


Unveiling the Role of In Situ Sulfidation and H2O Excess on H2S Decomposition to Carbon-Free H2 over Cobalt/Ceria Catalysts



Abstract
:1. Introduction
2. Results and Discussion
2.1. Characterization Studies
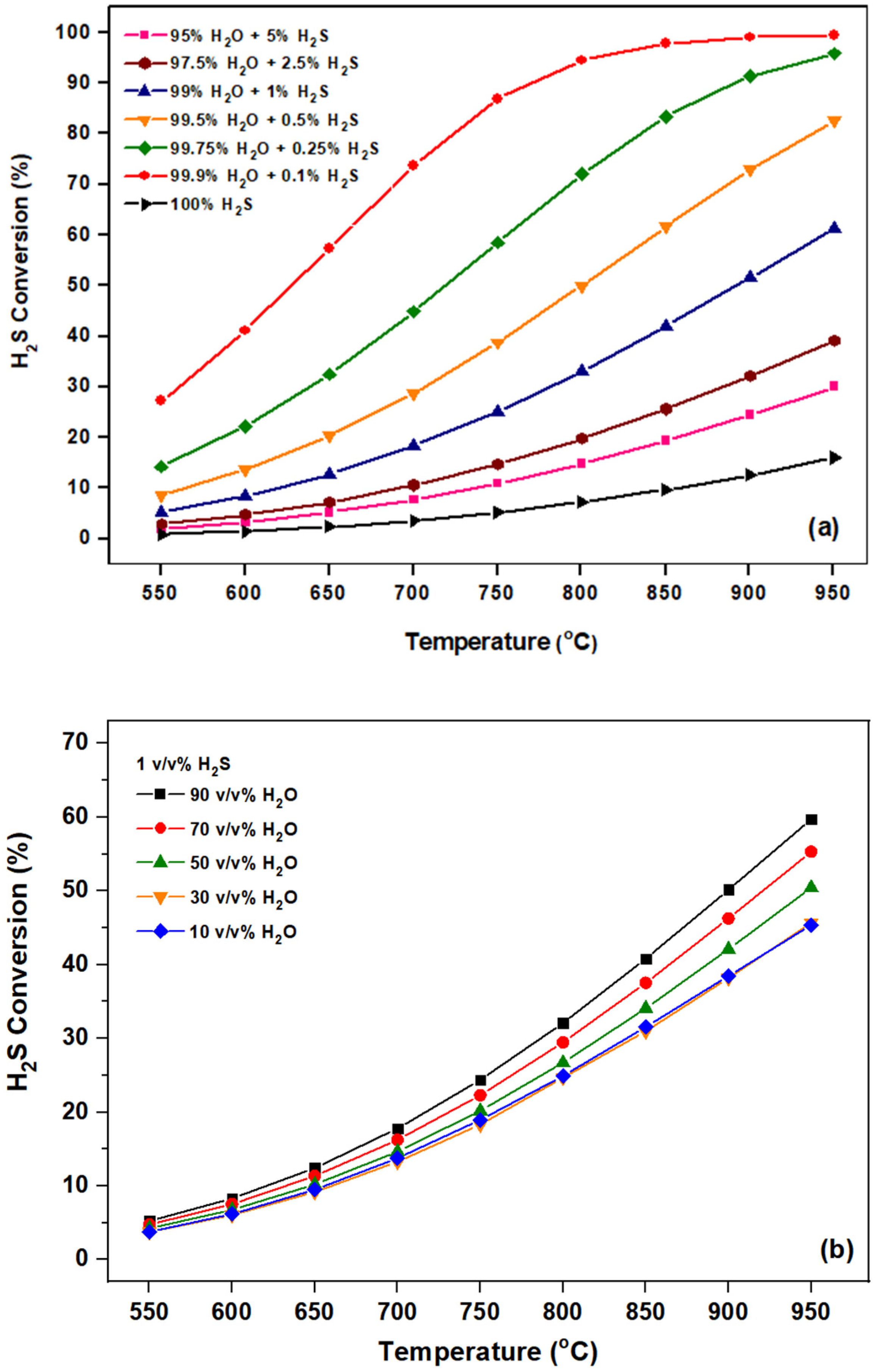
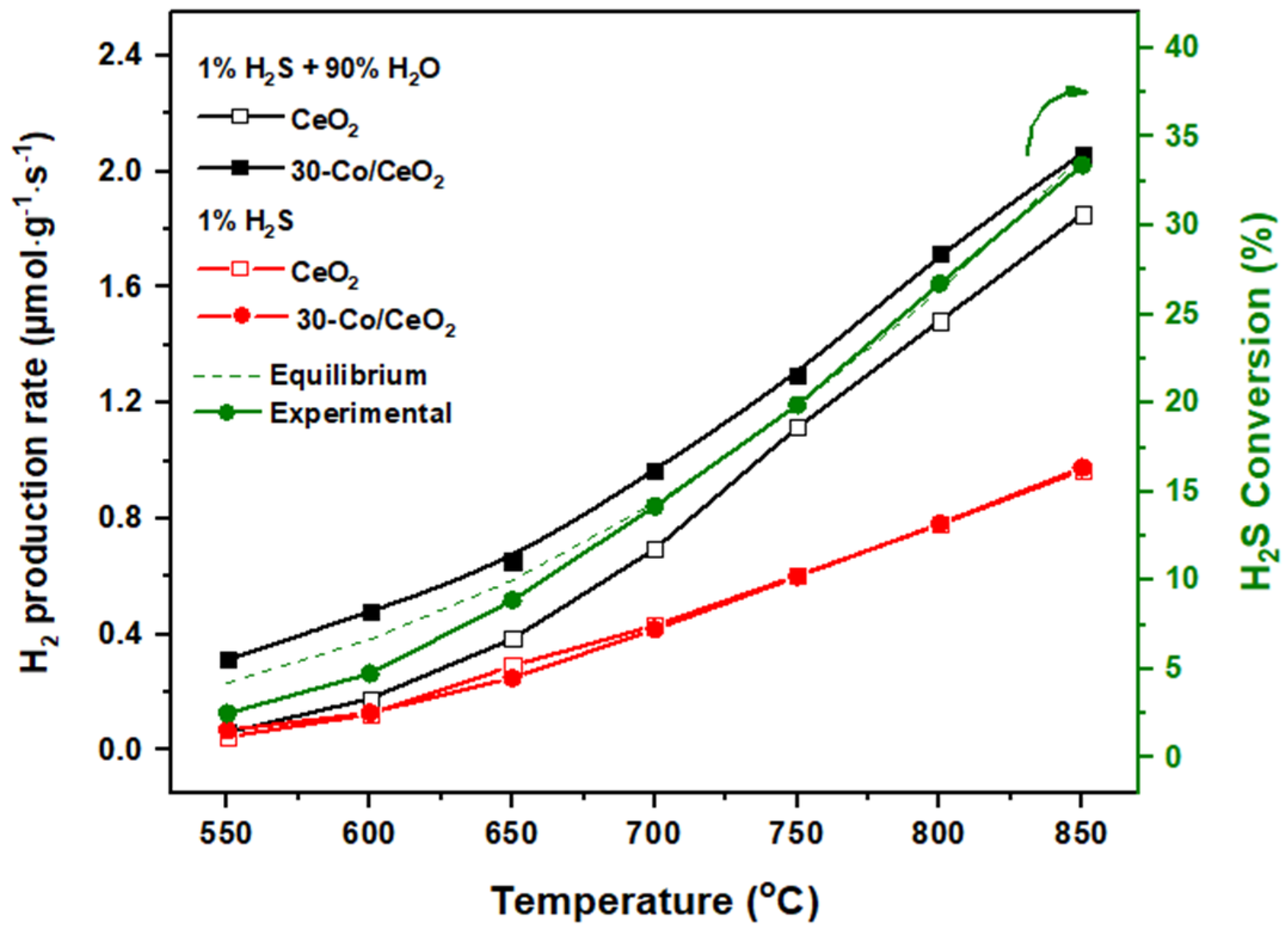
2.2. Catalytic Evaluation
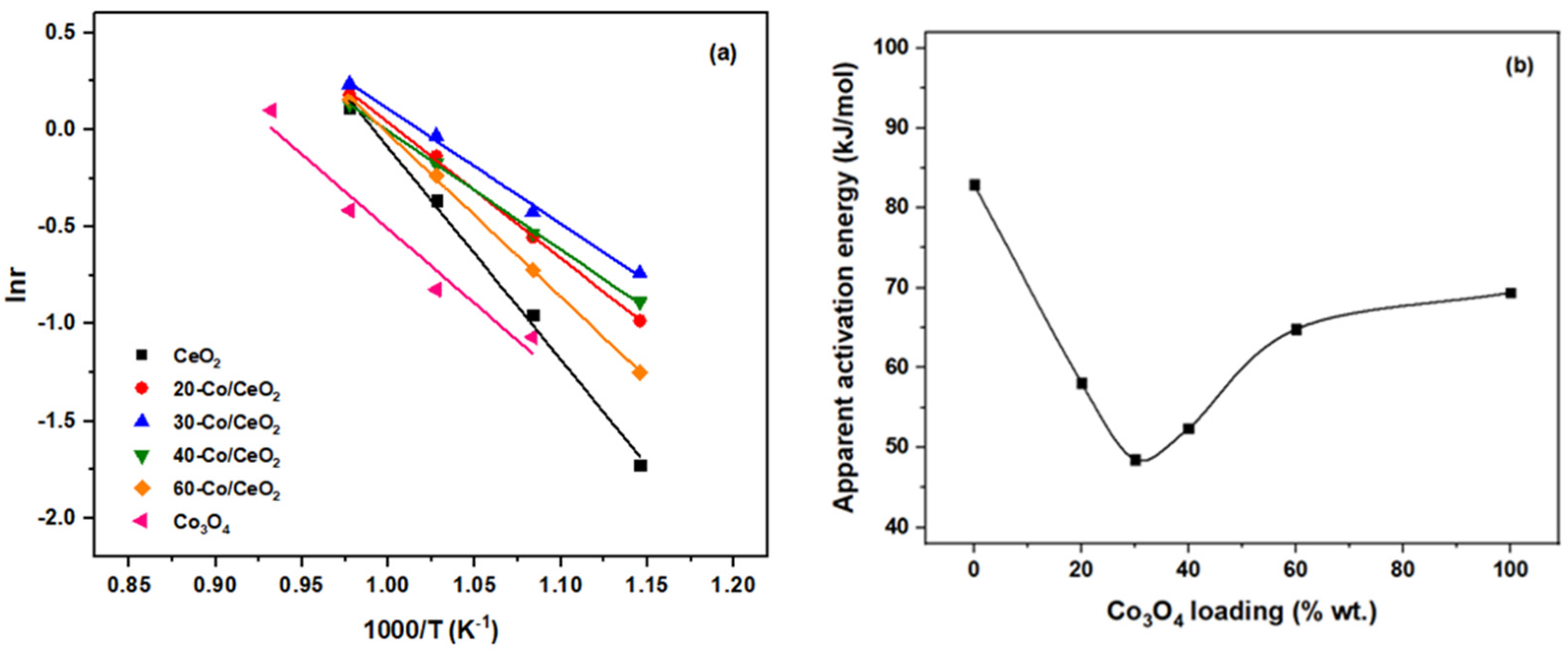
2.3. Apparent Activation Energies
2.4. Stability Tests
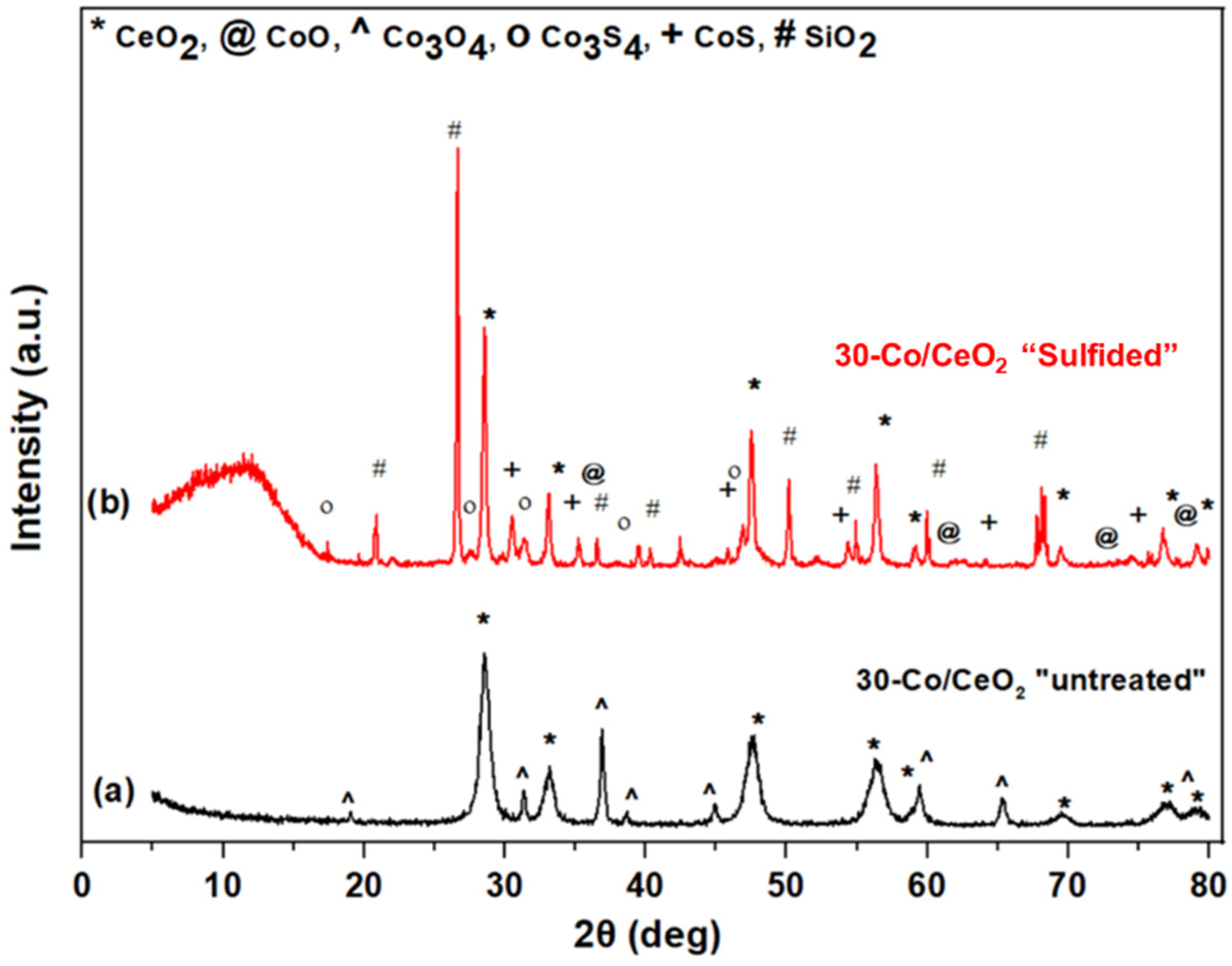
2.5. Spent Catalysts Characterization
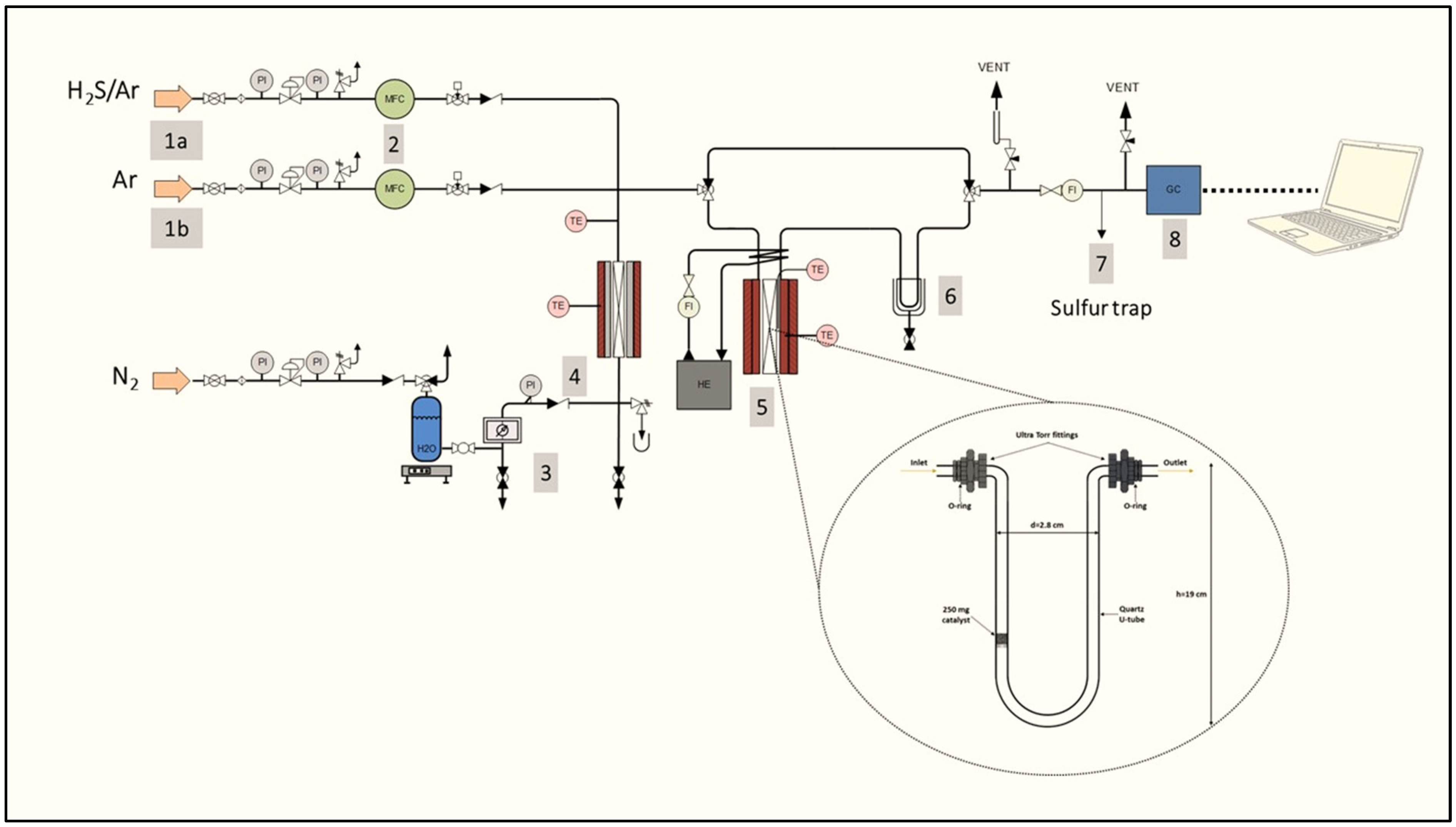
3. Materials and Methods
3.1. Materials Preparation
3.2. Materials Characterization
3.3. Catalytic Evaluation
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- IPCC. Climate Change 2021—The Physical Science Basis; Cambridge University Press: Cambridge, UK, 2021. [Google Scholar]
- Hulme, M. 1.5 °C and Climate Research after the Paris Agreement. Nat. Clim. Chang. 2016, 6, 222–224. [Google Scholar] [CrossRef] [Green Version]
- Depledge, J.; Saldivia, M.; Peñasco, C. Glass Half Full or Glass Half Empty?: The 2021 Glasgow Climate Conference. Clim. Policy 2022, 22, 147–157. [Google Scholar] [CrossRef]
- Capurso, T.; Stefanizzi, M.; Torresi, M.; Camporeale, S.M. Perspective of the Role of Hydrogen in the 21st Century Energy Transition. Energy Convers. Manag. 2022, 251, 114898. [Google Scholar] [CrossRef]
- Mac Dowell, N.; Sunny, N.; Brandon, N.; Herzog, H.; Ku, A.Y.; Maas, W.; Ramirez, A.; Reiner, D.M.; Sant, G.N.; Shah, N. The Hydrogen Economy: A Pragmatic Path Forward. Joule 2021, 5, 2524–2529. [Google Scholar] [CrossRef]
- Dincer, I.; Acar, C. Smart Energy Solutions with Hydrogen Options. Int. J. Hydrogen Energy 2018, 43, 8579–8599. [Google Scholar] [CrossRef]
- Yue, M.; Lambert, H.; Pahon, E.; Roche, R.; Jemei, S.; Hissel, D. Hydrogen Energy Systems: A Critical Review of Technologies, Applications, Trends and Challenges. Renew. Sustain. Energy Rev. 2021, 146, 111180. [Google Scholar] [CrossRef]
- International Energy Agency. Global Hydrogen Review 2021; International Energy Agency: Paris, France, 2021. [Google Scholar]
