Abstract
Pincer ligand supported RuII chloride complexes may be used for acetylene hydrochlorination as non-mercury molecular catalysts. Based on theoretical calculations, the catalytic mechanism and the interaction between catalysts and reactants has been evaluated, indicating that the (pincer)RuIICl2 platform supports electrophilic proton-ruthenation of C2H2. Energy decomposition studies further illustrate the electron-rich property of the RuII center, which can increase the negative charge of C2H2 via 4d-electron backdonation. Thus, the electrophilic reaction mechanism is favored due to lower energetic barriers. By improving the electron-donating ability of ligands, this lowering of energetic barriers can be enhanced. Therefore, non-mercury catalysts for acetylene hydrochlorination with milder reaction conditions and higher catalytic activity can be designed.
1. Introduction
Vinyl chloride monomer (VCM) is the major chemical intermediate leading to polyvinyl chloride (PVC), which is one of the most important polymers in use today with the production of over 40 million tons annually [1,2]. For industrial VCM production, acetylene hydrochlorination has been widely applied in countries where acetylene can be obtained economically from coal, using mercuric chloride (HgCl2) supported on activated carbon as the industrial heterogeneous catalyst [3,4]. However, the mercury-based catalyst has a number of serious problems, namely the facile volatility of mercuric chloride under the reaction temperature (170–180 °C), which results in mercury loss leading to deactivation of the catalyst and environmental poisoning. Moreover, the use of mercury-based catalysts will be forbidden by the Minamata Convention [5]. With the concerns associated with the severe mercury loss from the industrial units, it is necessary to produce a new, non-mercury heterogeneous catalyst for the acetylene hydrochlorination process. Over the past few decades, the catalytic performance of various metal chlorides for use in this reaction has been systematically investigated [6,7].
The Hutchings group have conducted pioneer work showing the possibility of a gold-based non-mercury commercial catalyst. They showed that the initial activity in catalytic acetylene hydrochlorination may be related to the standard reduction potential of metal cations [7]. It is observed that the potential of a gold-based catalyst leads to a well-performing catalyst. Up to now, their industrial gold-based catalyst has shown good performance in activity and stability [3,8]. However, as gold is one of the noble metals, there is interest in finding low-cost metals which have similar performance. Late-transition metals, such as group VIII, IB, and IIB metal compounds, are also expected to be effective catalysts. Among these compounds, recent studies have mainly focused on copper, ruthenium, and palladium as a single-metal catalyst [9,10].
Unlike the mercury loss that mainly leads to catalyst deactivation, non-mercury catalysts may undergo coke deposition or metal species agglutination (sintering) causing activity loss. The coke deposition phenomenon of non-mercury catalysts is thought to cause the acetylene polymerization-dehydrogenation process, with more acidic catalysts leading to a greater effect. On the other hand, the active metal ions may be reduced by acetylene and aggregate at the reaction temperature [3,11]. According to the existing research, there are two main ways to improve catalytic performance, and avoid sintering and coke deposition. Promoting the conversion from acetylene to VCM may effectively prevent the self-polymerization of acetylene. This is generally achieved by enhancing the adsorption and activation of hydrogen chloride on heterogeneous catalysts. The presence of support ligands is crucial for improving the stability of the metal ions and avoiding side reactions on the metal center. In addition, careful construction of the ligand support may improve the metal dispersion and prevent sintering [12].
The development of non-mercury catalysts for acetylene hydrochlorination is thus focused on engineering the supporting ligands through additive-based modification and generation of novel metal complexes. The support designing and modification aims to increase the electronic density, which is beneficial for the stronger metal-support interaction and the adsorption of HCl [12,13]. Importantly, if the additive or support is able to bind to the metal center as a ligand, the metal complex will be generated as a molecular system and the species can be highly dispersed on the support. For transition metal chlorides, the excess chloride anions were confirmed to be able to coordinate with metal centers, which may tune the catalytic performance for acetylene hydrochlorination, such as [PtCl4]2−, [PtCl6]2−, [PdCl4]2−, [AuCl4]−, [RuCl4]−, [CuClx]1−x (x = 2, 3 or 4) [14,15,16,17,18,19,20,21,22]. In addition to the Cl− anion, some organic and inorganic ligands have received increasing attention recently. S2O32− and SC(NH2)2, etc. are used for Au [3,4], N-alkylpyrrolidone (amides), amines, pyridines are used for Cu or Ru, where Ru allows wider applicability to neutral nitrogen-containing ligands, including azoles, guanidines, and N-heterocyclic carbenes [23,24,25,26,27,28,29,30,31,32]. The formation of these metal complexs allow for the depolymerization of the metal center from the metal chlorides cluster, and if the electronic effects and steric hindrance of the ligand are suitable, molecular complexes may be obtained allowing another avenue of exploration of “single-site” catalysts [33].
For this study, the ruthenium-based catalyst is investigated for its potential for high-performance-catalysis suggested by Kang et al. [34]. Beyond ruthenium trichloride RuCl3, the organometallic ruthenium catalysts may have a lower Lewis acidity that benefits acetylene hydrochlorination. Dérien et al. have reported the homogeneous hydrochlorination of substituted alkynes catalyzed by a RuII-complex supported by the pentamethylcyclopentadiene anion (Cp*) and phosphorus ligands at room temperature, showing a highly efficient method with wide applications in organic synthesis [35]. The mechanistic studies suggest that the RuII center shows π-backdonation to alkynes, which allows the electrophilic addition by HCl with a low energy barrier [36]. Alternatively, we would like to consider a neutral ligand here, rather than an ionic ligand like cyclopentadiene anion, to discuss the enhancement of ruthenium chloride catalysts. Phosphorus ligands are well matched for the group VIII metals, and the additional chelation effect enhances the stability of the complexes. In this work, the PNP pincer ligands PNPL1 and PNPL2 were considered for the generation of RuII-chloride complexes (Scheme 1).
