
De Novo Synthesis of Polysubstituted 3-Hydroxypyridines Via “Anti-Wacker”-Type Cyclization †

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
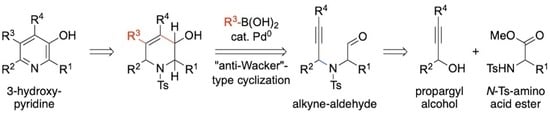
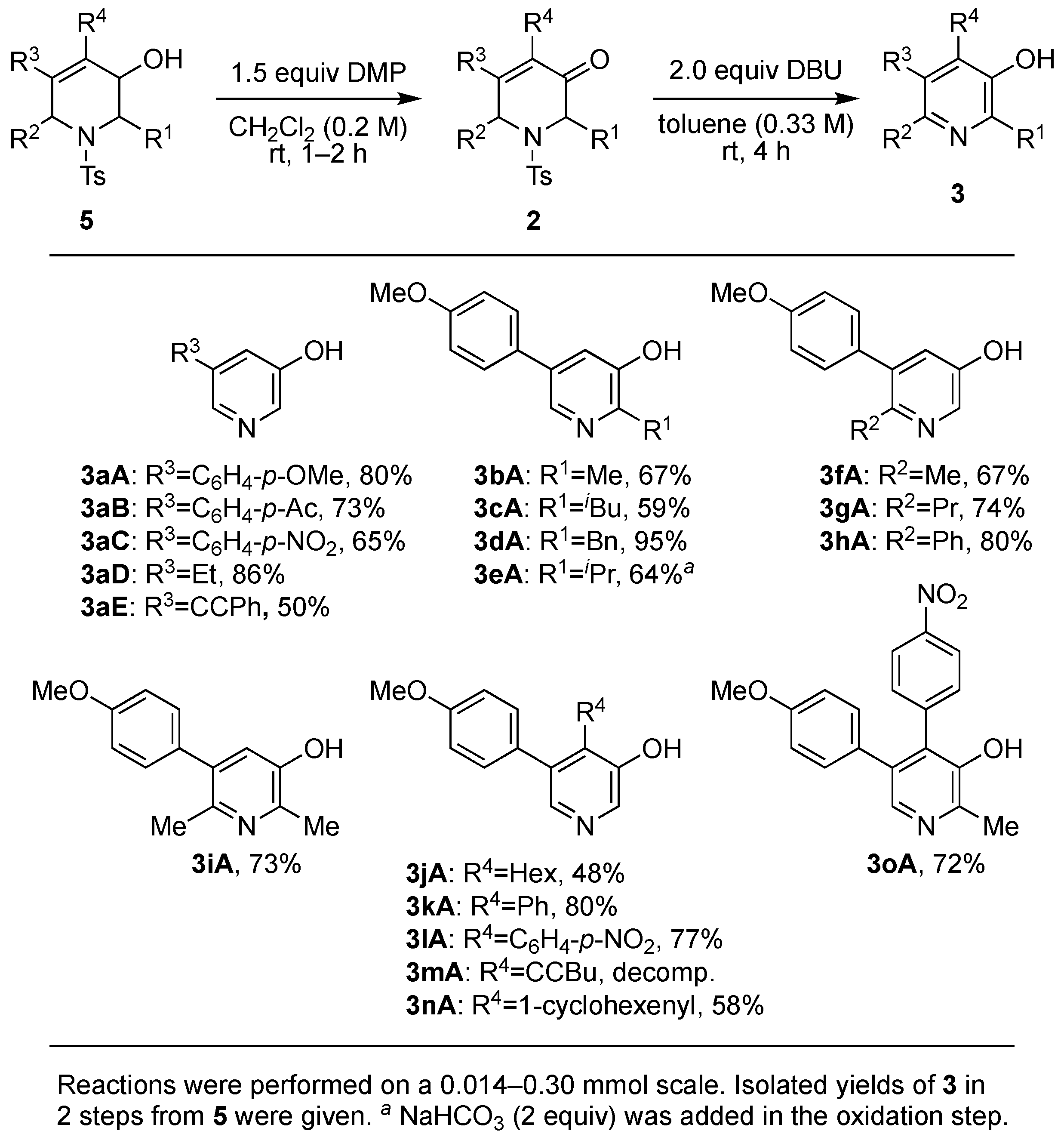
2. Results and Discussion
3. Materials and Methods
3.1. General Techniques
3.2. Materials
3.3. Methods
3.3.1. General Procedure for the Pd(PPh3)4-Catalyzed Arylative Cyclizations of Terminal Alkyne-Aldehyde 4a–i with Arylboronic Acid 7A–C
Procedure for 5-(4-methoxyphenyl)-1-tosyl-1,2,3,6-tetrahydropyridin-3-ol (5aA)
Procedure for 1-(4-(5-hydroxy-1-tosyl-1,2,5,6-tetrahydropyridin-3-yl)phenyl)ethan-1-one (5aB)
Procedure for 5-(4-nitrophenyl)-1-tosyl-1,2,3,6-tetrahydropyridin-3-ol (5aC)
Procedure for (2S,3S)-5-(4-methoxyphenyl)-2-methyl-1-tosyl-1,2,3,6-tetrahydropyridin-3-ol (5bA)
Procedure for 2-isobutyl-5-(4-methoxyphenyl)-1-tosyl-1,2,3,6-tetrahydropyridin-3-ol (5cA)
Procedure for (2R*, 3R*)-2-benzyl-5-(4-methoxyphenyl)-1-tosyl-1,2,3,6-tetrahydro-pyridin-3-ol (5dA)
Procedure for 2-isopropyl-5-(4-methoxyphenyl)-1-tosyl-1,2,3,6-tetrahydropyridin-3-ol (5eA)
Procedure for 5-(4-methoxyphenyl)-6-methyl-1-tosyl-1,2,3,6-tetrahydropyridin-3-ol (5fA)
Procedure for (3R*,6S*)-5-(4-methoxyphenyl)-6-propyl-1-tosyl-1,2,3,6-tetrahydropyridin-3-ol (5gA)
Procedure for (3R*,6S*)-5-(4-methoxyphenyl)-6-phenyl-1-tosyl-1,2,3,6-tetrahydropyridin-3-ol (5hA)
Procedure for (2S,6R)-5-(4-methoxyphenyl)-2,6-dimethyl-1-tosyl-1,2,3,6-tetrahydropyridin-3-ol (5iA) and (2S,5R)-4-((E)-4–methoxybenzylidene)-2,5-dimethyl-1-tosylpyrrolidin-3-ol (8iA)
3.3.2. Procedure for 5-ethyl-1-tosyl-1,2,3,6-tetrahydropyridin-3-ol (5aD)
3.3.3. Procedure for 5-(phenylethynyl)-1-tosyl-1,2,3,6-tetrahydropyridin-3-ol (5aE)
3.3.4. General Procedure for the Pd/PCy3-Catalyzed Arylative Cyclizations of Internal Alkyne-Aldehyde 4j–o with 7A
Procedure for 4-hexyl-5-(4-methoxyphenyl)-1-tosyl-1,2,3,6-tetrahydropyridin-3-ol (5jA) and (E)-4-(1-(4-methoxyphenyl)heptyli–dene)-1-tosylpyrrolidin-3-ol (8jA)
Procedure for 5-(4-methoxyphenyl)-4-phenyl-1-tosyl-1,2,3,6-tetrahydropyridin-3-ol (5kA)
Procedure for 5-(4-methoxyphenyl)-4-(4-nitrophenyl)-1-tosyl-1,2,3,6-tetrahydropyridin-3-ol (5lA)
Procedure for 4-(hex-1-ynyl)-5-(4-methoxyphenyl)-1-(toluene-4-sulfonyl)-1,2,3,6-tetrahydropyridin-3-ol (5mA)
Procedure for 4-(cyclohex-1-en-1-yl)-5-(4-methoxyphenyl)-1-tosyl-1,2,3,6-tetrahydropyridin-3-ol (5nA) and (E)-4-(cyclohex-1–en-1-yl(4-methoxyphenyl)methylene)-1-tosylpyrrolidin-3-ol (8nA)
Procedure for (2S,3S)-5-(4-methoxyphenyl)-2-methyl-4-(4-nitrophenyl)-1-tosyl-1,2,3,6-tetrahydropyridin-3-ol (5oA)