- Hermesmann, M.; Müller, T.E. Green, Turquoise, Blue, or Grey? Environmentally Friendly Hydrogen Production in Transforming Energy Systems. Prog. Energy Combust. Sci. 2022, 90, 100996. [Google Scholar] [CrossRef]
- Younas, M.; Shafique, S.; Hafeez, A.; Javed, F.; Rehman, F. An Overview of Hydrogen Production: Current Status, Potential, and Challenges. Fuel 2022, 316, 123317. [Google Scholar] [CrossRef]
- Nnabuife, S.G.; Ugbeh-Johnson, J.; Okeke, N.E.; Ogbonnaya, C. Present and Projected Developments in Hydrogen Production: A Technological Review. Carbon Capture Sci. Technol. 2022, 3, 100042. [Google Scholar] [CrossRef]
- Kayfeci, M.; Keçebaş, A.; Bayat, M. Hydrogen Production. In Solar Hydrogen Production; Elsevier: Amsterdam, The Netherland, 2019; pp. 45–83. [Google Scholar]
- Ishaq, H.; Dincer, I.; Crawford, C. A Review on Hydrogen Production and Utilization: Challenges and Opportunities. Int. J. Hydrogen Energy 2021, 47, 26238–26264. [Google Scholar] [CrossRef]
- De Crisci, A.G.; Moniri, A.; Xu, Y. Hydrogen from Hydrogen Sulfide: Towards a More Sustainable Hydrogen Economy. Int. J. Hydrogen Energy 2019, 44, 1299–1327. [Google Scholar] [CrossRef]
- Chan, Y.H.; Loy, A.C.M.; Cheah, K.W.; Chai, S.Y.W.; Ngu, L.H.; How, B.S.; Li, C.; Lock, S.S.M.; Wong, M.K.; Yiin, C.L.; et al. Hydrogen Sulfide (H2S) Conversion to Hydrogen (H2) and Value-Added Chemicals: Progress, Challenges and Outlook. Chem. Eng. J. 2023, 458, 141398. [Google Scholar] [CrossRef]
- Bahadori, A. Natural Gas Processing: Technology and Engineering Design; Elsevier: Amsterdam, The Netherlands, 2014; ISBN 9780080999715. [Google Scholar]
- Rodríguez, E.; Harvey, W.S.; Ásbjörnsson, E.J. Review of H2S Abatement Methods in Geothermal Plants. In Proceedings of the Thirty-Eighth Workshop on Geothermal Reservoir Engineering, Stanford, CA, USA, 11–13 February 2013. [Google Scholar]
- Deng, Q.; Wang, Q.; Liu, M.; Zhao, F. Geological Factors Controlling H2S in Coal Seams. In Mine Planning and Equipment Selection; Springer International Publishing: Cham, Switzerland, 2014; pp. 619–627. [Google Scholar]