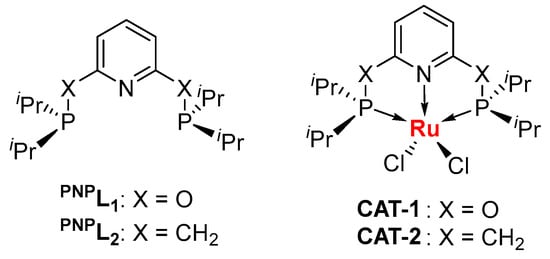
Scheme 1.
The pincer ligands used to modify the ruthenium(II) chloride as the catalysts for acety-lene hydrochlorination. iPr = CH(CH3)2 (isopropyl).
2. Results and Discussions
Complex (PNPL1)RuCl2 (CAT−1) and (PNPL2)RuCl2 (CAT−2) were selected for investigating the catalytic mechanism of acetylene hydrochlorination. The mechanism was discussed in Section 2.1 The interaction between catalysts and reactants and Section 2.2 The Gibbs free energy of intermediates and transition states.
2.1. The Interaction between Catalysts and Reactants
The optimized structures of the complex (PNPL1)RuCl2 (CAT−1) and (PNPL2)RuCl2 (CAT−2) show a quadrilateral cone coordination with a vacant ligand site as the active site. The most stable structures of the complexes interacting with C2H2 or HCl separately are presented in Figure 1. The adsorption Gibbs free energy (ΔGads) of HCl on CAT−1 and CAT−2 are +0.90 and −3.64 kcal/mol, respectively. While ΔGads of C2H2 adsorbed on CAT−1 and CAT−2 are −11.53 and −9.89 kcal/mol, respectively (Table 1). Thus, the adsorption of C2H2 by catalysts is more favored than HCl, which is consistent with previous studies [3,12,18,19]. The HCl adsorption capacity of CAT−2 is larger than CAT−1 due to its more negative ΔGads. Note that the basis-set superposition error (BSSE) under the counterpoise treatment should be included for calculating the adsorp−tion Gibbs free energy between catalysts and reactants. In the interacting structure, an HCl molecule coordinates its Cl atom to the RuII center. Meanwhile, the H atom of HCl is attracted by the Cl− anion of the catalyst via hydrogen bonding. These effects lead to the significant increase of the H-Cl bond length (~1.38 Å for HCl on CAT−1 and ~1.37 Å for HCl on CAT−2) compared to a free HCl (~1.29 Å). However, the Hirshfeld charge of HCl on CAT−1 and CAT−2 are −0.03 and −0.015, respectively. It implies that the catalyst does not donate electrons to activate HCl. Therefore, the metal-Cl (Cl in HCl) coordination is counterbalanced by the complex-H (H in HCl) coordination. This is consistent with the independent gradient model (IGM) analysis results (Figure 2), that the strong attraction was located in the blue area.
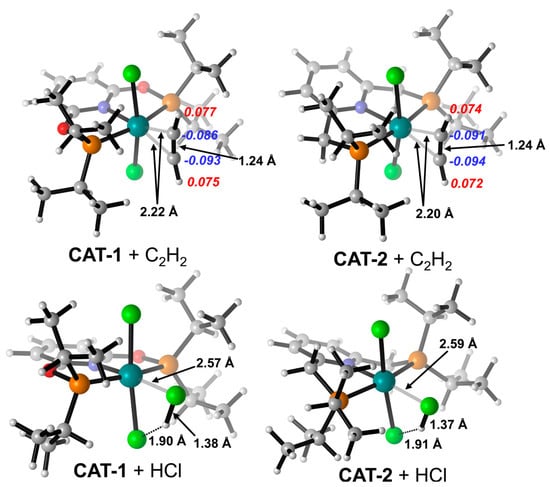
Figure 1.
Optimized structures for HCl and C2H2 adsorbed by CAT−1 and CAT−2. Key bond lengths (black numbers) and Hirshfeld charges of C2H2 (red and blue numbers in Italic font) were labeled. Ball colors: green (Cl), gray (C), white (H), blue (N), red (O), orange (P), cyan (Ru).

Table 1.
The adsorption Gibbs free energy (ΔGads) of HCl and C2H2 adsorbed on catalyst CAT−1 and CAT−2.
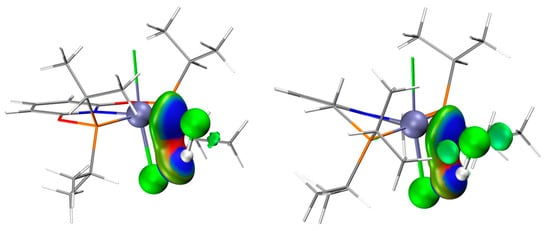
Figure 2.
IGM analysis of HCl adsorbed by CAT−1 (left) and CAT−2 (right). Ball colors: green (Cl), gray (C), white (H), blue (N), red (O), brown (P), cyan (Ru). Blue surface represents larger gradient value.