3.3.5. General Procedure for the Transformations of Tetrahydropyridine 5 into 3-Hydroxypyridine 3
Procedure for 5-(4-methoxyphenyl)pyridin-3-ol (3aA)
Procedure for 1-(4-(5-hydroxypyridin-3-yl)phenyl)ethan-1-one (3aB)
Procedure for 5-(4-nitrophenyl)pyridin-3-ol (3aC)
Procedure for 5-ethylpyridin-3-ol (3aD)
Procedure for 5-(phenylethynyl)pyridin-3-ol (3aE)
Procedure for 5-(4-methoxyphenyl)-2-methylpyridin-3-ol (3bA)
Procedure for 2-isobutyl-5-(4-methoxyphenyl)pyridin-3-ol (3cA)
Procedure for 2-benzyl-5-(4-methoxyphenyl)pyridin-3-ol (3dA)
Procedure for 2-isopropyl-5-(4-methoxyphenyl)pyridin-3-ol (3eA)
Procedure for 5-(4-methoxyphenyl)-6-methylpyridin-3-ol (3fA)
Procedure for 5-(4-methoxyphenyl)-6-propylpyridin-3-ol (3gA)
Procedure for 5-(4-methoxyphenyl)-6-phenylpyridin-3-ol (3hA)
Procedure for 5-(4-methoxyphenyl)-2,6-dimethylpyridin-3-ol (3iA)
Procedure for 4-hexyl-5-(4-methoxyphenyl)pyridin-3-ol (3jA)
Procedure for 5-(4-methoxyphenyl)-4-phenylpyridin-3-ol (3kA)
Procedure for 5-(4-methoxyphenyl)-4-(4-nitrophenyl)pyridin-3-ol (3lA)
Procedure for 4-(cyclohex-1-en-1-yl)-5-(4-methoxyphenyl)pyridin-3-ol (3nA)
Procedure for 5-(4-methoxyphenyl)-2-methyl-4-(4-nitrophenyl)pyridin-3-ol (3oA)
3.3.6. Procedure for 2-benzyl-3-(3-methoxyphenyl)-5-(4-methoxyphenyl)pyridine (10)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Schmidt, A. Biologically Active Mesomeric Betaines and Alkaloids, Derived from 3-Hydroxypyridine, Pyridin-N-oxide, Nicotinic Acid and Picolinic Acid: Three Types of Conjugation and Their Consequences. Curr. Org. Chem. 2004, 8, 653–670. [Google Scholar] [CrossRef]
- Baumann, M.; Baxendale, I.R. An overview of the synthetic routes to the best selling drugs containing 6-membered heterocycles. Beilstein J. Org. Chem. 2013, 9, 2265–2319. [Google Scholar] [CrossRef] [PubMed]
- de Ruiter, G.; Lahav, M.; van der Boom, M.E. Pyridine Coordination Chemistry for Molecular Assemblies on Surfaces. Acc. Chem. Res. 2014, 47, 3407–3416. [Google Scholar] [CrossRef] [PubMed]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Altaf, A.A.; Shahzad, A.; Gul, Z.; Rasool, N.; Badshah, A.; Lal, B.; Khan, E. A Review on the Medicinal Importance of Pyridine Derivatives. J. Drug Des. Med. Chem. 2015, 1, 1–11. [Google Scholar]
- Guan, A.-Y.; Liu, C.-L.; Sun, X.-F.; Xie, Y.; Wang, M.-A. Discovery of pyridine-based agrochemicals by using Intermediate Derivatization Methods. Bioorg. Med. Chem. 2016, 24, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Prachayasittikul, S.; Pingaew, R.; Worachartcheewan, A.; Sinthupoom, N.; Prachayasittikul, V.; Ruchirawat, S.; Prachayasittikul, V. Roles of Pyridine and Pyrimidine Derivatives as Privileged Scaffolds in Anticancer Agents. Mini-Rev. Med. Chem. 2017, 17, 869–901. [Google Scholar] [CrossRef] [PubMed]