- Corpas, F.J.; Palma, J.M. H2S Signaling in Plants and Applications in Agriculture. J. Adv. Res. 2020, 24, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, B.B.; Findlay, A.J.; Pellerin, A. The Biogeochemical Sulfur Cycle of Marine Sediments. Front. Microbiol. 2019, 10, 849. [Google Scholar] [CrossRef] [PubMed]
- King, M.J.; Davenport, W.G.; Moats, M.S. Sulfuric Acid Manufacture: Analysis, Control and Optimization; Elsevier: Amsterdam, The Netherlands, 2013; ISBN 978-0-08-098220-5. [Google Scholar]
- Kaiser, M.J.; de Klerk, A.; Gary, J.H.; Hwerk, G.E. Petroleum Refining: Technology, Economics, and Markets; CRC Press: Boca Raton, FL, USA, 2019; ISBN 9780429188893. [Google Scholar]
- Wiheeb, A.D.; Shamsudin, I.K.; Ahmad, M.A.; Murat, M.N.; Kim, J.; Othman, M.R. Present Technologies for Hydrogen Sulfide Removal from Gaseous Mixtures. Rev. Chem. Eng. 2013, 29, 449–470. [Google Scholar] [CrossRef]
- Ghahraloud, H.; Farsi, M.; Rahimpour, M.R. Hydrogen Production through Thermal Decomposition of Hydrogen Sulfide: Modification of the Sulfur Recovery Unit To Produce Ultrapure Hydrogen. Ind. Eng. Chem. Res. 2018, 57, 14114–14123. [Google Scholar] [CrossRef]
- Palma, V.; Vaiano, V.; Barba, D.; Colozzi, M.; Palo, E.; Barbato, L.; Cortese, S. H2 Production by Thermal Decomposition of H2S in the Presence of Oxygen. Int. J. Hydrogen Energy 2015, 40, 106–113. [Google Scholar] [CrossRef]
- Burra, K.R.G.; Bassioni, G.; Gupta, A.K. Catalytic Transformation of H2S for H2 Production. Int. J. Hydrogen Energy 2018, 43, 22852–22860. [Google Scholar] [CrossRef]
- Jiang, G.; Zhang, X.; Zhang, F.; Liu, Z.; Wang, Z.; Hao, Z.; Lin, C. Efficient Recovery of Hydrogen and Sulfur Resources over Non-Sulfide Based LaFexAl12-XO19 Hexaaluminate Catalysts by H2S Catalytic Decomposition. Appl. Catal. B Environ. 2020, 263, 118354. [Google Scholar] [CrossRef]
