

Recent Advances in Catalyst Design for Carboxylation Using CO2 as the C1 Feedstock

 and
and
Abstract
: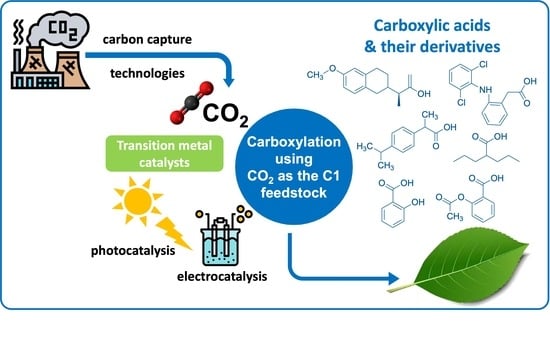
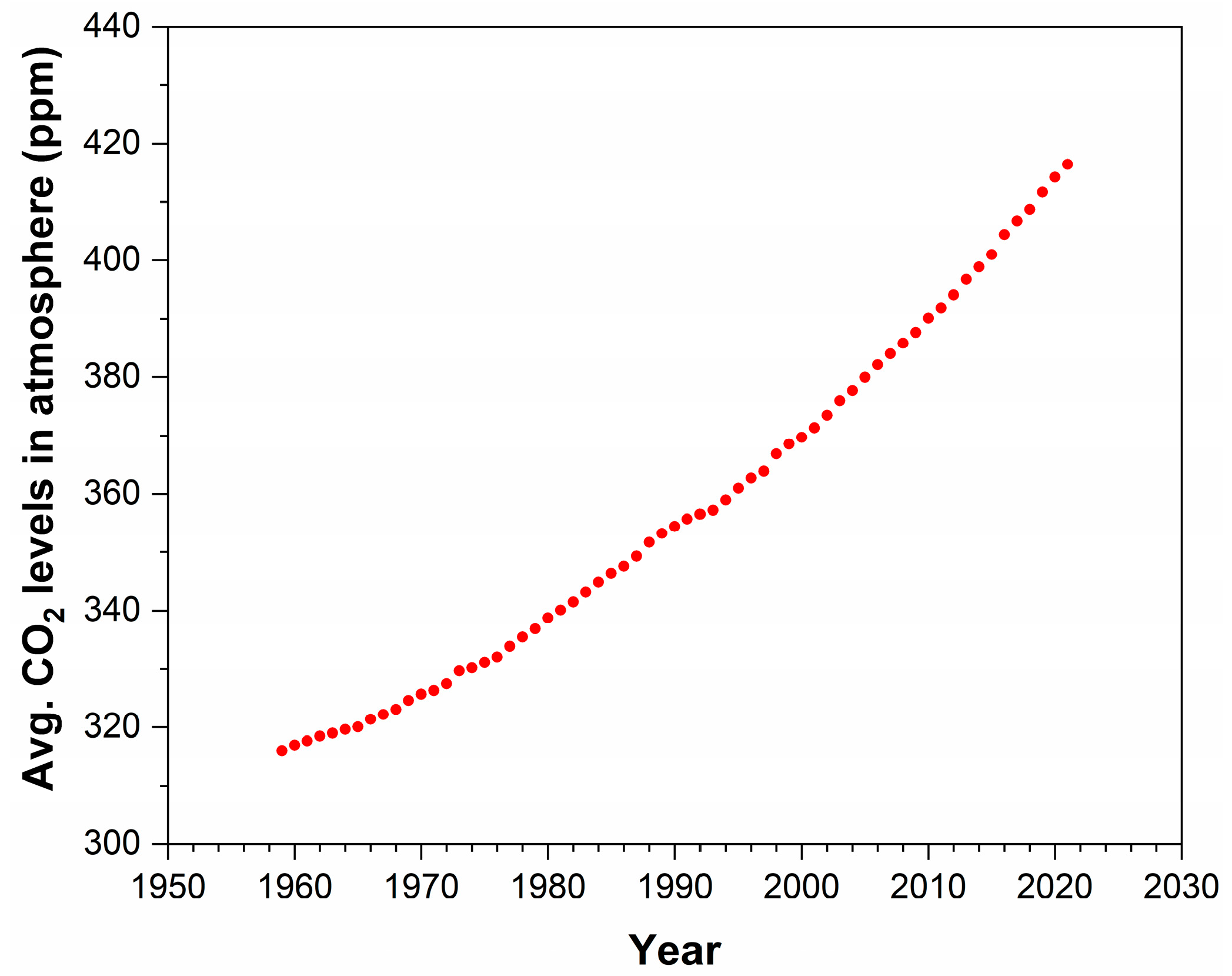
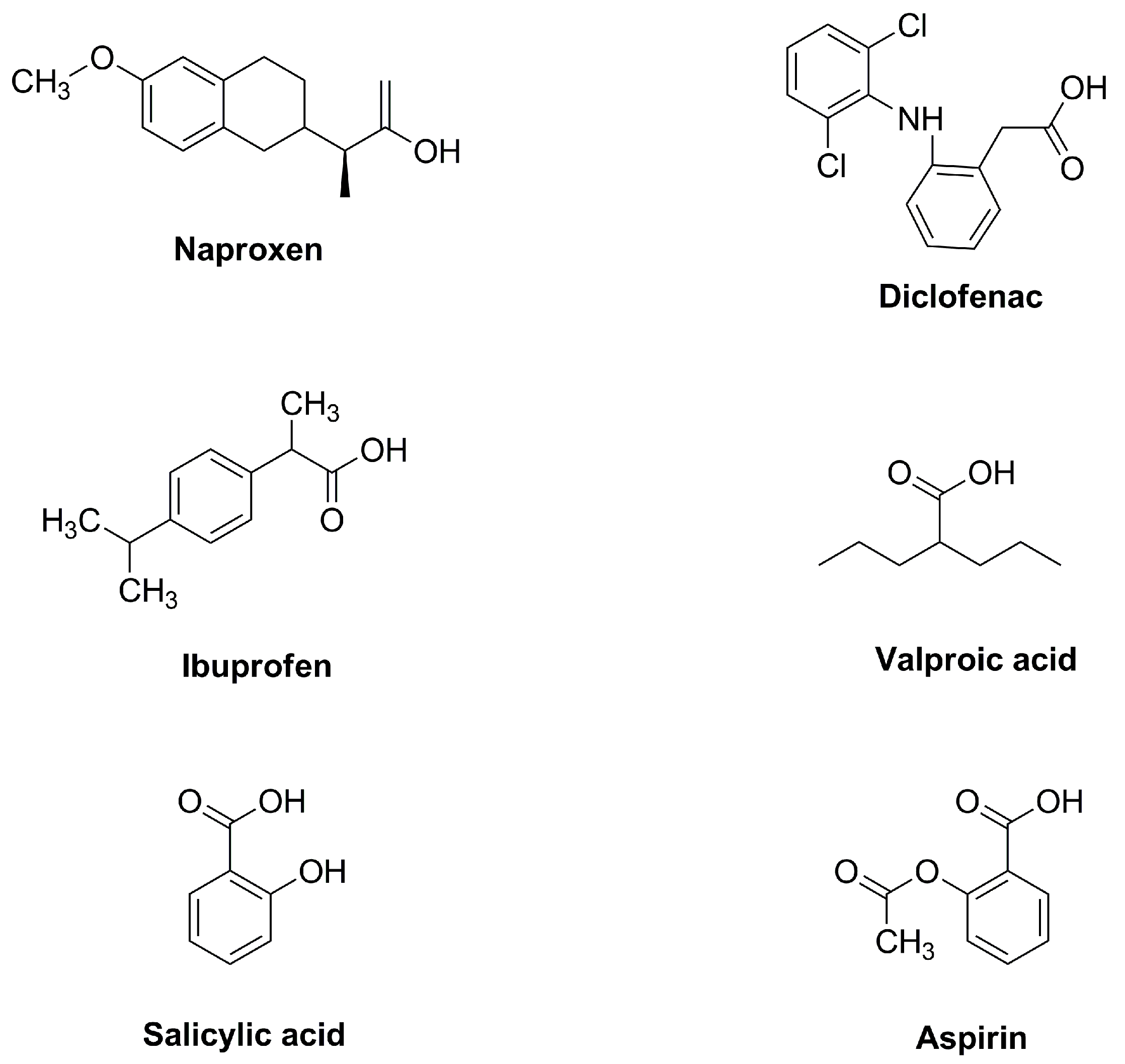
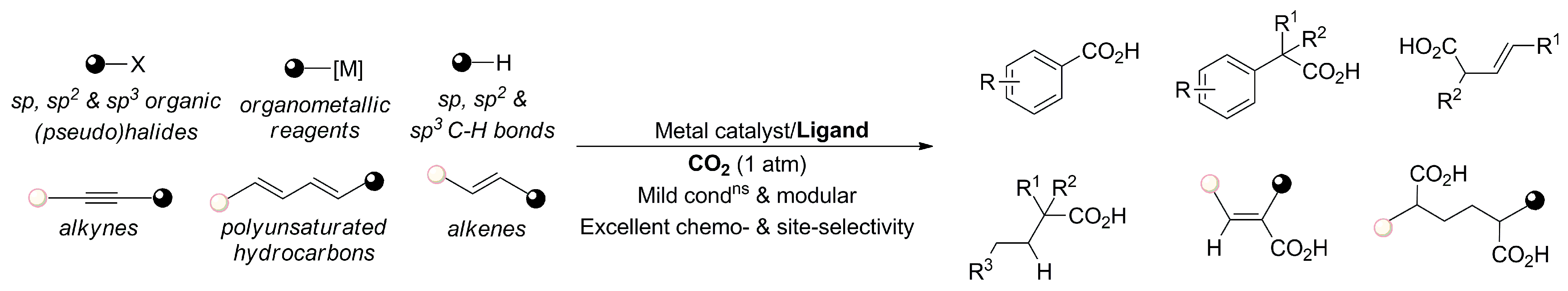
1. Introduction
2. Transition-Metal-Catalyzed Carboxylation
2.1. Carboxylation of C–X Bonds (Halogens and Oxygen)
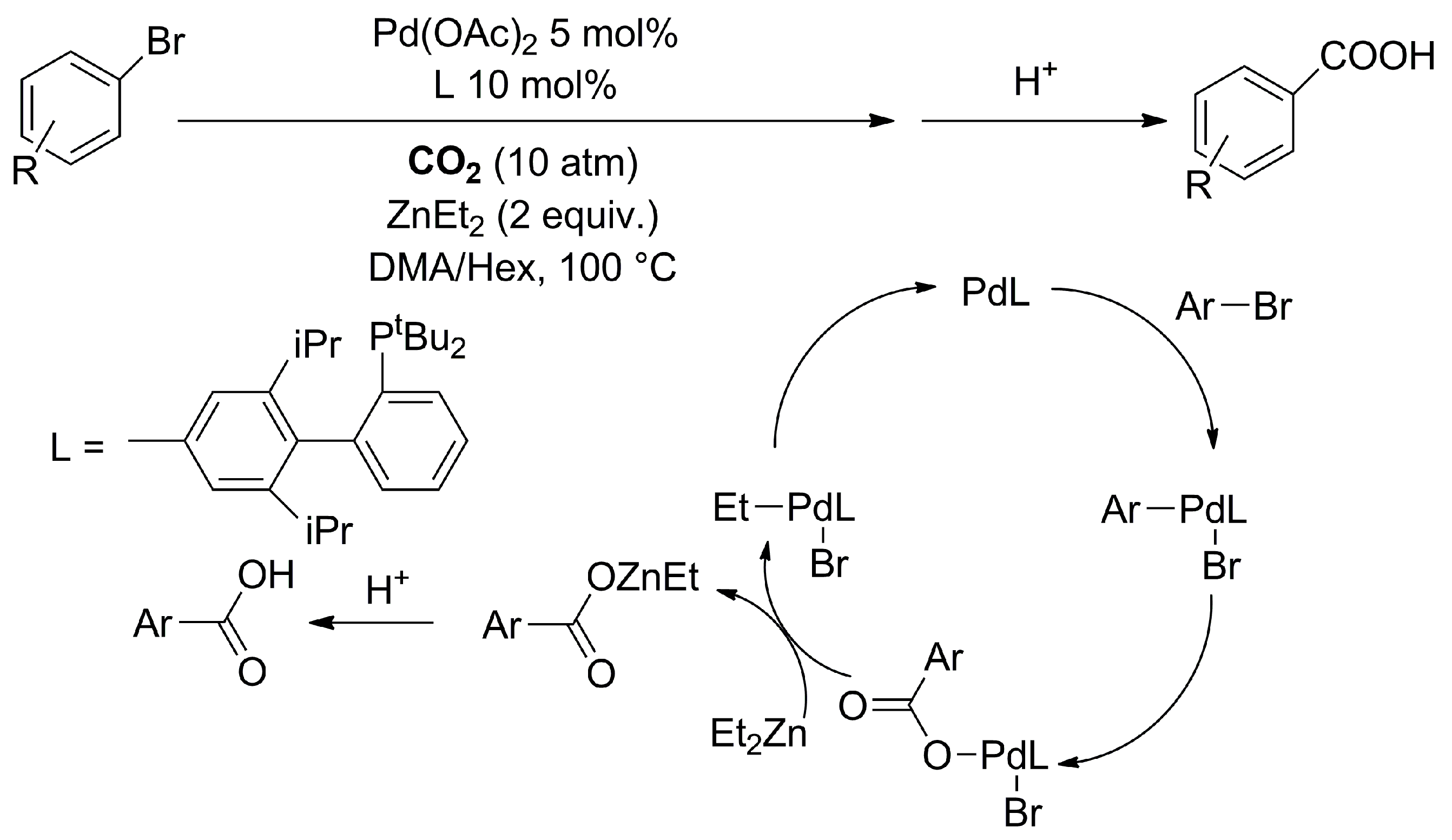
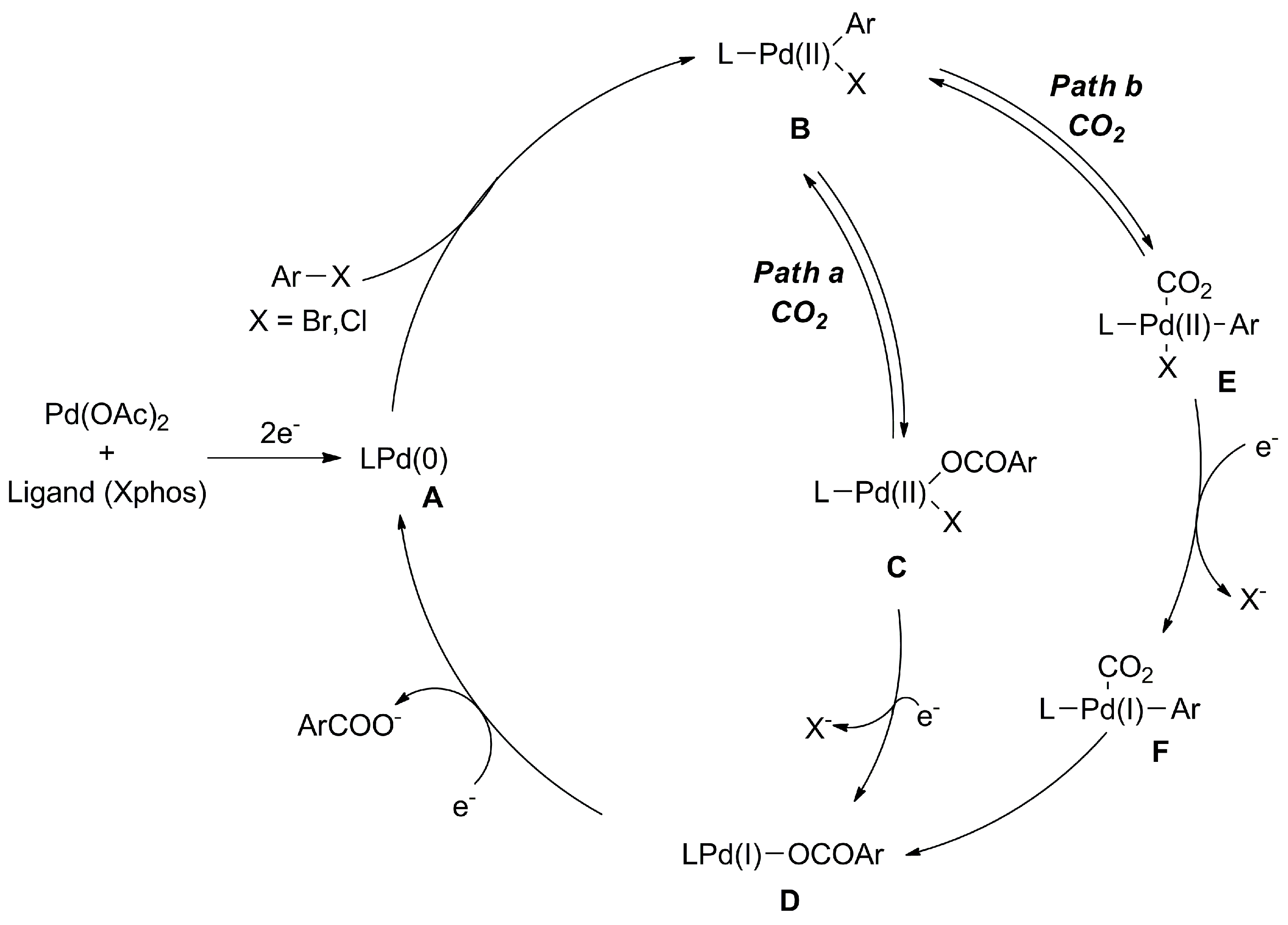
2.1.1. Pd-, Ni-, and Cu-Catalyzed Carboxylation of C-Halogens with CO2
2.1.2. Carboxylation of C–O Bonds with CO2 Catalyzed via Ni(II)
2.2. Carboxylation of Organometallic Compounds Employing Carbon Dioxide (CO2)
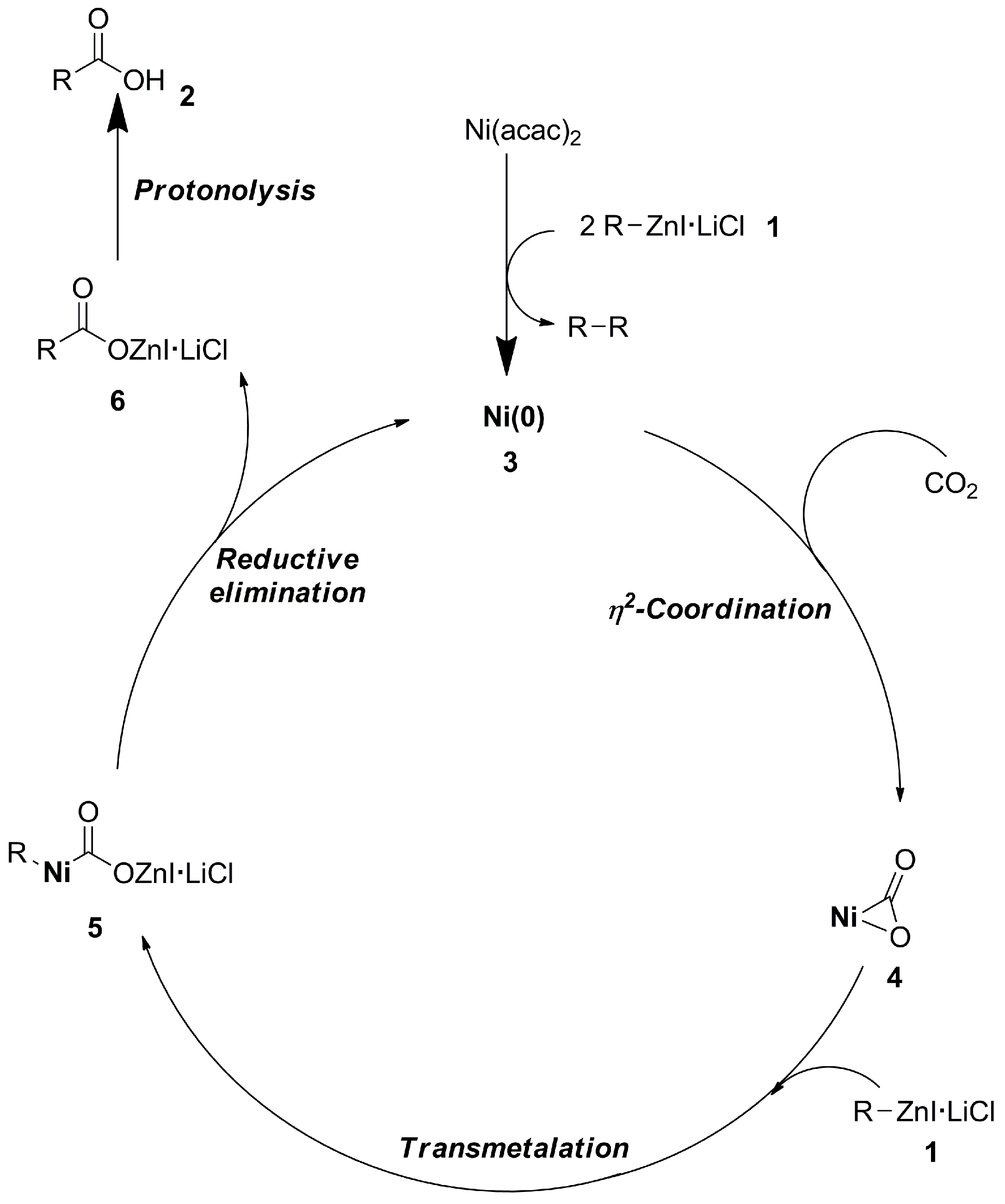
2.2.1. Nickel- and Palladium-Catalyzed Carboxylation of R-M (M = Sn, Zn) Reagents with CO2
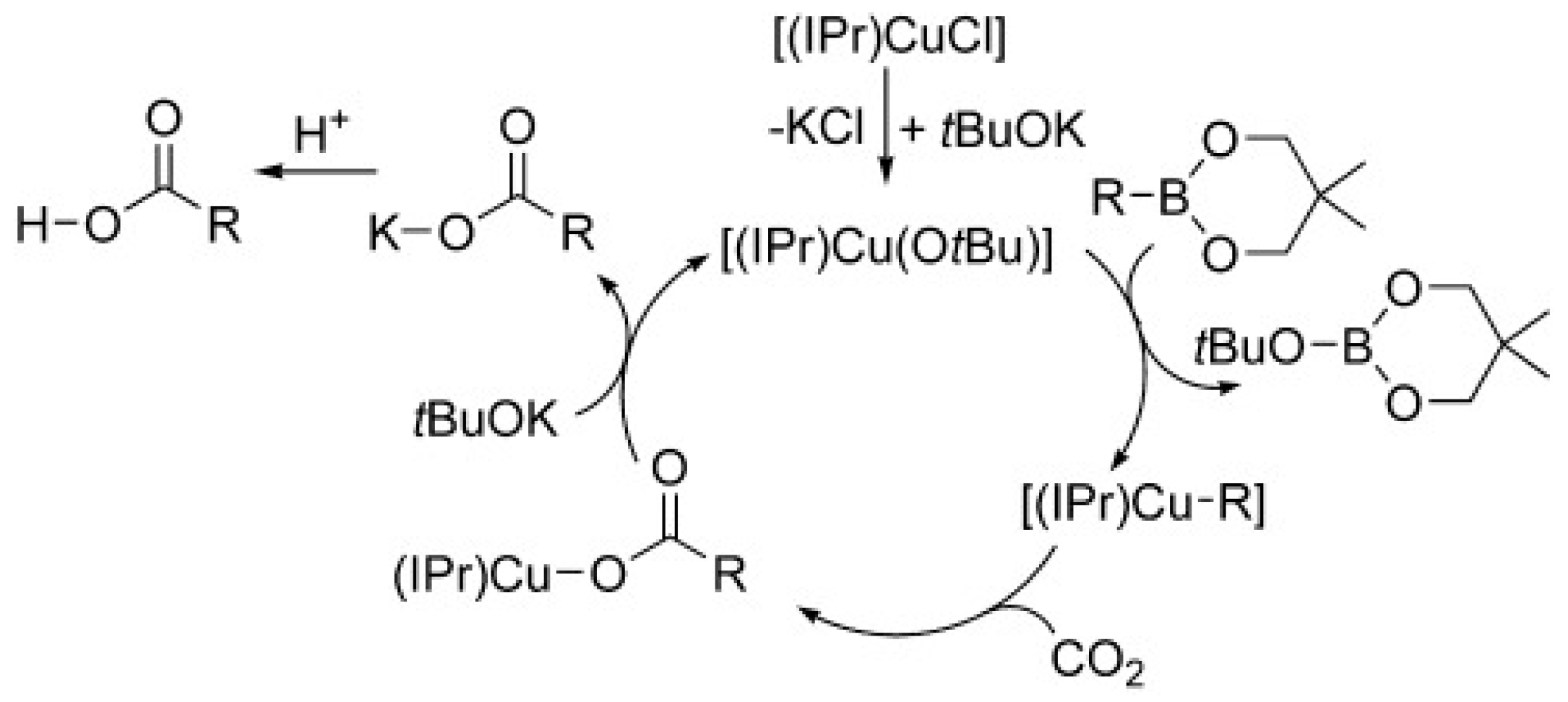
2.2.2. Copper (Cu)-, Palladium (Pd)-, and Rhodium (Rh)-Catalyzed Carboxylation of R B Reagents with CO2
2.3. Utilizing CO2 to Carboxylate C–H Bonds
2.3.1. Carboxylation sp1 C–H Bonds with CO2
Cu-Catalyzed Carboxylation of sp1 C–H Bonds
Ag-Catalyzed Carboxylation of sp1 C–H Bonds with CO2
2.3.2. Carboxylation of sp2 C–H Bonds with CO2
Cu- and Au-Catalyzed Carboxylation of sp2 C-H Bonds with CO2
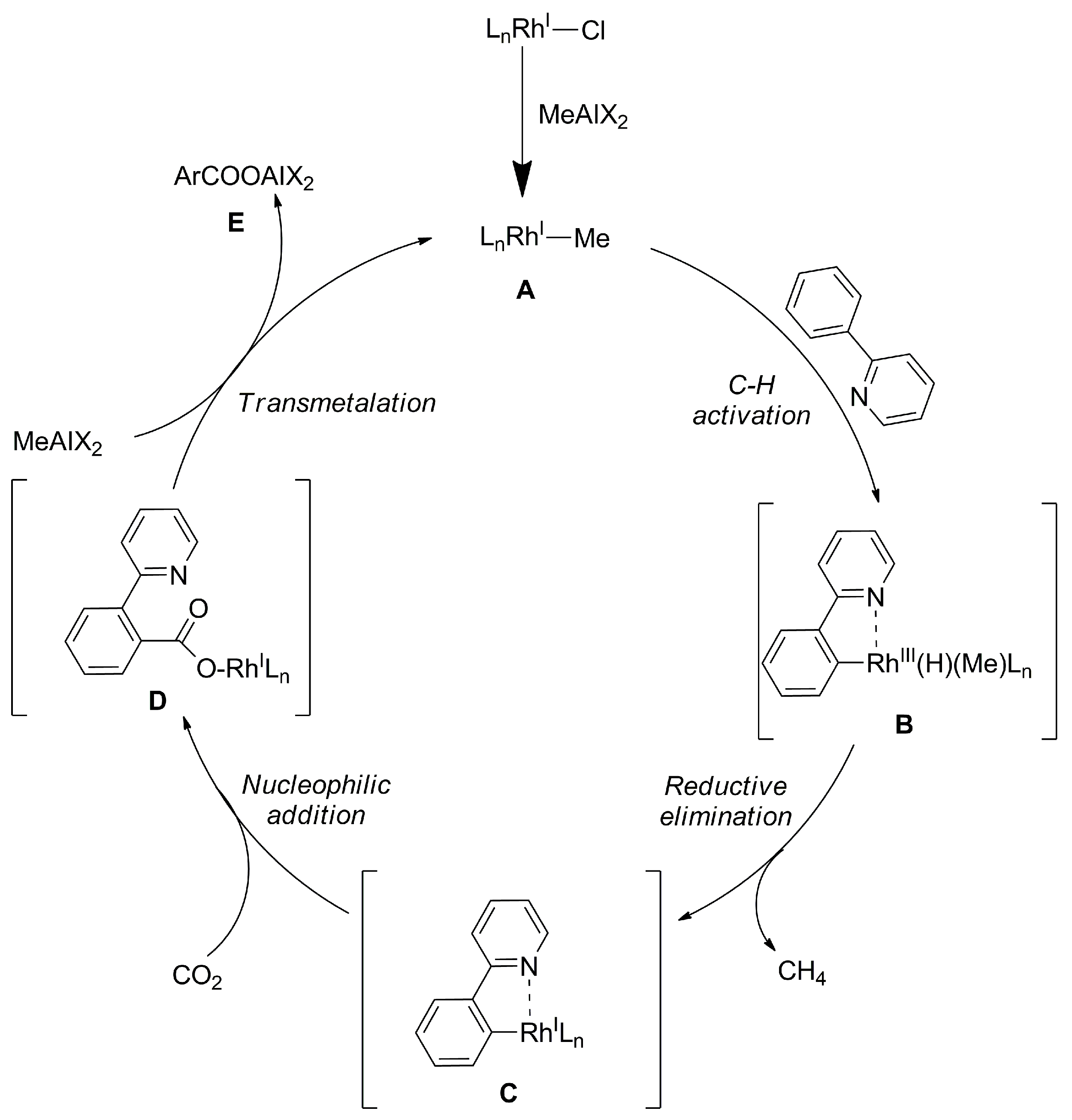
Carboxylation of sp2 C–H Bonds with CO2 Catalyzed by Pd and Rh
2.3.3. Carboxylation of sp3 C–H Bonds with CO2
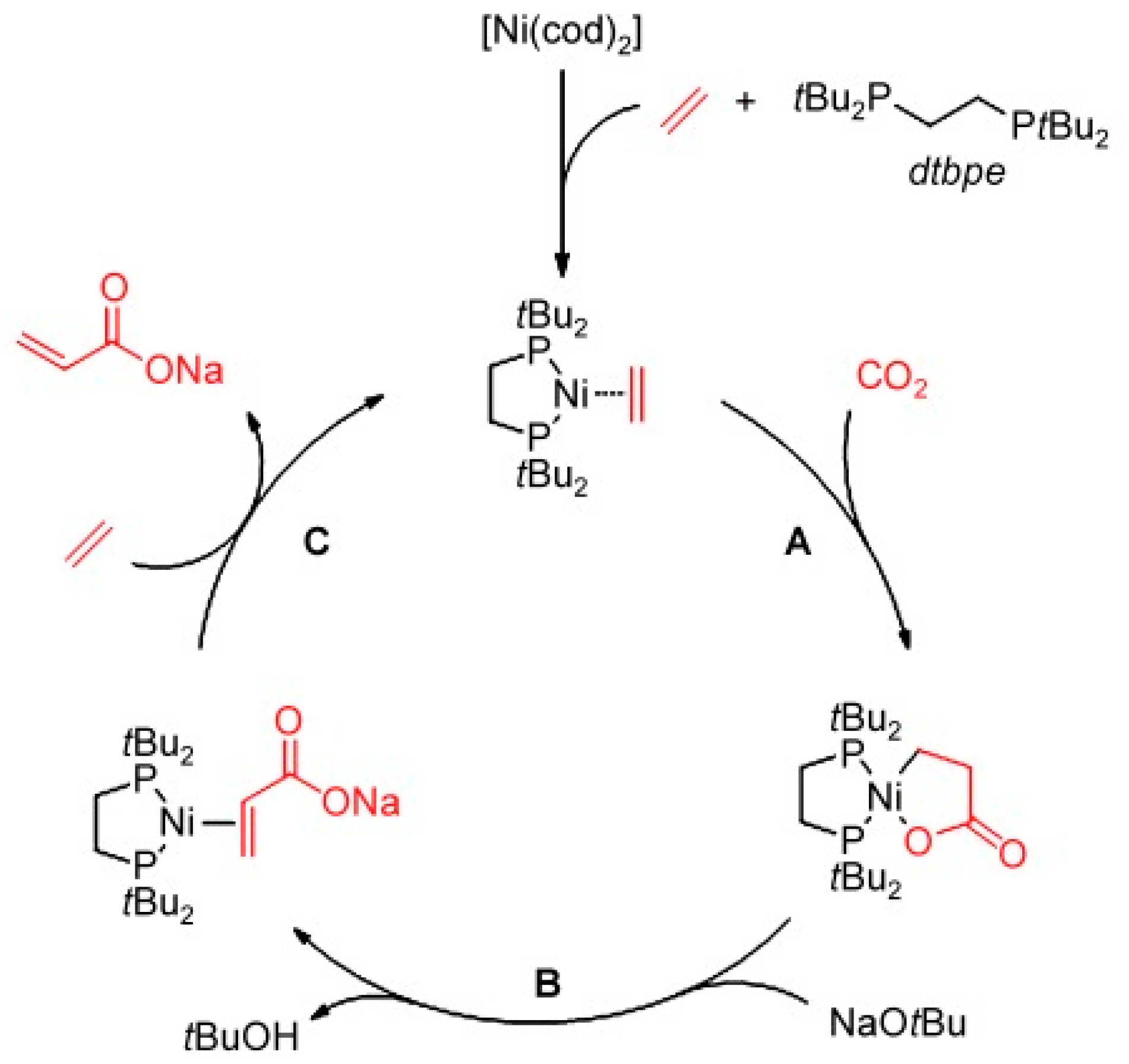
2.4. Carboxylation of Ethylene with CO2 to Acrylates
3. Photo-Catalytic Carboxylation
3.1. Carboxylation of C–Halogen Bonds
3.2. Carboxylation of C(sp3)–H–Bonds
3.3. Carboxylation of Alkenes
3.3.1. Hydrocarboxylation of Alkene
3.3.2. Difunctionalisation of Alkenes
3.4. Carboxylation of Alkynes
4. Electrochemical Carboxylation
4.1. Electrocarboxylation of Alkenes with CO2
4.2. Electrocarboxylation of Organic (Pseudo)halides with CO2
4.2.1. Electrocarboxylation of Organic Halides
Direct Carboxylation
Metal-Catalyzed Electrocarboxylation
Electrocarboxylation of Organic (Pseudo)halides
4.3. Electrocarboxylation of Aldehydes and Ketones with CO2
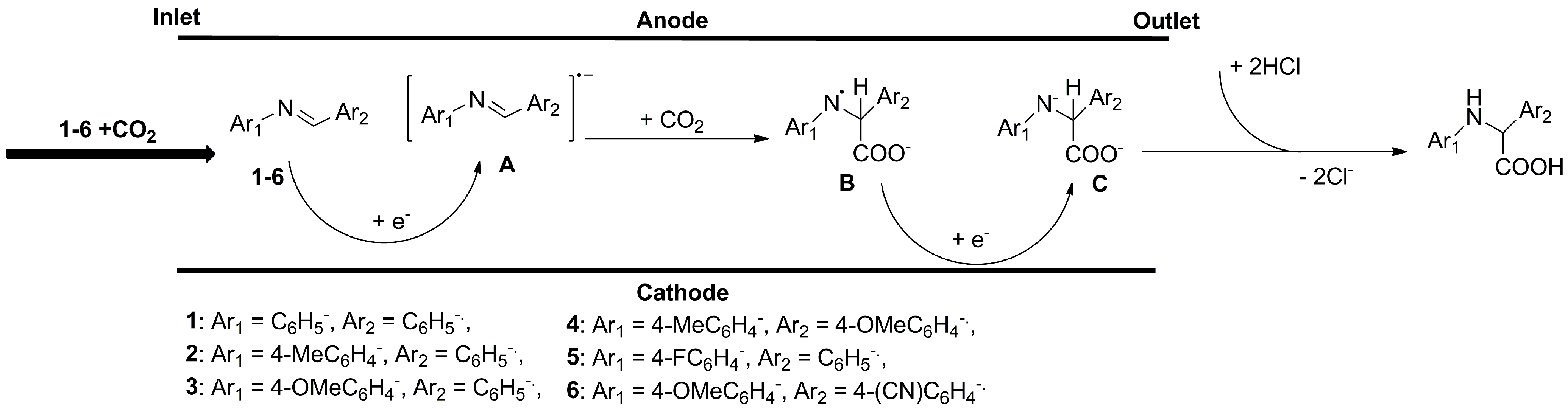
4.4. Electrocarboxylation of Imines with CO2

5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ritchie, H.; Roser, M.; Rosado, P. CO2 and Greenhouse Gas Emissions. Our World Data 2020. Available online: https://ourworldindata.org/co2-and-greenhouse-gas-emissions (accessed on 24 October 2023).
- Ritchie, H.; Roser, M.; Rosado, P. Energy. Our World Data 2022. Available online: https://ourworldindata.org/energy (accessed on 24 October 2023).
- Riduan, S.N.; Zhang, Y. Recent Developments in Carbon Dioxide Utilization under Mild Conditions. Dalton Trans. 2010, 39, 3347–3357. [Google Scholar] [CrossRef]
- Delarmelina, M.; Deshmukh, G.; Goguet, A.; Catlow, C.R.A.; Manyar, H. Role of Sulfation of Zirconia Catalysts in Vapor Phase Ketonization of Acetic Acid. J. Phys. Chem. C 2021, 125, 27578–27595. [Google Scholar] [CrossRef] [PubMed]
- Ethiraj, J.; Wagh, D.; Manyar, H. Advances in Upgrading Biomass to Biofuels and Oxygenated Fuel Additives Using Metal Oxide Catalysts. Energy Fuels 2022, 36, 1189–1204. [Google Scholar] [CrossRef]
- Keogh, J.; Deshmukh, G.; Manyar, H. Green Synthesis of Glycerol Carbonate via Transesterification of Glycerol Using Mechanochemically Prepared Sodium Aluminate Catalysts. Fuel 2022, 310, 122484. [Google Scholar] [CrossRef]
- Ralphs, K.; Collins, G.; Manyar, H.; James, S.L.; Hardacre, C. Selective Hydrogenation of Stearic Acid Using Mechanochemically Prepared Titania-Supported Pt and Pt–Re Bimetallic Catalysts. ACS Sustain. Chem. Eng. 2022, 10, 6934–6941. [Google Scholar] [CrossRef]
- Keogh, J.; Jeffrey, C.; Tiwari, M.S.; Manyar, H. Kinetic Analysis of Glycerol Esterification Using Tin Exchanged Tungstophosphoric Acid on K-10. Ind. Eng. Chem. Res. 2022, 62, 19095–19103. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, M.S.; Wagh, D.; Dicks, J.S.; Keogh, J.; Ansaldi, M.; Ranade, V.V.; Manyar, H.G. Solvent Free Upgrading of 5-Hydroxymethylfurfural (HMF) with Levulinic Acid to HMF Levulinate Using Tin Exchanged Tungstophosphoric Acid Supported on K-10 Catalyst. ACS Org. Inorg. Au 2023, 3, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Manyar, H. Synthesis of a Novel Redox Material UDCaT-3: An Efficient and Versatile Catalyst for Selective Oxidation, Hydroxylation and Hydrogenation Reactions. Adv. Synth. Catal. 2008, 350, 2286. [Google Scholar]
- Skillen, N.; Ralphs, K.; Craig, D.; McCalmont, S.; Muzio, A.F.V.; O’Rourke, C.; Manyar, H.; Robertson, P. Photocatalytic Reforming of Glycerol to H2 in a Thin Film Pt-TiO2 Recirculating Photo Reactor. J. Chem. Technol. Biotechnol. 2020, 95, 2619–2627. [Google Scholar] [CrossRef]
- Jakubek, T.; Ralphs, K.; Kotarba, A.; Manyar, H. Nanostructured Potassium-Manganese Oxides Decorated with Pd Nanoparticles as Efficient Catalysts for Low-Temperature Soot Oxidation. Catal. Lett. 2019, 149, 100–106. [Google Scholar] [CrossRef]