- Andersson, H.; Almqvist, F.; Olsson, R. Synthesis of 2-Substituted Pyridines via a Regiospecific Alkylation, Alkynylation, and Arylation of Pyridine N-Oxides. Org. Lett. 2007, 9, 1335–1337. [Google Scholar] [CrossRef]
- Do, H.-Q.; Kashif Khan, R.M.; Daugulis, O. A General Method for Copper-Catalyzed Arylation of Arene C-H Bonds. J. Am. Chem. Soc. 2008, 130, 15185–15192. [Google Scholar] [CrossRef]
- Li, M.; Hua, R. Gold(I)-catalyzed direct C–H arylation of pyrazine and pyridine with aryl bromides. Tetrahedron Lett. 2009, 50, 1478–1481. [Google Scholar] [CrossRef]
- Deng, J.Z.; Paone, D.V.; Ginnetti, A.T.; Kurihara, H.; Dreher, S.D.; Weissman, S.A.; Stauffer, S.R.; Burgey, C.S. Copper-Facilitated Suzuki Reactions: Application to 2-Heterocyclic Boronates. Org. Lett. 2009, 11, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Gøgsig, T.M.; Lindhardt, A.T.; Skrydstrup, T. Heteroaromatic Sulfonates and Phosphates as Electrophiles in Iron-Catalyzed Cross-Couplings. Org. Lett. 2009, 11, 4886–4888. [Google Scholar] [CrossRef] [PubMed]
- Wasa, M.; Worrell, B.T.; Yu, J.-Q. Pd0/PR3-Catalyzed Arylation of Nicotinic and Isonicotinic Acid Derivatives. Angew. Chem. Int. Ed. 2010, 49, 1275–1277. [Google Scholar] [CrossRef] [PubMed]
- Seiple, I.B.; Su, S.; Rodriguez, R.A.; Gianatassio, R.; Fujiwara, Y.; Sobel, A.L.; Baran, P.S. Direct C-H Arylation of Electron-Deficient Heterocycles with Arylboronic Acids. J. Am. Chem. Soc. 2010, 132, 13194–13196. [Google Scholar] [CrossRef]
- Berman, A.M.; Bergman, R.G.; Ellman, J.A. Rh(I)-Catalyzed Direct Arylation of Azines. J. Org. Chem. 2010, 75, 7863–7868. [Google Scholar] [CrossRef]
- Luzung, M.R.; Patel, J.S.; Yin, J. A Mild Negishi Cross-Coupling of 2-Heterocyclic Organozinc Reagents and Aryl Chlorides. J. Org. Chem. 2010, 75, 8330–8332. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Dixon, J.A.; O’Hara, F.; Funder, E.D.; Dixon, D.D.; Rodriguez, R.A.; Baxter, R.D.; Herlé, B.; Sach, N.; Collins, M.R.; et al. Practical and innate carbon–hydrogen functionalization of heterocycles. Nature 2012, 492, 95–100. [Google Scholar] [CrossRef]
- Dai, F.; Gui, Q.; Liu, J.; Yang, Z.; Chen, X.; Guo, R.; Tan, Z. Pd-catalyzed C3-selective arylation of pyridines with phenyl tosylates. Chem. Commun. 2013, 49, 4634–4636. [Google Scholar] [CrossRef]
- Liu, B.; Huang, Y.; Lan, J.; Song, F.; You, J. Pd-catalyzed oxidative C–H/C–H cross-coupling of pyridines with heteroarenes. Chem. Sci. 2013, 4, 2163–2167. [Google Scholar] [CrossRef]