As for C2H2 absorbed on CAT−1 and CAT−2, the attraction is dominated by π-interactions. C2H2 can coordinate its π-electrons to the metal center as a Lewis acid. Consequently, the electron density on C2H2 is reduced and C2H2 is more favored for nucleophilic attacks [37]. Indeed, C2H2 can be activated by CAT−1 and CAT−2 as its C-C bond is elongated slightly from 1.21 to 1.24 Å. It is noticeable that the Hirshfeld charge of C2H2 is remarkably negative as −0.027 for CAT−1 and −0.039 for CAT−2, respectively. The negative charges are populated on the C atoms of C2H2. This implies that the backdonation from RuII to C2H2 is superior in magnitude compared with the π-coordination of C2H2 to RuII. This will be further discussed in the following reaction mechanism section.
For the co-adsorption of C2H2 and HCl, possible interactions are discussed here, which are directly related to the subsequent reaction mechanism. We have found three stable adsorption minima in total, which correspond to the sequential adsorption of C2H2 and HCl. CAT−1 was taken as an example to illustrate. Figure 3 shows optimized structures and interacting energies with or without BSSE correction. The energies of the structures in the mechanism discussion below, including INT1, INT2 and INT3 in Figure 3, did not include BSSE. When C2H2 binds to the RuII center first, there are two adsorption sites for HCl, both of which are achieved by attracting the H atom of HCl via hydrogen bonding. Not only the Cl− anion of CAT−1 (in INT1, ΔG = −20.4 kcal/mol and ΔGads = −12.8 kcal/mol), but also the π-bond of C2H2 (in INT2, ΔG = −15.0 kcal/mol and ΔGads = −5.6 kcal/mol) is able to act as the hydrogen bond acceptor. If HCl binds to the metal center first, C2H2 can only find one stable adsorption site. Meanwhile, the π-bond of C2H2 interacts with the H atom of HCl via hydrogen bonding (INT3, ΔG = −4.7 kcal/mol and ΔGads = 2.6 kcal/mol), which could be also visualized by IGM analysis (Figure 3b). Interestingly, the H atom of C2H2 can form another weak hydrogen bond with the Cl atom of HCl in INT1. In the co-adsorption, the structures contain C2H2−RuII coordination is more favorable in energy, which is consistent with the larger ΔGads of individual C2H2. As for CAT−2, a similar conclusion has been found.
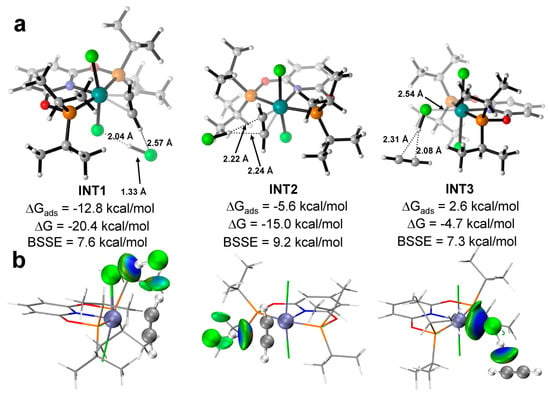
Figure 3.
(a) The optimized structure of the co-adsorption state as INT1, INT2 and INT3 with the key bond length (black numbers); (b) The IGM analysis of INT1, INT2 and INT3. Ball colors: green (Cl), gray (C), white (H), blue (N), red (O), brown (P), cyan (Ru).
2.2. The Gibbs Free Energy of Intermediates and Transition States
The pathway−1 of CAT−1 catalyzed acetylene hydrochlorination is shown in Figure 4. Firstly, we choose INT1 as the initial adsorption intermediate to investigate possible reaction pathways as INT1 is the dominating intermediate due to its strongest adsportion. In general, the activation by metal complexes of unsaturated hydrocarbons with respect to the nucleophilic attack has been theoretically revealed by Prof. Roald Hoffmann [38]. Indeed, we have found the chlororuthenation on the C2H2 adsorbed by CAT−1. The free energy barrier (ΔG≠) is quite high from INT1 to TS1, up to 43.3 kcal/mol (42.4 kcal/mol for CAT−2). This is significantly higher than the results reported [19,27,28,29,38]. Then, the H atom of the original HCl bonds with a Cl− anion of the catalyst to form another HCl molecule. It induced an oxidation addition on the RuII center. The H−Cl covalent bond was broken and a ruthenium-hydride (Ru-H) intermediate was formed. With no energy barrier, the −CH=CHCl group can be rapidly hydrogenated by Ru−H species to form INT6, where the VCM molecule is adsorbed by CAT−1. Finally, VCM is replaced by another C2H2 and HCl pair to form INT1 for the next catalytic cycle. The energy of INT1 (next catalytic cycle) is lower than INT6 (−45.9 kcal/mol vs. −38.7 kcal/mol), which is beneficial for releasing VCM. Note that VCM cannot desorb simultaneously due to its remarkably strong adsorption. Returning CAT−1 will cause an energy increase of 13.2 kcal/mol for the next catalytic cycle. Additionally, we have also considered the chlorination of C2H2 to generate cis-1,2-dichloroethylene (Z-C2H2Cl2). As expected, this side reaction is almost impossible for the very high energy barrier (ΔG≠ = 58.2 kcal/mol). This reflects that the metal center is extremely difficult to be reduced. Therefore, we expect it to well avoid the reduction of RuII and the aggregation of metal particles that causes the deactivation of catalysts in acetylene hydrochlorination.
