1. Introduction
Several CO
2 usage approaches have been implemented to reduce the impact of emitted greenhouse gases and to achieve a carbon neutral level. CO
2 utilization in chemical production gained significant attention in replacing traditionally used C1 sources such as phosgene or CO, which post more toxicity. However, the activation of CO
2 is challenging due to its thermodynamic stability, which requires appropriate catalysts, such as solid base catalysts, for instance, alkaline earth metal oxides. One of the useful reactions is CO
2 cycloaddition to epoxides, which form cyclic carbonates. The resultant cyclic carbonates could be applied as solvents [
1,
2,
3,
4,
5], monomers [
6,
7,
8,
9], electrolytes [
10,
11,
12,
13], and pharmaceuticals [
14,
15].
Recent applications of metal oxide clusters, namely, polyoxometalates, as base catalysts have been reported [
16,
17,
18,
19,
20,
21,
22]. One of the advantages of utilizing metal oxide clusters over bulk solid base catalysts is that it does not require the surface activation of catalysts [
23]. Up to now, the basicity of metal oxide clusters depends on the structures and the type of metal ions. The Lindqvist-type polyoxotungstate [W
6O
19]
2− shows the basicity with p
Ka value of 11.1 and defective Keggin-type Ge-incorporated polyoxotungstate [
γ-H
2GeW
10O
36]
6− exhibits high basicity with a p
Ka value of 21.9 [
24]. The series of group 5 metal polyoxometalates exhibit superior basicity to those of group 6 metal polyoxometalates, and the basicities of [Nb
10O
28]
6−, [Nb
6O
19]
8−, and [Ta
6O
19]
8− increase to a p
Ka value of 23.8 [
17,
18]. Recently, Uchida’s group reported that porous ionic crystals containing Nb/Ta were applied to Knoevenagel condensation reactions as base catalyst [
19]. Density functional theory (DFT) calculations reveal that the base strength of the clusters is related to the natural bond orbital (NBO) charges of the surface O atoms and the higher negativity of the NBO charges leads to stronger basicity [
16,
18]. We reported that Lindqvist-type polyoxometalates with group 5 metal ions (Nb, Ta) had higher negative NBO charges compared to group 6 metal ions (Mo, W) and [Nb
6O
19]
8− and [Ta
6O
19]
8− could activate CO
2 and worked as catalysts for CO
