
Computational Analysis of Structure–Activity Relationships in Highly Active Homogeneous Ruthenium−Based Water Oxidation Catalysts


Abstract
:
1. Introduction
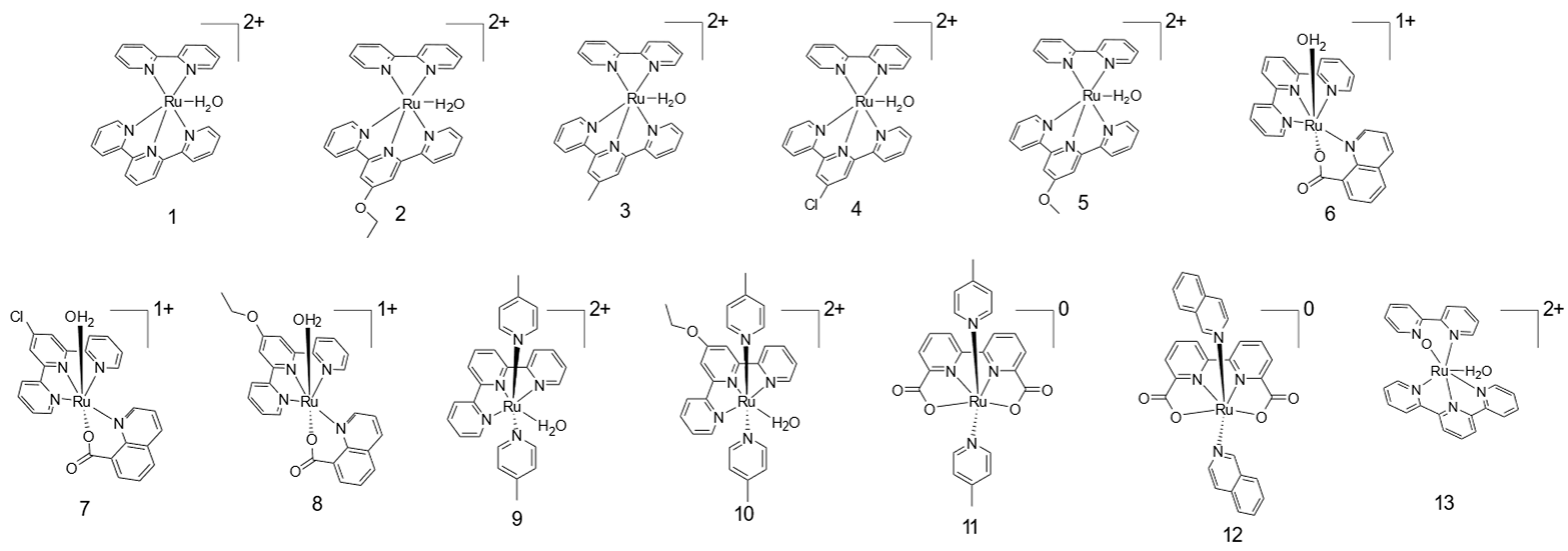
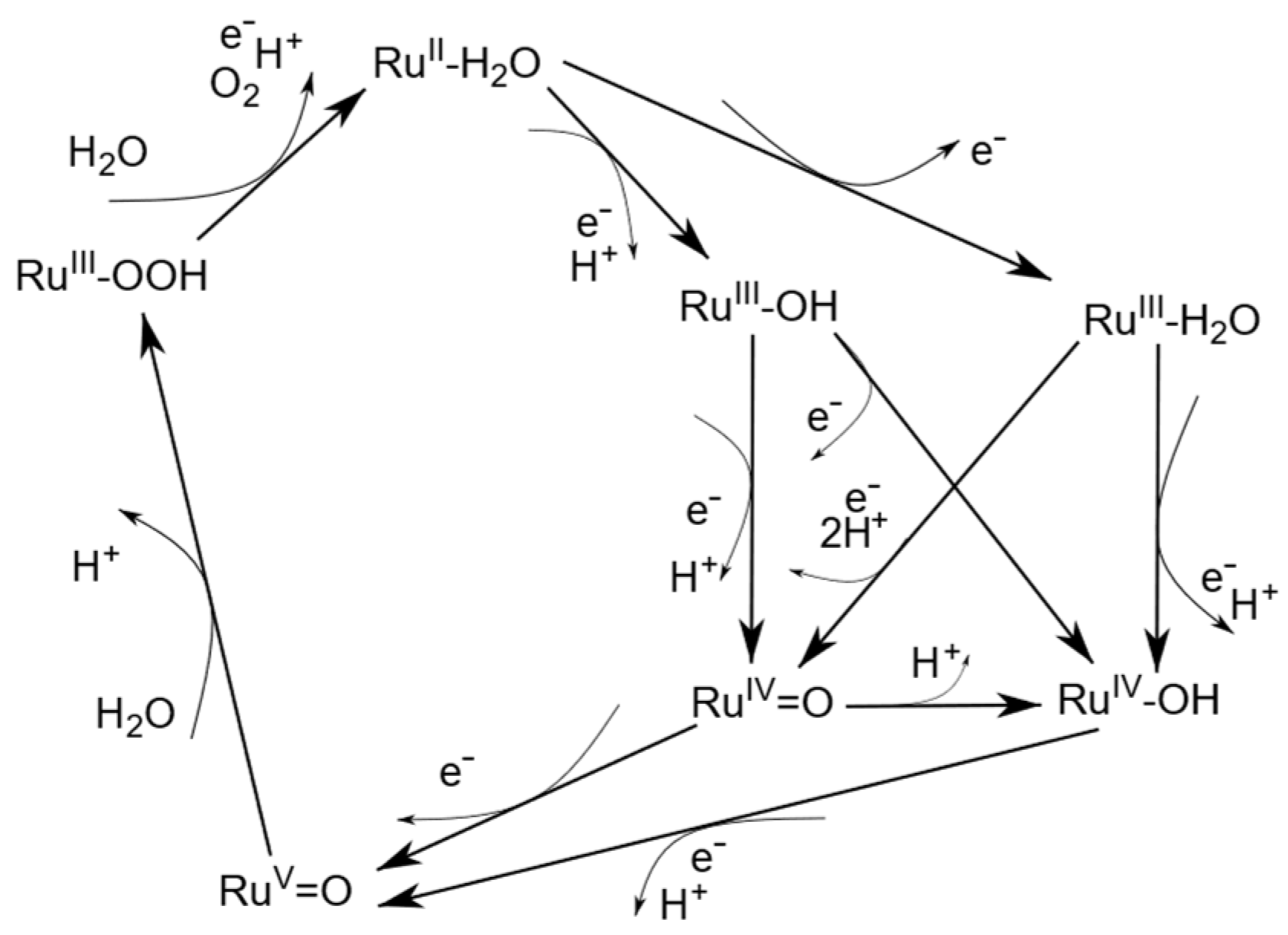
2. Results

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RuII/RuIII | RuIII/RuIV | RuIV/RuV | ||||||
|---|---|---|---|---|---|---|---|---|
| II−H2O/ III−H2O | II−H2O/ III−OH | III−H2O/ IV−OH | III−H2O/ IV = O | III−OH/ IV−OH | III−OH/ IV = O | IV−OH/ V = O | IV = O/ V = O | |
| Ru(EtoTpy)(4pic)2 | 1.30 | 0.69 | 1.26 | 0.32 | 1.87 | 0.94 | 1.29 | 2.23 |
| 0.97 V [44] | ||||||||
| Ru(Tpy)(4pic)2 | 1.38 | 0.70 | 1.42 | 0.30 | 2.09 | 0.97 | 1.21 | 2.33 |
| 1.00 V [44] | ||||||||
| Ru(tpy)(bpy) | 1.32 | 0.75 | 1.68 | 0.46 | 2.26 | 1.03 | 1.15 | 2.37 |
| 1.04 V [56] | 1.23 V [56], 1.39 V [57] | 1.60 V [58], 1.73 V [59], 1.80 V [60] | ||||||
| Ru(EtoTpy)(bpy) | 1.21 | 0.70 | 1.44 | 0.48 | 1.95 | 1.00 | 1.24 | 2.19 |
| 0.98 V [61] | 1.24 V [61] | |||||||
| Ru(TpyCl)(QC) | 0.68 | 0.47 | 1.25 | 0.75 | 1.45 | 0.96 | 1.30 | 1.79 |
| 0.61 V [*] | ||||||||
| Ru(EtoTpy)(QC) | 0.59 | 0.46 | 1.14 | 0.76 | 1.27 | 0.89 | 1.29 | 1.68 |
| 0.63 V [*] | 1.19 V [*] | 1.73 V [*] | ||||||
| Ru(Tpy)(QC) | 0.62 | 0.42 | 1.22 | 0.75 | 1.42 | 0.95 | 1.28 | 1.75 |
| 0.67 V [62], 0.82 V [*] | 1.20 V [62], 1.36 V [*] | 1.62 [62], 1.75 V [*] | ||||||
| Ru(TpyMeO)(bpy) | 1.23 | 1.00 | 1.48 | 0.52 | 1.70 | 0.74 | 1.22 | 2.18 |
| No Data | ||||||||
| Ru(Tpy−Me)(bpy) | 1.26 | 0.79 | 1.50 | 0.53 | 1.98 | 1.00 | 1.31 | 2.28 |
| No Data | ||||||||
| Ru(Tpy−Cl)(bpy) | 1.40 | 0.82 | 1.56 | 0.45 | 2.13 | 1.02 | 1.34 | 2.45 |
| No Data | ||||||||
| Ru(bda)(isoq)2 ** | 0.19 0.17 ** | 0.91 | 2.08 1.31 | 1.74 | 1.36 | 1.02 | 1.03 1.52 | 1.38 |
| 0.63 V [53] 1.06 | 1.09 V [53] | 1.27 V [53] | ||||||
| Ru(bda)(4pic)2 | 0.09 | 0.78 | 0.98 | 1.82 | 0.30 | 1.13 | 2.12 | 1.28 |
| 0.66 V [63] | 1.15 V [63] | 1.35 V [63] | ||||||
| Ru(tpy)(bpyNO) | 1.09 | 0.79 | 1.50 | 0.65 | 1.79 | 0.95 | 1.32 | 2.16 |
| 0.82 V [13] | 0.86 V [13] | |||||||
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lewis, N.S. Research opportunities to advance solar energy utilization. Science 2016, 351, aad1920. [Google Scholar] [CrossRef] [Green Version]