- Salisu, J.; Gao, N.; Quan, C.; Yanik, J.; Artioli, N. Co-Gasification of Rice Husk and Plastic in the Presence of CaO Using a Novel ANN Model-Incorporated Aspen plus Simulation. J. Energy Inst. 2023, 108, 101239. [Google Scholar] [CrossRef]
- Quan, C.; Zhang, G.; Gao, N.; Su, S.; Artioli, N.; Feng, D. Behavior Study of Migration and Transformation of Heavy Metals during Oily Sludge Pyrolysis. Energy Fuels 2022, 36, 8311–8322. [Google Scholar] [CrossRef]
- Byrne, E.L.; O’Donnell, R.; Gilmore, M.; Artioli, N.; Holbrey, J.D.; Swadźba-Kwaśny, M. Hydrophobic Functional Liquids Based on Trioctylphosphine Oxide (TOPO) and Carboxylic Acids. Phys. Chem. Chem. Phys. 2020, 22, 24744–24763. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, L.; Matarrese, R.; Kubiak, L.; Daturi, M.; Artioli, N.; Pompa, S.; Lietti, L. In-Depth Insights into N2O Formation over Rh- and Pt-Based LNT Catalysts. Catal. Today 2019, 320, 141–151. [Google Scholar] [CrossRef]
- Yilleng, M.T.; Gimba, E.C.; Ndukwe, G.I.; Bugaje, I.M.; Rooney, D.W.; Manyar, H.G. Batch to Continuous Photocatalytic Degradation of Phenol Using TiO2 and Au-Pd Nanoparticles Supported on TiO2. J. Environ. Chem. Eng. 2018, 6, 6382–6389. [Google Scholar] [CrossRef]
- O’Donnell, R.; Ralphs, K.; Grolleau, M.; Manyar, H.; Artioli, N. Doping Manganese Oxides with Ceria and Ceria Zirconia Using a One-Pot Sol–Gel Method for Low Temperature Diesel Oxidation Catalysts. Top. Catal. 2020, 63, 351–362. [Google Scholar] [CrossRef]
- Coney, C.; Hardacre, C.; Morgan, K.; Artioli, N.; York, A.P.E.; Millington, P.; Kolpin, A.; Goguet, A. Investigation of the oxygen storage capacity behaviour of three way catalysts using spatio-temporal analysis. Appl. Catal. B Environ. 2019, 258, 117918. [Google Scholar] [CrossRef]
- Lietti, L.; Righini, L.; Castoldi, L.; Artioli, N.; Forzatti, P. Labeled 15NO study on N2 and N2O formation over Pt-Ba/Al2O3 NSR catalysts. Top. Catal. 2013, 56, 7–13. [Google Scholar] [CrossRef]
- He, M.; Sun, Y.; Han, B. Green Carbon Science: Scientific Basis for Integrating Carbon Resource Processing, Utilization, and Recycling. Angew. Chem. Int. Ed. 2013, 52, 9620–9633. [Google Scholar] [CrossRef]
- Ye, J.-H.; Ju, T.; Huang, H.; Liao, L.-L.; Yu, D.-G. Radical Carboxylative Cyclizations and Carboxylations with CO2. Acc. Chem. Res. 2021, 54, 2518–2531. [Google Scholar] [CrossRef]
- Sakakura, T.; Choi, J.-C.; Yasuda, H. Transformation of Carbon Dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef] [PubMed]
- Maag, H. Prodrugs of Carboxylic Acids. In Prodrugs: Challenges and Rewards Part 1; Stella, V.J., Borchardt, R.T., Hageman, M.J., Oliyai, R., Maag, H., Tilley, J.W., Eds.; Biotechnology: Pharmaceutical Aspects; Springer: New York, NY, USA, 2007; pp. 703–729. ISBN 9780387497853. [Google Scholar]
- Ajvazi, N.; Stavber, S. Alcohols in direct carbon-carbon and carbon-heteroatom bond-forming reactions: Recent advances. Arkivoc 2018, part ii, 288–329. [Google Scholar] [CrossRef]
- Valera Lauridsen, J.M.; Cho, S.Y.; Bae, H.Y.; Lee, J.-W. CO2 (De)Activation in Carboxylation Reactions: A Case Study Using Grignard Reagents and Nucleophilic Bases. Organometallics 2020, 39, 1652–1657. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A. Key Issues in Carbon Dioxide Utilization as a Building Block for Molecular Organic Compounds in the Chemical Industry. In CO2 Conversion and Utilization; Song, C., Gaffney, A.F., Fujimoto, K., Eds.; American Chemical Society: Washington, DC, USA, 2002; Volume 809, pp. 54–70. ISBN 9780841237476. [Google Scholar]
- Wu, X.-F.; Fang, X.; Wu, L.; Jackstell, R.; Neumann, H.; Beller, M. Transition-Metal-Catalyzed Carbonylation Reactions of Olefins and Alkynes: A Personal Account. Acc. Chem. Res. 2014, 47, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Sun, C.-L.; Shi, Z.-J. Transition-Metal-Catalyzed C–C Bond Formation through the Fixation of Carbon Dioxide. Chem. Soc. Rev. 2011, 40, 2435–2452. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, Y.; Fujihara, T. Carbon Dioxide as a Carbon Source in Organic Transformation: Carbon–Carbon Bond Forming Reactions by Transition-Metal Catalysts. Chem. Commun. 2012, 48, 9956–9964. [Google Scholar] [CrossRef]
- Duong, H.A.; Huleatt, P.B.; Tan, Q.-W.; Shuying, E.L. Regioselective Copper-Catalyzed Carboxylation of Allylboronates with Carbon Dioxide. Org. Lett. 2013, 15, 4034–4037. [Google Scholar] [CrossRef]
- Yu, D.; Teong, S.P.; Zhang, Y. Transition Metal Complex Catalyzed Carboxylation Reactions with CO2. Coord. Chem. Rev. 2015, 293–294, 279–291. [Google Scholar] [CrossRef]
- Luan, Y.-X.; Ye, M. Transition Metal-Mediated or Catalyzed Hydrocarboxylation of Olefins with CO2. Tetrahedron Lett. 2018, 59, 853–861. [Google Scholar] [CrossRef]