2 fixation and conversion reactions [
18,
25]. The CO
2 was activated on the terminal O sites of metal oxide clusters, which are Lewis base sites, and activated CO
2 reacts with epoxides to form carbonates [
17,
18,
21]. The cycloaddition of CO
2 to epichlorohydrin proceeded on Keggin-type Na
16[SiNb
12O
40] [
21]. In the case of Lindqvist-type [M
6O
19]
8− (M = Nb, Ta), [Ta
6O
19]
8− showed higher activity for CO
2 fixation to styrene oxide (
SO) at 403 K than [Nb
6O
19]
8− [
18]. However, the styrene carbonate (
SC) selectivity of [Ta
6O
19]
8− was lower than 90% and byproducts were formed. In our previous study, Brønsted basicity was investigated using sodium salts of [Ta
xNb
6−xO
19]
8− as solid base catalyst in Knoevenagel condensation reactions and local symmetry of NbO
6 and TaO
6 units in the clusters affected base catalytic properties [
23]. In this study, tetrabutylammonium (TBA) salts of mixed metal oxide clusters [Ta
xNb
6−xO
19]
8− (TBA-Ta
xNb
6−x,
x = 0–6) were prepared and applied to CO
2 fixation to
SO to elucidate the Ta-substitution effect on the catalytic activities and selectivity. It was found that single-Ta-substituted TBA-Ta
1Nb
5 exhibited the highest
SC selectivity among TBA-Ta
xNb
6−x. We demonstrated that the high
SC selectivity was achieved by the selective adsorption of CO
2 on the terminal O
Ta without
SO activation under reaction conditions.
2. Results
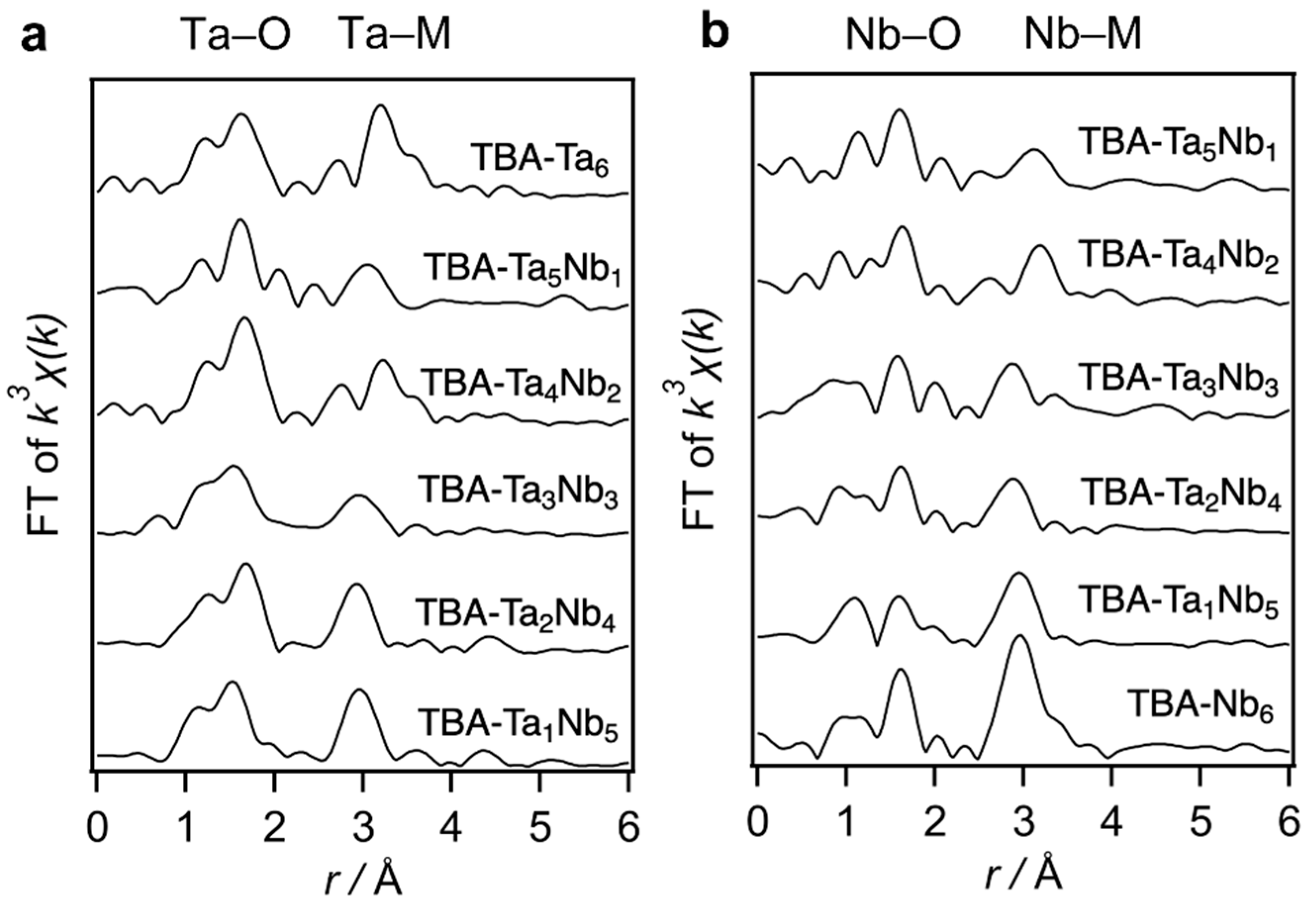
The fabricated TBA-Ta
xNb
6−x were characterized by X-ray absorption spectroscopy (XAS), electrospray ionization mass spectrometry (ESI–MS), Fourier-transformed infrared (FT-IR) in attenuated total reflectance (ATR) mode, and elemental analysis (
Figure 1,
Figure S1 and
Figure S2, and
Table S1, respectively). ESI–MS suggests that the various components of Ta–Nb mixed metal oxide clusters are contained in the TBA-Ta
xNb
6−x. Ta L
3-edge Fourier-transformed extended X-ray absorption fine structure (FT-EXAFS) of TBA-Ta
xNb
6−x indicates that peaks of Ta–M (M = Nb or Ta) shift to a longer length with increasing Ta content (
Figure 1a). A similar peak shift of Nb–M (M = Nb or Ta) is observed in the Nb K-edge FT-EXAFS spectra (
Figure 1b). Those indicate the Ta-substitution to Nb sites in [Ta
xNb
6−xO
19]
8−. Elemental analysis reveals that TBA/[Ta