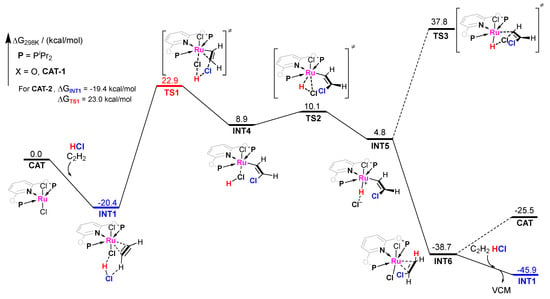
Figure 4.
Gibbs free energy profile for molecular complex CAT−1 catalyzed acetylene hydrochlorination mechanism pathway−1.
The pathway−2 of CAT−1 catalyzed acetylene hydrochlorination mechanism is shown in Figure 5. Since the energy barrier of pathway−1 is notably high, we continued to consider INT2 as a slightly less stable adsorption. When C2H2 is attached to CAT−1, the π-electrons of C2H2 can act as the hydrogen bond acceptor to interact with HCl. Unlike the traditional nucleophilic addition of unsaturated hydrocarbons activated by late transition metal complexes, the proton of HCl adds to the triple bond of C2H2 electrophilically via TS4 as the transition state, with an energy barrier of 18.2 kcal/mol. Compared with TS1 in Figure 4, TS4 is the more representative transition state for the electrophilic addition on adsorbed C2H2. The Hirshfeld charge of TS4 can support the electrophilic properties of this step (Figure 5). Interestingly, the potential energy surface of this step is so flat that the energy of intermediate INT7 is very close to that of TS4. In INT7, the C2H2 skeleton maintains the three-membered ring connection with the RuII center. However, the two C atoms in C2H2 show a clear difference. The distances of Ru and Cα and Cβ are 1.95 and 2.22 Å, respectively. So a more recognizable C−Ru bond is indicated for Cα of C2H2, even Cβ exhibits a similar coordination mode to the adsorbed C2H2 initially. The outer-sphere of INT7 is the Cl− anion, which could transfer the negative charge to Cα to form VCM via TS5 with ΔG = 11.9 kcal/mol. Therefore, ΔG≠ of the whole pathway−2 is 26.9 kcal/mol. Compared with pathway−1 (ΔG≠ = 43.3 kcal/mol), pathway−2 is more energetically favorable. When CAT−2 is used as the catalyst, ΔG≠ of the whole CAT−2 catalyzed pathway−2 is much lower, only 17.1 kcal/mol, which illustrates that CAT−2-catalyzed electrophilic addition activity performs much better than CAT−1. The difference between PNPL1 and PNPL2 is the part attached to the P atom (O atom or −CH2− group). Considering that the electronegativity of O atom is significantly greater than that of hydrocarbon groups, the electron donating ability of PNPL2 is stronger than that of PNPL1. So, this electrophilic proton-ruthenation mechanism is influenced by the electron density of the RuII center, which can be promoted by the ligands with stronger electron donating ability.
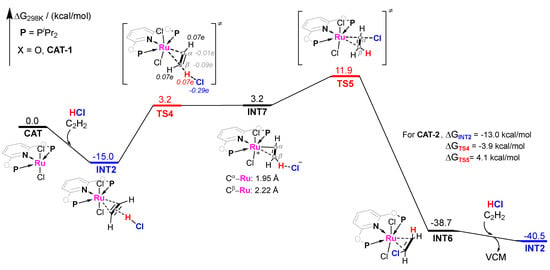
Figure 5.
Gibbs free energy profile for molecular complex CAT−1 catalyzed acetylene hydrochlorination mechanism pathway−2. The Hirshfeld charge of important parts was shown in Italic fonts.
The viewpoint in pathway−2 may be controversial with the general catalytic mechanism for acetylene hydrochlorination reported in several previous works [19,27,28,29,38]. However, supporting evidence also exists [39]. In order to give an in-depth understanding, we carried out an energy decomposition analysis (EDA) based on natural orbitals for chemical valence (NOCV) for C2H2 adsorbed on the catalyst. In Figure 6, the energy of orbital interaction between C2H2 and RuII can be divided into two parts for CAT−1: the σ-donation from C2H2 to RuII (−16.8 kcal/mol) and the π-backdonation from RuII to C2H2 (−21.3 kcal/mol). It indicates that the backdonation effect is stronger than the coordination effect, which causes more negative charges on C2H2 and illustrates a nucleophilic activation. In the total electron deformation density mapping, there is an increased electron density on the backside where C2H2 coordinates to RuII. Thus, C2H2 is more susceptible to acidic reagents, e.g., protonic acids, allowing an electrophilic addition mechanism. Additionally, when CAT−2 is used, the σ-donation and the π-backdonation contribute −38.1 and −43.8 kcal/mol to the orbital interaction energy, respectively, revealing the higher activity predicted by a lower energy barrier as studied above.
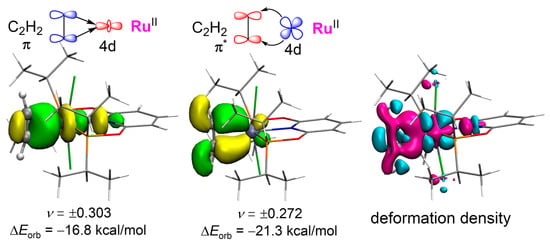
Figure 6.
