

Insights into the Three-Component Coupling Reactions of Aldehydes, Alkynes, and Amines Catalyzed by N-heterocyclic Carbene Silver: A DFT Study

 ,
,  , , ,
, , ,
Abstract
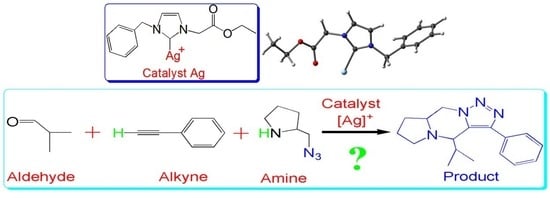
1. Introduction
2. Results and Discussion
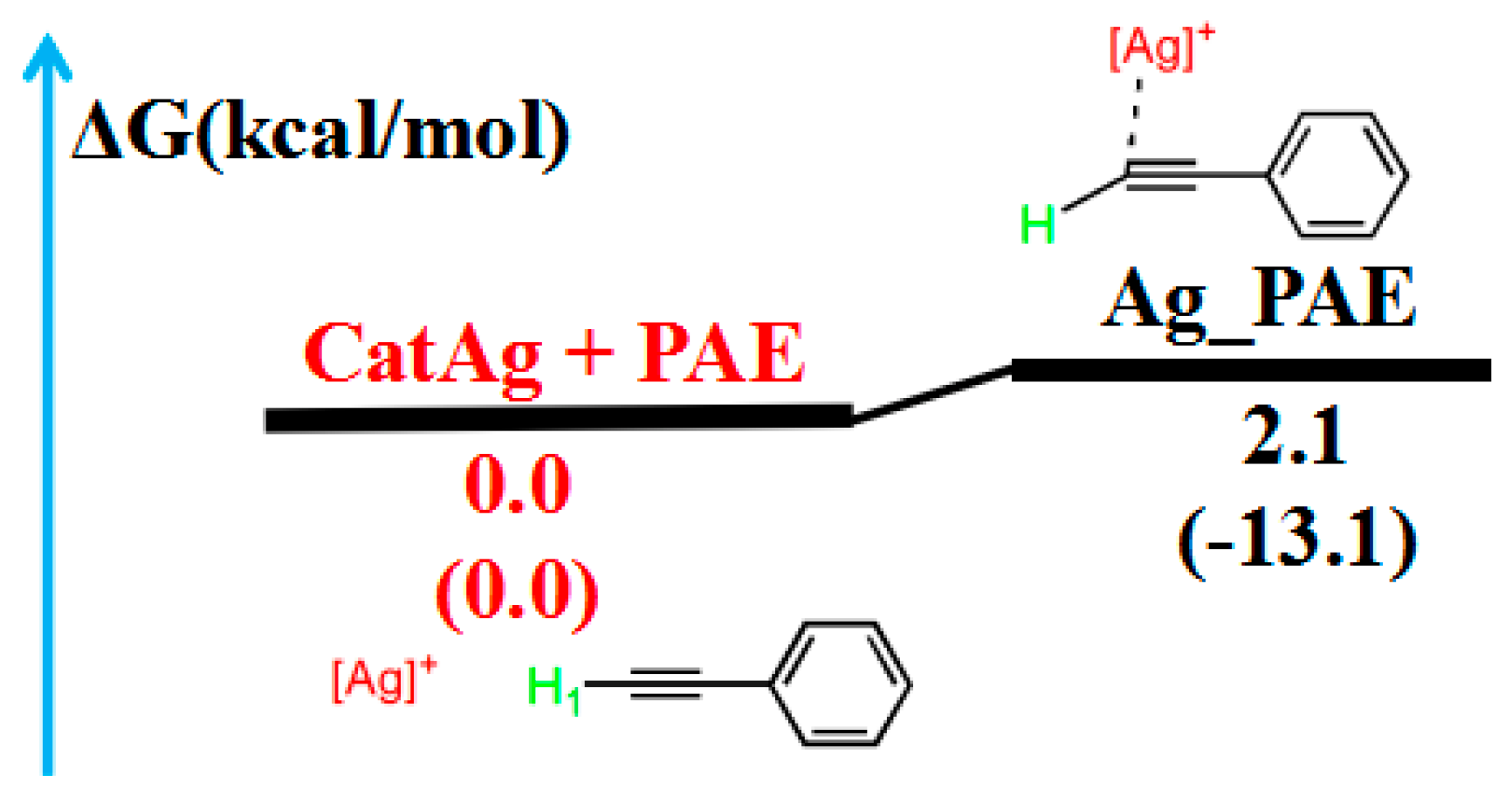
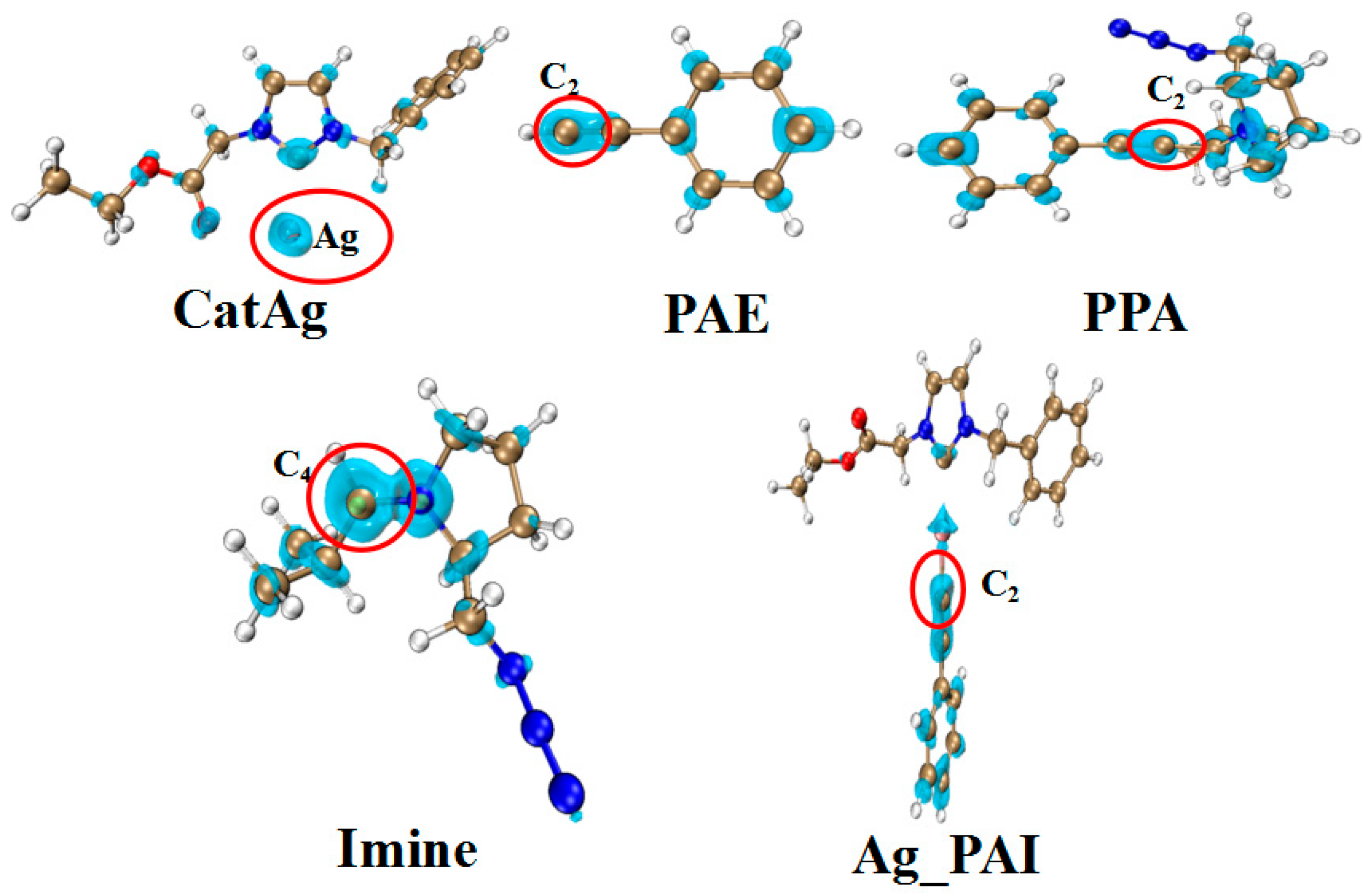
2.1. Addition Reaction between CatAg and PAE
2.2. Hydrogen Migration Reaction Involving Amine
2.3. Amine–Aldehyde Condensation Reaction
2.4. Imine Reaction with Ag_PAI
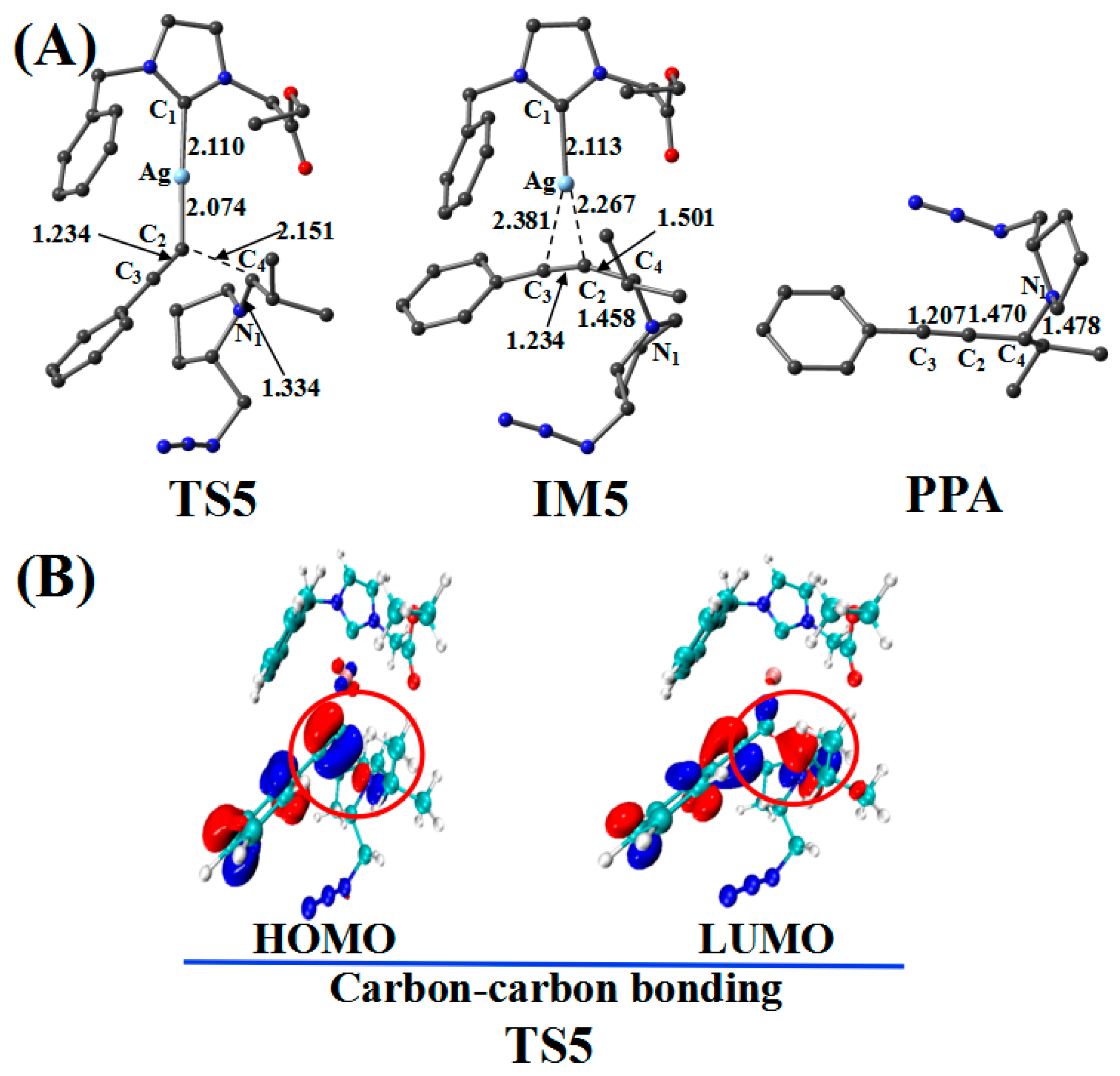
2.5. Cycloaddition Reaction
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Qiu, S.; Chen, W.; Li, D.; Chen, Y.; Niu, Y.; Wu, Y.; Lei, Y.; Wu, L.; He, W. The use of propargylamines to synthesize amino-1,2,3-triazoles via cycloaddition of azides with allenamines. Synthesis 2022, 54, 2175–2184. [Google Scholar]
- Jesin, I.; Nandi, G.C. Recent advances in the A3 coupling reactions and their applications. Eur. J. Org. Chem. 2019, 16, 2704–2720. [Google Scholar] [CrossRef]
- Ermolat’ev, D.S.; Bariwal, J.B.; Steenackers, H.P.L.; De Keersmaecker, S.C.J.; Van der Eycken, E.V. Concise and diversity-oriented route toward polysubstituted 2-aminoimidazole alkaloids and their analogues. Angew. Chem. Int. Ed. 2010, 49, 9465–9468. [Google Scholar] [CrossRef]