- Sakashita, S.; Takizawa, M.; Sugai, J.; Ito, H.; Yamamoto, Y. Tetrabutylammonium 2-Pyridyltriolborate Salts for Suzuki–Miyaura Cross-Coupling Reactions with Aryl Chlorides. Org. Lett. 2013, 15, 4308–4311. [Google Scholar] [CrossRef]
- Colombe, J.R.; Bernhardt, S.; Stathakis, C.; Buchwald, S.L.; Knochel, P. Synthesis of Solid 2-Pyridylzinc Reagents and Their Application in Negishi Reactions. Org. Lett. 2013, 15, 5754–5757. [Google Scholar] [CrossRef] [Green Version]
- Larionov, O.V.; Stephens, D.; Mfuh, A.; Chavez, G. Direct, Catalytic, and Regioselective Synthesis of 2-Alkyl-, Aryl-, and Alkenyl-Substituted N-Heterocycles from N-Oxides. Org. Lett. 2014, 16, 864–867. [Google Scholar] [CrossRef]
- Gao, G.-L.; Xia, W.; Jain, P.; Yu, J.-Q. Pd(II)-Catalyzed C3-Selective Arylation of Pyridine with (Hetero)arenes. Org. Lett. 2016, 18, 744–747. [Google Scholar] [CrossRef]
- Zeng, Y.; Zhang, C.; Yin, C.; Sun, M.; Fu, H.; Zheng, X.; Yuan, M.; Li, R.; Chen, H. Direct C−H Functionalization of Pyridine via a Transient Activator Strategy: Synthesis of 2,6-Diarylpyridines. Org. Lett. 2017, 19, 1970–1973. [Google Scholar] [CrossRef]
- Bull, J.A.; Mousseau, J.J.; Pelletier, G.; Charette, A.B. Synthesis of Pyridine and Dihydropyridine Derivatives by Regio- and Stereoselective Addition to N-Activated Pyridines. Chem. Rev. 2012, 112, 2642–2713. [Google Scholar] [CrossRef]
- Pomaranski, P.; Czarnocki, Z. Arylpyridines: A Review from Selective Synthesis to Atropisomerism. Synthesis 2019, 51, 587–611. [Google Scholar]
- Lu, J.-Y.; Keith, J.A.; Shen, W.-Z.; Schürmann, M.; Preut, H.; Jacob, T.; Arndt, H.-D. Regioselective De Novo Synthesis of Cyanohydroxypyridines with a Concerted Cycloaddition Mechanism. J. Am. Chem. Soc. 2008, 130, 13219–13221. [Google Scholar] [CrossRef]
- Sabot, C.; Oueis, E.; Brune, X.; Renard, P.-Y. Synthesis of polysubstituted 3-hydroxypyridines via the revisited hetero-Diels–Alder reaction of 5-alkoxyoxazoles with dienophiles. Chem. Commun. 2012, 48, 768–770. [Google Scholar] [CrossRef]
- Ishida, N.; Yuhki, T.; Murakami, M. Synthesis of Enantiopure Dehydropiperidinones from α-Amino Acids and Alkynes via Azetidin-3-ones. Org. Lett. 2012, 14, 3898–3901. [Google Scholar] [CrossRef]
- Barday, M.; Ho, K.Y.T.; Halsall, C.T.; Aïssa, C. Regioselective Synthesis of 3-Hydroxy-4,5-alkyl-Substituted Pyridines Using 1,3-Enynes as Alkynes Surrogates. Org. Lett. 2017, 19, 178–181. [Google Scholar]
- Erhardt, H.; Kunz, K.A.; Kirsch, S.F. Thermolysis of Geminal Diazides: Reagent-Free Synthesis of 3-Hydroxypyridines. Org. Lett. 2017, 19, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, T.J.; Basutto, J.A.; Bower, J.F.; Rathi, A. Heteroaromatic Synthesis via Olefin Cross-Metathesis: Entry to Polysubstituted Pyridines. Org. Lett. 2011, 13, 1036–1039. [Google Scholar] [CrossRef]
