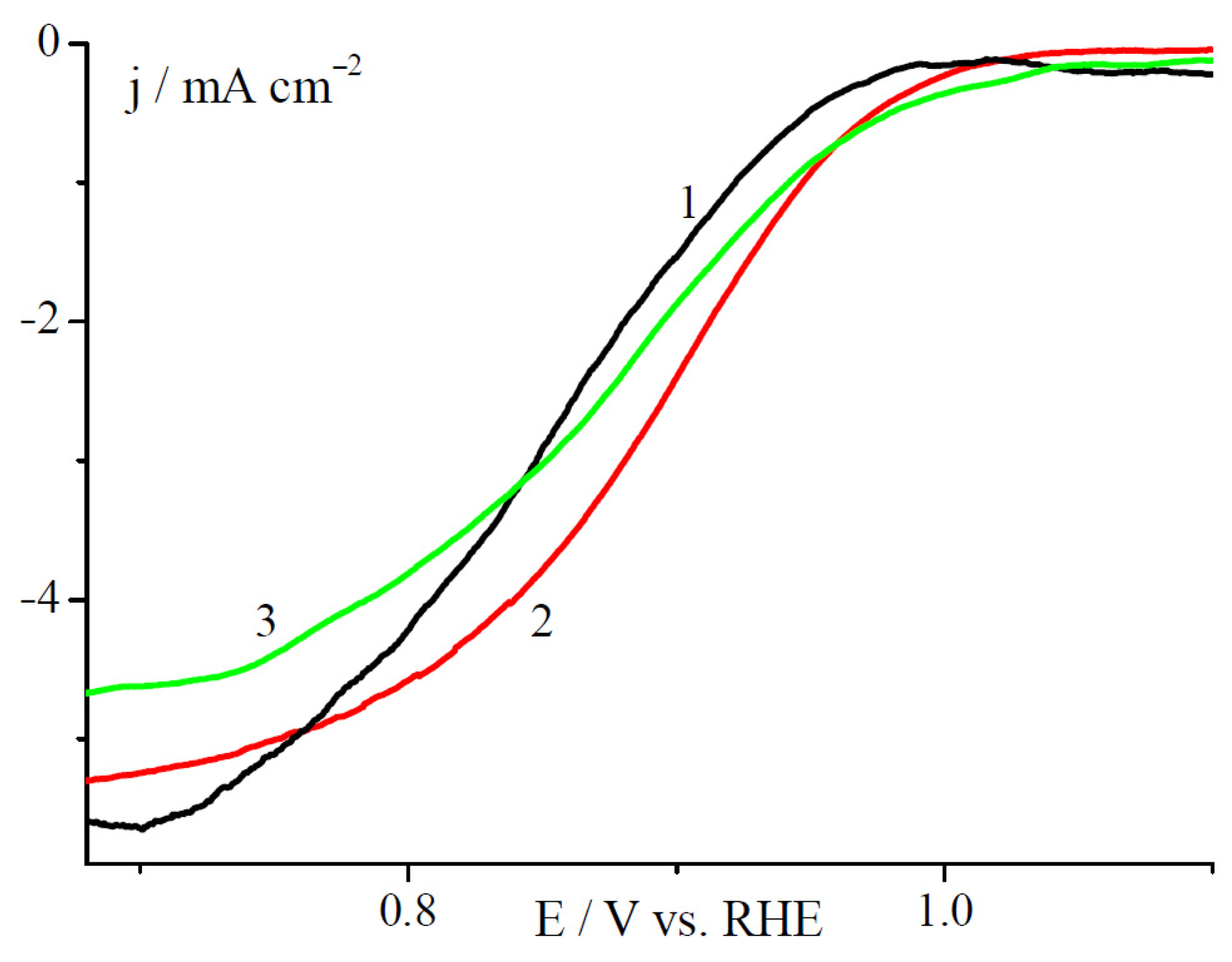
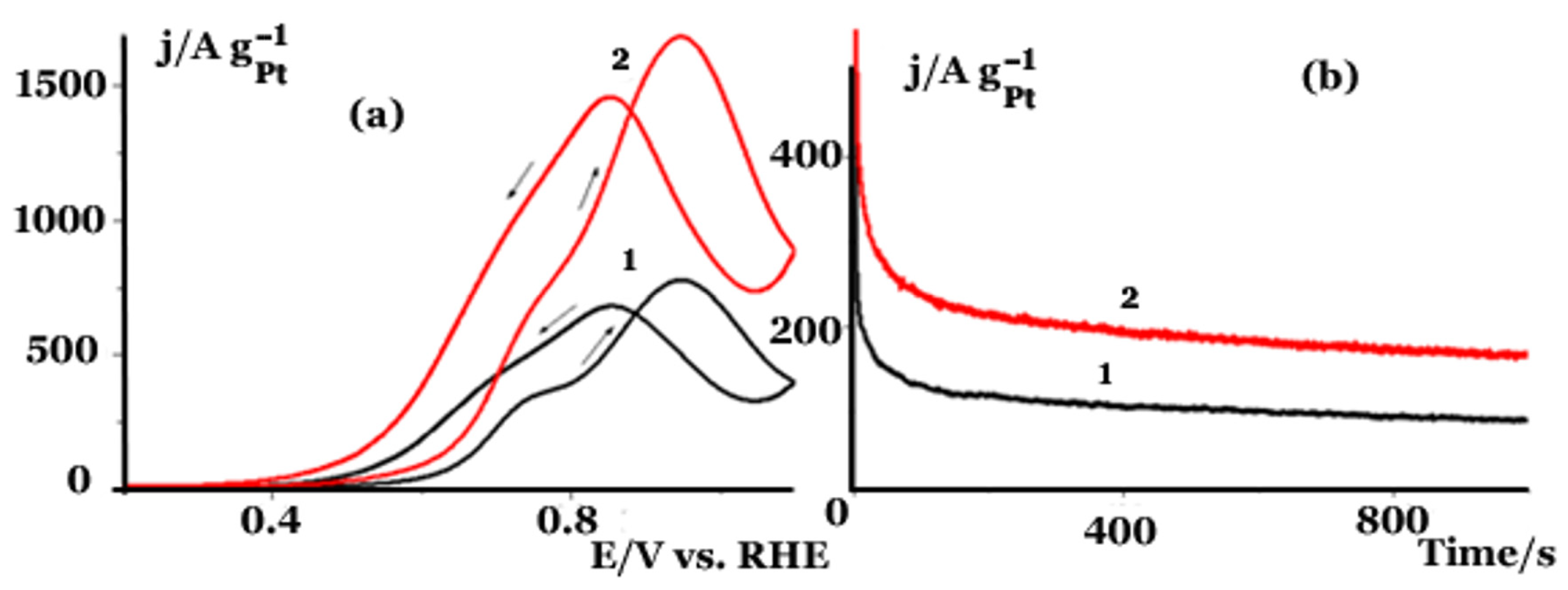
2.2. Electrochemical Studies in a Three Electrode Electrochemical Cell
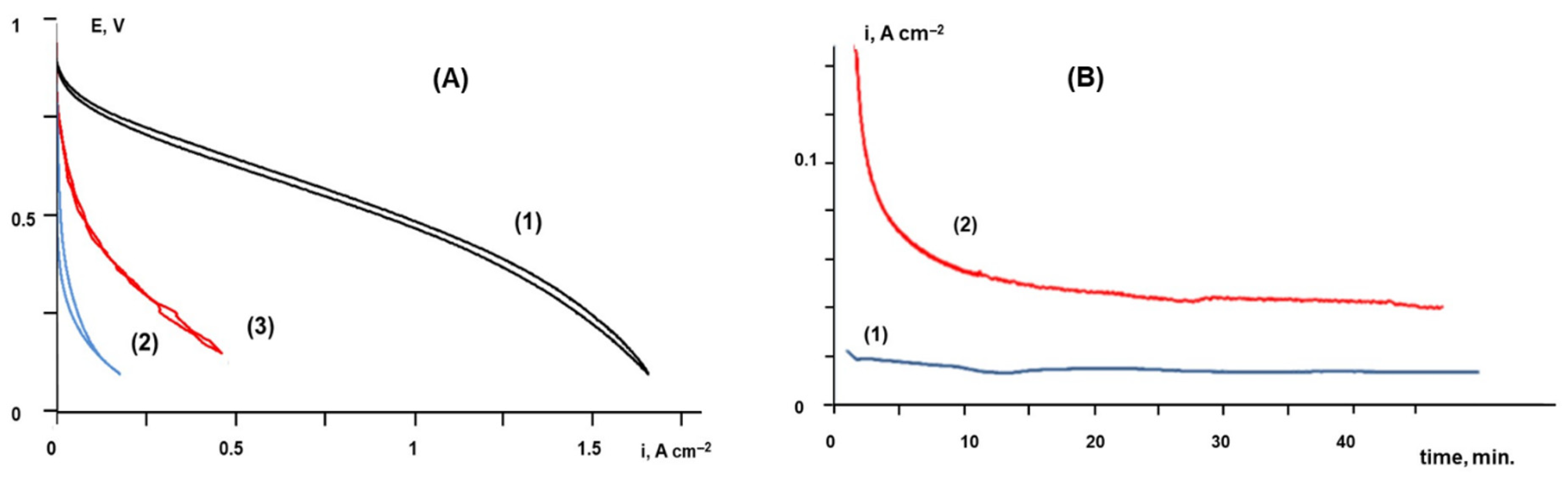
2.3. Tests of Membrane Electrode Assemblies
3. Materials and Methods
3.1. Preparation of Bimetallic PtMo/C, PtFe/C and Ternary PtMoSn/C Catalysts
3.2. Material Characterization
3.3. Electrochemical Measurements
3.4. Membrane Electrode Assembly (MEA) Fabrication and Testing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Carrette, L.; Friedrich, K.A.; Stimming, U. Fuel cells: Principles, types, fuels, and applications. ChemPhysChem 2000, 1, 162–193. [Google Scholar] [CrossRef]
- Neto, A.O.; Franco, E.G.; Arico, E.; Linardi, M.; Gonzalez, E.R. Electro-oxidation of methanol and ethanol on Pt–Ru/C and Pt–Ru–Mo/C electrocatalysts prepared by Bönnemann’s method. J. Eur. Ceram. Soc. 2003, 23, 2987–2992. [Google Scholar] [CrossRef]
- Zhou, W.; Zhou, Z.; Song, S.; Li, W.; Sun, G.; Tsiakaras, P.; Xin, Q. Pt based anode catalysts for direct ethanol fuel cells. Appl. Catal. B Environ. 2003, 46, 273–285. [Google Scholar] [CrossRef]
- Zhou, W.J.; Song, S.Q.; Li, W.Z.; Zhou, Z.H.; Sun, G.Q.; Xin, Q.; Douvartzides, S.; Tsiakaras, P. Direct ethanol fuel cells based on PtSn anodes: The effect of Sn content on the fuel cell performance. J. Power Sources 2005, 140, 50–58. [Google Scholar] [CrossRef]
- Vigier, F.; Rousseau, S.; Coutanceau, C.; Leger, J.M.; Lamy, C. Electrocatalysis for the direct alcohol fuel cell. Top. Catal. 2006, 40, 111–121. [Google Scholar] [CrossRef]
- Riberio, J.; Dos Anjos, D.M.; Kokoh, K.B.; Coutanceau, C.; Leger, J.M.; Olivi, P.; DeAndrade, A.R.; Tremiliosi-Filho, G. Carbon-supported ternary PtSnIr catalysts for direct ethanol fuel cell. Electrochim. Acta 2007, 52, 6997–7006. [Google Scholar] [CrossRef]
- Vigier, F.; Coutanceau, C.; Hahn, F.; Belgsir, E.M.; Lamy, C. On the mechanism of ethanol electro-oxidation on Pt and PtSn catalysts: Electrochemical and in situ IR reflectance spectroscopy studies. J. Electroanal. Chem. 2004, 563, 81–89. [Google Scholar] [CrossRef]