- Ganem, B. Strategies for innovation in multicomponent reaction design. Acc. Chem. Res. 2009, 42, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Zorba, L.P.; Vougioukalakis, G.C. The ketone-amine-alkyne (KA2) coupling reaction: Transition metal-catalyzed synthesis of quaternary propargylamines. Coord. Chem. Rev. 2021, 429, 213603. [Google Scholar] [CrossRef]
- Ramazani, A.; Ahankar, H.; Nafeh, T.Z.; Joo, W.S. Modern catalysts in A3—coupling reactions. Curr. Org. Chem. 2019, 23, 2783–2801. [Google Scholar] [CrossRef]
- McNally, J.J.; Youngman, M.A.; Dax, S.L. Mannich reactions of resin-bound substrates: 2. A versatile three-component solid-phase organic synthesis methodology. Tetrahedron Lett. 1998, 39, 967–970. [Google Scholar] [CrossRef]
- Dyatkin, A.B.; Rivero, R.A. The solid phase synthesis of complex propargylamines using the combination of sonogashira and mannich reactions. Tetrahedron Lett. 1998, 39, 3647–3650. [Google Scholar] [CrossRef]
- Li, C.-J.; Wei, C. Highly efficient grignard-type imine additions via C-H activation in water and under solvent-free conditions. Chem. Comm. 2002, 3, 268–269. [Google Scholar] [CrossRef]
- Shi, L.; Tu, Y.-Q.; Wang, M.; Zhang, F.-M.; Fan, C.-A. Microwave-promoted three-component coupling of aldehyde, alkyne, and amine via C-H activation catalyzed by copper in water. Org. Lett. 2004, 6, 1001–1003. [Google Scholar] [CrossRef]
- Yadav, J.S.; Subba Reddy, B.V.; Naveenkumar, V.; Srinivasa Rao, R.; Nagaiah, K. [bmim]PF6/CuBr: A novel and recyclable catalytic system for the synthesis of propargyl amines. New J. Chem. 2004, 28, 335–337. [Google Scholar] [CrossRef]
