

Chemoenzymatic Protocol for the Synthesis of Enantiopure β-Blocker (S)-Bisoprolol



Abstract
:1. Introduction
2. Results and Discussion
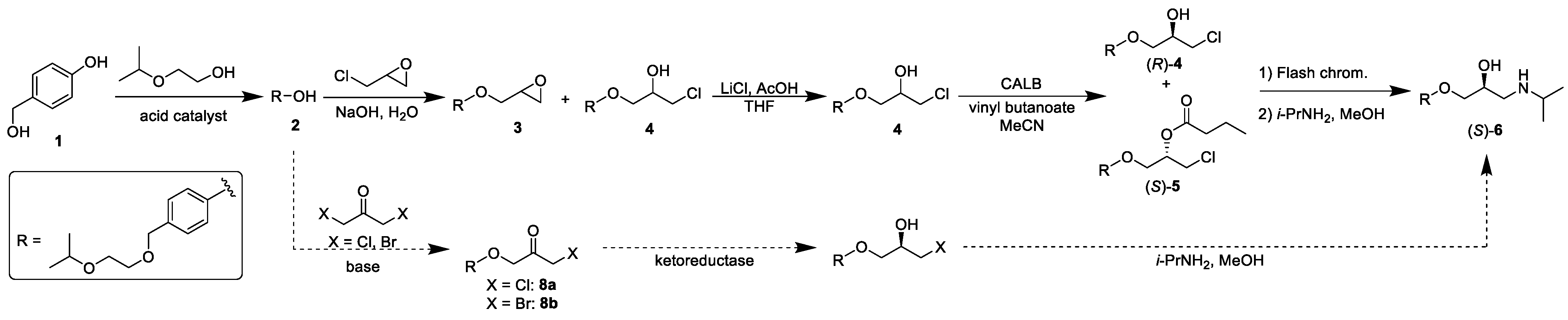
2.1. Synthesis of 4-((2-isopropoxyethoxy)methyl)phenol, 2
2.2. Synthesis of 1-chloro-3-(4-((2-isopropoxyethoxy)methyl)phenoxy)propan-2-ol, 4
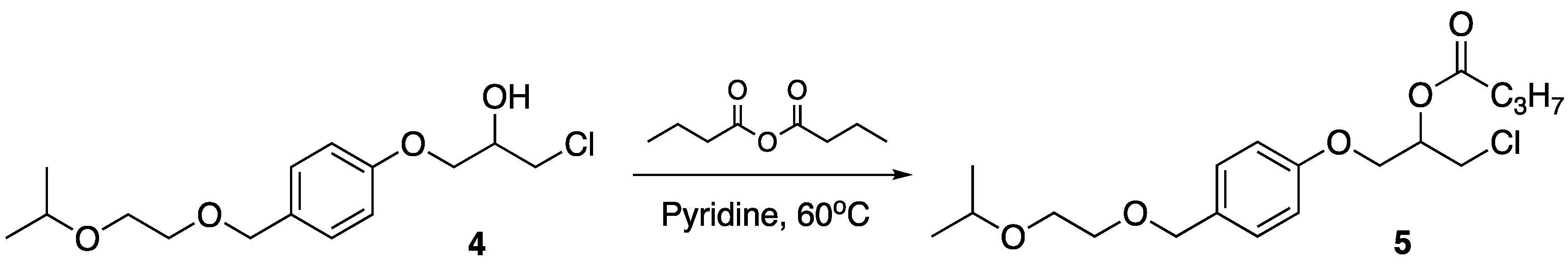
2.3. Synthesis of 1-chloro-3-(4-((2-isopropoxyethoxy)methyl)phenoxy)propan-2-yl butanoate, 5
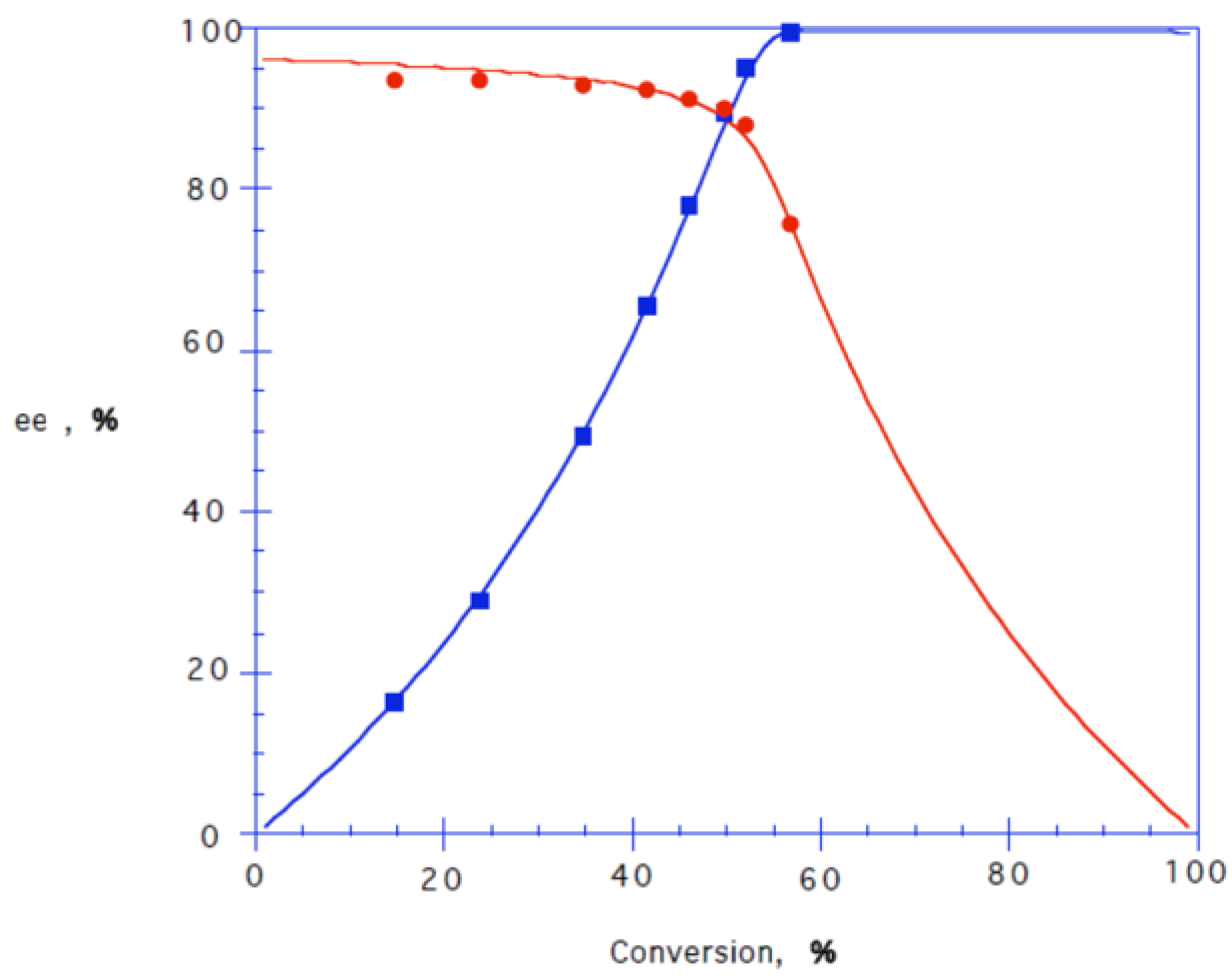
2.4. Synthesis of (R)-4 by CALB-Catalysed Kinetic Resolution of Chlorohydrin 4
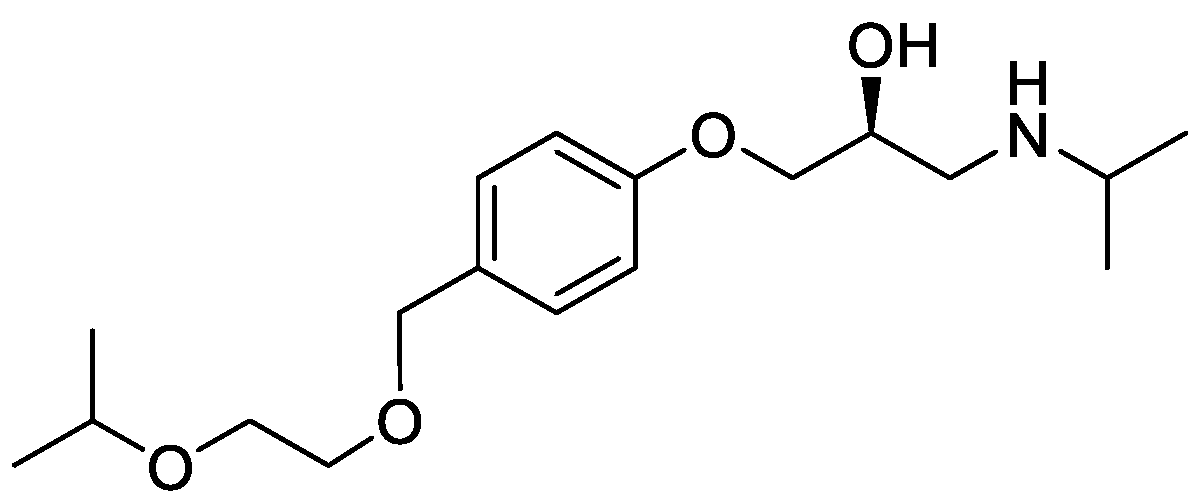
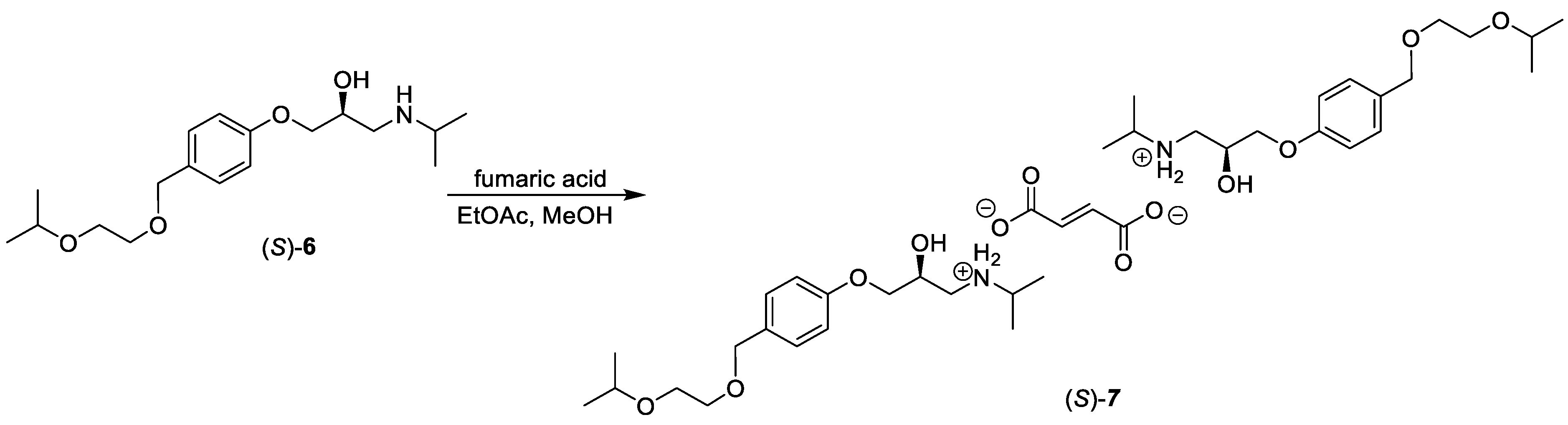
2.5. Synthesis of (S)-bisoprolol, (S)-6, and (S)-bisoprolol hemifumarate, (S)-7
2.6. Specific Rotation of (R)-4 and (S)-7
2.7. Unsuccessful Synthesis of 1-chloro-3-(4-((2-isopropoxyethoxy)methyl)phenoxy)propan-2-one, 8a, and 1-bromo-3-(4-((2-isopropoxyethoxy)methyl)phenoxy)propan-2-one, 8b
3. Materials and Methods
3.1. Chemicals and Solvents
3.2. TLC Analyses and Column Chromatography
3.3. Enzymes
3.4. Chiral HPLC Analyses
3.5. Optical Rotation
3.6. Absolute Configurations
3.7. NMR Analyses
3.8. Synthesis Protocols
3.8.1. Silica Sulfuric Acid Catalyst
3.8.2. 4-((2-Isopropoxyethoxy)methyl)phenol, 2
3.8.3. 1-Chloro-3-(4-((2-isopropoxyethoxy)methyl)phenoxy)propan-2-ol, 4