- Yi, Y.; Hang, W.; Xi, C. Recent Advance of Transition-Metal-Catalyzed Tandem Carboxylation Reaction of Unsaturated Hydrocarbons with Organometallic Reagents and CO2. Youji Huaxue 2021, 41, 80. [Google Scholar] [CrossRef]
- Ran, C.-K.; Liao, L.-L.; Gao, T.-Y.; Gui, Y.-Y.; Yu, D.-G. Recent Progress and Challenges in Carboxylation with CO2. Curr. Opin. Green. Sustain. Chem. 2021, 32, 100525. [Google Scholar] [CrossRef]
- Liu, X.-F.; Zhang, K.; Tao, L.; Lu, X.-B.; Zhang, W.-Z. Recent Advances in Electrochemical Carboxylation Reactions Using Carbon Dioxide. Green. Chem. Eng. 2022, 3, 125–137. [Google Scholar] [CrossRef]
- Tortajada, A.; Juliá-Hernández, F.; Börjesson, M.; Moragas, T.; Martin, R. Transition-Metal-Catalyzed Carboxylation Reactions with Carbon Dioxide. Angew. Chem. Int. Ed. 2018, 57, 15948–15982. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Caner, J.; Nishikawa, S.; Toriumi, N.; Iwasawa, N. Catalytic Direct Hydrocarboxylation of Styrenes with CO2 and H2. Nat. Commun. 2022, 13, 7584. [Google Scholar] [CrossRef] [PubMed]
- Stitt, M.; Schulze, D. Does Rubisco Control the Rate of Photosynthesis and Plant Growth? An Exercise in Molecular Ecophysiology. Plant Cell Environ. 1994, 17, 465–487. [Google Scholar] [CrossRef]
- Tazuke, S.; Kazama, S.; Kitamura, N. Reductive Photocarboxylation of Aromatic Hydrocarbons. J. Org. Chem. 1986, 51, 4548–4553. [Google Scholar] [CrossRef]
- Fan, Z.; Zhang, Z.; Xi, C. Light-Mediated Carboxylation Using Carbon Dioxide. ChemSusChem 2020, 13, 6201–6218. [Google Scholar] [CrossRef]
- Sun, G.-Q.; Zhang, W.; Liao, L.-L.; Li, L.; Nie, Z.-H.; Wu, J.-G.; Zhang, Z.; Yu, D.-G. Nickel-Catalyzed Electrochemical Carboxylation of Unactivated Aryl and Alkyl Halides with CO2. Nat. Commun. 2021, 12, 7086. [Google Scholar] [CrossRef]
- Yang, D.-T.; Zhu, M.; Schiffer, Z.J.; Williams, K.; Song, X.; Liu, X.; Manthiram, K. Direct Electrochemical Carboxylation of Benzylic C–N Bonds with Carbon Dioxide. ACS Catal. 2019, 9, 4699–4705. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Z.; Pan, D.; Wang, S.; Jia, K.; Ma, D.; Yang, G.; Xue, X.; Qiu, Y. Metal-Free Electrochemical Carboxylation of Organic Halides in the Presence of Catalytic Amounts of an Organomediator. Angew. Chem. Int. Ed. 2022, 61, e2022102012022. [Google Scholar] [CrossRef]
- Matthessen, R.; Fransaer, J.; Binnemans, K.; De Vos, D.E. Electrocarboxylation: Towards Sustainable and Efficient Synthesis of Valuable Carboxylic Acids. Beilstein J. Org. Chem. 2014, 10, 2484–2500. [Google Scholar] [CrossRef] [PubMed]
- Seyferth, D. Alkyl and Aryl Derivatives of the Alkali Metals: Useful Synthetic Reagents as Strong Bases and Potent Nucleophiles. 1. Conversion of Organic Halides to Organoalkali-Metal Compounds. Organometallics 2006, 25, 2–24. [Google Scholar] [CrossRef]
- Kingston, C.; Wallace, M.A.; Allentoff, A.J.; deGruyter, J.N.; Chen, J.S.; Gong, S.X.; Bonacorsi, S.; Baran, P.S. Direct Carbon Isotope Exchange through Decarboxylative Carboxylation. J. Am. Chem. Soc. 2019, 141, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Fujihara, T.; Nogi, K.; Xu, T.; Terao, J.; Tsuji, Y. Nickel-Catalyzed Carboxylation of Aryl and Vinyl Chlorides Employing Carbon Dioxide. J. Am. Chem. Soc. 2012, 134, 9106–9109. [Google Scholar] [CrossRef] [PubMed]
- Correa, A.; Martín, R. Palladium-Catalyzed Direct Carboxylation of Aryl Bromides with Carbon Dioxide. J. Am. Chem. Soc. 2009, 131, 15974–15975. [Google Scholar] [CrossRef] [PubMed]
- Osakada, K.; Sato, R.; Yamamoto, T. Nickel-Complex-Promoted Carboxylation of Haloarenes Involving Insertion of CO2 into NiII-C Bonds. Organometallics 1994, 13, 4645–4647. [Google Scholar] [CrossRef]
- Tortajada, A.; Duan, Y.; Sahoo, B.; Cong, F.; Toupalas, G.; Sallustrau, A.; Loreau, O.; Audisio, D.; Martin, R. Catalytic Decarboxylation/Carboxylation Platform for Accessing Isotopically Labeled Carboxylic Acids. ACS Catal. 2019, 9, 5897–5901. [Google Scholar] [CrossRef]
- Cerveri, A.; Giovanelli, R.; Sella, D.; Pedrazzani, R.; Monari, M.; Nieto Faza, O.; López, C.S.; Bandini, M. Enantioselective CO2 Fixation Via a Heck-Coupling/Carboxylation Cascade Catalyzed by Nickel. Chem.–A Eur. J. 2021, 27, 7657–7662. [Google Scholar] [CrossRef]
- Correa, A.; León, T.; Martin, R. Ni-Catalyzed Carboxylation of C(Sp2)– and C(Sp3)–O Bonds with CO2. J. Am. Chem. Soc. 2014, 136, 1062–1069. [Google Scholar] [CrossRef]
- Tran-Vu, H.; Daugulis, O. Copper-Catalyzed Carboxylation of Aryl Iodides with Carbon Dioxide. ACS Catal. 2013, 3, 2417–2420. [Google Scholar] [CrossRef]
- Yan, S.-S.; Wu, D.-S.; Ye, J.-H.; Gong, L.; Zeng, X.; Ran, C.-K.; Gui, Y.-Y.; Li, J.; Yu, D.-G. Copper-Catalyzed Carboxylation of C–F Bonds with CO2. ACS Catal. 2019, 9, 6987–6992. [Google Scholar] [CrossRef]
- Bew, S.P. Carboxylic Acids. In Comprehensive Organic Functional Group Transformations, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2005; pp. 19–125. ISBN 9780080446554. [Google Scholar]
