

Hydrogenation of Carbon Monoxide in the Liquid Phase: Influence of the Synthetic Methods on Characteristics and Activity of Hydrogenation Catalysts


Abstract
:
1. Introduction
2. Results and Discussion
2.1. X-ray Fluorescence Measurements of All Synthesised Catalysts
2.1.1. Pure Ruthenium Systems
2.1.2. Ruthenium-Promoted Nickel Systems
2.2. Powder X-ray Diffraction (XRD) and Scanning Electron Microscopy (SEM)
2.2.1. Pure Ruthenium Systems
2.2.2. Ruthenium-Promoted Nickel Systems
2.3. Temperature-Programmed Reduction (TPR) and Temperature-Programmed Desorption (TPD) Studies
2.3.1. Pure Ruthenium Systems (TPR)
2.3.2. Ruthenium-Promoted Nickel Systems (TPR)
2.3.3. Comparison of H2 Uptakes for Pure Ruthenium and Ruthenium-Nickel Systems (TPR)
2.3.4. Temperature-Programmed CO Desorption–Pure Ruthenium Systems (CO-TPD)
2.4. Catalyst Activity
3. Materials and Methods
3.1. Materials
3.2. Catalyst Synthesis
3.3. Catalyst Characterisation
3.3.1. X-ray Fluorescence Spectroscopy (XRF)
3.3.2. Powder X-ray Diffraction (PXRD)
3.3.3. Temperature-Programmed Desorption of CO (CO-TPD)
3.3.4. Temperature-Programmed Reduction (TPR)
3.4. Catalyst Activity Tests
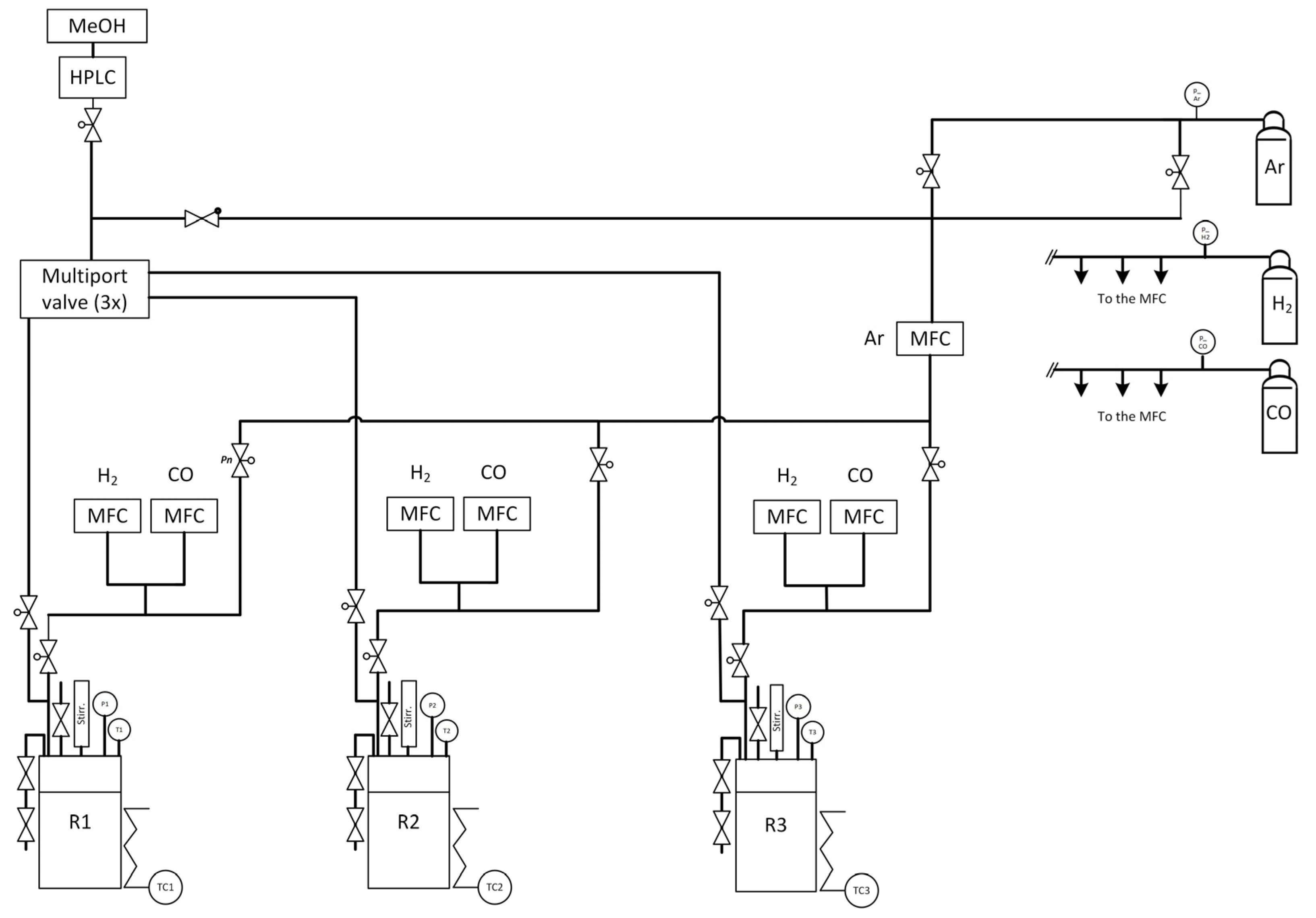
3.5. Process Analytics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jacob, E. C-1 Oxygenate als nachhaltige Kraftstoffe und deren günstige Eigenschaften. In Zukünftige Kraftstoffe: Energiewende des Transports als ein Weltweites Klimaziel; Maus, W., Ed.; Springer: Berlin/Heidelberg, Germany, 2019; pp. 155–180. [Google Scholar]
- Oestreich, D. Prozessentwicklung zur Gewinnung von Oxymethylenethern (OME) aus Methanol und Formaldehyd; KIT Scientific Publishing: Karlsruhe, Germany, 2017. [Google Scholar]
- Bahmanpour, A.M.; Hoadley, A.; Tanksale, A. Formaldehyde production via hydrogenation of carbon monoxide in the aqueous phase. Green Chem. 2015, 17, 3500–3507. [Google Scholar] [CrossRef]
- Bahmanpour Ali, M. Single-Step Conversion of Synthesis Gas into Formaldehyde; Monash University: Melbourne, Australia, 2016. [Google Scholar]
- Ahmad, W.; Chan, F.L.; Hoadley, A.; Wang, H.; Tanksale, A. Synthesis of oxymethylene dimethyl ethers (OMEn) via methanol mediated COx hydrogenation over Ru/BEA catalysts. Appl. Catal. B Environ. 2020, 269, 118765. [Google Scholar] [CrossRef]
- Ahmad, W.; Chan, F.L.; Chaffee, A.L.; Wang, H.; Hoadley, A.; Tanksale, A. Dimethoxymethane Production via Catalytic Hydrogenation of Carbon Monoxide in Methanol Media. ACS Sustain. Chem. Eng. 2020, 8, 2081–2092. [Google Scholar] [CrossRef]
- Remediakis, I.N.; Abild-Pedersen, F.; Nørskov, J.K. DFT Study of Formaldehyde and Methanol Synthesis from CO and H2 on Ni(111). J. Phys. Chem. B 2004, 108, 14535–14540. [Google Scholar] [CrossRef]
