
Computational Study on the Catalytic Performance of Single-Atom Catalysts Anchored on g-CN for Electrochemical Oxidation of Formic Acid

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
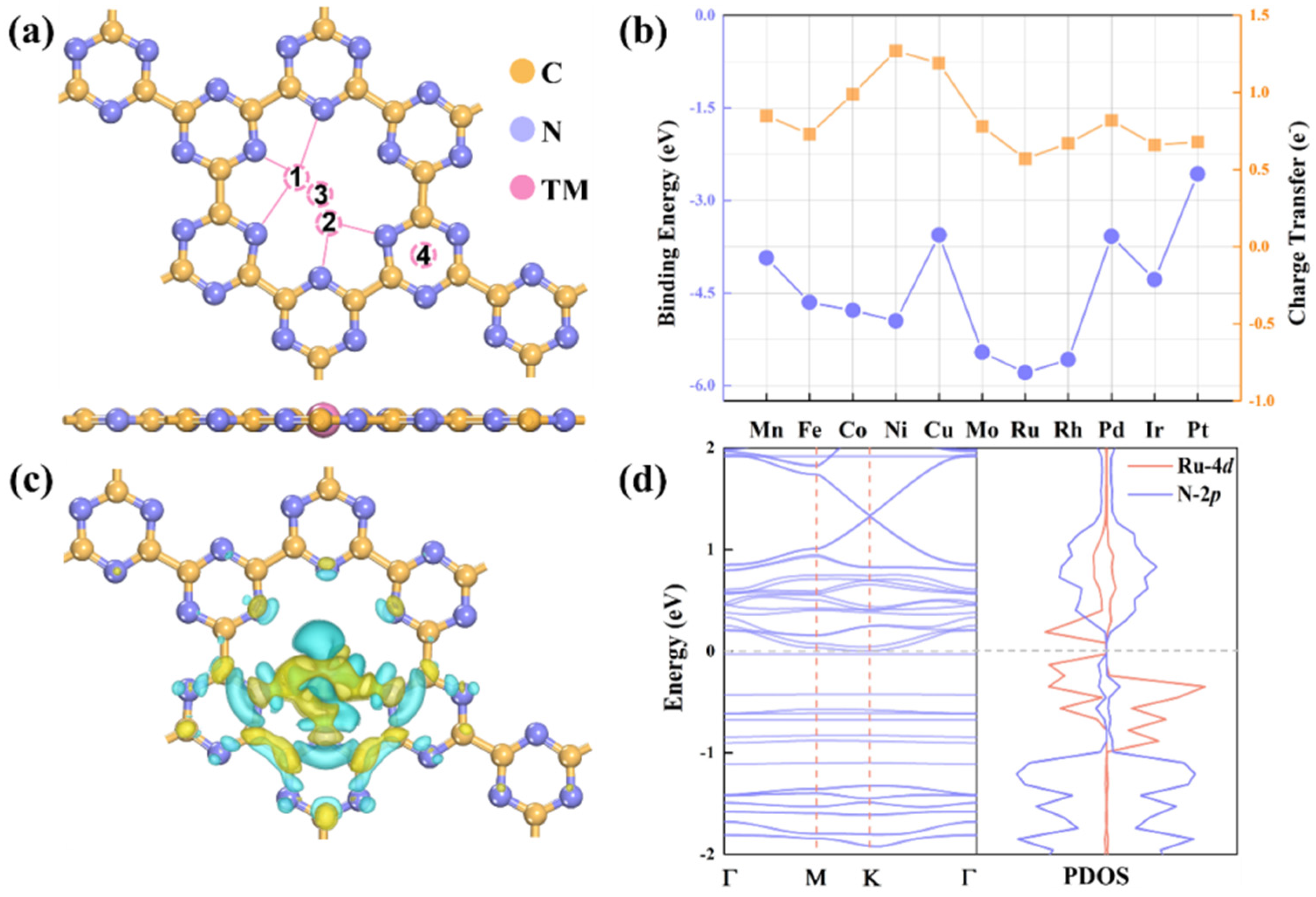
2.1. Structures, Stabilities, and Properties of TM/g-CN Catalysts
2.2. Catalytic Performance of TM/g-CN for the FAOR
2.3. Origin of Catalyst Activity
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McNicol, B.D.; Rand, D.A.J.; Williams, K.R. Direct methanol-air fuel cells for road transportation. J. Power Sources 1999, 83, 15–31. [Google Scholar] [CrossRef]
- Rice, C.; Ha, R.I.; Masel, R.I.; Waszczuk, P.; Wieckowski, A.; Barnard, T. Direct formic acid fuel cells. J. Power Sources 2002, 111, 83–89. [Google Scholar] [CrossRef]
- Singh, A.K.; Singh, S.; Kumar, A. Hydrogen energy future with formic acid: A renewable chemical hydrogen storage system. Catal. Sci. Technol. 2016, 6, 12–40. [Google Scholar] [CrossRef]
- Yu, X.; Pickup, P.G. Recent advances in direct formic acid fuel cells (DFAFC). J. Power Sources 2008, 182, 124–132. [Google Scholar] [CrossRef]
- Fang, Z.; Chen, W. Recent advances in formic acid electro-oxidation: From the fundamental mechanism to electrocatalysts. Nanoscale Adv. 2021, 3, 94–105. [Google Scholar] [CrossRef]
- Yang, X.; Meng, Q.; Wang, X.; Jin, Z.; Liu, C.; Ge, J.; Xing, W. A new pathway for formic acid electro-oxidation: The electro-chemically decomposed hydrogen as a reaction intermediate. J. Energy Chem. 2022, 71, 188–191. [Google Scholar] [CrossRef]
- Herrero, E.; Feliu, J. Understanding formic acid oxidation mechanism on platinum single crystal electrodes. Curr. Opin. Electrochem. 2018, 9, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Pramanick, B.; Kumar, T.; Halder, A.; Siril, P.F. Engineering the morphology of palladium nanostructures to tune their electrocatalytic activity in formic acid oxidation reactions. Nanoscale Adv. 2022, 4, 3109. [Google Scholar] [CrossRef]
- Tian, Q.; Zhu, Z.; Fu, B.; Li, Y. Kinetic study of formic acid electrochemical oxidation on supported Pd based electrocatalysts. J. Electrochem. Soc. 2018, 165, F1075–F1083. [Google Scholar] [CrossRef]
- Bai, J.; Jia, N.; Jin, P.; Chen, P.; Jiang, J.X.; Zeng, J.H.; Chen, Y. Metal-organic interface engineering for boosting the electroactivity of Pt nanodendrites for hydrogen production. J. Energy Chem. 2020, 51, 105–112. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, B.; Gao, Y.; Tang, Y.; Lu, T.; Xing, W.; Liu, C. Kinetic study of formic acid oxidation on carbon supported Pd electrocatalyst. J. Power Sources 2009, 192, 372–375. [Google Scholar] [CrossRef]
- Yang, Q.; Shi, L.; Yu, B.; Xu, J.; Wei, C.; Wang, Y.; Chen, H. Facile synthesis of ultrathin Pt-Pd nanosheets for enhanced formic acid oxidation and oxygen reduction reaction. J. Mater. Chem. A 2020, 8, 11460. [Google Scholar] [CrossRef]