3.8.4. Synthesis of 1-chloro-3-(4-((2-isopropoxyethoxy)methyl)phenoxy)propan-2-yl butanoate, 5, from 1-chloro-3-(4-((2-isopropoxyethoxy)methyl)phenoxy)propan-2-ol, 4
3.8.5. Synthesis of chlorohydrin (R)-4 and (S)-5 by CALB catalysed kinetic resolution of 1-chloro-3-(4-((2-isopropoxyethoxy)methyl)phenoxy)propan-2-ol, 4
3.8.6. (S)-Bisoprolol, (S)-6
3.8.7. (S)-Bisoprolol Hemifumarate, (S)-7
3.8.8. Attempts to Synthesize 1-chloro-3-(4-((2-isopropoxyethoxy)methyl)phenoxy)propan-2-one (8a)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Most Prescribed Beta Blockers in the U.S. Available online: https://www.ampliz.com/resources/beta-blockers-in-the-usa/ (accessed on 14 November 2022).
- Baker, J.G. The selectivity of beta-adrenoceptor antagonists at the human beta1, beta2 and beta3 adrenoceptors. Br. J. Pharmacol. 2005, 144, 317–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, A.; Lanc, R.; Begg, E.; Robson, R.; Sia, L.; Dukart, G.; Desjardins, R.; Yacobi, A. Dose Proportionality of Bisoprolol Enantiomers in Humans After Oral Administration of the Racemate. J. Clin. Pharmacol. 1994, 34, 829–836. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Zebeta (Bisoprolol fumarate) Tablets. July 2007. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2007/019982s014lbl.pdf (accessed on 1 November 2022).