- Munnik, P.; de Jongh, P.E.; de Jong, K.P. Recent Developments in the Synthesis of Supported Catalysts. Chem. Rev. 2015, 115, 6687–6718. [Google Scholar] [CrossRef]
- Swain, P.; Mallika, C.; Srinivasan, R.; Mudali, U.K.; Natarajan, R. Separation and recovery of ruthenium: A review. J. Radioanal. Nucl. Chem. 2013, 298, 781–796. [Google Scholar] [CrossRef]
- Mazzieri, V.; Coloma-Pascual, F.; González, M.; L’argentičre, P.; Fígoli, N. Preparation of Ru/Al2O3 Catalysts from RuCl3. React. Kinet. Catal. Lett. 2002, 76, 53–59. [Google Scholar] [CrossRef]
- Düllmann, C.E.; Eichler, B.; Eichler, R.; Gäggeler, H.W.; Türler, A. On the Stability and Volatility of Group 8 Tetroxides, MO4 (M = Ruthenium, Osmium, and Hassium (Z = 108)). J. Phys. Chem. B 2002, 106, 6679–6684. [Google Scholar] [CrossRef]
- Betancourt, P.; Rives, A.; Hubaut, R.; Scott, C.E.; Goldwasser, J. A study of the ruthenium–alumina system. Appl. Catal. A Gen. 1998, 170, 307–314. [Google Scholar] [CrossRef]
- Balcerzak, M.; Święcicka, E.; Balukiewicz, E. Determination of platinum and ruthenium in Pt and Pt–Ru catalysts with carbon support by direct and derivative spectrophotometry. Talanta 1999, 48, 39–47. [Google Scholar] [CrossRef]
- Balcerzak, M. Analytical Methods for the Determination of Ruthenium: The State of the Art. Crit. Rev. Anal. Chem. 2002, 32, 181–226. [Google Scholar] [CrossRef]
- Chang, X.-J.; Gong, B.-L.; Su, Z.-X.; Yang, D.; Luo, X.-Y. ICP-OES determination of traces of Ru, Au, V and Ti preconcentrated and separated by a new poly(epoxy-melamine) chelating resin from solutions. Fresenius J. Anal. Chem. 1998, 360, 736–739. [Google Scholar] [CrossRef]
- Suoranta, T.; Niemelä, M.; Perämäki, P. Comparison of digestion methods for the determination of ruthenium in catalyst materials. Talanta 2014, 119, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Wang, Z.; Cheng, H.; He, T.; Liu, Y.; Chen, K.; Hu, Z.; Liu, Y. Comparative Determination of Mass Fractions of Elements with Variable Chalcophile Affinities in Geological Reference Materials with and without HF-desilicification. Geostand. Geoanal. Res. 2020, 44, 501–521. [Google Scholar] [CrossRef]
- Grillo, F.; Van Bui, H.; Moulijn, J.A.; Kreutzer, M.T.; van Ommen, J.R. Understanding and Controlling the Aggregative Growth of Platinum Nanoparticles in Atomic Layer Deposition: An Avenue to Size Selection. J. Phys. Chem. Lett. 2017, 8, 975–983. [Google Scholar] [CrossRef] [Green Version]
- Grillo, F.; Soethoudt, J.; Marques, E.A.; de Martín, L.; Van Dongen, K.; van Ommen, J.R.; Delabie, A. Area-Selective Deposition of Ruthenium by Area-Dependent Surface Diffusion. Chem. Mater. 2020, 32, 9560–9572. [Google Scholar] [CrossRef]