xNb
6−xO
19]
8− ratio is 5~6.
The prepared clusters were employed in the catalytic CO
2 fixation to
SO.
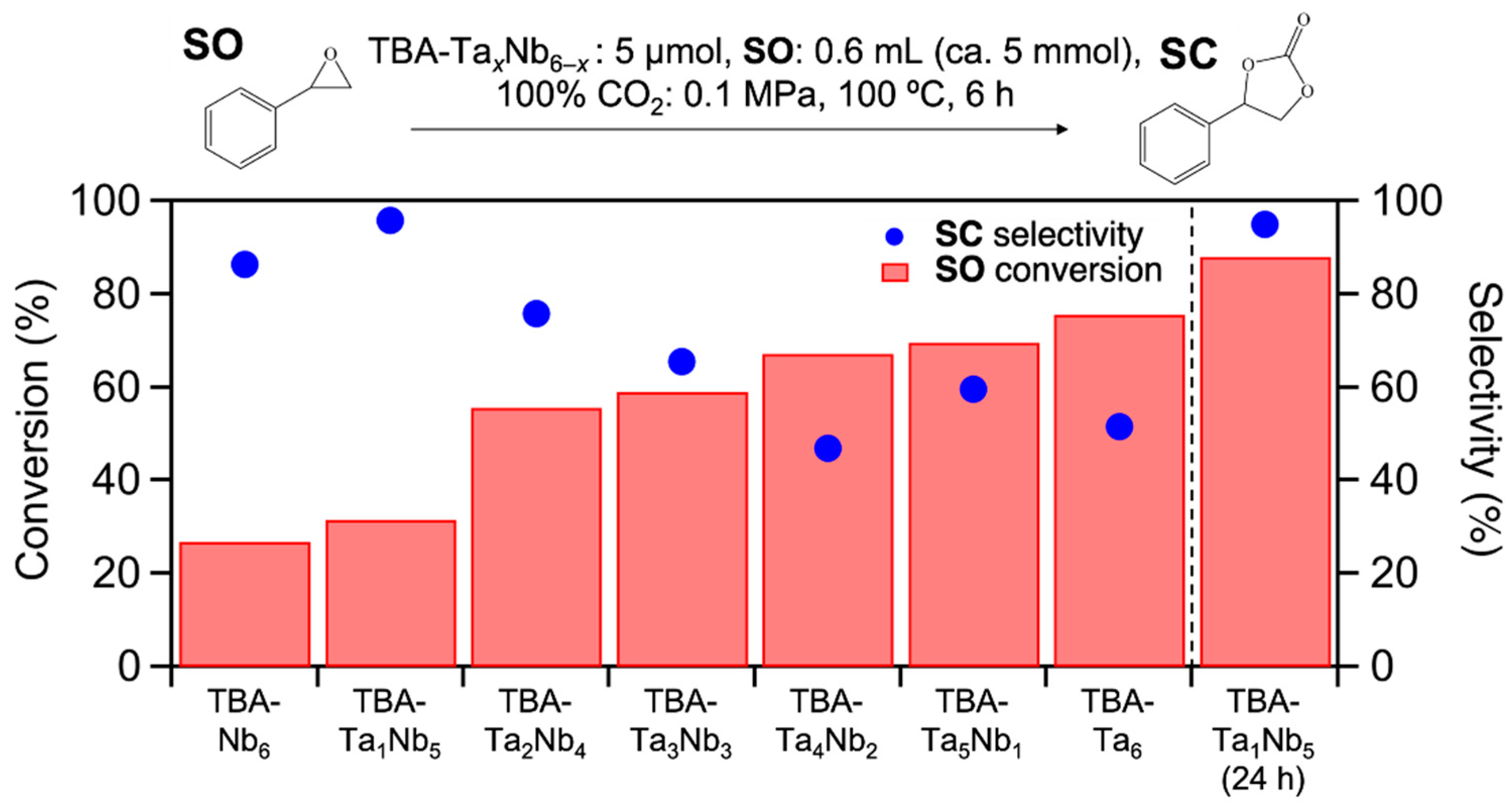
Figure 2 shows the
SO conversion and
SC selectivity for TBA-Ta
xNb
6−x catalysts. The
SO conversion gradually increases with incremental addition of Ta content and TBA-Ta
6 exhibits the highest
SO conversion among them. On the other hand, the trend of
SC selectivity for the composition of clusters differs from that of
SO conversation (
Figure 2). Interestingly, single-Ta-substituted TBA-Ta
1Nb
5 provides the highest
SC selectivity (95%) among the mixed metal oxide clusters. Further Ta-substitution decreases
SC selectivity and the
SC selectivity becomes constant in Ta-rich TBA-Ta
xNb
6−x catalysts (
x = 4–6). The number of TBA counteractions has a negligible impact on this reaction, although TBA/[Ta
xNb
6−xO
19]
8− ratio varies with the composition [
17]. The byproducts in this reaction over TBA-Ta
6 are polymers derived from the polymerization of
SO, because the
SO conversion is found for TBA-Ta
6 at 100 °C under N
2 atmosphere without CO
2 despite negligible
SO conversion for TBA-Nb
6, as shown in
Figure S3. The high
SC selectivity (95%) of TBA-Ta
1Nb
5 maintains a high
SO conversion (88%) for a 24 h reaction (
Figure 2). In addition, the >95% SC selectivity is only achieved by TBA-Ta
1Nb
5 among the TBA-Ta
xNb
6−x catalysts at >80% conversion (
Figure S4). Thus, the selective CO
2 fixation to
SO is achieved by the single Ta substitution to TBA-Nb
6.
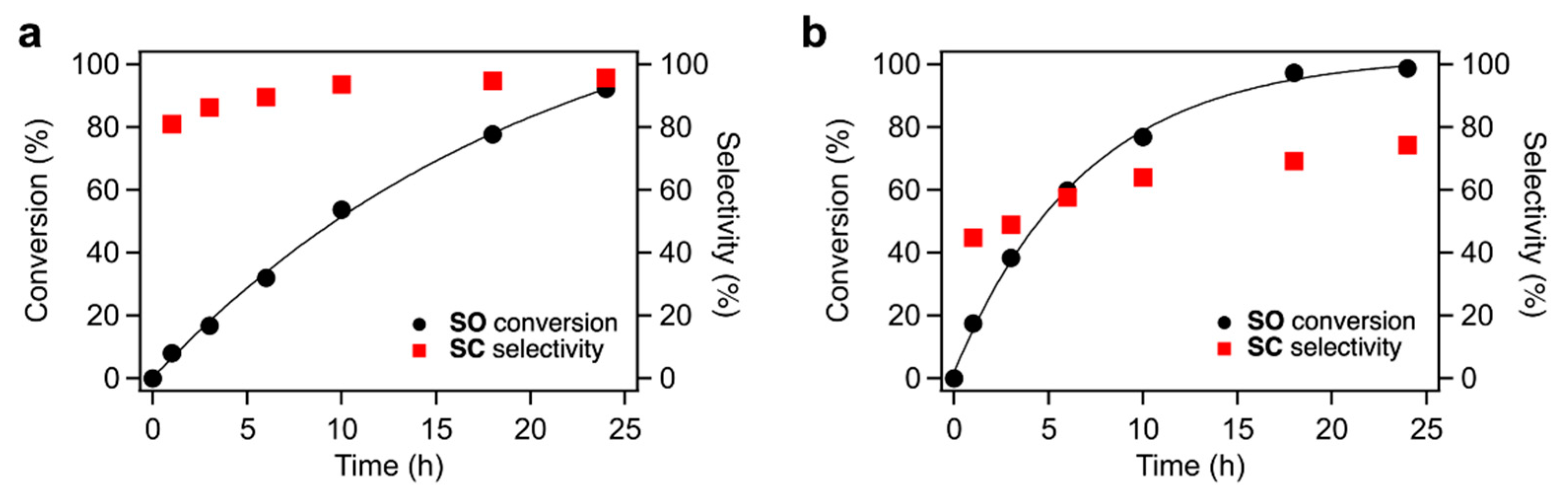
The time courses of CO
2 fixation to
SO over TBA-Ta
6, TBA-Ta
1Nb
5, and TBA-Nb
6 are shown in
Figure 3 and
Figure S5. The trends of conversion and selectivity depend on the composition of the clusters. TBA-Ta
6 exhibits the highest reaction rate among them, and
SC selectivity increases with reaction time. TBA-Ta
1Nb
5 and TBA-Nb
6 show high
SC selectivity at the initial stage of the reaction and the
SC selectivity is maintained at a high
SO conversion. Thus, TBA-Nb
6 and single-Ta-substituted TBA-Ta
1Nb
5 have the specific active sites for selective
SC formation. The increment in the
SC selectivity over TBA-Ta
6 is due to the consumption of
SO and suppression of undesired reactions during the reaction.