- Gust, D.; Moore, T.A.; Moore, A.L. Solar fuels via artificial photosynthesis. Acc. Chem. Res. 2009, 42, 1890–1898. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Chen, Z.; Brennaman, M.K.; Concepcion, J.J.; Patrocinio, A.O.T.; Iha, N.Y.M.; Meyer, T.J. Making solar fuels by artificial photosynthesis. Pure Appl. Chem. 2011, 83, 749–768. [Google Scholar] [CrossRef]
- Tachibana, Y.; Vayssieres, L.; Durrant, J.R. Artificial photosynthesis for solar water-splitting. Nat. Photonics 2012, 6, 511–518. [Google Scholar] [CrossRef]
- Lewis, N.S.; Nocera, D.G. Powering the planet: Chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [Google Scholar] [CrossRef] [Green Version]
- Nocera, D.G. Solar fuels and solar chemicals industry. Acc. Chem. Res. 2017, 50, 616–619. [Google Scholar] [CrossRef]
- Nocera, D.G. The Artificial Leaf. Acc. Chem. Res. 2012, 45, 767–776. [Google Scholar] [CrossRef]
- Kim, W.; Edri, E.; Frei, H. Hierarchical inorganic assemblies for artificial photosynthesis. Acc. Chem. Res. 2016, 49, 1634–1645. [Google Scholar] [CrossRef]
- Young, K.J.; Martini, L.A.; Milot, R.L.; Snoeberger III, R.C.; Batista, V.S.; Schmuttenmaer, C.A.; Crabtree, R.H.; Brudvig, G.W. Light-driven water oxidation for solar fuels. Coord. Chem. Rev. 2012, 256, 2503–2520. [Google Scholar] [CrossRef] [Green Version]
- Karkas, M.D.; Verho, O.; Johnston, E.V.; Åkermark, B. Artificial photosynthesis: Molecular systems for catalytic water oxidation. Chem. Rev. 2014, 114, 11863–12001. [Google Scholar] [CrossRef]
- Blakemore, J.D.; Crabtree, R.H.; Brudvig, G.W. Molecular catalysts for water oxidation. Chem. Rev. 2015, 115, 12974–13005. [Google Scholar] [CrossRef] [PubMed]
- Sivula, K.; Van De Krol, R. Semiconducting materials for photoelectrochemical energy conversion. Nat. Rev. Mater. 2016, 1, 15010. [Google Scholar] [CrossRef]
- Ravari, A.K.; Zhu, G.; Ezhov, R.; Pineda-Galvan, Y.; Page, A.; Weinschenk, W.; Yan, L.; Pushkar, Y. Unraveling the mechanism of catalytic water oxidation via de novo synthesis of reactive intermediate. J. Am. Chem. Soc. 2019, 142, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, M.V.; Sherman, B.D.; Xie, Y.; Wang, Y. Heterogeneous water oxidation catalysts for molecular anodes and photoanodes. Sol. RRL 2021, 5, 2000565. [Google Scholar] [CrossRef]
- Ye, S.; Ding, C.; Liu, M.; Wang, A.; Huang, Q.; Li, C. Water oxidation catalysts for artificial photosynthesis. Adv. Mater. 2019, 31, 1902069. [Google Scholar] [CrossRef]