- Linga Reddy, E.; Biju, V.M.; Subrahmanyam, C. Production of Hydrogen and Sulfur from Hydrogen Sulfide Assisted by Nonthermal Plasma. Appl. Energy 2012, 95, 87–92. [Google Scholar] [CrossRef]
- Dang, X.; Huang, J.; Kang, L.; Wu, T.; Zhang, Q. Research on Decomposition of Hydrogen Sulfide Using Nonthermal Plasma with Metal Oxide Catalysis. Energy Procedia 2012, 16, 856–862. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Bai, J.; Zhang, Y.; Zhou, C.; Wang, P.; Zha, L.; Li, J.; Simchi, A.; Zhou, B. High Yield of CO and Synchronous S Recovery from the Conversion of CO2 and H2S in Natural Gas Based on a Novel Electrochemical Reactor. Environ. Sci. Technol. 2021, 55, 14854–14862. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zhou, Y. Photochemical Decomposition of Hydrogen Sulfide. In Advanced Catalytic Materials-Photocatalysis and Other Current Trends; InTech: Rijeka, Croatia, 2016. [Google Scholar]
- Zaman, J.; Chakma, A. Production of Hydrogen and Sulfur from Hydrogen Sulfide. Fuel Process. Technol. 1995, 41, 159–198. [Google Scholar] [CrossRef]
- Chivers, T.; Lau, C. The Thermal Decomposition of Hydrogen Sulfide over Vanadium and Molybdenum Sulfides and Mixed Sulfide Catalysts in Quartz and Thermal Diffusion Column Reactors. Int. J. Hydrogen Energy 1987, 12, 235–243. [Google Scholar] [CrossRef]
- Kaloidas, V.; Papayannakos, N. Hydrogen Production from the Decomposition of Hydrogen Sulphide. Equilibrium Studies on the System H2S/H2/Si, (i = 1,…,8) in the Gas Phase. Int. J. Hydrogen Energy 1987, 12, 403–409. [Google Scholar] [CrossRef]
- Ipsakis, D.; Kraia, T.; Marnellos, G.E.; Ouzounidou, M.; Voutetakis, S.; Dittmeyer, R.; Dubbe, A.; Haas-Santo, K.; Konsolakis, M.; Figen, H.E.; et al. An Electrocatalytic Membrane-Assisted Process for Hydrogen Production from H 2 S in Black Sea: Preliminary Results. Int. J. Hydrogen Energy 2015, 40, 7530–7538. [Google Scholar] [CrossRef]
- Demirbas, A. Hydrogen Sulfide from the Black Sea for Hydrogen Production. Energy Sources Part A Recover. Util. Environ. Eff. 2009, 31, 1866–1872. [Google Scholar] [CrossRef]