- Li, Z.; Chen, Y.; Ji, S.; Tang, Y.; Chen, W.; Li, A.; Zhao, J.; Xiong, Y.; Wu, Y.; Gong, Y.; et al. Iridium single-atom catalyst on nitrogen-doped carbon for formic acid oxidation synthesized using a general host-guest strategy. Nat. Chem. 2020, 12, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Dong, J.; Huang, Z.-Q.; Xin, P.; Chen, W.; Wang, Y.; Li, Z.; Jin, Z.; Xing, W.; Zhuang, Z.; et al. Single-atom Rh/N-doped carbon electrocatalyst for formic acid oxidation. Nat. Nanotechnol. 2020, 15, 390. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jacob, T.; Gao, W. Progress of fundamental mechanism of formic acid decomposition and electrooxidation. J. Energy Chem. 2022, 70, 292–309. [Google Scholar] [CrossRef]
- Safdar Hossain, S.K.; Saleem, J.; Mudassir Ahmad Alwi, M.; Al-Odail, F.A.; Mozahar Hossain, M. Recent advances in anode electrocatalysts for direct formic acid fuel cells-part l-fundamentals and Pd based catalysts. Chem. Rec. 2022, 22, e202200045. [Google Scholar] [CrossRef]
- Shen, T.; Zhang, J.; Chen, K.; Deng, S.; Wang, D. Recent progress of palladium-based electrocatalysts for the formic acid oxidation reaction. Energy Fuels 2020, 34, 9137–9153. [Google Scholar] [CrossRef]
- Chang, J.; Feng, L.; Liu, C.; Xing, W.; Hu, X. An Effective Pd-Ni2P/C anode catalyst for direct formic acid fuel cells. Angew. Chem. Int. Ed. 2014, 53, 122–126. [Google Scholar] [CrossRef] [Green Version]
- Duchesne, P.N.; Li, Z.Y.; Deming, C.P.; Fung, V.; Zhao, X.; Yuan, J.; Regier, T.; Aldalbahi, A.; Almarhoon, Z.; Chen, S.; et al. Golden single-atomic-site platinum electrocatalysts. Nat. Mater. 2018, 17, 1033–1039. [Google Scholar] [CrossRef] [Green Version]
- Perales-Rondón, J.V.; Ferre-Vilaplana, A.; Feliu, J.M.; Herrero, E. Oxidation mechanism of formic acid on the bismuth adatom-modified Pt(111) surface. J. Am. Chem. Soc. 2014, 136, 13110–13113. [Google Scholar] [CrossRef]
- Xi, Z.; Li, J.; Su, D.; Muzzio, M.; Yu, C.; Li, Q.; Sun, S. Stabilizing CuPd nanoparticles via CuPd Coupling to WO2.72 nanorods in electrochemical oxidation of formic acid. J. Am. Chem. Soc. 2017, 139, 15191–15196. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, J.; Yin, H.; Song, R.; Tang, Z. “Raisin bun”-like nanocomposites of palladium clusters and porphyrin for superior formic acid Oxidation. Adv. Mater. 2013, 25, 2728–2732. [Google Scholar] [CrossRef] [PubMed]
- Park, I.-S.; Lee, K.-S.; Yoo, S.J.; Cho, Y.-H.; Sung, Y.-E. Electrocatalytic properties of Pd clusters on Au nanoparticles in formic acid electro-oxidation. Electrochim. Acta 2010, 55, 4339–4345. [Google Scholar] [CrossRef]
- Huang, X.; Tang, S.; Mu, X.; Dai, Y.; Chen, G.; Zhou, Z.; Ruan, F.; Yang, Z.; Zheng, N. Freestanding palladium nanosheets with plasmonic and catalytic properties. Nat. Nanotechnol. 2011, 6, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Jiang, B.; Miyamoto, N.; Kim, J.H.; Malgras, V.; Yamauchi, Y. Surfactant-directed synthesis of mesoporous Pd films with perpendicular mesochannels as efficient electrocatalysts. J. Am. Chem. Soc. 2015, 137, 11558–11561. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.Y.; Wu, H.B.; Yan, Y.; Lou, X.W.; Wang, X. Ultrathin and ultralong single-crystal platinum nanowire assemblies with highly stable electrocatalytic activity. J. Am. Chem. Soc. 2013, 135, 9480–9485. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, S.K.; Chen, Z.; Akl, D.F.; Mitchell, S.; Perez-Ramirez, J. Single-atom catalysts across the periodic table. Chem. Rev. 2020, 120, 11703–11809. [Google Scholar] [CrossRef] [PubMed]