- Thalluri, S.M.; Bai, L.; Lv, C.; Huang, Z.; Hu, X.; Liu, L. Strategies for semiconductor/electrocatalyst coupling toward solar-driven water splitting. Adv. Sci. 2020, 7, 1902102. [Google Scholar] [CrossRef] [Green Version]
- Craig, M.J.; Coulter, G.; Dolan, E.; Soriano-López, J.; Mates-Torres, E.; Schmitt, W.; García-Melchor, M. Universal scaling relations for the rational design of molecular water oxidation catalysts with near-zero overpotential. Nat. Commun. 2019, 10, 4993. [Google Scholar] [CrossRef] [Green Version]
- Hessels, J.; Detz, R.J.; Koper, M.T.M.; Reek, J.N.H. Rational design rules for molecular water oxidation catalysts based on scaling relationships. Chem. Eur. J. 2017, 23, 16413–16418. [Google Scholar] [CrossRef]
- Schilling, M.; Böhler, M.; Luber, S. Towards the rational design of the Py5-ligand framework for ruthenium-based water oxidation catalysts. Dalt. Trans. 2018, 47, 10480–10490. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Zhang, Y.; Ding, Y. Rationally designed/constructed CoOx/WO3 anode for efficient photoelectrochemical water oxidation. Acs Catal. 2017, 7, 1841–1845. [Google Scholar] [CrossRef]
- Gao, J.; Huang, X.; Cai, W.; Wang, Q.; Jia, C.; Liu, B. Rational Design of an Iridium–Tungsten Composite with an Iridium-Rich Surface for Acidic Water Oxidation. ACS Appl. Mater. Interfaces 2020, 12, 25991–26001. [Google Scholar] [CrossRef] [PubMed]
- Latimer, A.A.; Kulkarni, A.R.; Aljama, H.; Montoya, J.H.; Yoo, J.S.; Tsai, C.; Abild-Pedersen, F.; Studt, F.; Nørskov, J.K. Understanding trends in C–H bond activation in heterogeneous catalysis. Nat. Mater. 2017, 16, 225–229. [Google Scholar] [CrossRef]
- Liao, P.; Getman, R.B.; Snurr, R.Q. Optimizing open iron sites in metal–organic frameworks for ethane oxidation: A first-principles study. ACS Appl. Mater. Interfaces 2017, 9, 33484–33492. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Single-atom catalysts: A new frontier in heterogeneous catalysis. Acc. Chem. Res. 2013, 46, 1740–1748. [Google Scholar] [CrossRef] [PubMed]
- Flytzani-Stephanopoulos, M. Gold atoms stabilized on various supports catalyze the water–gas shift reaction. Acc. Chem. Res. 2014, 47, 783–792. [Google Scholar] [CrossRef]
- Abild-Pedersen, F.; Greeley, J.; Studt, F.; Rossmeisl, J.; Munter, T.R.; Moses, P.G.; Skulason, E.; Bligaard, T.; Nørskov, J.K. Scaling properties of adsorption energies for hydrogen-containing molecules on transition-metal surfaces. Phys. Rev. Lett. 2007, 99, 16105. [Google Scholar] [CrossRef] [Green Version]