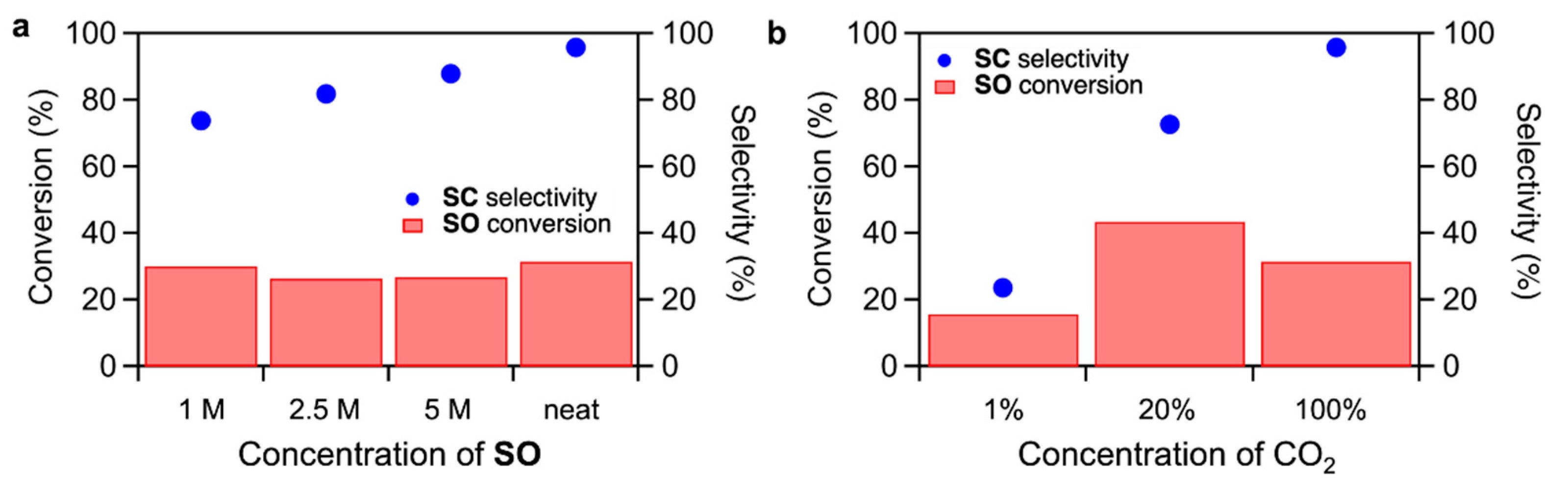
Effects of
SO concentration and CO
2 concentration on the CO
2 fixation to
SO were studied for TBA-Ta
1Nb
5 (
Figure 4). When increasing the
SO concentration in the dimethyl sulfoxide (DMSO) solution, the
SC selectivity slightly increases while maintaining the
SO conversion. On the other hand, the
SC selectivity dramatically decreases with decreasing CO
2 concentration (
Figure 4b), suggesting the CO
2 activation is a key step in the CO
2 fixation to
SO. We reported that CO
2 fixation to
SO proceeds due to the fact that the CO
2 is activated on the terminal O sites and activated CO
2 reacts with
SO to form
SC [
17,
18]. The rate determining step of CO
2 fixation to
SO is the nucleophilic attack of activated CO
2 to
SO. The above results could be explained by the reaction mechanism. The decrease in the
SC yield by reducing
SO concentration, as shown in
Figure 4a, is due to the inhibition of the reaction of activated CO
2 with
SO by a low
SO concentration. There are two reasons for the drastic decrease in
SC selectivity by reducing CO
2 concentration. One is the decrease in the amount of activated CO
2. The other is that
SO can be activated on TBA-Ta
1Nb
5 in low CO
2 concentration conditions. In fact, the
SO conversion of TBA-Ta
1Nb
5 under N
2 atmosphere without CO
2 is higher than that of TBA-Nb
6, as shown in
Figure S3, suggesting that the
SO activation occurs by single Ta-substitution at a low CO
2 concentration.
3. Discussion
We reported that the CO
2 fixation to
SO proceeded on the terminal O sites of TBA-Ta
6 because the terminal O sites, which have the negatively charged O, work as Lewis base sites [
18]. The CO
2 is activated on the terminal O sites and the activated CO
2 reacts with
SO to form
SC. The CO
2 adsorption on TBA-Nb
6, TBA-Ta
1Nb
5, and TBA-Ta
6 in
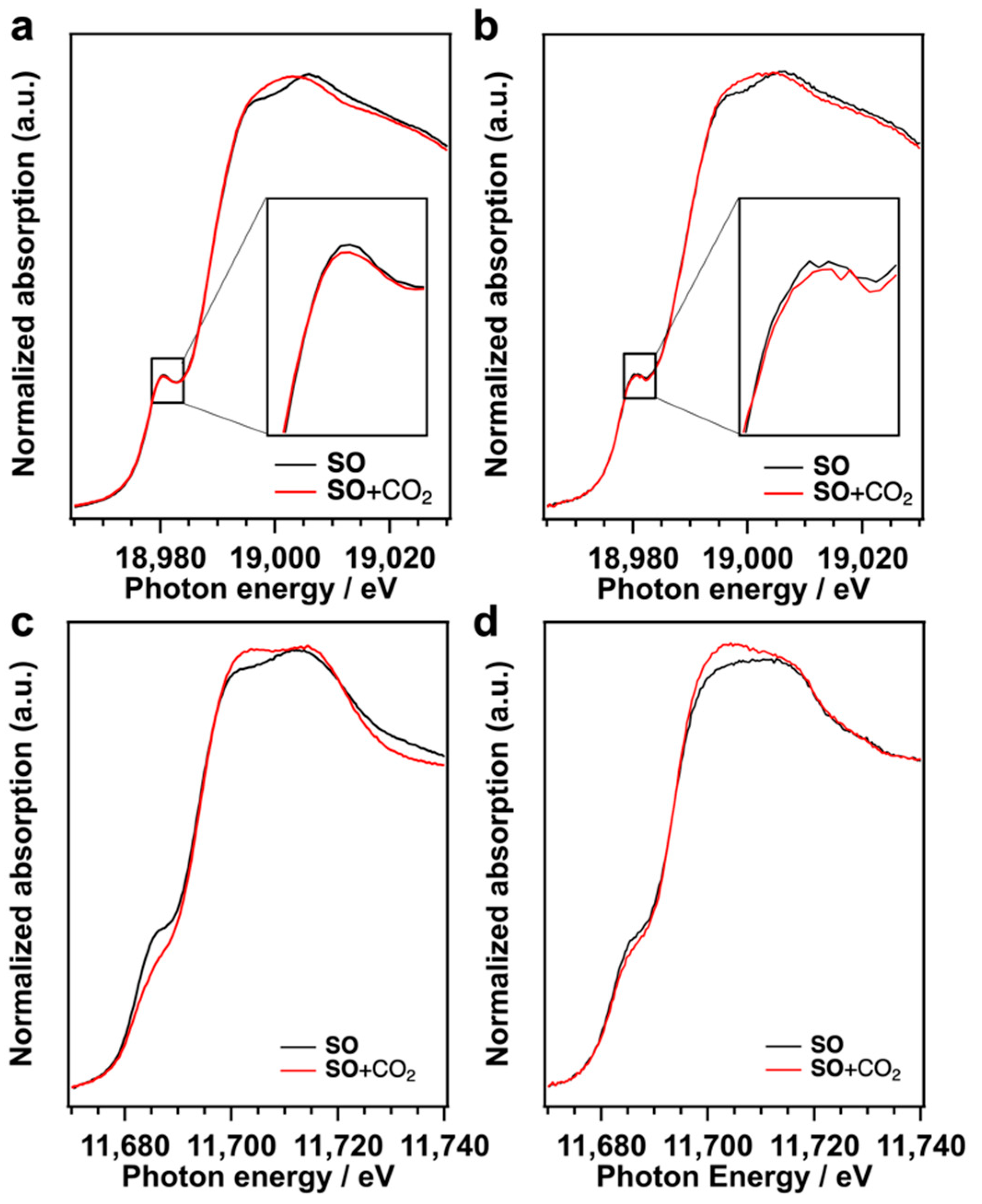
SO was examined by in situ XAFS measurements (
Figure 5). Nb K-edge XANES spectrum of TBA-Nb
6 in
SO exhibits a pre-edge peak at 18,980.5 eV assigned to electron excitation from 1
s to hybridized 4
d−5
p [
26,