- Bariwal, J.; Ermolat’ev, D.; Van der Eycken, E.V. Efficient microwave-assisted synthesis of secondary alkylpropargylamines by using A3-coupling with primary aliphatic amines. Chem. Eur. J. 2010, 16, 3281–3284. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Kumar, M.; Maurya, S.; Khanna, R.S. Regioselective synthesis of fused imidazo[1,2-a]pyrimidines via intramolecular C-N bond formation/6-endo-dig cycloisomerization. J. Org. Chem. 2014, 79, 6905–6912. [Google Scholar] [CrossRef]
- Bisai, V.; Suneja, A.; Singh, V.K. Asymmetric alkynylation/lactamization cascade: An expeditious entry to enantiomerically enriched isoindolinones. Angew. Chem. Int. Ed. 2014, 53, 10737–10741. [Google Scholar] [CrossRef]
- Trose, M.; Dell’Acqua, M.; Pedrazzini, T.; Pirovano, V.; Gallo, E.; Rossi, E.; Caselli, A.; Abbiati, G. [Silver(I)(pyridine-containing ligand)] complexes as unusual catalysts for A3-coupling reactions. J. Org. Chem. 2014, 79, 7311–7320. [Google Scholar] [CrossRef] [PubMed]
- Prakash, O.; Joshi, H.; Kumar, U.; Sharma, A.K.; Singh, A.K. Acridine based (S,N,S) pincer ligand: Designing silver(I) complexes for the efficient activation of A3 (aldehyde, alkyne and amine) coupling. Dalton Trans. 2015, 44, 1962–1968. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tzouras, N.V.; Nolan, S.P.; Bi, X. Silver N-heterocyclic carbenes: Emerging powerful catalysts. Trends Chem. 2021, 3, 674–685. [Google Scholar] [CrossRef]