- Nørskov, J.K.; Bligaard, T.; Rossmeisl, J.; Christensen, C.H. Towards the computational design of solid catalysts. Nat. Chem. 2009, 1, 37–46. [Google Scholar] [CrossRef]
- Wodrich, M.D.; Sawatlon, B.; Busch, M.; Corminboeuf, C. The genesis of molecular volcano plots. Acc. Chem. Res. 2021, 54, 1107–1117. [Google Scholar] [CrossRef]
- Cordova, M.; Wodrich, M.D.; Meyer, B.; Sawatlon, B.; Corminboeuf, C. Data-driven advancement of homogeneous nickel catalyst activity for aryl ether cleavage. Acs Catal. 2020, 10, 7021–7031. [Google Scholar] [CrossRef]
- Busch, M.; Wodrich, M.D.; Corminboeuf, C. Linear scaling relationships and volcano plots in homogeneous catalysis–revisiting the Suzuki reaction. Chem. Sci. 2015, 6, 6754–6761. [Google Scholar] [CrossRef] [Green Version]
- Mori-Sánchez, P.; Cohen, A.J.; Yang, W. Many-electron self-interaction error in approximate density functionals. J. Chem. Phys. 2006, 125, 201102. [Google Scholar] [CrossRef] [PubMed]
- Ruzsinszky, A.; Perdew, J.P.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E. Density functionals that are one-and two-are not always many-electron self-interaction-free, as shown for H2+, He2+, LiH+, and Ne2+. J. Chem. Phys. 2007, 126, 104102. [Google Scholar] [CrossRef] [PubMed]
- Haunschild, R.; Henderson, T.M.; Jimenez-Hoyos, C.A.; Scuseria, G.E. Many-electron self-interaction and spin polarization errors in local hybrid density functionals. J. Chem. Phys. 2010, 133, 134116. [Google Scholar] [CrossRef] [PubMed]
- Wodrich, M.D.; Busch, M.; Corminboeuf, C. Accessing and predicting the kinetic profiles of homogeneous catalysts from volcano plots. Chem. Sci. 2016, 7, 5723–5735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, M.J.; García-Melchor, M. High-throughput screening and rational design to drive discovery in molecular water oxidation catalysis. Cell Rep. Phys. Sci. 2021, 2, 100492. [Google Scholar] [CrossRef]
- Galán-Mascarós, J.R. Water oxidation at electrodes modified with earth-abundant transition-metal catalysts. ChemElectroChem 2015, 2, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.F.; Kaplan, C.; Sheats, J.E.; Robinson, D.M.; McCool, N.S.; Mezle, N.; Dismukes, G.C. What determines catalyst functionality in molecular water oxidation? dependence on ligands and metal nuclearity in cobalt clusters. Inorg. Chem. 2014, 53, 2113–2121. [Google Scholar] [CrossRef]