27]. This pre-edge peak intensity gives us the information on the distortion from NbO
6 octahedral (
Oh) symmetry. The pre-edge peak intensity decreases with the CO
2 introduction to TBA-Nb
6 in
SO, which indicates that the
Oh symmetry of NbO
6 in TBA-Nb
6 is improved by the CO
2 addition. Similar results were obtained for TBA-Ta
1Nb
5, as shown in
Figure 5b. Ta L
1-edge XANES spectra indicate that the pre-edge peak at 11,686 eV, which is assigned to electron transition from Ta 2
s orbitals to hybridized 5
d−6
p orbitals [
26,
27], decreases with the introduction of CO
2. This change of pre-edge peak intensity reveals that TaO
6 Oh symmetry is also improved by the CO
2 addition to TBA-Ta
6 and TBA-Ta
1Nb
5. These results suggest that the
Oh symmetry of TaO
6 unit increases, while NbO
6 symmetry is slightly improved in TBA-Ta
1Nb
5 by CO
2 adsorption. Actually, the optimized structure of [Ta
1Nb
5O
19]
8− with CO
2 adsorbed on the terminal O
Ta site has highly
Oh symmetric TaO
6 units compared to the bare [Ta
1Nb
5O
19]
8− (
Figure S6) CO
2 is also adsorbed on TBA-Ta
xNb
6−x in
SO solution. This structural change induced by CO
2 adsorption is also observed in FT-IR (
Figure S7). FT-IR spectra of TBA-Ta
1Nb
5 in DMSO solvent show the characteristic absorption band assignable to the stretching vibration between the metal and terminal O atoms in MO
6 units (M=O bond). The absorption band shifts to high energy, owing to the slight shrink in the O=Nb bond in NbO
6 units. Those results indicate that CO
2 is preferentially adsorbed on terminal O sites of TaO
6 unit and induces the structure change in TBA-Ta
xNb
6−x.
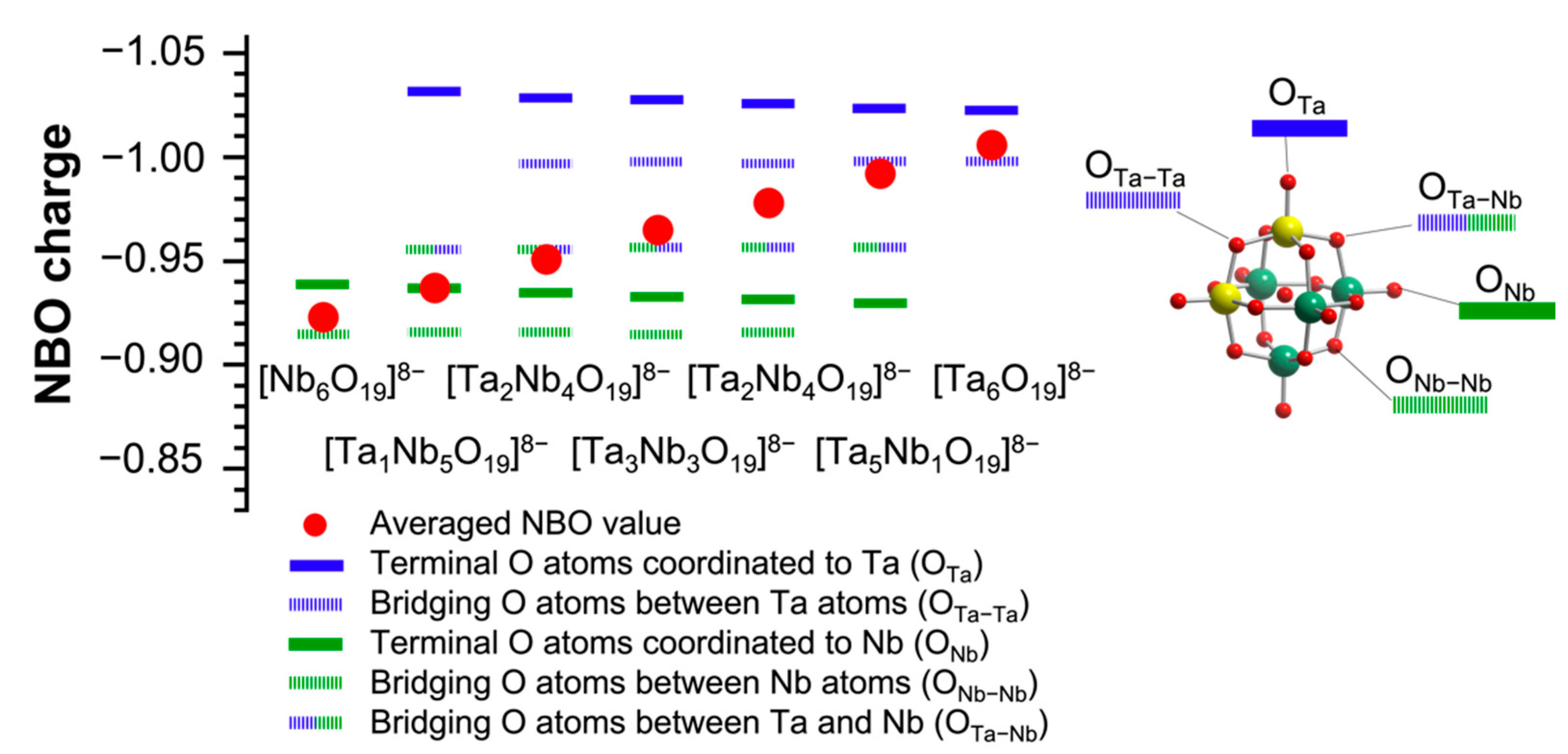
Next, NBO charges of the surface O atoms of TBA-Ta
xNb
6−x were calculated to elucidate the catalytic activity and selectivity of TBA-Ta
xNb
6−x for CO
2 fixation to
SO (
Figure 6). The average NBO charge was also evaluated in
Figure 6. The terminal O atoms coordinated to Ta (terminal O
Ta) have the most negative NBO charges (highest basicity) followed by the bridge O atom between Ta ions (bridge O
Ta–Ta) in TBA-Ta
xNb
6−x. The values of NBO charge of terminal O