Shape of the charge NOCV orbitals (isovalue = 0.05 a.u.) associated with the orbital interactions ΔEorb and the total electron deformation density for C2H2 adsorbed by CAT−1 (isovalue = 0.0025 a.u.). The eigenvalues (υ) of the fragment orbitals give the size of the charge migration. The color code for the charge transfer is pink → cyan.
Moreover, the pathway−3 of CAT−1 catalyzed acetylene hydrochlorination mechanism is shown in Figure 7. It shows the mechanism started at a relatively unstable adsorption state. A synergistic addition on C2H2 was approved in pathway-3. The H atom of HCl and the Cl− anion of the catalyst adds to the carbon-carbon triple bond simultaneously, which directly leads to the formation of a VCM molecule with ΔG≠ of 12.9 kcal/mol. Although a lower energy barrier compared with pathway-2 (ΔG≠ = 26.9 kcal/mol) emerged. The adsorption state is not stable enough. INT8 (ΔG = −20.8 kcal/mol) is prone to adjust its VCM adsorption structure to form a more stable intermediate INT6 (ΔG = −38.7 kcal/mol), rather than the VCM desorption under HCl and C2H2 (−38.7 kcal/mol vs. −30.2 kcal/mol). Similar conclusions were obtained for CAT−2 (ΔG≠ = 14.7 kcal/mol for pathway-3).
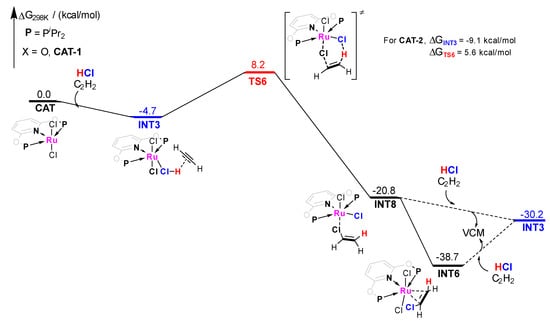
Figure 7.
Gibbs free energy profile for molecular complex CAT−1 catalyzed acetylene hydrochlorination mechanism pathway−3.
To further interpret the mechanistic preference beyond thermodynamic calculations, chemical kinetic evaluations are reported briefly. Figure 8 shows reaction rate constants versus the reaction temperature profile. All the possible intermediates and pathways were considered, including the formation of cis-1,2-dichloroethylene (Z-C2H2Cl2) as the side reaction. For CAT−1, the rate constant of pathway−3 is the largest despite the relatively unstable adsorption state, indicating that the acetylene hydrochlorination catalyzed by CAT−1 mainly occurs through the synergistic addition of acetylene. The rate constant of pathway−2 is more than two orders of magnitude lower than that of pathway−3. Due to the high energy barrier, the rate constant of pathway-1 is lower, while the rate of side reaction is so slow that it hardly occurs. Compared with CAT−1, rate constants of pathway−2 and pathway−3 catalyzed by CAT−2 are close. Pathway−2, which represents the electrophilic proton-metalation mechanism, plays a dominant role. Additionally, the maximum rate constant in Figure 8b is ~10 times that in Figure 8a. So, the RuII center supported by the stronger electron-donating ligand is the more active catalytic center, and more conducive to the electrophilic proton-metalation mechanism of acetylene hydrochlorination. In summary, we clarified several possible reaction mechanisms based on the thermodynamic calculation., Further chemical kinetic studies evaluated the dominant pathway.
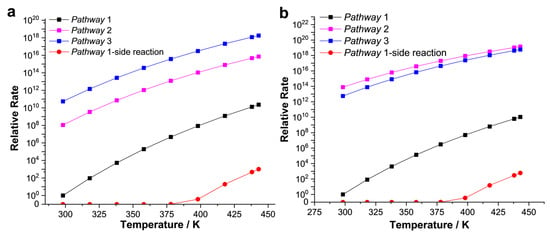
Figure 8.
Chemical kinetics studies for catalytic system CAT−1 (a) and CAT−2 (b). Reaction rate constants versus the reaction temperature were carried out via TST calculation. The relative rate unit of CAT−1 and CAT−2 are 5.3 × 10−30 s−1 and 2.3 × 10−29 s−1, respectively.
3. Conclusions
Above all, we have theoretically studied the PNP pincer ligand-supported RuII platform used for catalyzing acetylene hydrochlorination. Unlike the traditional nucleophilic chloro-metalation of C2H2, our study shows that the hydrochlorination of C2H2 is achieved via the electrophilic proton-metalation or synergistic addition when the metal center of the catalyst is electron-rich. As a late transition metal, ruthenium in +2 oxidation state has abundant 4d electrons to backdonate to the π-acidity ligand such as unsaturated hydrocarbon C2H2. When using strong σ-donors as ligands, the adsorbed C2H2 can be nucleophilicity activated, thus favoring the electrophilic pathway for its hydrochlorination. Many studies have shown that the promotion of acetylene hydrochlorination catalysts requires electron-rich modification to enhance the adsorption and activation of HCl. Because this can increase the concentration of Cl− in the system, which is conducive to the nucleophilic addition of adsorbed C2H2 [12,17,19,20,21,24]. In this work, we provide a new perspective to design ruthenium complex catalysts with higher potential activity, instead of changing the concentration to affect the chemical equilibrium but to establish different reaction mechanisms with more advantages in energy. Therefore, it is possible to study acetylene hydrochlorination under mild conditions or homogenously from our perspective, which can effectively avoid sintering and carbon deposition in industrial non-mercury heterogeneous catalysts.