- Lin, B.; Wang, R.; Lin, J.; Du, S.; Yu, X.; Wei, K. Preparation of chlorine-free alumina-supported ruthenium catalyst for ammonia synthesis base on RuCl3 by hydrazine reduction. Catal. Commun. 2007, 8, 1838–1842. [Google Scholar] [CrossRef]
- Rynkowski, J.M.; Paryjczak, T.; Lenik, M. Characterization of alumina supported nickel-ruthenium systems. Appl. Catal. A Gen. 1995, 126, 257–271. [Google Scholar] [CrossRef]
- Rynkowski, J.; Paryjczak, T.; Lenik, M. On the nature of oxidic nickel phases in NiO/γ-Al2O3 catalysts. Appl. Catal. A Gen. 1993, 106, 73–82. [Google Scholar] [CrossRef]
- Ping, D.; Wan, Y.; Zhao, X.; Geng, J.; Dong, X. Zr-promoted nickel-rich spinel-supported Ni catalysts with enhanced performance for selective CO methanation. Int. J. Energy Res. 2022, 46, 9128–9137. [Google Scholar] [CrossRef]
- Khalighi, R.; Bahadoran, F.; Panjeshahi, M.H.; Zamaniyan, A.; Tahouni, N. Effects of Nickel Aluminate Spinel (NiAl2O4) as Catalyst Support and Promoters (Ru, Rh) in Fischer-Tropsch Synthesis. ChemistrySelect 2020, 5, 7934–7940. [Google Scholar] [CrossRef]
- Hull, A.W. X-ray crystal analysis of thirteen common metals. Phys. Rev. 1921, 17, 571. [Google Scholar] [CrossRef] [Green Version]
- Wynblatt, P.; Gjostein, N.A. Supported metal crystallites. Prog. Solid State Chem. 1975, 9, 21–58. [Google Scholar] [CrossRef]
- Miranda, M.; Sasaki, J. The limit of application of the Scherrer equation. Acta Crystallogr. Sect. A Found. Adv. 2018, 74, 54–65. [Google Scholar] [CrossRef]
- Muniz, F.T.L.; Miranda, M.R.; dos Santos, C.M.; Sasaki, J.M. The Scherrer equation and the dynamical theory of X-ray diffraction. Acta Crystallogr. Sect. A Found. Adv. 2016, 72, 385–390. [Google Scholar] [CrossRef]
- Nandanwar, S.U.; Chakraborty, M.; Mukhopadhyay, S.; Shenoy, K.T. Stability of ruthenium nanoparticles synthesized by solvothermal method. Cryst. Res. Technol. 2011, 46, 393–399. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, Y.; Zheng, H.; Zhang, C.; Zhang, B.; Li, Y. Aqueous-phase hydrodeoxygenation of carboxylic acids to alcohols or alkanes over supported Ru catalysts. J. Mol. Catal. A Chem. 2011, 351, 217–227. [Google Scholar] [CrossRef]
- Hosokawa, S.; Kanai, H.; Utani, K.; Taniguchi, Y.-I.; Saito, Y.; Imamura, S. State of Ru on CeO2 and its catalytic activity in the wet oxidation of acetic acid. Appl. Catal. B Environ. 2003, 45, 181–187. [Google Scholar] [CrossRef]
- Sun, F.; Chen, L.; Weng, Y.; Wang, T.; Qiu, S.; Li, Q.; Wang, C.; Zhang, Q.; Ma, L. Transformation of biomass polyol into hydrocarbon fuels in aqueous medium over Ru-Mo/CNT catalyst. Catal. Commun. 2017, 99, 30–33. [Google Scholar] [CrossRef]