Ta hardly change with the Ta content. On the other hand, the NBO charge of the terminal O connecting to Nb (terminal O
Nb) has lower negativity than that of terminal O
Ta. The order of NBO charges in TBA-Ta
xNb
6−x is terminal O
Ta, bridge O
Ta–Ta, bridge O
Ta–Nb (Ta–O–Nb), terminal O
Nb, and bridge O
Nb–Nb (Nb–O–Nb). As a result, the average NBO charges of TBA-Ta
xNb
6−x gradually increase with increasing the Ta content. The catalytic activities of TBA-Ta
xNb
6−x, which increase with incremental addition of Ta content in
Figure 2, could be explained by the average NBO charges, indicating the increase in the active sites of terminal O
Ta by Ta substitution.
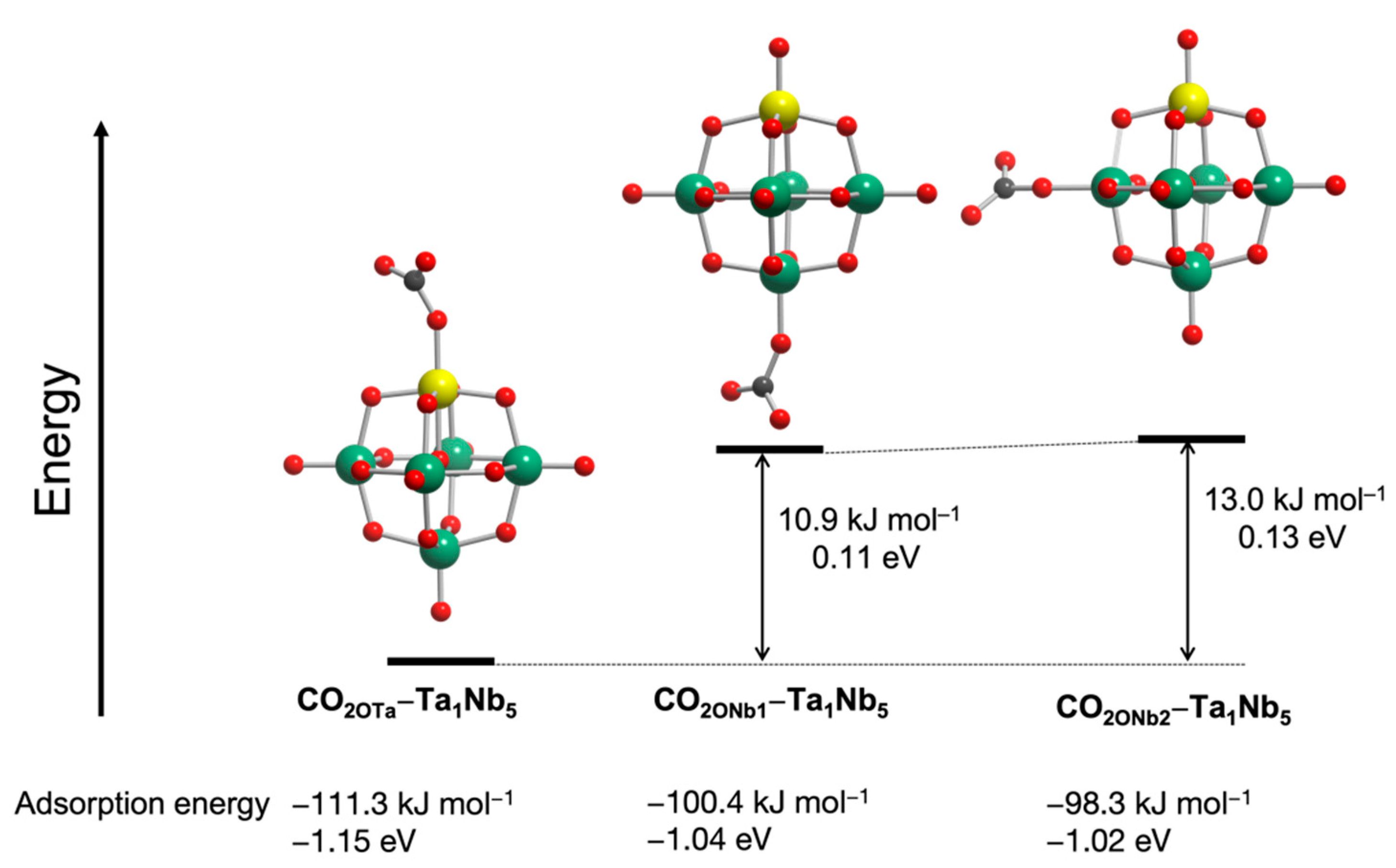
Finally, the CO
2 adsorption sites of [Ta
1Nb
5O
19]
8− were also predicted by DFT calculations. We reported that CO
2 was preferentially adsorbed on terminal O sites rather than bridge O sites [
18]. [Ta
1Nb
5O
19]
8− has three terminal O sites (see
Figure 7). To determine the CO
2 activation sites of [Ta
1Nb
5O
19]
8−, the CO
2 adsorption energy was calculated using three possible configurations (
Figure 7). Among the three structures, the lowest energy is found in a structure with CO
2 adsorbed on the terminal O
Ta site, which has the highest negative NBO charge among the surface oxygen atoms in [Ta
1Nb
5O
19]
8−. This result indicates that the CO
2 is preferentially adsorbed and activated on the terminal O
Ta. The
SO adsorption energy was also calculated to gain the insight into
SO activation sites (
Figure S8). The adsorption energy of
SO on O
Ta is lower than that of CO
2 on O
Ta in [Ta
1Nb
5O
19]
8−. In addition, adsorption energies reveal that
SO is more likely to be activated on O
Ta than O
Nb. These results suggest that CO
2 preferentially adsorbs on O
Ta site and it is unlikely that
SO activation occurs on O
Nb in TBA-Ta
1Nb
5. Therefore, high
SC selectivity is achieved in TBA-Ta
1Nb
5. The low
SC selectivity in Ta-rich TBA-Ta
xNb
6−x is explained that not only by the fact that CO
2 but also
SO is activated on O
Ta sites by competitive adsorption due to the large number of O
Ta adsorption sites.