- Abdelghafar, F.; Xu, X.; Shao, Z. Designing single-atom catalysts toward improved alkaline hydrogen evolution reaction. Mater. Rep. Energy 2022, 2, 100144. [Google Scholar] [CrossRef]
- Singh, B.; Sharma, V.; Gaikwad, R.P.; Fornasiero, P.; Zboril, R.; Gawande, M.B. Single-atom catalysts: A sustainable pathway for the advanced catalytic applications. Small 2021, 17, 2006473. [Google Scholar] [CrossRef]
- Wang, A.; Li, J.; Zhang, T. Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2018, 2, 65–81. [Google Scholar] [CrossRef]
- Xi, J.; Jung, H.S.; Xu, Y.; Xiao, F.; Bae, J.W.; Wang, S. Synthesis strategies, catalytic applications, and performance regulation of single-atom catalysts. Adv. Funct. Mater. 2021, 31, 2008318. [Google Scholar] [CrossRef]
- Yang, X.-F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Single-atom catalysts: A new frontier in heterogeneous catalysis. Acc. Chem. Res. 2013, 46, 1740–1748. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Wang, H.H.; Liu, J.; Liu, X.; Li, W.; Wang, Y. Research progress and application of single-atom catalysts: A Review. Molecules 2021, 26, 6501. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Q.; Wang, H.; Zhang, L.; Wilkinson, D.P.; Zhang, J. Recent progresses in oxygen reduction reaction electrocatalysts for electrochemical energy applications. Electrochem. Energy Rev. 2019, 2, 518–538. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yu, F.; Wu, K.; Xu, G.; Wu, C.; Liu, H.-K.; Dou, S.-X. Recent progress on Fe-based single/dual-atom catalysts for Zn-air batteries. Small 2022, 18, 2106635. [Google Scholar] [CrossRef]
- Ma, L.; Zhu, G.; Wang, D.; Chen, H.; Lv, Y.; Zhang, Y.; He, X.; Pang, H. Emerging metal single atoms in electrocatalysts and batteries. Adv. Funct. Mater. 2020, 30, 2003870. [Google Scholar] [CrossRef]
- Wang, Y.; Su, H.; He, Y.; Li, L.; Zhu, S.; Shen, H.; Xie, P.; Fu, X.; Zhou, G.; Feng, C.; et al. Advanced electrocatalysts with single-metal-atom active sites. Chem. Rev. 2020, 120, 12217–12314. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, B.; Jiang, Y.; Ma, T.; Pan, H.; Sun, W. Single-atom electrocatalysts for multi-electron reduction of CO2. Small 2021, 17, 2101443. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, J.; Gao, X.; Chu, W.; Gao, G.; Wang, L.-W. Recent advances in single-atom electrocatalysts supported on two-dimensional materials for the oxygen evolution reaction. J. Mater. Chem. A 2021, 9, 9979–9999. [Google Scholar] [CrossRef]
- Zhu, C.; Fu, S.; Shi, Q.; Du, D.; Lin, Y. Single-atom electrocatalysts. Angew. Chem. Int. Ed. 2017, 56, 13944–13960. [Google Scholar] [CrossRef]
- Han, A.; Zhang, Z.; Yang, J.; Wang, D.; Li, Y. Carbon-supported single-atom catalysts for formic acid oxidation and oxygen reduction reactions. Small 2021, 17, 2004500. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cao, C.; Hao, J.; Qiu, H.; Xu, Y.; Zhu, H. Self-assembled one-dimensional carbon nitride architectures. Diam. Relat. Mater. 2006, 15, 1593–1600. [Google Scholar] [CrossRef]