- Cattania, M.G.; Parmigiani, F.; Ragaini, V. A study of ruthenium catalysts on oxide supports. Surf. Sci. 1989, 211–212, 1097–1105. [Google Scholar] [CrossRef]
- Guczi, L.; Schay, Z.; Matusek, K.; Bogyay, I. Surface structure and selectivity control in the CO + H2 reaction over FeRu Bimetallic catalysts. Appl. Catal. 1986, 22, 289–309. [Google Scholar] [CrossRef]
- Pinheiro, A.N.; Valentini, A.; Sasaki, J.M.; Oliveira, A.C. Highly stable dealuminated zeolite support for the production of hydrogen by dry reforming of methane. Appl. Catal. A Gen. 2009, 355, 156–168. [Google Scholar] [CrossRef]
- Crisafulli, C.; Scirè, S.; Minicò, S.; Solarino, L. Ni–Ru bimetallic catalysts for the CO2 reforming of methane. Appl. Catal. A Gen. 2002, 225, 1–9. [Google Scholar] [CrossRef]
- Arif, N.N.M.; Abidin, S.Z.; Osazuwa, O.U.; Vo, D.-V.N.; Azizan, M.T.; Taufiq-Yap, Y.H. Hydrogen production via CO2 dry reforming of glycerol over ReNi/CaO catalysts. Int. J. Hydrogen Energy 2019, 44, 20857–20871. [Google Scholar] [CrossRef]
- Moreno, A.Á.; Ramirez-Reina, T.; Ivanova, S.; Roger, A.-C.; Centeno, M.Á.; Odriozola, J.A. Bimetallic Ni–Ru and Ni–Re catalysts for dry reforming of methane: Understanding the synergies of the selected promoters. Front. Chem. 2021, 9, 694976. [Google Scholar] [CrossRef] [PubMed]
- Andraos, S.; Abbas-Ghaleb, R.; Chlala, D.; Vita, A.; Italiano, C.; Laganà, M.; Pino, L.; Nakhl, M.; Specchia, S. Production of hydrogen by methane dry reforming over ruthenium-nickel based catalysts deposited on Al2O3, MgAl2O4, and YSZ. Int. J. Hydrogen Energy 2019, 44, 25706–25716. [Google Scholar] [CrossRef]
- Low, G.G.; Bell, A.T. Studies of CO desorption and reaction with H2 on alumina-supported Ru. J. Catal. 1979, 57, 397–405. [Google Scholar] [CrossRef]
- Low, G.G. Temperature-Programmed Desorption and Reaction of CO and H2 on Alumina-Supported Ruthenium Catalyst; U.S. Department of Energy Office of Scientific and Technical Information: Oak Ridge, TN, USA, 1978. [Google Scholar]
- Kang, J.; Zhang, S.; Zhang, Q.; Wang, Y. Ruthenium Nanoparticles Supported on Carbon Nanotubes as Efficient Catalysts for Selective Conversion of Synthesis Gas to Diesel Fuel. Angew. Chem. Int. Ed. 2009, 48, 2565–2568. [Google Scholar] [CrossRef]
- Yang, E.; Jang, E.J.; Lee, J.G.; Yoon, S.; Lee, J.; Musselwhite, N.; Somorjai, G.A.; Kwak, J.H.; An, K. Acidic effect of porous alumina as supports for Pt nanoparticle catalysts in n-hexane reforming. Catal. Sci. Technol. 2018, 8, 3295–3303. [Google Scholar] [CrossRef]
- Kaiser, D.; Beckmann, L.; Walter, J.; Bertau, M. Conversion of Green Methanol to Methyl Formate. Catalysts 2021, 11, 869. [Google Scholar] [CrossRef]