- Ozturk, M.; Midilli, A.; Dincer, I. Effective Use of Hydrogen Sulfide and Natural Gas Resources Available in the Black Sea for Hydrogen Economy. Int. J. Hydrogen Energy 2021, 46, 10697–10707. [Google Scholar] [CrossRef]
- Ryann, A.; Perkins, N.J. The Black Sea: Dynamics, Ecology, and Conservation; Nova Science Publishers, Inc.: London, UK, 2011. [Google Scholar]
- Kraia, T.; Wachowski, S.; Vøllestad, E.; Strandbakke, R.; Konsolakis, M.; Norby, T.; Marnellos, G.E. Electrochemical Performance of Co3O4/CeO2 Electrodes in H2S/H2O Atmospheres in a Proton-Conducting Ceramic Symmetrical Cell with BaZr0.7Ce0.2Y0.1O3 Solid Electrolyte. Solid State Ionics 2017, 306, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Petrov, K.; Baykara, S.Z.; Ebrasu, D.; Gulin, M.; Veziroglu, A. An Assessment of Electrolytic Hydrogen Production from H 2 S in Black Sea Waters. Int. J. Hydrogen Energy 2011, 36, 8936–8942. [Google Scholar] [CrossRef]
- Guldal, N.O.; Figen, H.E.; Baykara, S.Z. Perovskite Catalysts for Hydrogen Production from Hydrogen Sulfide. Int. J. Hydrogen Energy 2018, 43, 1038–1046. [Google Scholar] [CrossRef]
- Guldal, N.O.; Figen, H.E.; Baykara, S.Z. Production of Hydrogen from Hydrogen Sulfide with Perovskite Type Catalysts: LaMO3. Chem. Eng. J. 2017, 313, 1354–1363. [Google Scholar] [CrossRef]
- Lykaki, M.; Pachatouridou, E.; Iliopoulou, E.; Carabineiro, S.A.C.; Konsolakis, M. Impact of the Synthesis Parameters on the Solid State Properties and the CO Oxidation Performance of Ceria Nanoparticles. RSC Adv. 2017, 7, 6160–6169. [Google Scholar] [CrossRef] [Green Version]
- Konsolakis, M. The Role of Copper–Ceria Interactions in Catalysis Science: Recent Theoretical and Experimental Advances. Appl. Catal. B Environ. 2016, 198, 49–66. [Google Scholar] [CrossRef]
- Xu, Y.; Mofarah, S.S.; Mehmood, R.; Cazorla, C.; Koshy, P.; Sorrell, C.C. Design Strategies for Ceria Nanomaterials: Untangling Key Mechanistic Concepts. Mater. Horizons 2021, 8, 102–123. [Google Scholar] [CrossRef] [PubMed]