- Stoschitzky, K.; Lindner, W.; Zernig, G. Racemic beta-blockers—Fixed combinations of different drugs. J. Clin. Bas. Cardiol. 1998, 1, 15–19. [Google Scholar]
- Jun, Y.; Xian, Z.; Ying-ming, W.; Hong-wei, A.N. Research in synthesis of chiral drug bisoprolol fumarate. J. Hebei Univ. Sci. Technol. 2010, 31, 317–320. [Google Scholar]
- Fumin, Z. Preparation Method of Chiral Bisoprolol Fumarate. Chinese Patent CN112194587A, 8 January 2021. [Google Scholar]
- Soloviev, D.V.; Matarrese, M.; Moresco, R.M.; Todde, S.; Bonasera, T.A.; Sudati, F.; Simonelli, P.; Magni, F.; Colombo, D.; Carpinelli, A.; et al. Asymmetric synthesis and preliminary evaluation of (R)- and (S)-[11C]bisoprolol, a putative beta1-selective adrenoceptor radioligand. Neurochem. Int. 2001, 38, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Kitaori, K.; Furukawa, Y.; Yoshimoto, H.; Otera, J. CsF in organic synthesis. The first and convenient synthesis of enantiopure bisoprolol by use of glycidyl nosylate. Tetrahedron Lett. 1998, 39, 3173–3176. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Woodley, J.M. Role of Biocatalysis in Sustainable Chemistry. Chem. Rev. 2018, 118, 801–838. [Google Scholar] [CrossRef] [PubMed]
- Lund, I.T.; Bøckmann, P.L.; Jacobsen, E.E. Highly enantioselective CALB-catalyzed kinetic resolution of building blocks for β-blocker atenolol. Tetrahedron 2016, 72, 7288–7292. [Google Scholar] [CrossRef]