- Černohorský, M. The ratio method for absolute measurements of lattice parameters with cylindrical cameras. Acta Crystallogr. 1960, 13, 823–826. [Google Scholar] [CrossRef]
- Lejus, A. Formation at high temperature of nonstoichiometric spinels and of derived phases in several oxide systems based on alumina and in the system alumina–aluminum nitride. Rev. Int. Hautes Temp. Refract 1964, 1, 53–95. [Google Scholar]
- Hull, A.W. A new method of X-ray crystal analysis. Phys. Rev. 1917, 10, 661. [Google Scholar] [CrossRef]
- Papoulis, A. Proceedings; Polytechnic Institute of Brooklyn Brooklyn, Interscience Publishers: New York, NY, USA, 1967; p. 87. [Google Scholar]
- Spanjers, C.S.; Beach, C.A.; Jones, A.J.; Dauenhauer, P.J. Increasing flame ionization detector (FID) sensitivity using post-column oxidation–methanation. Anal. Methods 2017, 9, 1928–1934. [Google Scholar] [CrossRef]
- Jorgensen, A.D.; Picel, K.C.; Stamoudis, V.C. Prediction of gas chromatography flame ionization detector response factors from molecular structures. Anal. Chem. 1990, 62, 683–689. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | Precursor | Precursor Concentration [mM] | Final Catalyst | ||
|---|---|---|---|---|---|
| Ru | Ni | Ru | Ni | ||
| Ru-NO-1 | * | - | 5 | - | Ru/γ-Al2O3 1 wt% |
| Ru-NO-3 | * | - | 15 | - | Ru/γ-Al2O3 3 wt% |
| Ru-NO-5 | * | - | 24 | - | Ru/γ-Al2O3 5 wt% |
| RuNi-NO-ir. | * | ‡ | 20 | 90 | RuNi/γ-Al2O3 |
| RuNi-NO-icr. | * | ‡ | 30 | 80 | RuNi/γ-Al2O3 |
| Ru-Cl-1 | ° | - | 5 | - | Ru/γ-Al2O3 1 wt% |
| Ru-Cl-3 | ° | - | 15 | - | Ru/γ-Al2O3 3 wt% |
| Ru-Cl-5 | ° | - | 24 | - | Ru/γ-Al2O3 5 wt% |
| RuNi-Cl-ir. | ° | ‡ | 20 | 90 | RuNi/γ-Al2O3 4 wt% |
| RuNi-Cl-icr. | ° | ‡ | 20 | 80 | RuNi/γ-Al2O3 6 wt% |
| Catalyst | Ru (ic) a | Cl (ic) a | Ru (icr) b | Cl (icr) b |
|---|---|---|---|---|
| Ru-NO-1 | 0.28 | - | 0.08 | - |
| Ru-NO-3 | 1.05 | - | 0.52 | - |
| Ru-NO-5 | 1.60 | - | 0.67 | - |
| Ru-Cl-1 | 0.47 | 0.11 | 0.15 | 0.10 |
| Ru-Cl-3 | 1.44 | 0.12 | 0.60 | 0.12 |
| Ru-Cl-5 | 2.58 | 0.18 | 1.82 | 0.19 |
| Catalyst | Calcined | Reduced | ||||
|---|---|---|---|---|---|---|
| Ru | Ni | Cl | Ru | Ni | Cl | |
| RuNi-NO-ir. | - | - | - | 0.79 | 10.92 | - |
| RuNi-NO-icr. | 2.13 | 10.90 | - | 1.96 | 11.14 | - |
| RuNi-Cl-ir. | - | - | - | 1.42 | 11.16 | 1.06 |
| RuNi-Cl-icr. | 1.19 | 10.34 | 0.516 | 1.14 | 10.78 | 0.49 |
| Catalyst | d (Ru0) [nm] Average Crystallite Size |
|---|---|
| Ru-NO-1-icr. | 54 ± 2 |
| Ru-NO-3-icr. | 33 ± 4 |
| Ru-NO-5-icr. | 30 ± 3 |
| Ru-Cl-1-icr. | 44 ± 3 |
| Ru-Cl-3-icr. | 37 ± 3 |
| Ru-Cl-5-icr. | 31 ± 1 |