- Kamdar, J.M.; Grotjahn, D.B. An overview of significant achievements in ruthenium-based molecular water oxidation catalysis. Molecules 2019, 24, 494. [Google Scholar] [CrossRef] [Green Version]
- Matheu, R.; Garrido-Barros, P.; Gil-Sepulcre, M.; Ertem, M.Z.; Sala, X.; Gimbert-Suriñach, C.; Llobet, A. The development of molecular water oxidation catalysts. Nat. Rev. Chem. 2019, 3, 331–341. [Google Scholar] [CrossRef]
- Zeng, Q.; Lewis, F.W.; Harwood, L.M.; Hartl, F. Role of ligands in catalytic water oxidation by mononuclear ruthenium complexes. Coord. Chem. Rev. 2015, 304, 88–101. [Google Scholar] [CrossRef]
- Moonshiram, D.; Alperovich, I.; Concepcion, J.J.; Meyer, T.J.; Pushkar, Y. Experimental demonstration of radicaloid character in a RuV= O intermediate in catalytic water oxidation. Proc. Natl. Acad. Sci.USA 2013, 110, 3765–3770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moonshiram, D.; Pineda-Galvan, Y.; Erdman, D.; Palenik, M.; Zong, R.; Thummel, R.; Pushkar, Y. Uncovering the Role of Oxygen Atom Transfer in Ru-Based Catalytic Water Oxidation. J. Am. Chem. Soc. 2016, 138, 15605–15616. [Google Scholar] [CrossRef] [PubMed]
- Ertem, M.Z.; Concepcion, J.J. Oxygen Atom Transfer as an Alternative Pathway for Oxygen–Oxygen Bond Formation. Inorg. Chem. 2020, 59, 5966–5974. [Google Scholar] [CrossRef]
- Patel, J.; Bury, G.; Ravari, A.K.; Ezhov, R.; Pushkar, Y. Systematic Influence of Electronic Modification of Ligands on the Catalytic Rate of Water Oxidation by a Single-Site Ru-Based Catalyst. ChemSusChem 2022, 15, e202101657. [Google Scholar] [CrossRef] [PubMed]
- Erdman, D.; Pineda-Galvan, Y.; Pushkar, Y. Mechanistic analysis of water oxidation catalyst cis-[Ru(bpy)2(H2O)2]2+: Effect of dimerization. Catalysts 2017, 7, 39. [Google Scholar] [CrossRef] [Green Version]
- Ezhov, R.; Karbakhsh Ravari, A.; Page, A.; Pushkar, Y. Water oxidation catalyst cis-[Ru (bpy)(5,5′-dcbpy)(H2O)2]2+ and Its Stabilization in Metal–Organic Framework. ACS Catal. 2020, 10, 5299–5308. [Google Scholar] [CrossRef]
- Bucci, A.; Menendez Rodriguez, G.; Bellachioma, G.; Zuccaccia, C.; Poater, A.; Cavallo, L.; Macchioni, A. An alternative reaction pathway for iridium-catalyzed water oxidation driven by cerium ammonium nitrate (can). ACS Catal. 2016, 6, 4559–4563. [Google Scholar] [CrossRef] [Green Version]