- Blindheim, F.H.; Hansen, M.B.; Evjen, S.; Zhu, W.; Jacobsen, E.E. Chemoenzymatic Synthesis of Synthons as Precursors for Enantiopure Clenbuterol and Other β2-Agonists. Catalysts 2018, 8, 516–528. [Google Scholar] [CrossRef] [Green Version]
- Gundersen, M.A.; Austli, G.B.; Løvland, S.S.; Hansen, M.B.; Rødseth, M.; Jacobsen, E.E. Lipase Catalyzed Synthesis of Enantiopure Precursors and Derivatives for β-Blockers Practolol, Pindolol and Carteolol. Catalysts 2021, 11, 503–518. [Google Scholar] [CrossRef]
- Verho, O.; Bäckvall, J.-E. Chemoenzymatic Dynamic Kinetic Resolution: A Powerful Tool for the Preparation of Enantiomerically Pure Alcohols and Amines. J. Am. Chem. Soc. 2015, 137, 3996–4009. [Google Scholar] [CrossRef] [PubMed]
- Morthala, R.R.; Gharpure, M.; Jagtap, A.; Mhaskar, M.; Krishnmurthy, D. An Improved Process for the Preparation of Bisoprolol and its Intermediate. International Patent WO2016135616A1, 1 September 2016. [Google Scholar]
- Anthonsen, H.W.; Hoff, B.H.; Anthonsen, T. Calculation of enantiomer ratio and equilibrium constants in biocatalytic ping-pong bi-bi resolutions. Tetrahedron Asymmetry 1996, 7, 2633–2638. [Google Scholar] [CrossRef]