- Ebert, G.W.; Juda, W.L.; Kosakowski, R.H.; Ma, B.; Dong, L.; Cummings, K.E.; Phelps, M.V.B.; Mostafa, A.E.; Luo, J. Carboxylation and Esterification of Functionalized Arylcopper Reagents. J. Org. Chem. 2005, 70, 4314–4317. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.W. Protecting-Group-Free Synthesis. Synthesis 2006, 2006, 3531–3541. [Google Scholar] [CrossRef]
- Shi, M.; Nicholas, K.M. Palladium-Catalyzed Carboxylation of Allyl Stannanes. J. Am. Chem. Soc. 1997, 119, 5057–5058. [Google Scholar] [CrossRef]
- Johansson, R.; Wendt, O.F. Insertion of CO2 into a Palladiumallyl Bond and a Pd(ii) Catalysed Carboxylation of Allyl Stannanes. Dalton Trans. 2007, 4, 488–492. [Google Scholar] [CrossRef]
- Wu, J.; Hazari, N. Palladium Catalyzed Carboxylation of Allylstannanes and Boranes Using CO2. Chem. Commun. 2011, 47, 1069–1071. [Google Scholar] [CrossRef] [PubMed]
- Hruszkewycz, D.P.; Wu, J.; Hazari, N.; Incarvito, C.D. Palladium(I)-Bridging Allyl Dimers for the Catalytic Functionalization of CO2. J. Am. Chem. Soc. 2011, 133, 3280–3283. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, H.; Jang, M.; Hirano, K.; Yorimitsu, H.; Oshima, K. Nickel-Catalyzed Carboxylation of Organozinc Reagents with CO2. Org. Lett. 2008, 10, 2681–2683. [Google Scholar] [CrossRef]
- Yeung, C.S.; Dong, V.M. Beyond Aresta’s Complex: Ni- and Pd-Catalyzed Organozinc Coupling with CO2. J. Am. Chem. Soc. 2008, 130, 7826–7827. [Google Scholar] [CrossRef]
- Shibasaki, M.; Kanai, M. Asymmetric Synthesis of Tertiary Alcohols and α-Tertiary Amines via Cu-Catalyzed C−C Bond Formation to Ketones and Ketimines. Chem. Rev. 2008, 108, 2853–2873. [Google Scholar] [CrossRef]
- Evano, G.; Blanchard, N.; Toumi, M. Copper-Mediated Coupling Reactions and Their Applications in Natural Products and Designed Biomolecules Synthesis. Chem. Rev. 2008, 108, 3054–3131. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, C.; Krause, N.; Lipshutz, B.H. CuH-Catalyzed Reactions. Chem. Rev. 2008, 108, 2916–2927. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, J.; Zheng, G. Designing Copper-Based Catalysts for Efficient Carbon Dioxide Electroreduction. Adv. Mater. 2021, 33, 2005798. [Google Scholar] [CrossRef] [PubMed]
- Nitopi, S.; Bertheussen, E.; Scott, S.B.; Liu, X.; Engstfeld, A.K.; Horch, S.; Seger, B.; Stephens, I.E.L.; Chan, K.; Hahn, C.; et al. Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte. Chem. Rev. 2019, 119, 7610–7672. [Google Scholar] [CrossRef] [PubMed]
- Ukai, K.; Aoki, M.; Takaya, J.; Iwasawa, N. Rhodium(I)-Catalyzed Carboxylation of Aryl- and Alkenylboronic Esters with CO2. J. Am. Chem. Soc. 2006, 128, 8706–8707. [Google Scholar] [CrossRef]
- Ohishi, T.; Nishiura, M.; Hou, Z. Carboxylation of Organoboronic Esters Catalyzed by N-Heterocyclic Carbene Copper(I) Complexes. Angew. Chem. Int. Ed. 2008, 47, 5792–5795. [Google Scholar] [CrossRef] [PubMed]
- Ohmiya, H.; Tanabe, M.; Sawamura, M. Copper-Catalyzed Carboxylation of Alkylboranes with Carbon Dioxide: Formal Reductive Carboxylation of Terminal Alkenes. Org. Lett. 2011, 13, 1086–1088. [Google Scholar] [CrossRef] [PubMed]
- Hay, A.S. Oxidative Coupling of Acetylenes. II. J. Org. Chem. 1962, 27, 3320–3321. [Google Scholar] [CrossRef]
- Fukue, Y.; Oi, S.; Inoue, Y. Direct Synthesis of Alkyl 2-Alkynoates from Alk-1-Ynes, CO2, and Bromoalkanes Catalysed by Copper(I) or Silver(I) Salt. J. Chem. Soc. Chem. Commun. 1994, 18, 2091. [Google Scholar] [CrossRef]
- Zhang, W.-Z.; Li, W.-J.; Zhang, X.; Zhou, H.; Lu, X.-B. Cu(I)-Catalyzed Carboxylative Coupling of Terminal Alkynes, Allylic Chlorides, and CO2. Org. Lett. 2010, 12, 4748–4751. [Google Scholar] [CrossRef]
- Yu, D.; Zhang, Y. Copper- and Copper– N. -Heterocyclic Carbene-Catalyzed C–H Activating Carboxylation of Terminal Alkynes with CO2 at Ambient Conditions. Proc. Natl. Acad. Sci. USA 2010, 107, 20184–20189. [Google Scholar] [CrossRef]
- Yang, L.; Yuan, Y.; Wang, H.; Zhang, N.; Hong, S. Theoretical Insights into Copper(i)–NHC-Catalyzed C–H Carboxylation of Terminal Alkynes with CO2: The Reaction Mechanisms and the Roles of NHC. RSC Adv. 2014, 4, 32457–32466. [Google Scholar] [CrossRef]
- Trivedi, M.; Singh, G.; Kumar, A.; Rath, N.P. 1,1′-Bis(Di-Tert-Butylphosphino)Ferrocene Copper(i) Complex Catalyzed C–H Activation and Carboxylation of Terminal Alkynes. Dalton Trans. 2015, 44, 20874–20882. [Google Scholar] [CrossRef]
- Bondarenko, G.N.; Dvurechenskaya, E.G.; Magommedov, E.S.; Beletskaya, I.P. Copper(0) Nanoparticles Supported on Al2O3 as Catalyst for Carboxylation of Terminal Alkynes. Catal. Lett. 2017, 147, 2570–2580. [Google Scholar] [CrossRef]