- Wang, J.; Hao, D.; Ye, J.; Umezawa, N. Determination of crystal structure of graphitic carbon nitride: Ab initio evolutionary search and experimental validation. Chem. Mater. 2017, 29, 2694–2707. [Google Scholar] [CrossRef]
- Di Liberto, G.; Tosoni, S.; Pacchioni, G. Z-Scheme versus type-II junction in g-C3N4/TiO2 and g-C3N4/SrTiO3/TiO2 heterostructures. Catal. Sci. Technol. 2021, 11, 3589–3598. [Google Scholar] [CrossRef]
- Vilé, G.; Di Liberto, G.; Tosoni, S.; Sivo, A.; Ruta, V.; Nachtegaal, M.; Clark, A.H.; Agnoli, S.; Zou, Y.; Savateev, A.; et al. Azide-alkyne click chemistry over a heterogeneous copper-based single-atom catalyst. ACS Catal. 2022, 12, 2947–2958. [Google Scholar] [CrossRef]
- Barlocco, I.; Cipriano, L.A.; Di Liberto, G.; Pacchioni, G. Does the oxygen evolution reaction follow the classical OH *, O *, OOH * path on single atom catalysts? J. Catal. 2023, 417, 351–359. [Google Scholar] [CrossRef]
- Niu, H.; Zhang, Z.; Wang, X.; Wan, X.; Shao, C.; Guo, Y. Theoretical insights into the mechanism of selective nitrate-to-ammonia electroreduction on single-atom catalysts. Adv. Funct. Mater. 2021, 31, 2008533. [Google Scholar] [CrossRef]
- Niu, H.; Wang, X.; Shao, C.; Zhang, Z.; Guo, Y. Computational screening single-atom catalysts supported on g-CN for N2 reduction: High activity and selectivity. ACS Sustain. Chem. Eng. 2020, 8, 13749–13758. [Google Scholar] [CrossRef]
- Lv, X.; Wei, W.; Huang, B.; Dai, Y.; Frauenheim, T. High-throughput screening of synergistic transition metal dual-atom catalysts for efficient nitrogen fixation. Nano Lett. 2021, 21, 1871–1878. [Google Scholar] [CrossRef]
- Lv, X.; Wei, W.; Li, F.; Huang, B.; Dai, Y. Metal-free B@g-CN: Visible/infrared light-driven single atom photocatalyst enables spontaneous dinitrogen reduction to ammonia. Nano Lett. 2019, 19, 6391–6399. [Google Scholar] [CrossRef]
- Lv, X.; Wei, W.; Wang, H.; Huang, B.; Dai, Y. Holey graphitic carbon nitride (g-CN) supported bifunctional single atom electrocatalysts for highly efficient overall water splitting. Appl. Catal. B Environ. 2020, 264, 118521. [Google Scholar] [CrossRef]
- Wang, S.; Wei, W.; Lv, X.; Huang, B.; Dai, Y. W supported on g-CN manifests high activity and selectivity for N2 electroreduction to NH3. J. Mater. Chem. A 2020, 8, 1378–1385. [Google Scholar] [CrossRef]
- Besharat, F.; Ahmadpoor, F.; Nezafat, Z.; Nasrollahzadeh, M.; Manwar, N.R.; Fornasiero, P.; Gawande, M.B. Advances in carbon nitride-based materials and their electrocatalytic applications. ACS Catal. 2022, 12, 5605–5660. [Google Scholar] [CrossRef]
- Mahmood, J.; Lee, E.K.; Jung, M.; Shin, D.; Jeon, I.Y.; Jung, S.M.; Choi, H.J.; Seo, J.M.; Bea, S.Y.; Sohn, S.D.; et al. Nitrogenated holey two-dimensional structures. Nat. Commun. 2015, 6, 6486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, W.; Keith, J.A.; Anton, J.; Jacob, T. Theoretical elucidation of the competitive electro-oxidation mechanisms of formic acid on Pt(111). J. Am. Chem. Soc. 2010, 132, 18377–18385. [Google Scholar] [CrossRef]