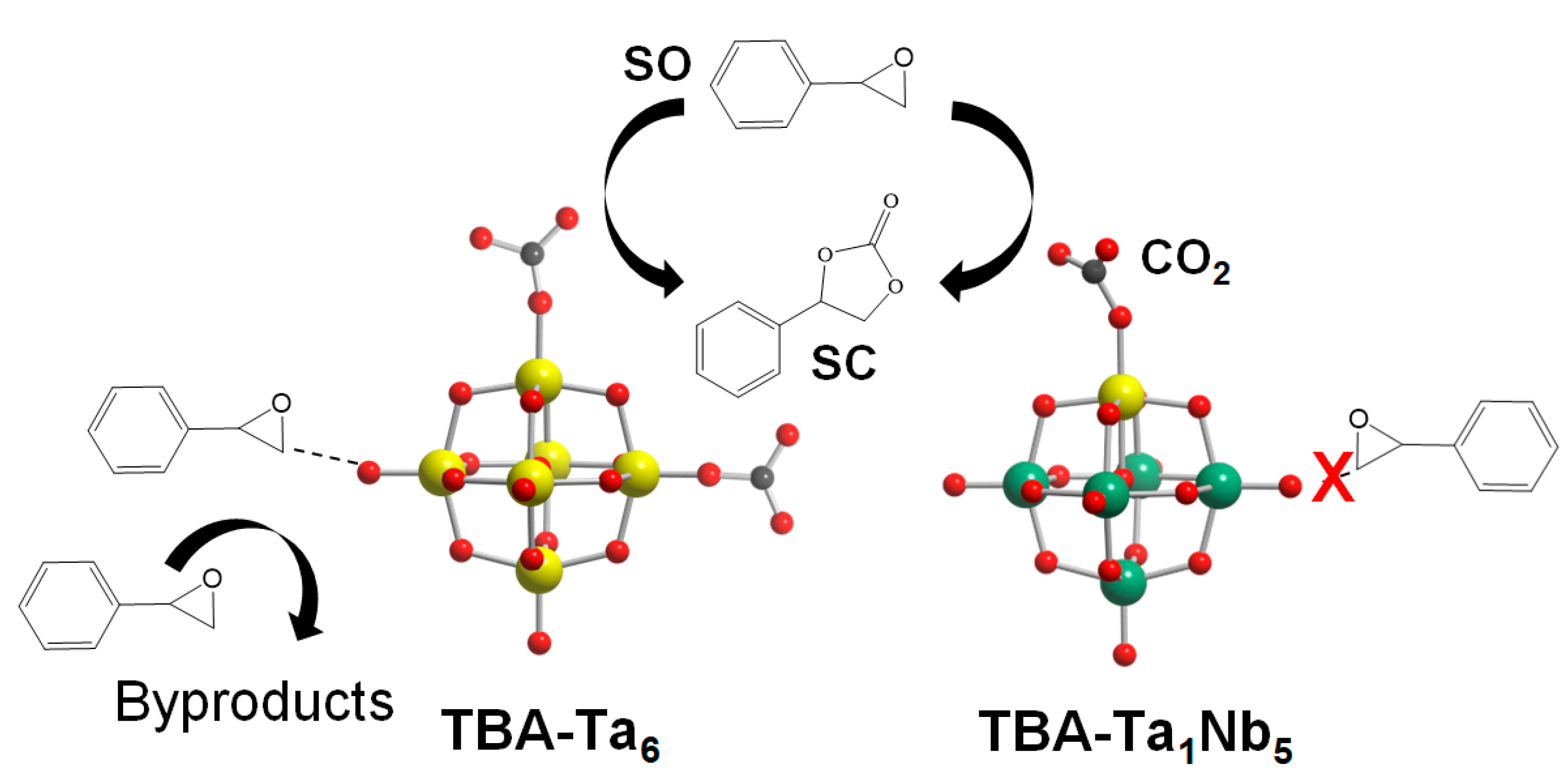
The reaction mechanism of TBA-Ta
xNb
6−x for CO
2 fixation to
SO is discussed. In the case of TBA-Nb
6, CO
2 is adsorbed on the terminal O
Nb sites and the activated CO
2 reacts nucleophilically with
SO to form
SC. The low catalytic activity of TBA-Nb
6 for CO
2 fixation to
SO is due to the weak Lewis base strength (low negativity in NBO charges) of terminal O
Nb compared with terminal O
Ta of other TBA-Ta
xNb
6−x. The
SO activation hardly occurs on TBA-Nb
6, as shown in
Figure S3, which is one of the reasons why TBA-Nb
6 shows high
SC selectivity. The
SO conversion gradually increases with Ta substitution amount, as shown in
Figure 2. This can be explained by the increase in active terminal O
Ta sites. On the other hand, the
SC selectivity decreases for high Ta content of TBA-Ta
xNb
6−x (
x ≥ 2). The low
SC selectivity is due to the
SO activation on the surface of TBA-Ta
xNb
6−x (
x ≥ 2), as shown in
Scheme 1. In fact, TBA-Ta
6 exhibits the highest
SO conversion among TBA-Ta
6, TBA-Ta
1Nb
5, and TBA-Nb
6 in the absence of CO
2 accompanied with a viscosity increase (
Figure S3). The sharp contrast in
SO conversion in the absence of CO
2 conditions for TBA-Ta
6 and TBA-Nb
6 clearly indicates that terminal O
Ta and/or bridge O
Ta−Ta can activate
SO. On the other hand, TBA-Ta
1Nb
5 exhibits the highest
SC selectivity among TBA-Ta
xNb
6−x despite having a terminal O
Ta site. The DFT calculation (
Figure 7) and CO
2 concentration dependence on CO
2 fixation to
SO (
Figure 4b) reveal that the single terminal O
Ta in [Ta
1Nb
5O
19]
8− preferentially adsorbs CO
2 at 100% CO
2 conditions and
SO is not activated on terminal O
Nb, bridge O
Nb−Nb, and bridge O
Ta−Nb (
Scheme 1). We conclude that the selective CO
2 activation at the terminal O
Ta in TBA-Ta
1Nb
5 without
SO activation is a crucial factor for high
SC selectivity in the CO
2 fixation to
SO.