- Wendling, T.; Risto, E.; Krause, T.; Gooßen, L.J. Salt-Free Strategy for the Insertion of CO2 into C−H Bonds: Catalytic Hydroxymethylation of Alkynes. Chem.–A Eur. J. 2018, 24, 6019–6024. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, W.-Z.; Ren, X.; Zhang, L.-L.; Lu, X.-B. Ligand-Free Ag(I)-Catalyzed Carboxylation of Terminal Alkynes with CO2. Org. Lett. 2011, 13, 2402–2405. [Google Scholar] [CrossRef]
- Arndt, M.; Risto, E.; Krause, T.; Gooßen, L.J. C–H Carboxylation of Terminal Alkynes Catalyzed by Low Loadings of Silver(I)/DMSO at Ambient CO2 Pressure. ChemCatChem 2012, 4, 484–487. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, K.H.; Hong, S.H. Carbon Dioxide Capture and Use: Organic Synthesis Using Carbon Dioxide from Exhaust Gas. Angew. Chem. Int. Ed. 2014, 53, 771–774. [Google Scholar] [CrossRef]
- Liu, C.; Luo, Y.; Zhang, W.; Qu, J.; Lu, X. DFT Studies on the Silver-Catalyzed Carboxylation of Terminal Alkynes with CO2: An Insight into the Catalytically Active Species. Organometallics 2014, 33, 2984–2989. [Google Scholar] [CrossRef]
- Gaillard, S.; Cazin, C.S.J.; Nolan, S.P. N-Heterocyclic Carbene Gold(I) and Copper(I) Complexes in C–H Bond Activation. Acc. Chem. Res. 2012, 45, 778–787. [Google Scholar] [CrossRef]
- Boogaerts, I.I.F.; Fortman, G.C.; Furst, M.R.L.; Cazin, C.S.J.; Nolan, S.P. Carboxylation of N–H/C–H Bonds Using N-Heterocyclic Carbene Copper(I) Complexes. Angew. Chem. Int. Ed. 2010, 49, 8674–8677. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Cheng, J.; Ohishi, T.; Hou, Z. Copper-Catalyzed Direct Carboxylation of C–H Bonds with Carbon Dioxide. Angew. Chem. Int. Ed. 2010, 49, 8670–8673. [Google Scholar] [CrossRef] [PubMed]
- Inomata, H.; Ogata, K.; Fukuzawa, S.; Hou, Z. Direct C–H Carboxylation with Carbon Dioxide Using 1,2,3-Triazol-5-Ylidene Copper(I) Complexes. Org. Lett. 2012, 14, 3986–3989. [Google Scholar] [CrossRef] [PubMed]
- Boogaerts, I.I.F.; Nolan, S.P. Carboxylation of C−H Bonds Using N.-Heterocyclic Carbene Gold(I) Complexes. J. Am. Chem. Soc. 2010, 132, 8858–8859. [Google Scholar] [CrossRef]
- Sasano, K.; Takaya, J.; Iwasawa, N. Palladium(II)-Catalyzed Direct Carboxylation of Alkenyl C–H Bonds with CO2. J. Am. Chem. Soc. 2013, 135, 10954–10957. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, H.; Takaya, J.; Iwasawa, N. Rhodium(I)-Catalyzed Direct Carboxylation of Arenes with CO2 via Chelation-Assisted C−H Bond Activation. J. Am. Chem. Soc. 2011, 133, 1251–1253. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, S.; Sekine, K.; Ishida, T.; Yamada, T. C–C Bond Formation with Carbon Dioxide Promoted by a Silver Catalyst. Angew. Chem. Int. Ed. 2012, 51, 6989–6992. [Google Scholar] [CrossRef]
- Sekine, K.; Takayanagi, A.; Kikuchi, S.; Yamada, T. Silver-Catalyzed C–C Bond Formation with Carbon Dioxide: Significant Synthesis of Dihydroisobenzofurans. Chem. Commun. 2013, 49, 11320–11322. [Google Scholar] [CrossRef]
- Hoberg, H.; Minato, M. Palladium-Catalysed Dimerization of Isoprene with Carbon Dioxide in the Presence of Organotin Ethoxide and 1,8-Diazabicyclo[5.4.0]Undec-7-Ene (DBU). J. Organomet. Chem. 1991, 406, C25–C281991. [Google Scholar] [CrossRef]
- Lejkowski, M.L.; Lindner, R.; Kageyama, T.; Bódizs, G.É.; Plessow, P.N.; Müller, I.B.; Schäfer, A.; Rominger, F.; Hofmann, P.; Futter, C.; et al. The First Catalytic Synthesis of an Acrylate from CO2 and an Alkene—A Rational Approach. Chem.–A Eur. J. 2012, 18, 14017–14025. [Google Scholar] [CrossRef]
- Plessow, P.N.; Schäfer, A.; Limbach, M.; Hofmann, P. Acrylate Formation from CO2 and Ethylene Mediated by Nickel Complexes: A Theoretical Study. Organometallics 2014, 33, 3657–3668. [Google Scholar] [CrossRef]
- Hou, J.; Li, J.; Wu, J. Recent Development of Light-Mediated Carboxylation Using CO2 as the Feedstock. Asian J. Org. Chem. 2018, 7, 1439–1447. [Google Scholar] [CrossRef]
- Shimomaki, K.; Murata, K.; Martin, R.; Iwasawa, N. Visible-Light-Driven Carboxylation of Aryl Halides by the Combined Use of Palladium and Photoredox Catalysts. J. Am. Chem. Soc. 2017, 139, 9467–9470. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Wang, S.; König, B. Carboxylation of Aromatic and Aliphatic Bromides and Triflates with CO2 by Dual Visible-Light–Nickel Catalysis. Angew. Chem. Int. Ed. 2017, 56, 13426–13430. [Google Scholar] [CrossRef] [PubMed]
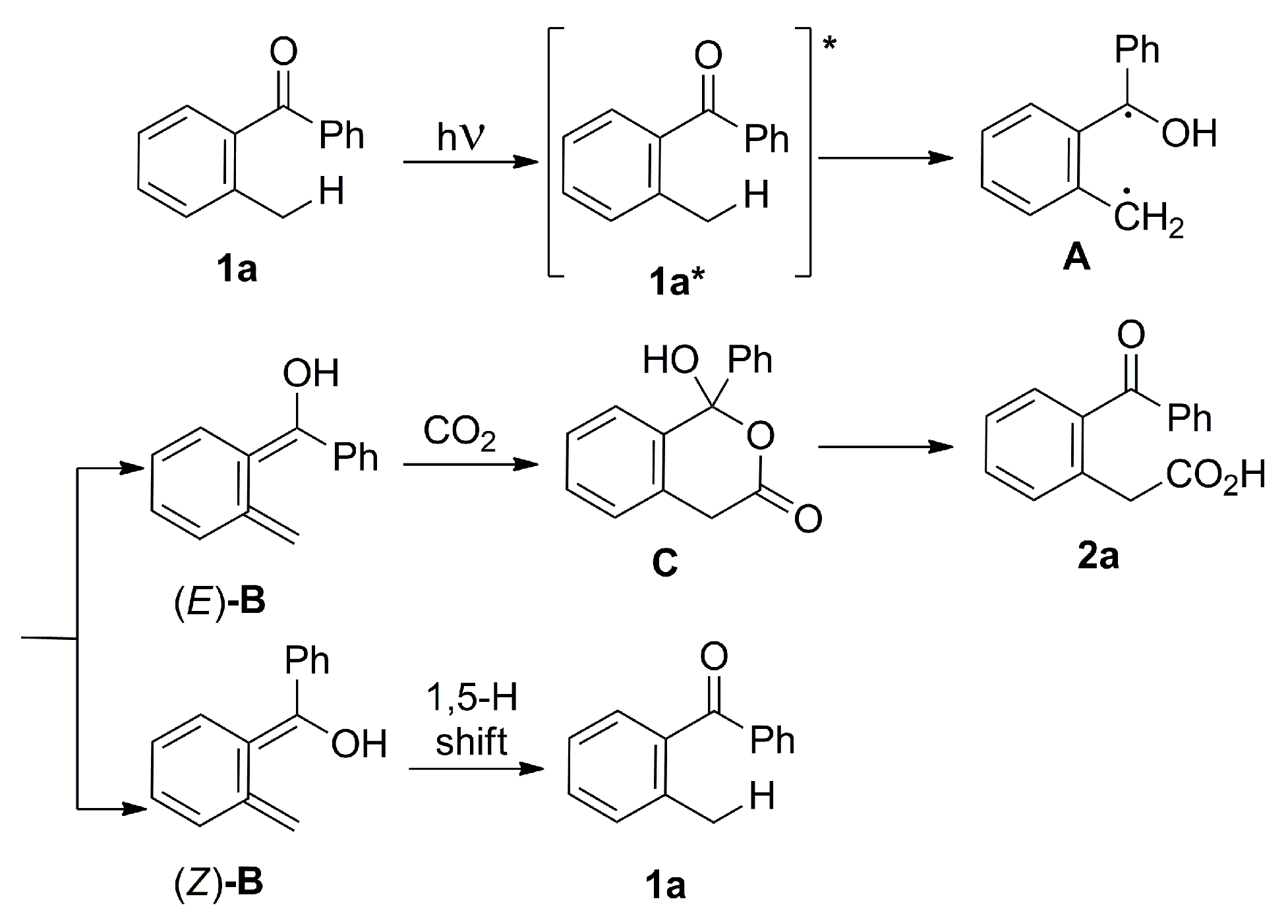
- Masuda, Y.; Ishida, N.; Murakami, M. Light-Driven Carboxylation of o-Alkylphenyl Ketones with CO2. J. Am. Chem. Soc. 2015, 137, 14063–14066. [Google Scholar] [CrossRef] [PubMed]
- Ishida, N.; Masuda, Y.; Uemoto, S.; Murakami, M. A Light/Ketone/Copper System for Carboxylation of Allylic C−H Bonds of Alkenes with CO2. Chem.–A Eur. J. 2016, 22, 6524–6527. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.M.; Johnson, J.B.; Rovis, T. Nickel-Catalyzed Reductive Carboxylation of Styrenes Using CO2. J. Am. Chem. Soc. 2008, 130, 14936–14937. [Google Scholar] [CrossRef]