- Antolini, E. Catalysts for direct ethanol fuel cells. J. Power Sources 2007, 170, 1–12. [Google Scholar] [CrossRef]
- Zhou, W.J.; Li, W.Z.; Song, S.Q.; Zhou, Z.H.; Jiang, L.H.; Sun, G.Q.; Xin, Q.; Poulianitis, K.; Kontou, S.; Tsiakaras, P. Bi- and tri-metallic Pt-based anode catalysts for direct ethanol fuel cells. J. Power Sources 2004, 131, 217–223. [Google Scholar] [CrossRef]
- Dos Anjos, D.; Kokoh, K.; Leger, J.M.; Andrade, A.R.; Olivi, P.; Tremiliosi-Filho, G.J. Electrocatalytic oxidation of ethanol on Pt–Mo bimetallic electrodes in acid medium. Appl. Electrochem. 2006, 36, 1391–1397. [Google Scholar] [CrossRef]
- Akhairi, M.A.F.; Kamarudin, S.K. Catalysts in direct ethanol fuel cell (DEFC): An overview. Int. J. Hydrogen Energy 2016, 41, 4214–4228. [Google Scholar] [CrossRef]
- Hoogers, G.; Thompsett, D. Catalysis in proton exchange membrane fuel cell technology. Cattech 1999, 3, 106–124. [Google Scholar]
- Mukerjee, S.; Urian, R.C. Bifunctionality in Pt alloy nanocluster electrocatalysts for enhanced methanol oxidation and CO tolerance in PEM fuel cells: Electrochemical and in situ synchrotron spectroscopy. Electrochim. Acta 2002, 47, 3219–3231. [Google Scholar] [CrossRef]
- Massong, H.; Wang, H.; Samjeske, G.; Baltruschat, H. The co-catalytic effect of Sn, Ru and Mo decorating steps of Pt(111) vicinal electrode surfaces on the oxidation of CO. Electrochim. Acta 2000, 46, 701–707. [Google Scholar] [CrossRef]
- Gotz, M.; Wendt, H. Binary and ternary anode catalyst formulations including the elements W, Sn and Mo for PEMFCs operated on methanol or reformate gas. Electrochim. Acta 1998, 43, 3637–3644. [Google Scholar] [CrossRef]
- Park, K.W.; Choi, J.-H.; Lee, S.-A.; Pak, C.; Chang, H.; Sung, Y.E. PtRuRhNi nanoparticle electrocatalyst for methanol electrooxidation in direct methanol fuel cell. J. Catal. 2004, 224, 236–242. [Google Scholar] [CrossRef]
- Buchwalter, P.; Rose, J.; Braunstein, P. Multimetallic catalysis based on heterometallic complexes and clusters. Chem. Rev. 2015, 115, 28–126. [Google Scholar] [CrossRef]