4. Materials and Methods
General Methods: Geometry optimizations, frequency calculations, and thermal corrections to the free energy (from 298 to 443 K) were performed at B3LYP-D3(BJ) level for all the intermediates and transition states. The exchange-correlation functional is B3LYP for all calculations. D3(BJ) stands for the dispersion correction of Grimme’s dispersion method with Becke–Johnson damping. Def2-SVP basis set was assigned to short-period elements and MDF10 was used for ruthenium center [40,41,42,43,44,45]. Counterpoise corrections were considered for basis-set superposition errors (BSSE) in adsorption Gibbs free energy (ΔGads) calculations [46]. Since the fragmentation method is not unique, BSSE correction is only used on ΔGads. All calculations were performed using the Gaussian 09 package [47]. The computed structures were illustrated using CYL View [48].
To visualize the secondary interactions between catalyst and reactants, the independent gradient model (IGM) raised by Hénon group was used based on the optimized structures [49]. This method was implemented by Multiwfn, and the figure was presented by VMD [50,51].
The interaction between different species can be analyzed by means of an energy decomposition analysis (EDA) together with the natural orbitals for chemical valence (NOCV) method [52,53]. For this analysis, the intrinsic interaction energy (ΔΕint) between two fragments can be divided into for energy components (Equation (1)).
where the ΔEelstat, ΔEPauli, ΔEorb and ΔEdisp represent the electrostatic interaction, the Pauli repulsion, the orbital mixing/charge transfer, and the dispersion effect between two fragments, respectively. Importantly, the orbital term ΔEorb can be further decomposed into contributions from each irreducible representation of the point group of the interacting system (Equation (2)).
ΔΕint = ΔEelstat + ΔEPauli + ΔEorb + ΔEdisp
The combination of the EDA with NOCV enables the partition of the total orbital interactions into pairwise contributions of the orbital interactions which is very vital to get a complete picture of the chemical interaction. The charge deformation Δρk(r), resulting from the mixing of the orbital pairs ψk(r) and ψ-k(r) of the interacting fragments presents the amount and the shape of the charge flow due to the orbital interactions (Equation (3)), and the associated energy term ΔEorb provides with the size of stabilizing orbital energy originated from such interaction (Equation (4)).
The EDA-NOCV analysis was performed by ORCA 5.0.2 [54] under the same level as the general calculation, where the basis set was transformed by using the fch2mkl module in the MOKIT package [55].
The reaction rate constants (k) of all discussed reactions were calculated using transition state theory (TST) [56,57].
where h, kB and R are the Planck, Boltzmann, and ideal gas constants, respectively. T represents the reaction temperature. ΔG≠ is the activation free energy. The relative rate constant was used in kinetics studies of this work.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal13010031/s1, Figure S1. The Gibbs free energy profile for molecular complex CAT−2 catalyzed acetylene hydro-chlorination mechanism pathway-1; Figure S2. The Gibbs free energy profile for molecular complex CAT−1 catalyzed acetylene hydrochlorination mechanism pathway-2; Figure S3. The Gibbs free energy profile for molecular complex CAT−2 catalyzed acetylene hydrochlorination mechanism pathway-3; The xyz coordinates for intermediates and transition states (unit: Angstrom).
Author Contributions
Conceptualization, B.W.; methodology, X.W. and Y.L.; validation, X.W. and Y.L.; formal analysis, J.Z.; investigation, X.W.; resources, T.W.; data curation, X.W.; writing—original draft preparation, X.W.; writing—review and editing, B.W. and T.W.; visualization, J.Z.; supervision, B.W. and T.W.; project administration, F.W. and X.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no funding.
Data Availability Statement
The computationally optimized structures are available in the Supplementary Materials.
Acknowledgments
We gratefully thank Ryan A. Beck for providing language editing helps.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Saeki, Y.; Emura, T. Technical progresses for PVC production. Prog. Polym. Sci. 2002, 27, 2055–2131. [Google Scholar] [CrossRef]
- Lin, R.; Amrute, A.P.; Pérez-Ramírez, J. Halogen-Mediated Conversion of Hydrocarbons to Commodities. Chem. Rev. 2017, 117, 4182. [Google Scholar] [CrossRef] [PubMed]
- Johnston, P.; Carthey, N.; Hutchings, G.J. Discovery, Development, and Commercialization of Gold Catalysts for Acetylene Hydrochlorination. J. Am. Chem. Soc. 2015, 137, 14548–14557. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.J.; Miedziak, P.J.; Brett, G.L.; Hutchings, G.J. Vinyl chloride monomer production catalyzed by gold: A review. Chin. J. Catal. 2016, 37, 1600–1607. [Google Scholar] [CrossRef]
- Minamata Convention on Mercury. Available online: https://www.mercuryconvention.org/ (accessed on 24 September 2017).