- Murata, K.; Numasawa, N.; Shimomaki, K.; Takaya, J.; Iwasawa, N. Construction of a Visible Light-Driven Hydrocarboxylation Cycle of Alkenes by the Combined Use of Rh(I) and Photoredox Catalysts. Chem. Commun. 2017, 53, 3098–3101. [Google Scholar] [CrossRef]
- Meng, Q.-Y.; Wang, S.; Huff, G.S.; König, B. Ligand-Controlled Regioselective Hydrocarboxylation of Styrenes with CO2 by Combining Visible Light and Nickel Catalysis. J. Am. Chem. Soc. 2018, 140, 3198–3201. [Google Scholar] [CrossRef]
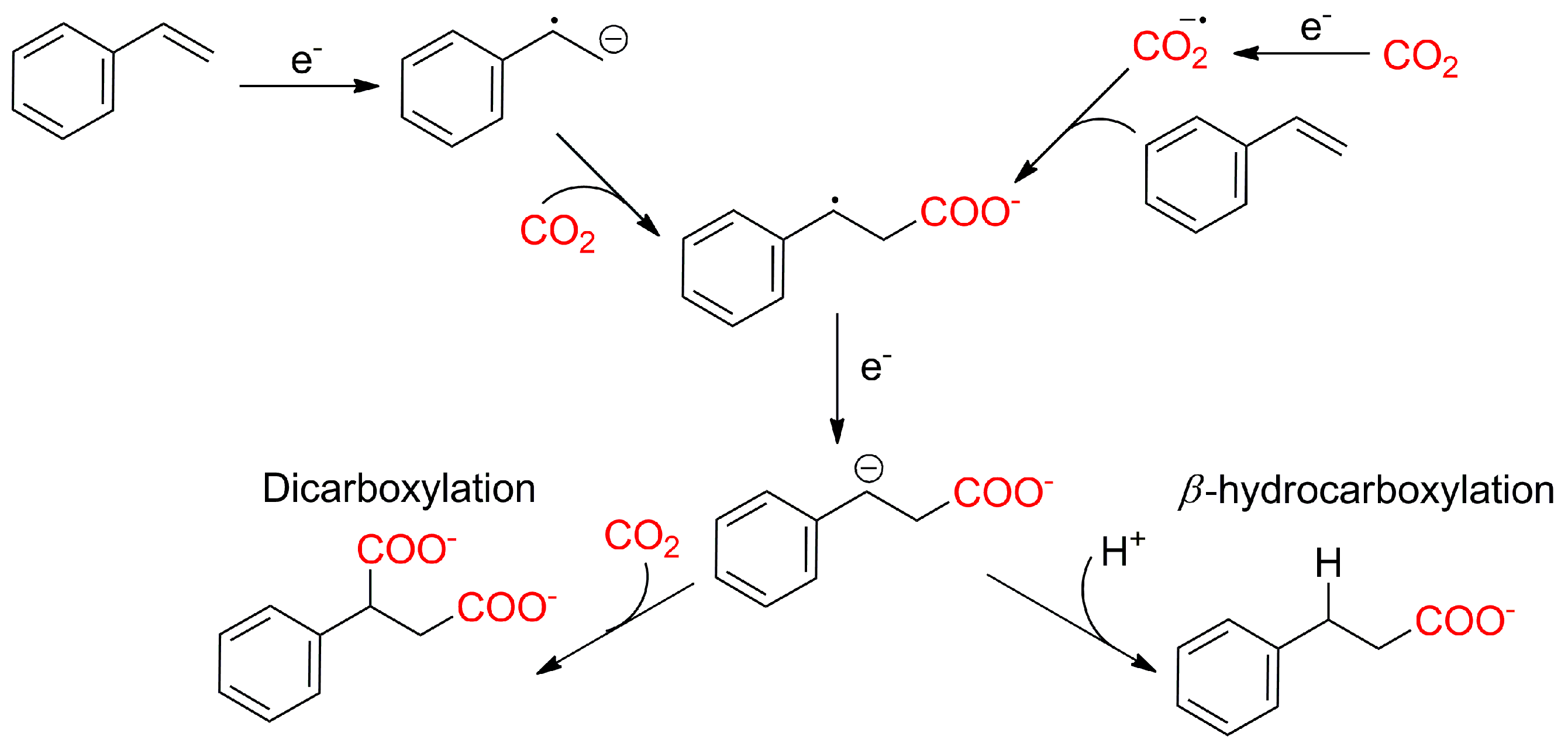
- Seo, H.; Liu, A.; Jamison, T.F. Direct β-Selective Hydrocarboxylation of Styrenes with CO2 Enabled by Continuous Flow Photoredox Catalysis. J. Am. Chem. Soc. 2017, 139, 13969–13972. [Google Scholar] [CrossRef]
- Yatham, V.R.; Shen, Y.; Martin, R. Catalytic Intermolecular Dicarbofunctionalization of Styrenes with CO2 and Radical Precursors. Angew. Chem. Int. Ed. 2017, 56, 10915–10919. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Miao, M.; Huang, H.; Yan, S.; Yin, Z.; Zhou, W.; Yu, D. Visible-Light-Driven Iron-Promoted Thiocarboxylation of Styrenes and Acrylates with CO2. Angew. Chem. Int. Ed. 2017, 56, 15416–15420. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Ee, A.; Feng, W.; Xu, J.-H.; Zhao, Y.; Wu, J. Visible-Light-Driven Alkyne Hydro-/Carbocarboxylation Using CO2 via Iridium/Cobalt Dual Catalysis for Divergent Heterocycle Synthesis. J. Am. Chem. Soc. 2018, 140, 5257–5263. [Google Scholar] [CrossRef] [PubMed]
- Bertuzzi, G.; Cerveri, A.; Lombardi, L.; Bandini, M. Tandem Functionalization-Carboxylation Reactions of π-Systems with CO2. Chin. J. Chem. 2021, 39, 3116–3126. [Google Scholar] [CrossRef]
- Lin, Q.; Li, L.; Luo, S. Asymmetric Electrochemical Catalysis. Chem.–A Eur. J. 2019, 25, 10033–10044. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Luo, S. Tailoring Radicals by Asymmetric Electrochemical Catalysis. Org. Chem. Front. 2020, 7, 2997–3000. [Google Scholar] [CrossRef]
- Kim, Y.; Park, G.D.; Balamurugan, M.; Seo, J.; Min, B.K.; Nam, K.T. Electrochemical β-Selective Hydrocarboxylation of Styrene Using CO2 and Water. Adv. Sci. 2020, 7, 1900137. [Google Scholar] [CrossRef]
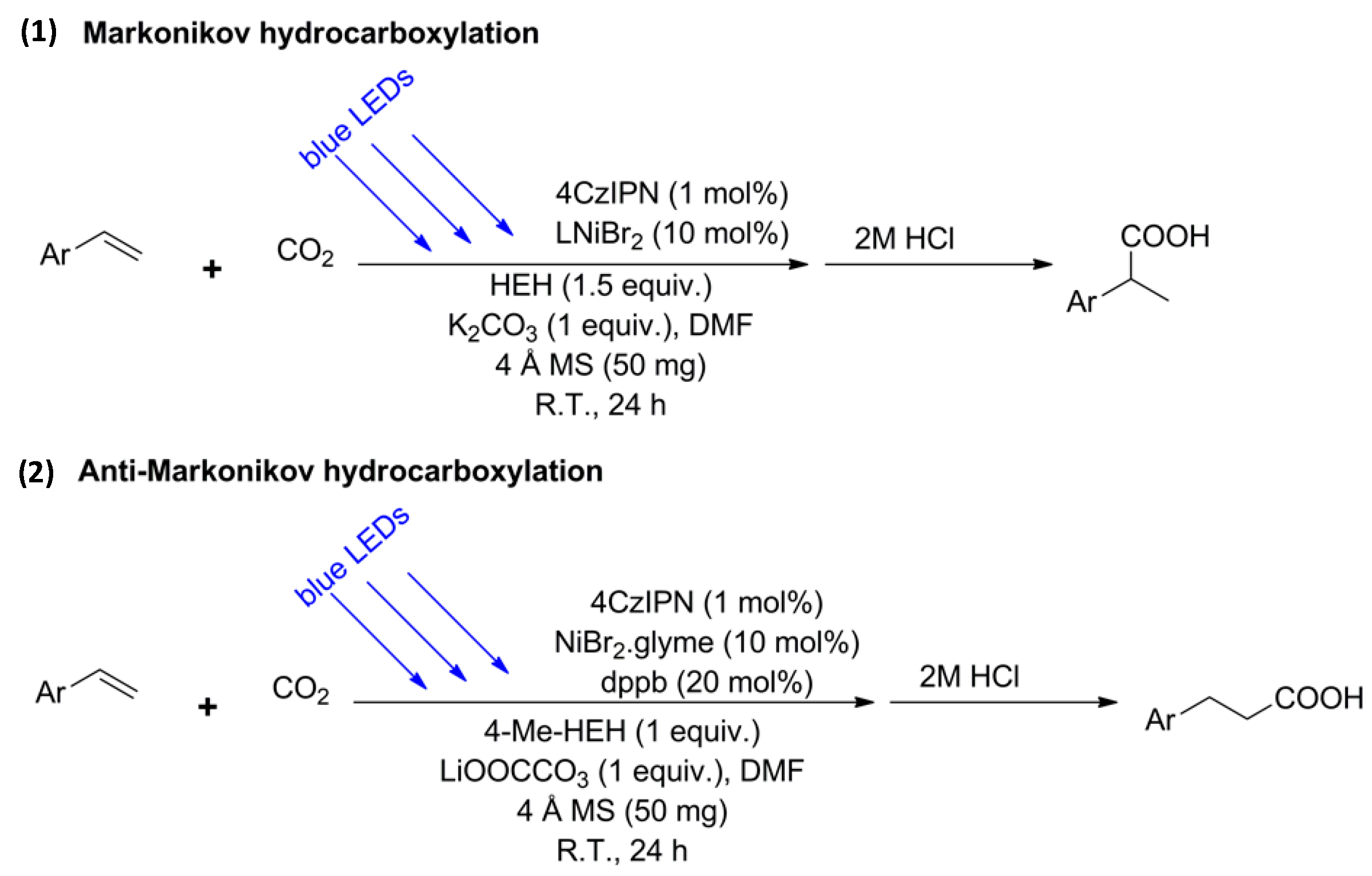
- Alkayal, A.; Tabas, V.; Montanaro, S.; Wright, I.A.; Malkov, A.V.; Buckley, B.R. Harnessing Applied Potential: Selective β-Hydrocarboxylation of Substituted Olefins. J. Am. Chem. Soc. 2020, 142, 1780–1785. [Google Scholar] [CrossRef]
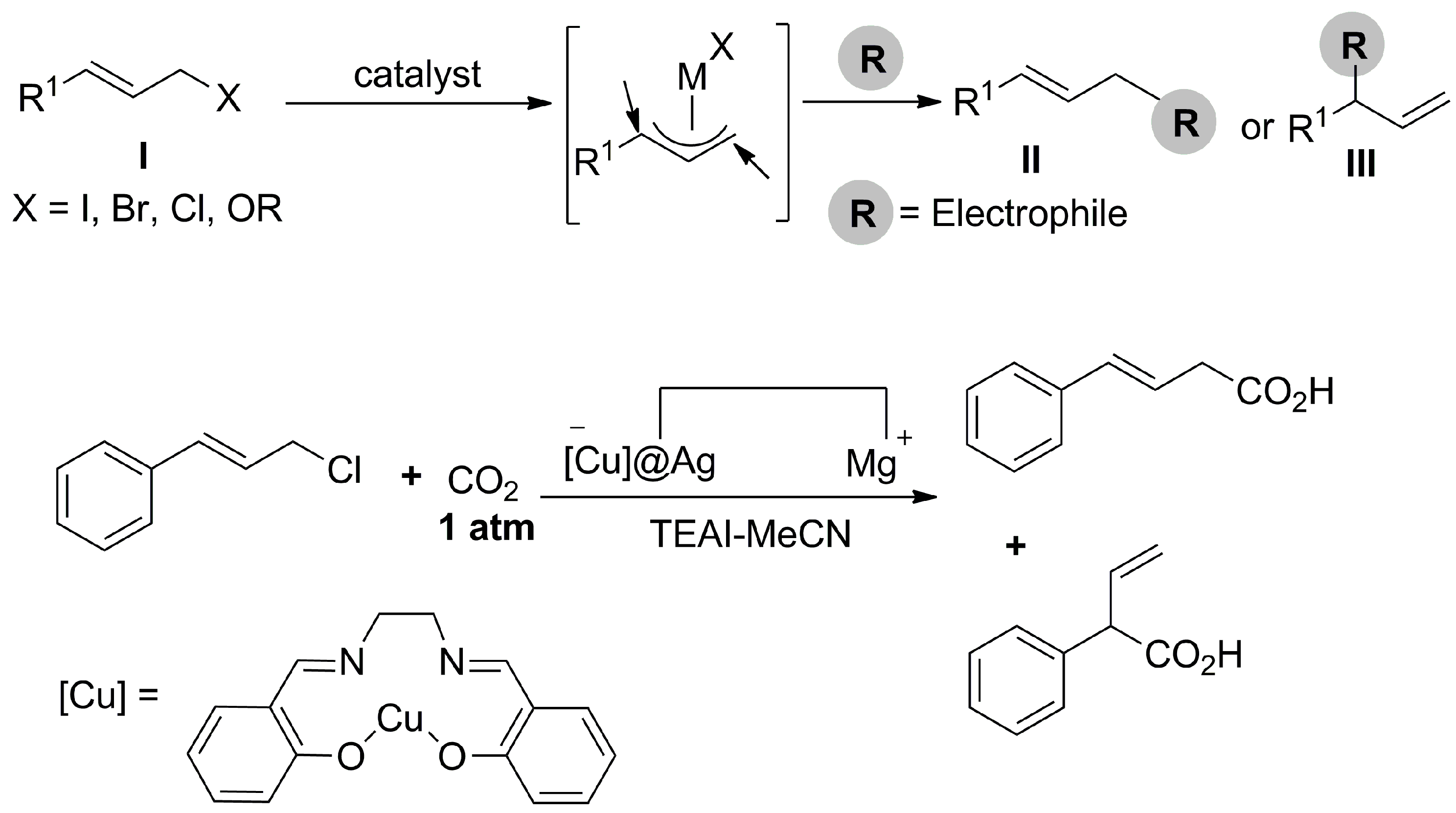
- Sheta, A.M.; Mashaly, M.A.; Said, S.B.; Elmorsy, S.S.; Malkov, A.V.; Buckley, B.R. Selective α,δ-Hydrocarboxylation of Conjugated Dienes Utilizing CO2 and Electrosynthesis. Chem. Sci. 2020, 11, 9109–9114. [Google Scholar] [CrossRef]
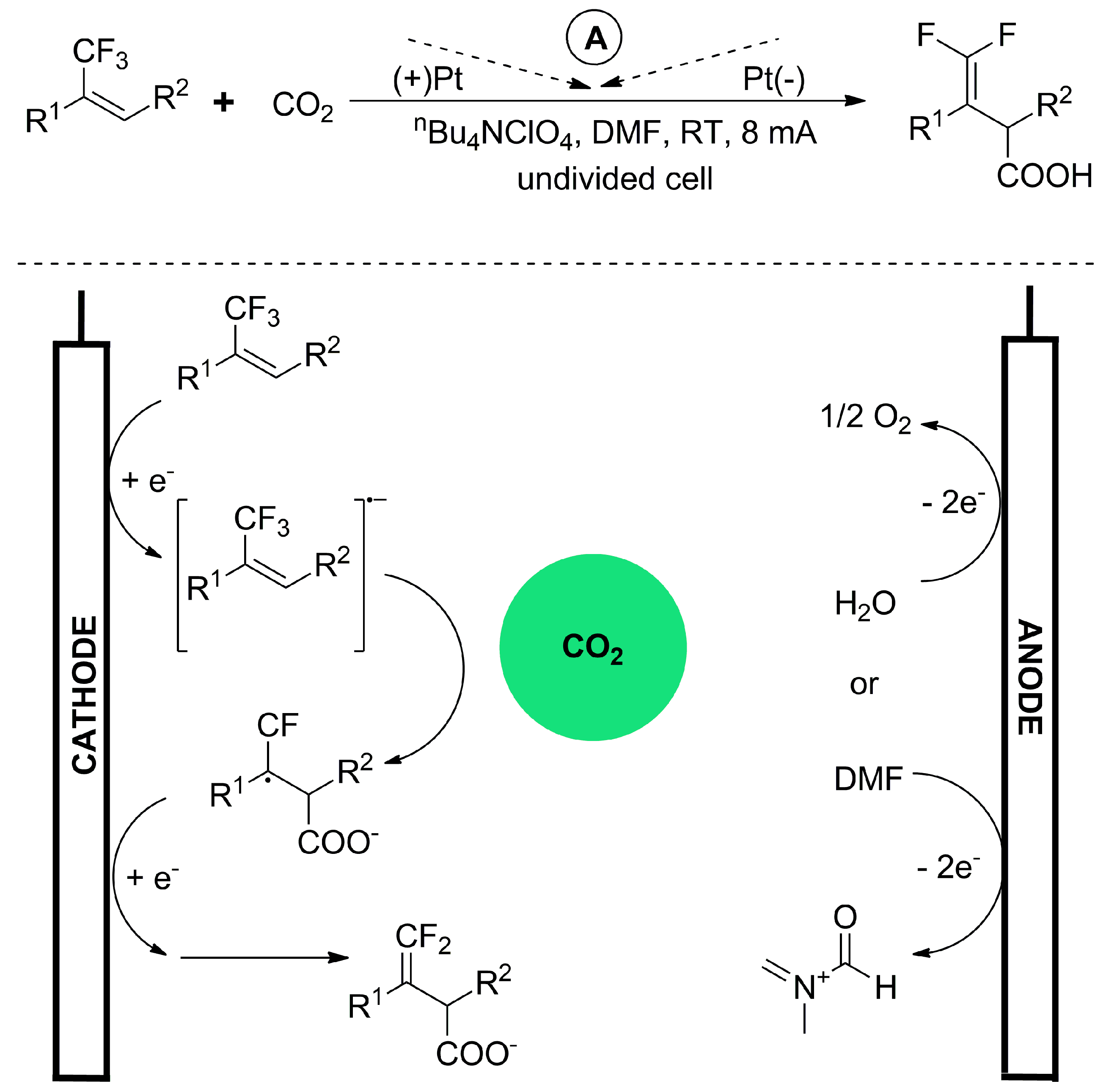
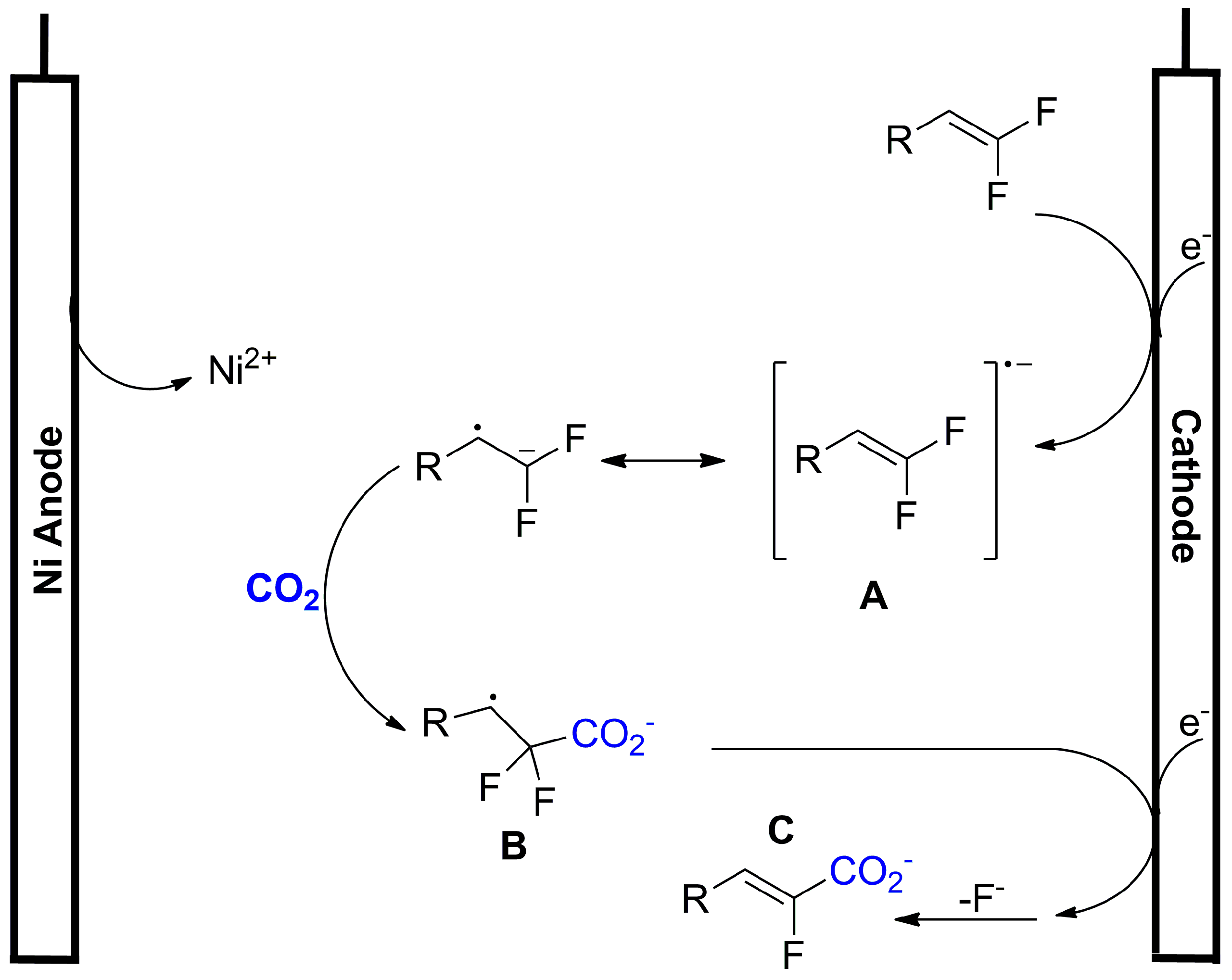
- Gao, X.-T.; Zhang, Z.; Wang, X.; Tian, J.-S.; Xie, S.-L.; Zhou, F.; Zhou, J. Direct Electrochemical Defluorinative Carboxylation of α-CF 3 Alkenes with Carbon Dioxide. Chem. Sci. 2020, 11, 10414–10420. [Google Scholar] [CrossRef]
- Zhang, W.; Lin, S. Electroreductive Carbofunctionalization of Alkenes with Alkyl Bromides via a Radical-Polar Crossover Mechanism. J. Am. Chem. Soc. 2020, 142, 20661–20670. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; He, D.; Chen, R.; Gao, T.; Wang, Y.; Chen, H.; Zhang, Y.; Wang, D. Understanding Photoelectrochemical Kinetics in a Model CO2 Fixation Reaction. Phys. Chem. Chem. Phys. 2019, 21, 17517–17520. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Tian, K.; He, D.; Gao, T.; Yang, G.; Xu, J.; Chen, H.; Wang, D.; Zhang, Y. Carboxylation of α,β-Unsaturated Ketones by CO2 Fixation through Photoelectro-Chemistry. ACS Appl. Energy Mater. 2020, 3, 5813–5818. [Google Scholar] [CrossRef]
- Quan, Y.; Yu, R.; Zhu, J.; Guan, A.; Lv, X.; Yang, C.; Li, S.; Wu, J.; Zheng, G. Efficient Carboxylation of Styrene and Carbon Dioxide by Single-Atomic Copper Electrocatalyst. J. Colloid. Interface Sci. 2021, 601, 378–384. [Google Scholar] [CrossRef]
- Lan, Y.-C.; Wang, H.; Wu, L.-X.; Zhao, S.-F.; Gu, Y.-Q.; Lu, J.-X. Electroreduction of Dibromobenzenes on Silver Electrode in the Presence of CO2. J. Electroanal. Chem. 2012, 664, 33–38. [Google Scholar] [CrossRef]
- Zhang, J.; Niu, D.; Lan, Y.; Wang, H.; Zhang, G.; Lu, J. Electrocatalytic Carboxylation of Arylic Bromides at Silver Cathode in the Presence of Carbon Dioxide. Synth. Commun. 2011, 41, 3720–3727. [Google Scholar] [CrossRef]
- Mena, S.; Sanchez, J.; Guirado, G. Electrocarboxylation of 1-Chloro-(4-Isobutylphenyl)Ethane with a Silver Cathode in Ionic Liquids: An Environmentally Benign and Efficient Way to Synthesize Ibuprofen. RSC Adv. 2019, 9, 15115–15123. [Google Scholar] [CrossRef]
- Mena, S.; Gallardo, I.; Guirado, G. Electrocatalytic Processes for the Valorization of CO2: Synthesis of Cyanobenzoic Acid Using Eco-Friendly Strategies. Catalysts 2019, 9, 413. [Google Scholar] [CrossRef]
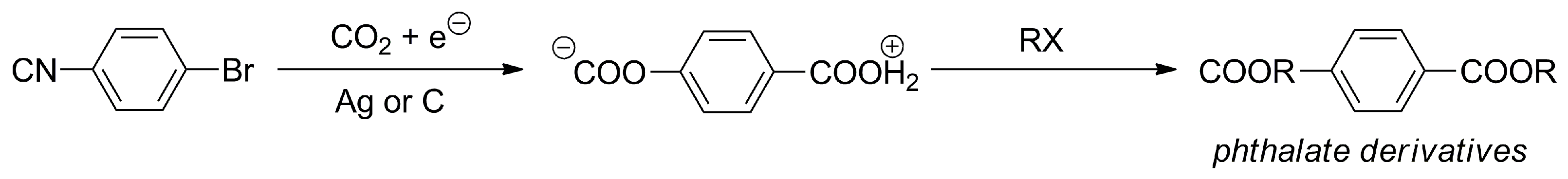