4. Materials and Methods
TBA salts of [Ta
xNb
6−xO
19]
8− (TBA-Ta
xNb
6−x,
x = 0–6) were prepared by microwave-assisted hydrothermal synthesis (Biotage Initiator
+ 400 W) using Ta
2(x/6)Nb
2(1−x/6)O
5·
nH
2O as the precursors. First, Na
3Ta
x/6Nb
1−x/6O
4 were prepared by modified solid-state reaction method according to the reported procedures [
23,
28]. M
2O
5 (M = Ta or Nb), Na
2C
2O
4, and (NH
2)
2CO at a molar ratio between (Ta + Nb):Na:(NH
2)
2CO of 1:1:4 was ground to fine powder prior to calcination at 773 K for 4 h to obtain NaTa
x/6Nb
1−x/6O
3. NaTa
x/6Nb
1−x/6O
3 was mixed with Na
2C
2O
4, and (NH
2)
2CO at a molar ratio between NaTa
x/6Nb
1−x/6O
3:Na:(NH
2)
2CO of 1:1:3 followed by calcination at 1173 K for 4 h. The resulting powder, Na
3Ta
x/6Nb
1−x/6O
4, was characterized by XRD (Rigaku Miniflex) having diffraction patterns corresponding to the references (
Figure S9). Na
3Ta
x/6Nb
1−x/6O
4 was dissolved in water and 1 M HCl was added until pH of the supernatant reached 1 or less. The white precipitate was collected by centrifugation and washed with pure water until the pH of the supernatant became neutral. After drying in vacuum and oven, the Ta
2(x/6)Nb
2(1−x/6)O
5·
nH
2O was obtained. Then, 10% tetrabutylammonium hydroxide (TBAOH) aqueous solution was added to Ta
2(x/6)Nb
2(1−x/6)O
5·
nH
2O. The mixture was reacted using microwave-assisted hydrothermal synthesis at 180 °C for 5−15 min. The resultant product was washed with hexane to obtain TBA
6H
2[Ta
xNb
6−xO
19]. The fabricated clusters were characterized by ESI–MS (
Figure S1) (Bruker, MicroOTOFII-ST1), Fourier-transformed infrared spectrometry (JASCO, FT/IR-4700) equipped with attenuated total reflectance-infrared spectroscopy (JASCO, ATR-PRO ONE) (
Figure S2), elemental analysis (
Table S1), and X-ray absorption fine structure (XAFS) analysis (BL01B1, SPring-8) (
Figure 1). XAFS spectra were recorded in transmittance mode using ionization chambers as detectors at room temperature. Si(111) double-crystal monochromator was used to obtain the incident X-ray beam for Ta L
1- and L
3-edges XAFS. In the case of Nb K-edge XAFS measurements, Si(311) double-crystal monochromator was employed. The data were analyzed using xTunes software [
29]. The XANES spectra were extracted as the extended X-ray absorption fine structure (EXAFS) after normalization at edge height. The EXAFS spectra in the
k range 3.0–14.0 Å
−1 were Fourier-transformed into
r space to obtain FT-EXAFS spectra. The illustrations of Ta
xNb
6−xO
19 were computed using VESTA [
30].
In general, CO
2 fixation to
SO over TBA-Ta
xNb
6−x were carried out using 5 µmol of catalyst,
SO (0.6 mL, ca. 5 mmol), 100% CO
2 (0.1 MPa) at 100 °C for 6 h using biphenyl as an internal standard. The product solutions were analyzed using gas chromatography equipped with flame ionization detector (GC-FID, Shimadzu, GC-2014 with column Restex, Rtx-1) and gas chromatography–mass spectrometry (GC–MS, Shimadzu, GCMS-QP2010 SE with column Agilent, DB-1MS). Time course of CO
2 fixation to
SO reactions were carried out using 10 µmol of catalyst,
SO (1.2 mL, ca. 10 mmol), 100% CO
2 (0.1 MPa) at 100 °C for 24 h. Small amount of solution (ca. 20 µL) was drawn to measure at specified reaction times. The peak areas from GC-FID chromatograms were used to calculate with this formula:
where
Sub. = substrate (
SO),
IS = internal standard (biphenyl),
Pro. = product (
SC),
ECN = equivalent carbon number, subscripted 0 = initial value before reaction.
The DFT calculations were conducted using Gaussian 16 program as previously reported [
18]. The structural optimization for [Ta
xNb
6−xO
19]
8− was performed by B3LYP with the solvation effect of DMSO using PCM (dielectric constant = 46.826). LanL2DZ basis sets were employed for Ta and Nb atoms and 6−31 + G(d) basis sets for O and C atoms to investigate the effect of the composition of the clusters on the NBO charge of O atoms and the adsorption energies of CO
2 on [Ta
1Nb
5O
19]
8−.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