- Kraia, T.; Kaklidis, N.; Konsolakis, M.; Marnellos, G.E. Hydrogen Production by H2S Decomposition over Ceria Supported Transition Metal (Co, Ni, Fe and Cu) Catalysts. Int. J. Hydrogen Energy 2019, 44, 9753–9762. [Google Scholar] [CrossRef]
- Jha, A.; Jeong, D.W.; Lee, Y.L.; Nah, I.W.; Roh, H.S. Enhancing the Catalytic Performance of Cobalt Oxide by Doping on Ceria in the High Temperature Water-Gas Shift Reaction. RSC Adv. 2015, 5, 103023–103029. [Google Scholar] [CrossRef]
- Liu, Z.; Li, J.; Buettner, M.; Ranganathan, R.V.; Uddi, M.; Wang, R. Metal-Support Interactions in CeO2- and SiO2-Supported Cobalt Catalysts: Effect of Support Morphology, Reducibility, and Interfacial Configuration. ACS Appl. Mater. Interfaces 2019, 11, 17035–17049. [Google Scholar] [CrossRef]
- Varvoutis, G.; Lykaki, M.; Papista, E.; Carabineiro, S.A.C.; Psarras, A.C.; Marnellos, G.E.; Konsolakis, M. Effect of Alkali (Cs) Doping on the Surface Chemistry and CO2 Hydrogenation Performance of CuO/CeO2 Catalysts. J. CO2 Util. 2021, 44, 101408. [Google Scholar] [CrossRef]
- Díez-Ramírez, J.; Sánchez, P.; Kyriakou, V.; Zafeiratos, S.; Marnellos, G.E.; Konsolakis, M.; Dorado, F. Effect of Support Nature on the Cobalt-Catalyzed CO2 Hydrogenation. J. CO2 Util. 2017, 21, 562–571. [Google Scholar] [CrossRef]
- Parastaev, A.; Muravev, V.; Osta, E.H.; Kimpel, T.F.; Simons, J.F.M.; van Hoof, A.J.F.; Uslamin, E.; Zhang, L.; Struijs, J.J.C.; Burueva, D.B.; et al. Breaking Structure Sensitivity in CO2 Hydrogenation by Tuning Metal–Oxide Interfaces in Supported Cobalt Nanoparticles. Nat. Catal. 2022, 5, 11. [Google Scholar] [CrossRef]
- Liu, H.; Xu, S.; Zhou, G.; Xiong, K.; Jiao, Z.; Wang, S. CO2 Hydrogenation to Methane over Co/KIT-6 Catalysts: Effect of Co Content. Fuel 2018, 217, 570–576. [Google Scholar] [CrossRef]
- Al-Musa, A.; Al-Saleh, M.; Ioakeimidis, Z.C.; Ouzounidou, M.; Yentekakis, I.V.; Konsolakis, M.; Marnellos, G.E. Hydrogen Production by Iso-Octane Steam Reforming over Cu Catalysts Supported on Rare Earth Oxides (REOs). Int. J. Hydrogen Energy 2014, 39, 1350–1363. [Google Scholar] [CrossRef]