- Reche, I.; Mena, S.; Gallardo, I.; Guirado, G. Electrocarboxylation of Halobenzonitriles: An Environmentally Friendly Synthesis of Phthalate Derivatives. Electrochim. Acta 2019, 320, 134576. [Google Scholar] [CrossRef]
- Bazzi, S.; Le Duc, G.; Schulz, E.; Gosmini, C.; Mellah, M. CO2 Activation by Electrogenerated Divalent Samarium for Aryl Halide Carboxylation. Org. Biomol. Chem. 2019, 17, 8546–8550. [Google Scholar] [CrossRef]
- Xie, S.-L.; Gao, X.-T.; Wu, H.-H.; Zhou, F.; Zhou, J. Direct Electrochemical Defluorinative Carboxylation of Gem -Difluoroalkenes with Carbon Dioxide. Org. Lett. 2020, 22, 8424–8429. [Google Scholar] [CrossRef]
- Medvedev, J.J.; Medvedeva, X.V.; Li, F.; Zienchuk, T.A.; Klinkova, A. Electrochemical CO2 Fixation to α-Methylbenzyl Bromide in Divided Cells with Nonsacrificial Anodes and Aqueous Anolytes. ACS Sustain. Chem. Eng. 2019, 7, 19631–19639. [Google Scholar] [CrossRef]
- Corbin, N.; Yang, D.-T.; Lazouski, N.; Steinberg, K.; Manthiram, K. Suppressing Carboxylate Nucleophilicity with Inorganic Salts Enables Selective Electrocarboxylation without Sacrificial Anodes. Chem. Sci. 2021, 12, 12365–12376. [Google Scholar] [CrossRef] [PubMed]
- Jiao, K.-J.; Li, Z.-M.; Xu, X.-T.; Zhang, L.-P.; Li, Y.-Q.; Zhang, K.; Mei, T.-S. Palladium-Catalyzed Reductive Electrocarboxylation of Allyl Esters with Carbon Dioxide. Org. Chem. Front. 2018, 5, 2244–2248. [Google Scholar] [CrossRef]
- Bazzi, S.; Schulz, E.; Mellah, M. Electrogenerated Sm(II)-Catalyzed CO 2 Activation for Carboxylation of Benzyl Halides. Org. Lett. 2019, 21, 10033–10037. [Google Scholar] [CrossRef]
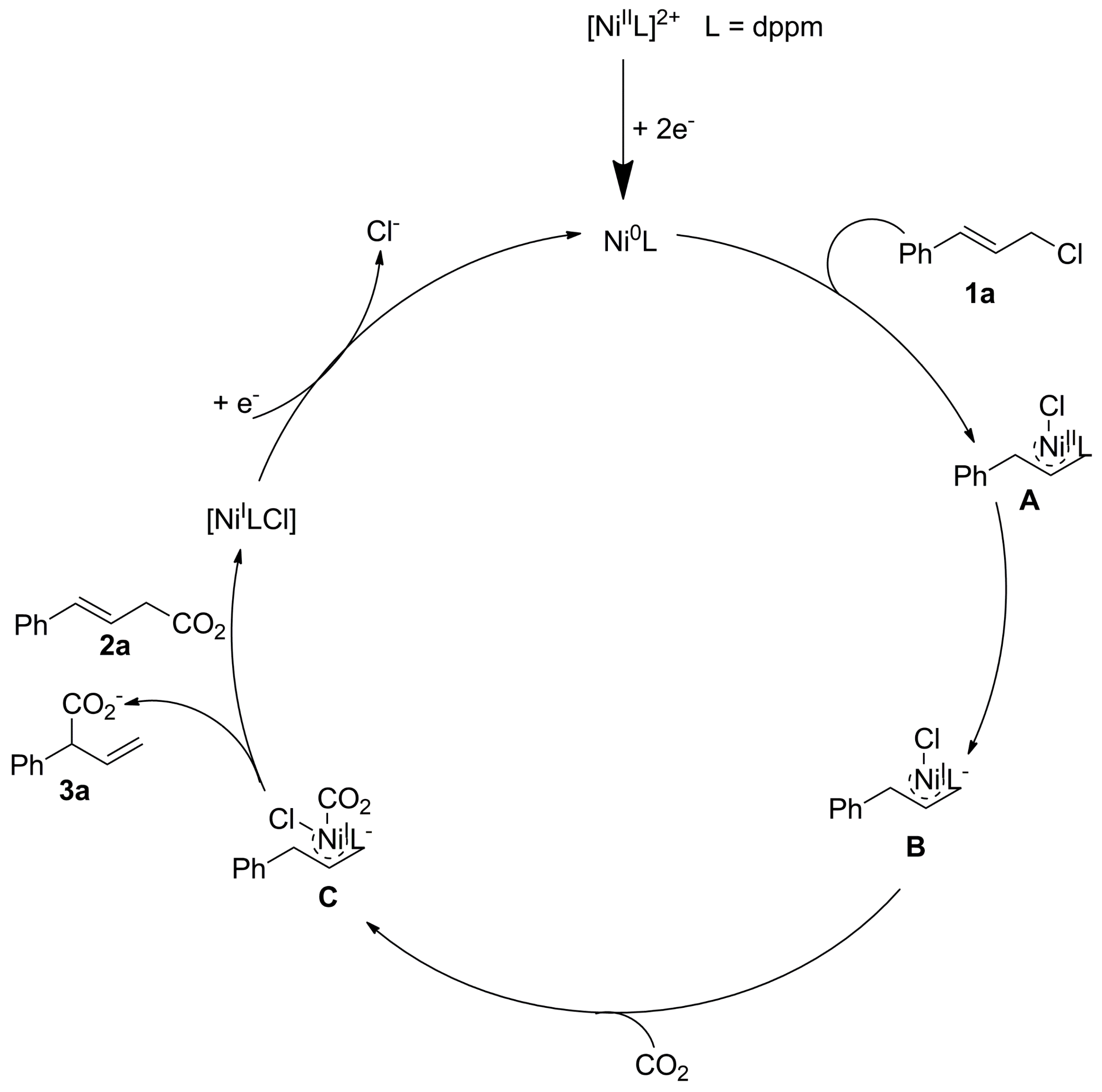
- Wu, L.-X.; Zhao, Y.-G.; Guan, Y.-B.; Wang, H.; Lan, Y.-C.; Wang, H.; Lu, J.-X. Silver Encapsulated Copper Salen Complex: Efficient Catalyst for Electrocarboxylation of Cinnamyl Chloride with CO2. RSC Adv. 2019, 9, 32628–32633. [Google Scholar] [CrossRef]
- Ang, N.W.J.; Oliveira, J.C.A.; Ackermann, L. Electroreductive Cobalt-Catalyzed Carboxylation: Cross-Electrophile Electrocoupling with Atmospheric CO2. Angew. Chem. Int. Ed. 2020, 59, 12842–12847. [Google Scholar] [CrossRef]
- Wu, L.-X.; Deng, F.-J.; Wu, L.; Wang, H.; Chen, T.; Guan, Y.-B.; Lu, J.-X. Nickel-Catalyzed Electrocarboxylation of Allylic Halides with CO2. New J. Chem. 2021, 45, 13137–13141. [Google Scholar] [CrossRef]
- Mohammadzadeh, S.; Zare, H.R.; Khoshro, H.; Ghobadi, K.; Benvidi, A. The Electrochemical Behavior of 4-Nitrobenzyl Bromide and Its Catalytic Activity for Reduction of CO2 in the Acetonitrile Solvent at the Cu/Pd/rGO/GCE Surface. Electrochim. Acta 2020, 352, 136483. [Google Scholar] [CrossRef]
- Senboku, H.; Sakai, K.; Fukui, A.; Sato, Y.; Yamauchi, Y. Efficient Synthesis of Mandel Acetates by Electrochemical Carboxylation of Benzal Diacetates. ChemElectroChem 2019, 6, 4158–4164. [Google Scholar] [CrossRef]
- Zhong, J.-S.; Yang, Z.-X.; Ding, C.-L.; Huang, Y.-F.; Zhao, Y.; Yan, H.; Ye, K.-Y. Desulfonylative Electrocarboxylation with Carbon Dioxide. J. Org. Chem. 2021, 86, 16162–16170. [Google Scholar] [CrossRef] [PubMed]
- Senboku, H.; Minemura, Y.; Suzuki, Y.; Matsuno, H.; Takakuwa, M. Synthesis of N-Boc-α-Amino Acids from Carbon Dioxide by Electrochemical Carboxylation of N -Boc-α-Aminosulfones. J. Org. Chem. 2021, 86, 16077–16083. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.-L.; Zhang, Z.-X.; Zhang, J.-J.; Li, S.-M.; Wang, H.; Lu, J.-X. Ordered Mesoporous Carbon Embedded with Cu Nanoparticle Materials for Electrocatalytic Synthesis of Benzyl Methyl Carbonate from Benzyl Alcohol and Carbon Dioxide. ACS Omega 2020, 5, 3498–3503. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Sohal, H.S.; Singh, B. Synthesis of α-Hydroxycarboxylic Acids from Various Aldehydes and Ketones by Direct Electrocarboxylation: A Facile, Efficient and Atom Economy Protocol. Asian J. Chem. 2021, 33, 839–845. [Google Scholar] [CrossRef]
- Tian, K.; Chen, R.; Xu, J.; Yang, G.; Xu, X.; Zhang, Y. Understanding the Photo- and Electro-Carboxylation of o-Methylbenzophenone with Carbon Dioxide. Catalysts 2020, 10, 664. [Google Scholar] [CrossRef]
- Blaskovich, M.A.T. Unusual Amino Acids in Medicinal Chemistry. J. Med. Chem. 2016, 59, 10807–10836. [Google Scholar] [CrossRef]
- Koshechko, V.G.; Titov, V.E.; Bondarenko, V.N.; Pokhodenko, V.D. Electrochemical Carboxylation of Fluorocontaining Imines with Preparation of Fluorinated N-Phenylphenylglycines. J. Fluor. Chem. 2008, 129, 701–706. [Google Scholar] [CrossRef]
- Qu, Y.; Tsuneishi, C.; Tateno, H.; Matsumura, Y.; Atobe, M. Green Synthesis of α-Amino Acids by Electrochemical Carboxylation of Imines in a Flow Microreactor. React. Chem. Eng. 2017, 2, 871–875. [Google Scholar] [CrossRef]
- Naito, Y.; Nakamura, Y.; Shida, N.; Senboku, H.; Tanaka, K.; Atobe, M. Integrated Flow Synthesis of α-Amino Acids by In Situ Generation of Aldimines and Subsequent Electrochemical Carboxylation. J. Org. Chem. 2021, 86, 15953–15960. [Google Scholar] [CrossRef]