- Cesari, C.; Shon, J.-H.; Zacchini, S.; Berben, L.A. Metal carbonyl clusters of groups 8–10: Synthesis and catalysis. Chem. Soc. Rev. 2021, 50, 9503–9539. [Google Scholar] [CrossRef]
- Li, Z.; Gao, R.; Feng, M.; Deng, Y.-P.; Xiao, D.; Zheng, Y.; Zhao, Z.; Luo, D.; Liu, Y.; Zhang, Z.; et al. Modulating metal–organic frameworks as advanced oxygen electrocatalysts. Adv. Energy Mater. 2021, 16, 2003291. [Google Scholar] [CrossRef]
- Virovets, A.V.; Peresypkina, E.; Scheer, M. Structural chemistry of giant metal based supramolecules. Chem. Rev. 2021, 121, 14485–14554. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, L. Macrocycle-encircled polynuclear metal clusters: Controllable synthesis, reactivity studies, and applications. Acc. Chem. Res. 2018, 51, 2535–2545. [Google Scholar] [CrossRef]
- Yang, H.; Vogel, W.; Lamy, C.; Alonso-Vante, N. Structure and Electrocatalytic Activity of Carbon-Supported Pt−Ni Alloy Nanoparticles Toward the Oxygen Reduction Reaction. J. Phys. Chem. B 2004, 108, 11024–11034. [Google Scholar] [CrossRef]
- Kritsanaviparkporn, E.; Baena-Moreno, F.M.; Reina, T.R. Catalytic Converters for Vehicle Exhaust: Fundamental Aspects and Technology Overview for Newcomers to the Field. Chemistry 2021, 3, 630–646. [Google Scholar] [CrossRef]
- Jeong, S.-H.; Jin, H.C.; Kim, Y.; Lee, T.-W. Silver-Based Nanoparticles for Surface Plasmon Resonance in Organic Optoelectronics. Part. Part. Syst. Char. 2015, 32, 164–175. [Google Scholar] [CrossRef]
- Hedayatnasa, Z.; Abnisa, F.; Daud, W.M.A.W. Review on magnetic nanoparticles for magnetic nanofluid hyperthermia application. Mater. Des. 2017, 123, 174–196. [Google Scholar] [CrossRef]
- Ahamed, M.; Akhtar, M.J.; Khan, M.A.M.; Alhadlaq, H.A. A Novel Green Preparation of Ag/RGO Nanocomposites with Highly Effective Anticancer Performance. Polymers 2021, 13, 3350. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Alonso-Vante, N.; Leger, J.-M.; Lamy, C. Tailoring, structure, and activity of carbon-supported nanosized Pt−Cr alloy electrocatalysts for oxygen reduction in pure and methanol-containing electrolytes. J. Phys. Chem. B 2004, 108, 1938–1947. [Google Scholar] [CrossRef]