- Donohoe, T.J.; Bower, J.F.; Baker, D.B.; Basutto, J.A.; Chan, L.M.K.; Gallagher, P. Synthesis of 2,4,6-trisubstituted pyridines via an olefin cross-metathesis/Heck–cyclisation–elimination sequence. Chem. Commun. 2011, 47, 10611–10613. [Google Scholar] [CrossRef]
- Chen, M.Z.; Micalizio, G.C. Three-Component Coupling Sequence for the Regiospecific Synthesis of Substituted Pyridines. J. Am. Chem. Soc. 2012, 134, 1352–1356. [Google Scholar] [CrossRef] [PubMed]
- Henry, G.D. De novo synthesis of substituted pyridines. Tetrahedron 2004, 60, 6043–6061. [Google Scholar] [CrossRef]
- Heller, B.; Hapke, M. The fascinating construction of pyridine ring systems by transition metal-catalysed [2 + 2 + 2] cycloaddition reactions. Chem. Soc. Rev. 2007, 36, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Groenendaal, B.; Ruijter, E.; Orru, R.V.A. 1-Azadienes in cycloaddition and multicomponent reactions towards N-heterocycles. Chem. Commun. 2008, 5474–5489. [Google Scholar] [CrossRef]
- Hill, M.D. Recent Strategies for the Synthesis of Pyridine Derivatives. Chem. Eur. J. 2010, 16, 12052–12062. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, T.J.; Bower, J.F.; Chan, L.K.M. Olefin cross-metathesis for the synthesis of heteroaromatic compounds. Org. Biomol. Chem. 2012, 10, 1322–1328. [Google Scholar] [CrossRef]
- Allais, C.; Grassot, J.-M.; Rodriguez, J.; Constantieux, T. Metal-Free Multicomponent Syntheses of Pyridines. Chem. Rev. 2014, 114, 10829–10868. [Google Scholar] [CrossRef]
- Wang, Q.; Wan, C.; Gu, Y.; Zhang, J.; Gao, L.; Wang, Z. A metal-free decarboxylative cyclization from natural a-amino acids to construct pyridine derivatives. Green Chem. 2011, 13, 578–581. [Google Scholar] [CrossRef]
- Xiang, J.-C.; Wang, M.; Cheng, Y.; Wu, A.-X. Molecular Iodine-Mediated Chemoselective Synthesis of Multisubstituted Pyridines through Catabolism and Reconstruction Behavior of Natural Amino Acids. Org. Lett. 2016, 18, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.-C.; Cheng, Y.; Wang, Z.-X.; Ma, J.-T.; Wang, M.; Tang, B.-C.; Wu, Y.-D.; Wu, A.-X. Oxidative Trimerization of Amino Acids: Selective Synthesis of 2,3,5-Trisubstituted Pyridines. Org. Lett. 2017, 19, 2997–3000. [Google Scholar] [CrossRef]
- Tilley, J.W.; Zawoiski, S. A Convenient Palladium-Catalyzed Coupling Approach to 2,5-Disubstituted Pyridines. J. Org. Chem. 1988, 53, 386–390. [Google Scholar] [CrossRef]
- Vyvyan, J.R.; Dell, J.A.; Ligon, T.J.; Motanic, K.K.; Wall, H.S. Suzuki–Miyaura Cross-Coupling of 3-Pyridyl Triflates with Alk-1-enyl-2-pinacol Boronates. Synthesis 2010, 3637–3644. [Google Scholar] [CrossRef]