- Ghasemi, K.; Darroudi, M.; Rahimi, M.; Rouh, H.; Gupta, A.R.; Cheng, C.; Amini, A. Magnetic AgNPs/Fe3O4@chitosan/PVA nanocatalyst for fast one-pot green synthesis of propargylamine and triazole derivatives. New J. Chem. 2021, 45, 16119–16130. [Google Scholar] [CrossRef]
- Xin, N.; Jing, X.; Zhang, C.-G.; Peng, X.; Liu, J.; Wang, Q.; Wang, W.; Cao, J.; Tao, M. N-heterocyclic carbene silver complex modified polyacrylonitrile fiber/MIL-101(Cr) composite as efficient chiral catalyst for three-component coupling reaction. Nanomaterials 2022, 12, 4175. [Google Scholar] [CrossRef]
- Cao, J.; Li, P.; Xu, G.; Tao, M.; Ma, N.; Zhang, W. Cooperative N-heterocyclic carbene Au and amino catalysis for continuous synthesis of secondary propargylamines in a fiber supported hydrophilic microenvironment. Chem. Eng. J. 2018, 349, 456–465. [Google Scholar] [CrossRef]
- Srinivas, V.; Koketsu, M. Synthesis of indole-2-, 3-, or 5-substituted propargylamines via gold(III)-catalyzed three component reaction of aldehyde, alkyne, and amine in aqueous medium. Tetrahedron 2013, 69, 8025–8033. [Google Scholar] [CrossRef]
- Huang, J.-L.; Gray, D.G.; Li, C.-J. A3-coupling catalyzed by robust Au nanoparticles covalently bonded to HS-functionalized cellulose nanocrystalline films. Beilstein J. Org. Chem. 2013, 9, 1388–1396. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-B.; Zhao, Y.; Liao, Y. Aldehyde-alkyne-amine (A3) coupling catalyzed by a highly efficient dicopper complex. RSC Adv. 2015, 5, 37737–37741. [Google Scholar] [CrossRef]