- Banoth, L.; Banerjee, U.C. New chemical and chemo-enzymatic synthesis of (RS)-, (R)-, and (S)-esmolol. Arab. J. Chem. 2017, 10, S3603–S3613. [Google Scholar] [CrossRef] [Green Version]
- Jacobsen, E.E.; Hoff, B.H.; Anthonsen, T. Enantiopure derivatives of 1,2-alkanediols: Substrate requirements of lipase B from Candida antarctica. Chirality 2000, 12, 654–659. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ee (%) | Yield (%) | Specific Rotation | E-Value |
|---|---|---|---|---|
| (R)-4 | 99 | 44 | (c 1.0, MeOH) | 52 |
| (S)-7 | 96 | 99 | (c 1.0, MeOH) | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bocquin, L.; Jacobsen, E.E. Chemoenzymatic Protocol for the Synthesis of Enantiopure β-Blocker (S)-Bisoprolol. Catalysts 2023, 13, 54. https://doi.org/10.3390/catal13010054
Bocquin L, Jacobsen EE. Chemoenzymatic Protocol for the Synthesis of Enantiopure β-Blocker (S)-Bisoprolol. Catalysts. 2023; 13(1):54. https://doi.org/10.3390/catal13010054
Chicago/Turabian StyleBocquin, Lucas, and Elisabeth Egholm Jacobsen. 2023. "Chemoenzymatic Protocol for the Synthesis of Enantiopure β-Blocker (S)-Bisoprolol" Catalysts 13, no. 1: 54. https://doi.org/10.3390/catal13010054
APA StyleBocquin, L., & Jacobsen, E. E. (2023). Chemoenzymatic Protocol for the Synthesis of Enantiopure β-Blocker (S)-Bisoprolol. Catalysts, 13(1), 54. https://doi.org/10.3390/catal13010054