- Adesina, A.A.; Meeyoo, V.; Foulds, G. Thermolysis of Hydrogen Sulphide in an Open Tubular Reactor. Int. J. Hydrogen Energy 1995, 20, 777–783. [Google Scholar] [CrossRef]
- Shi, X.; Yu, Y.; Xue, L.; He, H. Effect of Sulfur Poisoning on Co3O4/CeO2 Composite Oxide Catalyst for Soot Combustion. Chinese J. Catal. 2014, 35, 1504–1510. [Google Scholar] [CrossRef]
- Lykaki, M.; Papista, E.; Kaklidis, N.; Carabineiro, S.; Konsolakis, M. Ceria Nanoparticles’ Morphological Effects on the N2O Decomposition Performance of Co3O4/CeO2 Mixed Oxides. Catalysts 2019, 9, 233. [Google Scholar] [CrossRef] [Green Version]
- Carabineiro, S.A.C.; Chen, X.; Konsolakis, M.; Psarras, A.C.; Tavares, P.B.; Órfão, J.J.M.; Pereira, M.F.R.; Figueiredo, J.L. Catalytic Oxidation of Toluene on Ce–Co and La–Co Mixed Oxides Synthesized by Exotemplating and Evaporation Methods. Catal. Today 2015, 244, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Parastaev, A.; Muravev, V.; Huertas Osta, E.; van Hoof, A.J.F.; Kimpel, T.F.; Kosinov, N.; Hensen, E.J.M. Boosting CO2 Hydrogenation via Size-Dependent Metal–Support Interactions in Cobalt/Ceria-Based Catalysts. Nat. Catal. 2020, 3, 526–533. [Google Scholar] [CrossRef]
- Hegde, R.I.; White, J.M. Chemisorption and Decomposition of H2S on Rh(100). J. Phys. Chem. 1986, 90, 296–300. [Google Scholar] [CrossRef]
- Zagoruiko, A.; Mikenin, P. Decomposition of Hydrogen Sulfide into Elements in the Cyclic Chemisorption-Catalytic Regime. Catal. Today 2021, 378, 176–188. [Google Scholar] [CrossRef]
- Blanchard, J.; Achouri, I.; Abatzoglou, N. H 2 S Poisoning of NiAl2O4/Al2O3-YSZ Catalyst during Methane Dry Reforming. Can. J. Chem. Eng. 2016, 94, 650–654. [Google Scholar] [CrossRef]
- Dou, X.; Veksha, A.; Chan, W.P.; Oh, W.-D.; Liang, Y.N.; Teoh, F.; Mohamed, D.K.B.; Giannis, A.; Lisak, G.; Lim, T.-T. Poisoning Effects of H2S and HCl on the Naphthalene Steam Reforming and Water-Gas Shift Activities of Ni and Fe Catalysts. Fuel 2019, 241, 1008–1018. [Google Scholar] [CrossRef]