- Garcia, B.L.; Captain, B.; Adams, R.D.; Hangria, A.B.; Midgley, P.A.; Thomas, J.M.; Weidner, J.W. Bimetallic cluster provides a higher activity electrocatalyst for methanol oxidation. Cluster Sci. 2007, 18, 121–130. [Google Scholar] [CrossRef]
- Grinberg, V.A.; Maiorova, N.A.; Pasynskii, A.A.; Emets, V.V.; Shiryaev, A.A.; Vysotskii, V.V.; Gerasimov, V.K.; Matveev, V.V.; Nizhnikovskii, E.A.; Andreev, V.N. Nanostructured catalysts for direct electrooxidation of dimethyl ether based on Bi- and trimetallic Pt–Ru and Pt–Ru–Pd alloys prepared from coordination compounds. Russ. J. Coord. Chem. 2017, 43, 206–212. [Google Scholar] [CrossRef]
- Grinberg, V.A.; Mayorova, N.A.; Pasynskii, A.A.; Shiryaev, A.A.; Vysotskii, V.V.; Stolarov, I.P.; Yakushev, I.A.; Cherkashina, N.V.; Vargaftik, M.N.; Zubavichus, Y.V.; et al. Nanosized catalysts of oxygen reduction reaction prepared on the base of bimetallic cluster compounds. Electrochim. Acta 2019, 299, 886–893. [Google Scholar] [CrossRef]
- Grinberg, V.A.; Emets, V.V.; Modestov, A.D.; Mayorova, N.A.; Shiryaev, A.A.; Vysotskii, V.V.; Stolyarov, I.P.; Pasynskii, A.A. Nanoscale catalysts of oxygen reduction based on bimetallic clusters in hydrogen-air fuel cell operating conditions. Prot. Met. Phys. Chem. Surf. 2019, 55, 277–282. [Google Scholar] [CrossRef]
- Mayorova, N.A.; Modestov, A.D.; Grinberg, V.A.; Shiryaev, A.A.; Shapovalov, S.S.; Nickolsky, M.S.; Stolyarov, I.P. Nanoscale catalyst based on a heterometallic carboxylatecomplex of platinum and iron for hydrogen-air fuel cells. Mater. Chem. Phys. 2021, 259, 123968. [Google Scholar] [CrossRef]
- Grinberg, V.A.; Kulova, T.L.; Mayorova, N.A.; Dobrokhotova, Z.V.; Pasynskii, A.A.; Skundin, A.M.; Khazova, O.A. Nanostructured catalysts for cathodes of oxygen-hydrogen fuel cells. Russ. J. Electrochem. 2007, 43, 75–84. [Google Scholar] [CrossRef]
- Grinberg, V.A.; Mayorova, N.A.; Pasynskii, A.A. A Cluster Pt-Sn-Catalyst for the Ethanol Direct Oxidation. Russ. J. Electrochem. 2009, 45, 1321–1326. [Google Scholar] [CrossRef]