| Catalyst | H2 Uptake [µmol/g] |
|---|---|
| Ru-NO-1-ic. | 70 |
| Ru-NO-3-ic. | 150 |
| Ru-NO-5-ic. | 290 |
| RuNi-NO-ic. (LT) * | 230 |
| RuNi-NO-ic. (HT) ° | 500 |
| Ru-Cl-1-ic. | 70 |
| Ru-Cl-3-ic. | 210 |
| Ru-Cl-5-ic. | 320 |
| RuNi-Cl-ic. (LT) * | 170 |
| RuNi-Cl-ic. (HT) ° | 500 |
| Catalyst | Monolayer CO Uptake [µmol/g] | Active Surface Area [m2/g] | Desorption Maxima [°C] | Dispersion [%] |
|---|---|---|---|---|
| Ru-NO-3-icr. | 1.43 | 0.053 | 80/291/671 | 1.8 |
| Ru-Cl-3-icr. | - | - | 83/325/686 | - |
| Treatment | Procedure | Remark |
|---|---|---|
| 1 | Gas: 5% H2/Ar Flow: 50 mL/min Ramp: 30 °C ➔ 450 °C Rate: 10 K/min Hold 300 min | Reduction |
| 2 | Gas: Ar Flow: 50 mL/min Temp.: 450 °C Hold: 60 min Cool down to 30 °C Hold 10 min | Purging after reduction |
| 3 | Gas: 25% CO/He Puls (15 times) Gas: He Flow: 10 mL/min Temp.: 30 °C | Pulse titration |
| 4 | Gas: He Flow: 50 mL/min Temp.: 30 °C Hold: 15 min | Purging after saturation |
| 5 | Gas: He Flow: 30 mL/min Ramp: 3 K/min Hold: 60 min | Temperature-programmed desorption (TPD) |
| Treatment | Procedure | Remark |
|---|---|---|
| 1 | Gas: Ar Flow: 30 mL/min Ramp: 30 °C ➔ 200 °C Rate: 10 K/min Hold 15 min | Drying at 200 °C/purging |
| 2 | Gas: Ar Flow: 30 mL/min Ramp: 200 °C ➔ 30 °C Rate: 10 K/min Hold 20 min | Back to 30 °C/purging |
| 3 | Gas: 10% H2/Ar Flow: 30 mL/min Temp.:30 °C; Hold 5 min Ramp: 30 °C ➔ 850 °C Rate: 5 K/min Hold 10 min | Temperature-programmed reduction (TPR) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheikh, K.A.; Drexler, R.; Zevaco, T.A.; Sauer, J.; Bender, M. Hydrogenation of Carbon Monoxide in the Liquid Phase: Influence of the Synthetic Methods on Characteristics and Activity of Hydrogenation Catalysts. Catalysts 2023, 13, 482. https://doi.org/10.3390/catal13030482
Sheikh KA, Drexler R, Zevaco TA, Sauer J, Bender M. Hydrogenation of Carbon Monoxide in the Liquid Phase: Influence of the Synthetic Methods on Characteristics and Activity of Hydrogenation Catalysts. Catalysts. 2023; 13(3):482. https://doi.org/10.3390/catal13030482
Chicago/Turabian StyleSheikh, Kalim A., Ricki Drexler, Thomas A. Zevaco, Jörg Sauer, and Michael Bender. 2023. "Hydrogenation of Carbon Monoxide in the Liquid Phase: Influence of the Synthetic Methods on Characteristics and Activity of Hydrogenation Catalysts" Catalysts 13, no. 3: 482. https://doi.org/10.3390/catal13030482
APA StyleSheikh, K. A., Drexler, R., Zevaco, T. A., Sauer, J., & Bender, M. (2023). Hydrogenation of Carbon Monoxide in the Liquid Phase: Influence of the Synthetic Methods on Characteristics and Activity of Hydrogenation Catalysts. Catalysts, 13(3), 482. https://doi.org/10.3390/catal13030482