- Wachter, P.; Gaber, C.; Raic, J.; Demuth, M.; Hochenauer, C. Experimental Investigation on H2S and SO2 Sulphur Poisoning and Regeneration of a Commercially Available Ni-Catalyst during Methane Tri-Reforming. Int. J. Hydrogen Energy 2021, 46, 3437–3452. [Google Scholar] [CrossRef]
- Iqbal, S.; Amjad, A.; Javed, M.; Alfakeer, M.; Mushtaq, M.; Rabea, S.; Elkaeed, E.B.; Pashameah, R.A.; Alzahrani, E.; Farouk, A.-E. Boosted Spatial Charge Carrier Separation of Binary ZnFe2O4/S-g-C3N4 Heterojunction for Visible-Light-Driven Photocatalytic Activity and Antimicrobial Performance. Front. Chem. 2022, 10, 894. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | N2 Porosimetry | XRD Analysis | |||
|---|---|---|---|---|---|
| BET Surface Area (m2/g) | Pore Volume (cm3/g) | Average Pore Diameter (nm) | Crystal Phase | Crystallite Size (nm) | |
| CeO2 a | 71.5 | 0.27 | 15.4 | CeO2 | 11.0 |
| 20-Co/CeO2 a | 33.4 | 0.13 | CeO2 | 10.2 | |
| 16.0 | Co3O4 | 37.7 | |||
| 30-Co/CeO2 | 44.9 | 0.21 | CeO2 | 10.4 | |
| 18.7 | Co3O4 | 37.9 | |||
| 40-Co/CeO2 | 28.4 | 0.10 | CeO2 | 10.5 | |
| 14.7 | Co3O4 | 41.7 | |||
| 60-Co/CeO2 | 15.1 | 0.07 | CeO2 | 10.5 | |
| 19.3 | Co3O4 | 42.2 | |||
| Co3O4 | 2.9 | 0.01 | Co3O4 | 50.7 | |
| 17.8 | CoO | 52.7 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kraia, T.; Varvoutis, G.; Marnellos, G.E.; Konsolakis, M. Unveiling the Role of In Situ Sulfidation and H2O Excess on H2S Decomposition to Carbon-Free H2 over Cobalt/Ceria Catalysts. Catalysts 2023, 13, 504. https://doi.org/10.3390/catal13030504
Kraia T, Varvoutis G, Marnellos GE, Konsolakis M. Unveiling the Role of In Situ Sulfidation and H2O Excess on H2S Decomposition to Carbon-Free H2 over Cobalt/Ceria Catalysts. Catalysts. 2023; 13(3):504. https://doi.org/10.3390/catal13030504
Chicago/Turabian StyleKraia, Tzouliana, Georgios Varvoutis, George E. Marnellos, and Michalis Konsolakis. 2023. "Unveiling the Role of In Situ Sulfidation and H2O Excess on H2S Decomposition to Carbon-Free H2 over Cobalt/Ceria Catalysts" Catalysts 13, no. 3: 504. https://doi.org/10.3390/catal13030504