- Kiyonori, S. The vapor-phase hidrochlorination of acetylene over metal chlorides supported on activated carbon. Chem. Lett. 1975, 4, 219–220. [Google Scholar]
- Hutchings, G.J. Vapor phase hydrochlorination of acetylene: Correlation of catalytic activity of supported metal chloride catalysts. J. Catal. 1985, 96, 292–295. [Google Scholar] [CrossRef]
- Malta, G.; Freakley, S.J.; Kondrat, S.A.; Hutchings, G.J. Acetylene hydrochlorination using Au/carbon: A journey towards single site catalysis. Chem. Commun. 2017, 53, 11733. [Google Scholar] [CrossRef]
- Zhu, M.; Wang, Q.; Chen, K.; Wang, Y.; Huang, C.; Dai, H.; Yu, F.; Kang, L.; Dai, B. Development of a Heterogeneous Non-Mercury Catalyst for Acetylene Hydrochlorination. ACS Catal. 2015, 5, 5306–5316. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, N.; Li, W.; Dai, B. Progress on cleaner production of vinyl chloride monomers over non-mercury catalysts. Front. Chem. Sci. Eng. 2011, 5, 514–520. [Google Scholar] [CrossRef]
- Conte, M.; Carley, A.F.; Heirene, C.; Willock, D.J.; Johnston, P.; Herzing, A.A.; Kiely, C.J.; Hutchings, G.J. Hydrochlorination of acetylene using a supported gold catalyst: A study of the reaction mechanism. J. Catal. 2007, 250, 231–239. [Google Scholar] [CrossRef]
- Zhong, J.; Xu, Y.; Liu, Z. Heterogeneous non-mercury catalysts for acetylene hydrochlorination: Progress, challenges, and opportunities. Green Chem. 2018, 20, 2412–2427. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, L.; Zhang, Y.; Zhang, L.; Zan, X. Progress and Challenges of Mercury-Free Catalysis for Acetylene Hydrochlorination. Catalysts 2020, 10, 1218. [Google Scholar] [CrossRef]
- Mitchenko, S.A.; Krasnyakova, T.V.; Mitchenko, R.S.; Korduban, A.N. Acetylene catalytic hydrochlorination over powder catalyst prepared by pre-milling of K2PtCl4 salt. J. Mol. Cat. A Chem. 2007, 275, 101–108. [Google Scholar] [CrossRef]
- Mitchenko, S.A.; Khomutov, E.V.; Shubin, A.A.; Shul’ga, Y.M. Catalytic hydrochlorination of acetylene by gaseous HCl on the surface of mechanically pre-activated K2PtCl6 salt. J. Mol. Cat. A Chem. 2004, 212, 345–352. [Google Scholar] [CrossRef]
- Mitchenko, S.A.; Krasnyakova, T.V.; Zhikharev, I.V. Catalytic hydrochlorination of acetylene on mechanochemically-activated K2PdCl4. Theor. Exp. Chem. 2008, 44, 316–319. [Google Scholar] [CrossRef]
- Zhao, J.; Gu, S.; Xu, X.; Zhang, T.; Yu, Y.; Di, X.; Ni, J.; Pan, Z.; Li, X. Supported ionic-liquid-phase-stabilized Au(iii) catalyst for acetylene hydrochlorination. Catal. Sci. Technol. 2016, 6, 3263–3270. [Google Scholar] [CrossRef]
- Li, H.; Wu, B.; Wang, J.; Wang, F.; Zhang, X.; Wang, G.; Li, H. Efficient and stable Ru(III)-choline chloride catalyst system with low Ru content for non-mercury acetylene hydrochlorination. Chin. J. Catal. 2018, 39, 1770–1781. [Google Scholar] [CrossRef]
- Ren, Y.; Wu, B.; Wang, F.; Li, H.; Lv, G.; Sun, M.; Zhang, X. Chlorocuprate(i) ionic liquid as an efficient and stable Cu-based catalyst for hydrochlorination of acetylene. Catal. Sci. Technol. 2019, 9, 2868–2878. [Google Scholar] [CrossRef]
- Zhai, Y.; Zhao, J.; Di, X.; Di, S.; Wang, B.; Yue, Y.; Sheng, G.; Lai, H.; Guo, L.; Wang, H.; et al. Carbon-supported perovskite-like CsCuCl3 nanoparticles: A highly active and cost-effective heterogeneous catalyst for the hydrochlorination of acetylene to vinyl chloride. Catal. Sci. Technol. 2018, 8, 2901–2908. [Google Scholar] [CrossRef]
- Wang, Y.; Nian, Y.; Zhang, J.; Li, W.; Han, Y. MOMTPPC improved Cu-based heterogeneous catalyst with high efficiency for acetylene hydrochlorination. Mol. Catal. 2019, 479, 110612. [Google Scholar] [CrossRef]
- Wang, B.; Jiang, Z.; Wang, T.; Tang, Q.; Yu, M.; Feng, T.; Tian, M.; Chang, R.; Yue, Y.; Pan, Z.; et al. Controllable Synthesis of Vacancy-Defect Cu Site and Its Catalysis for the Manufacture of Vinyl Chloride Monomer. ACS Catal. 2021, 11, 11016–11028. [Google Scholar] [CrossRef]
- Han, Y.; Wang, Y.; Wang, Y.; Hu, Y.; Nian, Y.; Li, W.; Zhang, J. Pyrrolidone ligand improved Cu-based catalysts with high performance for acetylene hydrochlorination. Appl. Organomet. Chem. 2021, 35, e6066. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, T.; Liu, Y.; Li, W.; Zhang, H.; Zhang, J. Phosphine-oxide organic ligand improved Cu-based catalyst for acetylene hydrochlorination. Appl. Catal. A Gen. 2022, 630, 118461. [Google Scholar] [CrossRef]
- Chen, X.; Liu, X.; Hu, R.; Xu, C. Complexation effect between Cu-based catalyst and DESMP in acetylene hydration. Mol. Catal. 2022, 531, 112659. [Google Scholar] [CrossRef]