- Dengel, A.C.; Griffith, W.P. Studies on transition-metal oxo and nitrido complexes. 12. Synthesis, spectroscopic properties, and reactions of stable ruthenium (V) and osmium (V) oxo complexes containing. alpha.-hydroxy carboxylate and. alpha.-amino carboxylate ligands. Inorg. Chem. 1991, 30, 869–871. [Google Scholar] [CrossRef]
- Planas, N.; Vigara, L.; Cady, C.; Miró, P.; Huang, P.; Hammarstrom, L.; Styring, S.; Leidel, N.; Dau, H.; Haumann, M. Electronic structure of oxidized complexes derived from cis-[RuII (bpy)2(H2O)2]2+ and its photoisomerization mechanism. Inorg. Chem. 2011, 50, 11134–11142. [Google Scholar] [CrossRef]
- Pineda-Galvan, Y.; Ravari, A.K.; Shmakov, S.; Lifshits, L.; Kaveevivitchai, N.; Thummel, R.; Pushkar, Y. Detection of the site protected 7-coordinate RuV= O species and its chemical reactivity to enable catalytic water oxidation. J. Catal. 2019, 375, 1–7. [Google Scholar] [CrossRef]
- Lebedev, D.; Pineda-Galvan, Y.; Tokimaru, Y.; Fedorov, A.; Kaeffer, N.; Copéret, C.; Pushkar, Y. The Key RuV=O Intermediate of Site-Isolated Mononuclear Water Oxidation Catalyst Detected by in Situ X-ray Absorption Spectroscopy. J. Am. Chem. Soc. 2018, 140, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Levin, N.; Casadevall, C.; Cutsail III, G.E.; Lloret-Fillol, J.; DeBeer, S.; Rüdiger, O. XAS and EPR in situ observation of Ru (V) oxo intermediate in a Ru water oxidation complex. ChemElectroChem 2022, 9, e202101271. [Google Scholar] [PubMed]
- Duan, L.; Bozoglian, F.; Mandal, S.; Stewart, B.; Privalov, T.; Llobet, A.; Sun, L. A molecular ruthenium catalyst with water-oxidation activity comparable to that of photosystem II. Nat. Chem. 2012, 4, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Pushkar, Y.; Moonshiram, D.; Purohit, V.; Yan, L.; Alperovich, I. Spectroscopic Analysis of Catalytic Water Oxidation by [RuII(bpy)(tpy) H2O]2+ Suggests That RuV O Is Not a Rate-Limiting Intermediate. J. Am. Chem. Soc. 2014, 136, 11938–11945. [Google Scholar] [CrossRef]
- Jurss, J.W.; Concepcion, J.C.; Norris, M.R.; Templeton, J.L.; Meyer, T.J. Surface catalysis of water oxidation by the blue ruthenium dimer. Inorg. Chem. 2010, 49, 3980–3982. [Google Scholar] [CrossRef]
- Wasylenko, D.J.; Ganesamoorthy, C.; Henderson, M.A.; Koivisto, B.D.; Osthoff, H.D.; Berlinguette, C.P. Electronic modification of the [RuII (tpy)(bpy)(OH2)]2+ scaffold: Effects on catalytic water oxidation. J. Am. Chem. Soc. 2010, 132, 16094–16106. [Google Scholar] [CrossRef]