- Gao, W.; Song, E.H.; Jiang, Q.; Jacob, T. Revealing the active intermediates in the oxidation of formic acid on Au and Pt(111). Chem.-Eur. J. 2014, 20, 11005–11012. [Google Scholar] [CrossRef] [PubMed]
- Karamad, M.; Hansen, H.A.; Rossmeisl, J.; Nørskov, J.K. Mechanistic pathway in the electrochemical reduction of CO2 on RuO2. ACS Catal. 2015, 5, 4075–4081. [Google Scholar] [CrossRef]
- Sakong, S.; Gross, A. The Importance of the Electrochemical environment in the electro-oxidation of methanol on Pt(111). ACS Catal. 2016, 6, 5575–5586. [Google Scholar] [CrossRef]
- Sui, L.; An, W.; Feng, Y.; Wang, Z.; Zhou, J.; Hur, S.H. Bimetallic Pd-based surface alloys promote electrochemical oxidation of formic acid: Mechanism, kinetics and descriptor. J. Power Sources 2020, 451, 227830. [Google Scholar] [CrossRef]
- Medford, A.J.; Vojvodic, A.; Hummelshøj, J.S.; Voss, J.; Abild-Pedersen, F.; Studt, F.; Bligaard, T.; Nilsson, A.; Nørskov, J.K. From the Sabatier principle to a predictive theory of transition-metal heterogeneous catalysis. J. Catal. 2015, 328, 36–42. [Google Scholar] [CrossRef]
- Hu, S.; Li, W.-X. Sabatier principle of metal-support interaction for design of ultrastable metal nanocatalysts. Science 2021, 374, 1360–1365. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Rodriguez, J.A. Catalysts for hydrogen evolution from the [NiFe] hydrogenase to the Ni2P(001) surface: The importance of ensemble effect. J. Am. Chem. Soc. 2005, 127, 14871–14878. [Google Scholar] [CrossRef] [PubMed]
- Delley, B. Hardness conserving semilocal pseudopotentials. Phys. Rev. B 2002, 66, 155125. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Govind, N.; Petersen, M.; Fitzgerald, G.; King-Smith, D.; Andzelm, J. A generalized synchronous transit method for transition state location. Comput. Mater. Sci. 2003, 28, 250–258. [Google Scholar] [CrossRef]
- Peterson, A.A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Nørskov, J.K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 2010, 3, 1311–1315. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Logadottir, A.; Nørskov, J.K. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 2005, 319, 178–184. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Klamt, A.; Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 1993, 2, 799–805. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qadeer, A.; Yang, M.; Liu, Y.; Cai, Q.; Zhao, J. Computational Study on the Catalytic Performance of Single-Atom Catalysts Anchored on g-CN for Electrochemical Oxidation of Formic Acid. Catalysts 2023, 13, 187. https://doi.org/10.3390/catal13010187
Qadeer A, Yang M, Liu Y, Cai Q, Zhao J. Computational Study on the Catalytic Performance of Single-Atom Catalysts Anchored on g-CN for Electrochemical Oxidation of Formic Acid. Catalysts. 2023; 13(1):187. https://doi.org/10.3390/catal13010187
Chicago/Turabian StyleQadeer, Abdul, Meiqi Yang, Yuejie Liu, Qinghai Cai, and Jingxiang Zhao. 2023. "Computational Study on the Catalytic Performance of Single-Atom Catalysts Anchored on g-CN for Electrochemical Oxidation of Formic Acid" Catalysts 13, no. 1: 187. https://doi.org/10.3390/catal13010187