- Bera, M.K.; Hommes, P.; Reissig, H.-U. In Search of Oligo(2-thienyl)-Substituted Pyridine Derivatives: A Modular Approach to Di-, Tri- and Tetra(2-thienyl)pyridines. Chem. Eur. J. 2011, 17, 11383–11843. [Google Scholar] [CrossRef]
- Doebelin, C.; Wagner, P.; Bihel, F.; Humbert, N.; Kenfack, C.A.; Mely, Y.; Bourguignon, J.-J.; Schmitt, M. Fully Regiocontrolled Polyarylation of Pyridine. J. Org. Chem. 2014, 79, 908–918. [Google Scholar] [CrossRef]
- Zhang, E.; Tang, J.; Li, S.; Wu, P.; Moses, J.E.; Sharpless, K.B. Chemoselective Synthesis of Polysubstituted Pyridines from Heteroaryl Fluorosulfates. Chem. Eur. J. 2016, 22, 5692–5697. [Google Scholar] [CrossRef]
- Asako, T.; Hayashi, W.; Amaike, K.; Suzuki, S.; Itami, K.; Muto, K.; Yamaguchi, J. Synthesis of multiply arylated pyridines. Tetrahedron 2017, 73, 3669–3676. [Google Scholar]
- Donohoe, T.J.; Fishlock, L.P.; Basutto, J.A.; Bower, J.F.; Procopiou, P.A.; Thompson, A.L. Synthesis of substituted pyridines and pyridazines via ring closing metathesis. Chem. Commun. 2009, 3008–3010. [Google Scholar] [CrossRef]
- Yoshida, K.; Kawagoe, F.; Hayashi, K.; Horiuchi, S.; Imamoto, T.; Yanagisawa, A. Synthesis of 3-Hydroxypyridines Using Ruthenium-Catalyzed Ring-Closing Olefin Metathesis. Org. Lett. 2009, 11, 515–518. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Ueno, T.; Kondo, Y. Palladium(0)-Catalyzed Alkylative Cyclization of Alkynals and Alkynones: Remarkable trans-Addition of Organoboronic Reagents. J. Am. Chem. Soc. 2006, 128, 1406–1407. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Ito, K.; Ueno, T.; Shiraishi, M.; Kondo, Y.; Doi, T. Palladium(0)-Catalyzed Anti-Selective Addition-Cyclizations of Alkynyl Electrophiles. Chem. Eur. J. 2022, e202203068. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Nakamura, S.; Tomida, A.; Doi, T. Scalable Total Syntheses and Structure–Activity Relationships of Haouamines A, B, and Their Derivatives as Stable Formate Salts. Chem. Eur. J. 2020, 26, 12528–12532. [Google Scholar] [CrossRef]
- Boger, D.L.; Brotherton, C.E.; Panek, J.S.; Yohannes, D. Direct Introduction of Nitriles via Use of Unstable Reissert Intermediates: Convenient Procedures for the Preparation of 2-Cyanoquinolines and 1-Cyanoisoquinolines. J. Org. Chem. 1984, 49, 4056–4058. [Google Scholar] [CrossRef]
- Chinchilla, R.; Nájera, C. The Sonogashira Reaction: A Booming Methodology in Synthetic Organic Chemistry. Chem. Rev. 2007, 107, 874–922. [Google Scholar] [CrossRef]