- Peng, F.; Zhou, C.; Wang, H.; Yu, H.; Liang, J.; Yang, J. The role of RuO2 in the electrocatalytic oxidation of methanol for direct methanol fuel cell. Catal. Commun. 2009, 10, 533–537. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Fachini, E.R.; Cabrera, C.R. Electrochemically codeposited platinum/molybdenum oxide electrode for catalytic oxidation of methanol in acid solution. Electrochem. Solid-State Lett. 1999, 2, 437–439. [Google Scholar] [CrossRef]
- Lee, E.; Murthy, A.; Manthiram, A. Effect of Mo addition on the electrocatalytic activity of Pt–Sn–Mo/C for direct ethanol fuel cells. Electrochim. Acta 2011, 56, 1611–1618. [Google Scholar] [CrossRef]
- Xiong, L.; Manthiram, A. Effect of atomic ordering on the catalytic activity of carbon supported PtM (M = Fe, Co, Ni, and Cu) Alloys for Oxygen Reduction in PEMFCs. J. Electrochem. Soc. 2005, 152, A697–A703. [Google Scholar] [CrossRef]
- Gao, X.; Chen, S.; Deng, J.; Ibraheem, S.; Li, J.; Zhou, Q.; Lan, H.; Zou, X.; Wei, Z. High temperature self-assembly one-step synthesis of a structurally ordered PtFe catalyst for the oxygen reduction reaction. Chem. Commun. 2019, 55, 12028–12031. [Google Scholar] [CrossRef]
- Crabb, E.M.; Marshall, R.; Thompsett, D. Carbon monoxide electro-oxidation properties of carbon-supported PtSn catalysts prepared using surface organometallic chemistry. J. Electrochem. Soc. 2000, 147, 4440. [Google Scholar] [CrossRef]
- Wang, Y.; Fachini, E.R.; Cruz, G.; Zhu, Y.; Ishikawa, Y.; Colucci, J.A.; Cabrera, C.R. Effect of surface composition of electrochemically codeposited platinum/molybdenum oxide on methanol oxidation. J. Electrochem. Soc. 2001, 148, C222. [Google Scholar] [CrossRef]
- Wang, Z.-B.; Yin, G.-P.; Lin, Y.-G. Synthesis and characterization of PtRuMo/C nanoparticle electrocatalyst for direct ethanol fuel cell. J. Power Sources 2007, 170, 242–250. [Google Scholar] [CrossRef]
- Ohi, J.M.; Vanderborgh, N.; Ahmed, S.; Kumar, R.; Papadius, D.; Rockward, T. Hydrogen Fuel Quality Specifications for Polymer Electrolyte Fuel Cells in Road Vehicles Report to The Safety, Codes and Standards Program; Fuel Cell Technologies Office, U.S. Department of Energy: Washington, DC, USA, 2016. Available online: https://www.energy.gov/sites/prod/files/2016/11/f34/fcto_h2_fuel_quality_specs_pem_fc_road_vehicles.pdf (accessed on 22 February 2022).