- Takeuchi, K.J.; Thompson, M.S.; Pipes, D.W.; Meyer, T.J. Redox and spectral properties of monooxo polypyridyl complexes of ruthenium and osmium in aqueous media. Inorg. Chem. 1984, 23, 1845–1851. [Google Scholar] [CrossRef]
- Concepcion, J.J.; Jurss, J.W.; Norris, M.R.; Chen, Z.; Templeton, J.L.; Meyer, T.J. Catalytic water oxidation by single-site ruthenium catalysts. Inorg. Chem. 2010, 49, 1277–1279. [Google Scholar] [CrossRef]
- Wasylenko, D.J.; Ganesamoorthy, C.; Koivisto, B.D.; Henderson, M.A.; Berlinguette, C.P. Insight into water oxidation by mononuclear polypyridyl Ru catalysts. Inorg. Chem. 2010, 49, 2202–2209. [Google Scholar] [CrossRef]
- Wasylenko, D.J.; Ganesamoorthy, C.; Henderson, M.A.; Berlinguette, C.P. Unraveling the roles of the acid medium, experimental probes, and terminal oxidant, (NH4)2[Ce(NO3)6], in the study of a homogeneous water oxidation catalyst. Inorg. Chem. 2011, 50, 3662–3672. [Google Scholar] [CrossRef]
- Yagi, M.; Tajima, S.; Komi, M.; Yamazaki, H. Highly active and tunable catalysts for O 2 evolution from water based on mononuclear ruthenium (II) monoaquo complexes. Dalt. Trans. 2011, 40, 3802–3804. [Google Scholar] [CrossRef]
- Hoque, M.A.; Chowdhury, A.D.; Maji, S.; Benet-Buchholz, J.; Ertem, M.Z.; Gimbert-Suriñach, C.; Lahiri, G.K.; Llobet, A. Synthesis, characterization, and water oxidation activity of isomeric Ru complexes. Inorg. Chem. 2021, 60, 5791–5803. [Google Scholar] [CrossRef]
- Timmer, B.J.J.; Kravchenko, O.; Liu, T.; Zhang, B.; Sun, L. Off-Set Interactions of Ruthenium–bda Type Catalysts for Promoting Water-Splitting Performance. Angew. Chem. 2021, 133, 14625–14632. [Google Scholar] [CrossRef]
- Busch, M.; Fabrizio, A.; Luber, S.; Hutter, J.; Corminboeuf, C. Exploring the limitation of molecular water oxidation catalysts. J. Phys. Chem. C 2018, 122, 12404–12412. [Google Scholar] [CrossRef]
- Xu, X.; Xu, H.; Cheng, D. Design of high-performance MoS 2 edge supported single-metal atom bifunctional catalysts for overall water splitting via a simple equation. Nanoscale 2019, 11, 20228–20237. [Google Scholar] [CrossRef]
- Piqué, O.; Illas, F.; Calle-Vallejo, F. Designing water splitting catalysts using rules of thumb: Advantages, dangers and alternatives. Phys. Chem. Chem. Phys. 2020, 22, 6797–6803. [Google Scholar] [CrossRef]