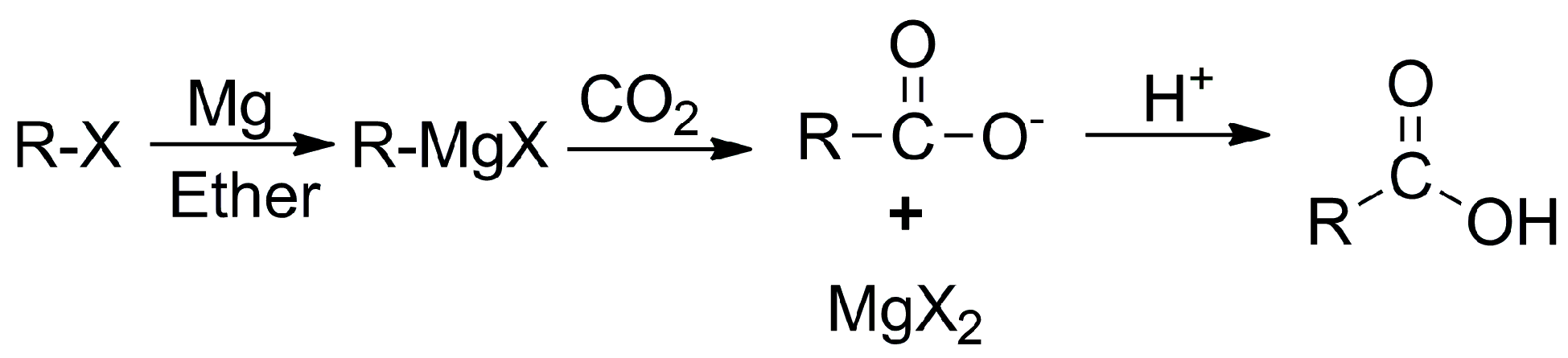

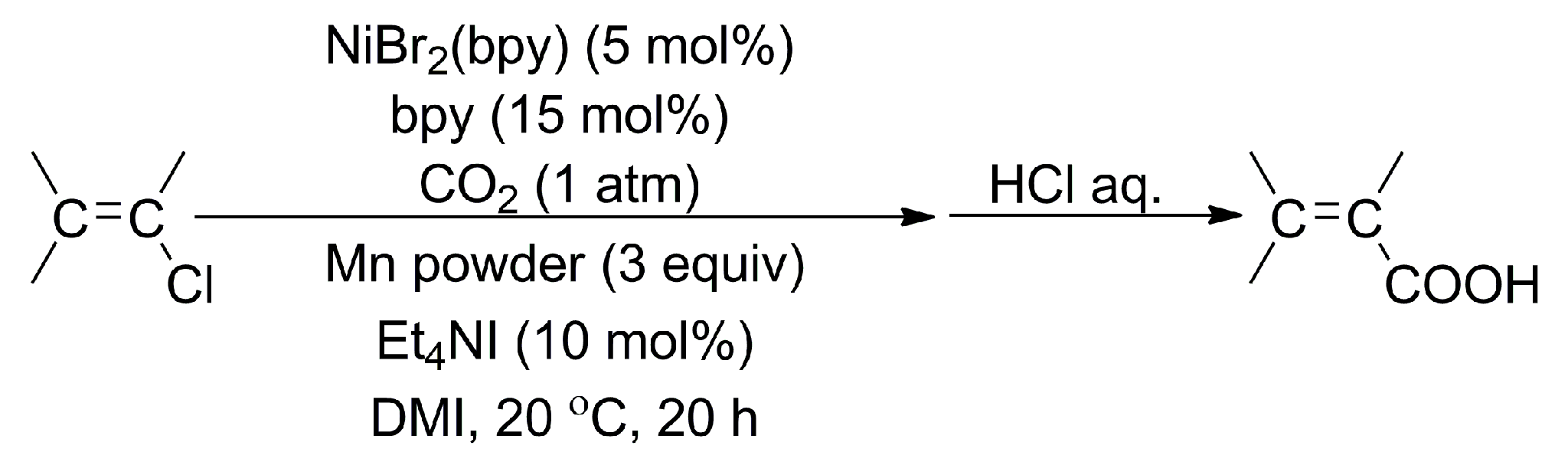







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Catalyst Element | Reactant | Reaction Conditions | Product | Reference |
|---|---|---|---|---|---|
| 1 | Ni cathode and Mg anode | Styrene | Bu4NBF4 as the electrolyte, water as the proton source, and DMF as the solvent | 65% β-hydrocarboxylated styrene | [112] |
| 2 | Carbon cathode and carbon anode | Styrene and di- and tr-substituted olefins | CO2 (1 atm), Et4NI (0.5 equiv), TEOA (1.0 equiv), DMF, single compartment cell, 10 V, 3.5 h rt, and Electrasyn 2.0 | β-hydrocarboxylated alkenes as a major regioisomer | [113] |
| 3 | Stainless steel cathode and carbon anode | Conjugated dienes | CO2 (1 atm), Et4NI (0.5 equiv.), TEOA (1.0 equiv.), DMF, single compartment cell, 10 V (60–100 mA), and 5 h rt | α,δ-hydrocarboxylated products | [114] |
| 4 | Pt cathode and Pt anode | α-CF3 alkenes | nBu4NClO4 (0.07 M), CO2 bubbling in DMF (7 mL), and 4–7 h | γ-carboxylated-fluorine-containing carboxylic acids | [115] |
| 5 | Graphite cathode and Mg anode | Alkenes and alkyl bromides | TBAPF6 (1.1 equiv), DMF (4.2 mL), CO2 balloon, Mg plate and graphite plate, undivided cell (ElectraSyn), constant current i = 10 mA, 4 F/mol, and 22 °C | α-substituent phenylacetic acids | [116] |
| 6 | Ag/AgBr as a reference electrode, Al as a counter electrode, and p-type Si nanowires as a working electrode | Trans-stilbene | 0.5 M of Bu4NBr and 20.0 mL of acetonitrile with CO2 bubbling | 2,3-diphenylpropanoic acid | [117] |
| 7 | SiNWs as a cathode, Al as an anode, and Ag/AgBr as a reference electrode | α,β-unsaturated ketones | Bu4NBr (2.0 mmol) in MeCN (20 mL) with CO2 bubbling | β-carboxyl ketones | [118] |
| 8 | Nitrogen-coordinated single-atomic Cu catalyst and Ag/AgI as a reference electrode | Styrene | Anhydrous acetonitrile, tetrabutylammonium bromide, and CO2 | Phenylsuccinic acid | [119] |
| # | Catalyst Element | Reactant | Reaction Conditions | Product | Reference |
|---|---|---|---|---|---|
| 1 | Pd as a catalyst, Pt electrode, and Mg rod as a sacrificial anode | Homostyrenyl acetates | DPPPh (5.3 mol.%), Et4NOTs (0.07 M) and EtOH (1.0 equiv.), DMF (6 mL), and CO2 (1 atm) | α-aryl carboxylic acids, up to 95% yield, up to 67% ee | [129] |
| 2 | Sm(II) anode and stainless steel cathode | Benzyl halides | nBu4NX (1 equiv) in CH3CN (50 mL), TMSCl additive, and dry ice | Phenylacetic acids, up to 98% yield | [130] |
| 3 | [Cu]@Ag as a cathode and a Mg anode | Cinnamyl chloride | tetraethyl ammonium iodide (TEAI), CH3CN, and CO2 | β,γ-unsaturated carboxylic acids, up to 98% yield | [131] |
| 4 | Cobalt(II) acetate catalyst and Mg and Ni foil electrodes | Allylic chlorides | PPh3, nBu4NPF6, DMF, and CO2 | Styrylacetic acids | [132] |
| 5 | Ni(II) catalyst, Mg anode, and Ag cathode | Allylic halides | tetraethyl ammonium iodide (TEAI), DMF, and CO2 | β,γ-unsaturated carboxylic acids, up to 96% yield | [133] |
| 6 | 4-Nitrobenzyl bromide as an autocatalyst and a Cu/Pd/rGO/GCE electrode | 4-Nitrobenzyl bromide | TBAP (0.05 M), CH3CN, and CO2 | 4-nitrophenylacetic acid | [134] |
| 7 | Pt plate cathode and Mg rod anode | Benzal diacetates | Bu4NBF4, DMF or CH3CN, and CO2 | Mandel acetates and 2-acetoxy-2-arylacetic acids | [135] |
| 8 | Mg anode and Pt cathode | Organic sulfones | TBAI, DMF, and CO2 | 2-phenylacetic acids, 90–99% yields | [136] |
| 9 | Pt plate cathode and Mg rod anode | N-Boc-α-aminosulfones | Bu4NBF4, DMF, and CO2 | N-Boc-α-amino acids, up to 87% yields | [137] |
| 10 | Cu/ordered mesoporous carbons and graphite anode | Benzyl alcohol | TBAI, CH3CN, and CO2 (1 atm) | Benzyl methyl carbonate, 69.7% yield | [138] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, S.R.; Mazumdar, N.J.; Centeno-Pedrazo, A.; Deka, D.; Artioli, N.; Manyar, H. Recent Advances in Catalyst Design for Carboxylation Using CO2 as the C1 Feedstock. Catalysts 2023, 13, 1489. https://doi.org/10.3390/catal13121489
Shah SR, Mazumdar NJ, Centeno-Pedrazo A, Deka D, Artioli N, Manyar H. Recent Advances in Catalyst Design for Carboxylation Using CO2 as the C1 Feedstock. Catalysts. 2023; 13(12):1489. https://doi.org/10.3390/catal13121489
Chicago/Turabian StyleShah, Sagarkumar Rajendrakumar, Nayan Jyoti Mazumdar, Ander Centeno-Pedrazo, Dhanapati Deka, Nancy Artioli, and Haresh Manyar. 2023. "Recent Advances in Catalyst Design for Carboxylation Using CO2 as the C1 Feedstock" Catalysts 13, no. 12: 1489. https://doi.org/10.3390/catal13121489
APA StyleShah, S. R., Mazumdar, N. J., Centeno-Pedrazo, A., Deka, D., Artioli, N., & Manyar, H. (2023). Recent Advances in Catalyst Design for Carboxylation Using CO2 as the C1 Feedstock. Catalysts, 13(12), 1489. https://doi.org/10.3390/catal13121489