- Ralph, T.R.; Hogarth, M.P. Catalysis for low temperature fuel cells. Part I: The cathode challenges. Platinum Metals Rev. 2002, 46, 3–14. [Google Scholar]
- Ralph, T.R.; Hogarth, M.P. Catalysis for low temperature fuel cells. Part II: The Anode Challenges. Platinum Metals Rev. 2002, 46, 117. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pt/C | PtFe/C | PtMo/C | PtMoSn/C | |
|---|---|---|---|---|
| Metals ratio in precursor | 1:1 0.5:0.5 | 1:1 0.5:0.5 | 1:1:1 0.33:0.33:0.33 | |
| Metals ratio in catalyst | 0.49:0.51 | 0.4:0.6 | 0.25:0.37:0.38 | |
| SH, m2gPt−1 | 98.5 | 111.6 | 146 | 139 |
| jO20.9 V, mA cmgeom−2 | 1.55 | 2.4 | 1.85 | |
| jO20.9 V, mA cmecsa−2 | 0.055 | 0.097 | 0.058 | |
| jO20.9 V, mA mg−1 | 54.3 | 108.3 | 86.3 | |
| jC2H5OH0.6 V, A g−1 | 88 | 168 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shapovalov, S.S.; Mayorova, N.A.; Modestov, A.D.; Shiryaev, A.A.; Egorov, A.V.; Grinberg, V.A. Pt-Mo/C, Pt-Fe/C and Pt-Mo-Sn/C Nanocatalysts Derived from Cluster Compounds for Proton Exchange Membrane Fuel Cells. Catalysts 2022, 12, 255. https://doi.org/10.3390/catal12030255
Shapovalov SS, Mayorova NA, Modestov AD, Shiryaev AA, Egorov AV, Grinberg VA. Pt-Mo/C, Pt-Fe/C and Pt-Mo-Sn/C Nanocatalysts Derived from Cluster Compounds for Proton Exchange Membrane Fuel Cells. Catalysts. 2022; 12(3):255. https://doi.org/10.3390/catal12030255
Chicago/Turabian StyleShapovalov, Sergey S., Natalia A. Mayorova, Alexander D. Modestov, Andrei A. Shiryaev, Alexander V. Egorov, and Vitali A. Grinberg. 2022. "Pt-Mo/C, Pt-Fe/C and Pt-Mo-Sn/C Nanocatalysts Derived from Cluster Compounds for Proton Exchange Membrane Fuel Cells" Catalysts 12, no. 3: 255. https://doi.org/10.3390/catal12030255
APA StyleShapovalov, S. S., Mayorova, N. A., Modestov, A. D., Shiryaev, A. A., Egorov, A. V., & Grinberg, V. A. (2022). Pt-Mo/C, Pt-Fe/C and Pt-Mo-Sn/C Nanocatalysts Derived from Cluster Compounds for Proton Exchange Membrane Fuel Cells. Catalysts, 12(3), 255. https://doi.org/10.3390/catal12030255