- Pushkar, Y.; Davis, K.M.; Palenik, M.C. Model of the oxygen evolving complex which is highly predisposed to O–O bond formation. J. Phys. Chem. Lett. 2018, 9, 3525–3531. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Govindasamy, P.; Gunasekaran, S.; Srinivasan, S. Molecular geometry, conformational, vibrational spectroscopic, molecular orbital and Mulliken charge analysis of 2-acetoxybenzoic acid. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 130, 329–336. [Google Scholar] [CrossRef]
- Du, X.; Zhang, X.; Yang, Z.; Gong, Y. Water oxidation catalysis beginning with CuCo2S4: Investigation of the true electrochemically driven catalyst. Chem. Asian J. 2018, 13, 266–270. [Google Scholar] [CrossRef]
- Shi, J.; Guo, Y.; Xie, F.; Chen, Q.; Zhang, M. Redox-Active Ligand Assisted Catalytic Water Oxidation by a RuIV= O Intermediate. Angew. Chem. 2020, 132, 4029–4037. [Google Scholar] [CrossRef]
- Ezhov, R.; Ravari, A.; Bury, G.; Smith, P.; Pushkar, Y. Formation of CoIV=O intermediate at the Boundary of the “Oxo-wall” Induces Water Oxidation. Res. Sq. 2020, 1–21. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M. Theoretical studies of O-O bond formation in photosystem II. Inorg. Chem. 2008, 47, 1779–1786. [Google Scholar] [CrossRef]
- Coggins, M.K.; Zhang, M.; Chen, Z.; Song, N.; Meyer, T.J. Single-Site Copper (II) Water Oxidation Electrocatalysis: Rate Enhancements with HPO42− as a Proton Acceptor at pH 8. Angew. Chem. Int. Ed. 2014, 53, 12226–12230. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bury, G.; Pushkar, Y. Computational Analysis of Structure–Activity Relationships in Highly Active Homogeneous Ruthenium−Based Water Oxidation Catalysts. Catalysts 2022, 12, 863. https://doi.org/10.3390/catal12080863
Bury G, Pushkar Y. Computational Analysis of Structure–Activity Relationships in Highly Active Homogeneous Ruthenium−Based Water Oxidation Catalysts. Catalysts. 2022; 12(8):863. https://doi.org/10.3390/catal12080863
Chicago/Turabian StyleBury, Gabriel, and Yulia Pushkar. 2022. "Computational Analysis of Structure–Activity Relationships in Highly Active Homogeneous Ruthenium−Based Water Oxidation Catalysts" Catalysts 12, no. 8: 863. https://doi.org/10.3390/catal12080863
APA StyleBury, G., & Pushkar, Y. (2022). Computational Analysis of Structure–Activity Relationships in Highly Active Homogeneous Ruthenium−Based Water Oxidation Catalysts. Catalysts, 12(8), 863. https://doi.org/10.3390/catal12080863