- Wang, B.; Jin, C.; Shao, S.; Yue, Y.; Zhang, Y.; Wang, S.; Chang, R.; Zhang, H.; Zhao, J.; Li, X. Electron-deficient Cu site catalyzed acetylene hydrochlorination. Green Energy Environ. 2022; in press. [Google Scholar] [CrossRef]
- Li, J.; Zhang, H.; Cai, M.; Li, L.; Li, Y.; Zhao, R.; Zhang, J. Enhanced catalytic performance of activated carbon-supported ru-based catalysts for acetylene hydrochlorination by azole ligands. Appl. Catal. A Gen. 2020, 592, 117431. [Google Scholar] [CrossRef]
- Li, Y.; Wang, F.; Hu, J.; Sun, M.; Wang, J.; Zhang, X. A study on the rules of ligands in highly efficient Ru–amide/AC catalysts for acetylene hydrochlorination. Catal. Sci. Technol. 2021, 11, 7347–7358. [Google Scholar] [CrossRef]
- Li, Y.; Wang, F.; Wu, B.; Wang, X.; Sun, M.; Zhang, Z.; Zhang, X. Competing on the same stage: Ru-based catalysts modified by basic ligands and organic chlorine salts for acetylene hydrochlorination. Catal. Sci. Technol. 2022, 12, 5086–5096. [Google Scholar] [CrossRef]
- Cai, M.; Zhang, H.; Man, B.; Li, J.; Li, L.; Li, Y.; Xie, D.; Deng, R.; Zhang, J. Synthesis of a vinyl chloride monomer via acetylene hydrochlorination with a ruthenium-based N-heterocyclic carbene complex catalyst. Catal. Sci. Technol. 2020, 10, 3552–3560. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, M.; Dai, B. Effect of Phosphorus Ligand on Cu-Based Catalysts for Acetylene Hydrochlorination. ACS Sustain. Chem. Eng. 2019, 7, 6170–6177. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, C.; Qiao, X.; Guan, Q.; Li, W. Regulating the coordination environment of Ru single-atom catalysts and unravelling the reaction path of acetylene hydrochlorination. Green Energy Environ. 2022; in press. [Google Scholar] [CrossRef]
- Samantaray, M.K.; D’Elia, V.; Pump, E.; Falivene, L.; Harb, M.; Ould Chikh, S.; Cavallo, L.; Basset, J.-M. The Comparison between Single Atom Catalysis and Surface Organometallic Catalysis. Chem. Rev. 2020, 120, 734–813. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Kang, L.; Su, Y.; Zhang, S.; Dai, B. MClx (M = Hg, Au, Ru; x = 2, 3) catalyzed hydrochlorination of acetylene—A density functional theory study. Can. J. Chem. 2013, 91, 120–125. [Google Scholar] [CrossRef]
- Dérien, S.; Klein, H.; Bruneau, C. Selective Ruthenium-Catalyzed Hydrochlorination of Alkynes: One-Step Synthesis of Vinylchlorides. Angew. Chem. Int. Ed. 2015, 54, 12112–12115. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Jiang, J.-l.; Yu, H.-Z.; Fu, Y. Mechanistic Study on the Ruthenium-Catalyzed Terminal Alkyne Hydrochlorination. Organometallics 2017, 36, 523–529. [Google Scholar] [CrossRef]
- Eisenstein, O.; Hoffmann, R. Activation of a coordinated olefin toward nucleophilic attack. J. Am. Chem. Soc. 1980, 102, 6148–6149. [Google Scholar] [CrossRef]
- Zhao, C.; Zhang, X.; He, Z.; Guan, Q.; Li, W. Demystifying the mechanism of NMP ligands in promoting Cu-catalyzed acetylene hydrochlorination: Insights from a density functional theory study. Inorg. Chem. Front. 2020, 7, 3204–3216. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, T.; Jia, Y.; Zhang, J.; Han, Y. Single-Atom Ruthenium Catalytic Sites for Acetylene Hydrochlorination. J. Phys. Chem. Lett. 2021, 12, 7350–7356. [Google Scholar] [CrossRef]
- Becke, A.D. Becke’s three parameter hybrid method using the LYP correlation functional. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. Energy-adjusted abinitio pseudopotentials for the first row transition elements. J. Chem. Phys. 1987, 86, 866–872. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Rev. E.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Legault, C.Y. CYLview, 1.0b. Available online: http://www.cylview.org (accessed on 1 January 2009).
- Lefebvre, C.; Rubez, G.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Hénon, E. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 2017, 19, 17928–17936. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Michalak, A.; Mitoraj, M.; Ziegler, T. Bond Orbitals from Chemical Valence Theory. J. Phys. Chem. A 2008, 112, 1933–1939. [Google Scholar] [CrossRef]
- Mitoraj, M.P.; Michalak, A.; Ziegler, T. A Combined Charge and Energy Decomposition Scheme for Bond Analysis. J. Chem. Theory Comput. 2009, 5, 962–975. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Zou, J. MOKIT Program. Available online: https://gitlab.com/jxzou/mokit (accessed on 25 October 2022).
- Upadhyay, S.K. Chemical Kinetics and Reaction Dynamics; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar] [CrossRef]
- El-Nahas, A.M.; Mangood, A.H.; Takeuchi, H.; Taketsugu, T. Thermal Decomposition of 2-Butanol as a Potential Nonfossil Fuel: A Computational Study. J. Phys. Chem. A 2011, 115, 2837–2846. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).