- Radhika, S.; Harry, N.A.; Neetha, M.; Anikumar, G. Recent trends and applications of the Cadiot–Chodkiewicz reaction. Org. Biomol. Chem. 2019, 17, 9081–9094. [Google Scholar] [CrossRef] [PubMed]
- Bürgi, H.B.; Duntz, J.D.; Lehn, J.M.; Wipff, G. Stereochemistry of Reaction Paths at Carbonyl Centres. Tetrahedron 1974, 30, 1563–1572. [Google Scholar] [CrossRef]
- Gilmore, K.; Alabugin, I.V. Cyclizations of Alkynes: Revisiting Baldwin’s Rules for Ring Closure. Chem. Rev. 2011, 111, 6513–6556. [Google Scholar] [CrossRef]
- Salimbeni, A.; Canevotti, R.; Paleari, F.; Bonaccorsi, F.; Renzetti, A.R.; Belvisi, L.; Bravi, G.; Scolastico, C. Nonpeptide Angiotensin II Receptor Antagonists. Synthesis, in Vitro Activity, and Molecular Modeling Studies of N-[(Heterobiaryl)methylimidazole. J. Med. Chem. 1994, 37, 3928–3938. [Google Scholar] [CrossRef]
- Shibata, N.; Tsuchiya, T.; Hashimoto, Y.; Morita, N.; Ban, S.; Tamura, O. Thiyl radical-mediated cyclization of ω-alkynyl O-tert-butyldiphenylsilyloximes. Org. Biomol. Chem. 2017, 15, 3025–3034. [Google Scholar] [CrossRef]
- Takahashi, K.; Honda, T. Diastereoselective Syntheses of Functionalized Five-Membered Carbocycles and Heterocycles by a SmI2-Promoted Intramolecular Coupling of Bromoalkynes and α,β-Unsaturated Esters. Org. Lett. 2010, 12, 3026–3029. [Google Scholar] [CrossRef]
- Padín, D.; Cambeiro, F.; Fañanás-Mastral, M.; Varela, J.; Saá, A.C. [2 + 1] Cycloaddition of Catalytic Ruthenium Vinyl Carbenes: A Stereoselective Controlled Access to (Z)- and (E)-Vinyl Epoxypyrrolidines. ACS Catal. 2017, 7, 992–996. [Google Scholar] [CrossRef]
- Ordóñez, M.; De la Cruz-Cordero, R.; Fernández-Zertuche, M.; Muñoz-Hernández, M.A.; García-Barradas, O. Diastereoselective reduction of dimethyl γ-[(N-p-toluenesulfonyl)amino]-β-ketophosphonates derived from amino acids. Tetrahedron Asymmetry 2004, 15, 3035–3043. [Google Scholar] [CrossRef]
- Morales, S.; Guijarro, F.G.; Ruano, J.L.G.; Cid, M.B. A General Aminocatalytic Method for the Synthesis of Aldimines. J. Am. Chem. Soc. 2014, 136, 1082–1089. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ito, K.; Doi, T.; Tsukamoto, H. De Novo Synthesis of Polysubstituted 3-Hydroxypyridines Via “Anti-Wacker”-Type Cyclization. Catalysts 2023, 13, 319. https://doi.org/10.3390/catal13020319
Ito K, Doi T, Tsukamoto H. De Novo Synthesis of Polysubstituted 3-Hydroxypyridines Via “Anti-Wacker”-Type Cyclization. Catalysts. 2023; 13(2):319. https://doi.org/10.3390/catal13020319
Chicago/Turabian StyleIto, Kazuya, Takayuki Doi, and Hirokazu Tsukamoto. 2023. "De Novo Synthesis of Polysubstituted 3-Hydroxypyridines Via “Anti-Wacker”-Type Cyclization" Catalysts 13, no. 2: 319. https://doi.org/10.3390/catal13020319