- Trang, T.T.T.; Ermolat’ev, D.S.; Van der Eycken, E.V. Facile and diverse microwave-assisted synthesis of secondary propargylamines in water using CuCl/CuCl2. RSC Adv. 2015, 5, 28921–28924. [Google Scholar] [CrossRef]
- Neshat, A.; Gholinejad, M.; Afrasi, M.; Mastrorilli, P.; Todisco, S.; Gilanchi, S.; Osanlou, F. Heterocyclic thiolates and phosphine ligands in copper-catalyzed synthesis of propargylamines in water. Appl. Organomet. Chem. 2021, 35, e6180. [Google Scholar] [CrossRef]
- Yadav, J.S.; Subba Reddy, B.V.; Hara Gopal, A.V.; Patil, K.S. InBr3-catalyzed three-component reaction: A facile synthesis of propargyl amines. Tetrahedron Lett. 2009, 50, 3493–3496. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, P.; Wang, M.; Wang, L. Indium-catalyzed highly efficient three-component coupling of aldehyde, alkyne, and amine via C-H bond activation. J. Org. Chem. 2009, 74, 4364–4367. [Google Scholar] [CrossRef]
- Li, P.; Zhang, Y.; Wang, L. Iron-catalyzed ligand-free three-component coupling reactions of aldehydes, terminal alkynes, and amines. Chem. Eur. J. 2009, 15, 2045–2049. [Google Scholar] [CrossRef]
- Chen, W.-W.; Nguyen, R.V.; Li, C.-J. Iron-catalyzed three-component coupling of aldehyde, alkyne, and amine under neat conditions in air. Tetrahedron Lett. 2009, 50, 2895–2898. [Google Scholar] [CrossRef]
- Huo, X.; Liu, J.; Wang, B.; Zhang, H.; Yang, Z.; She, X.; Xi, P. A one-step method to produce graphene-Fe3O4 composites and their excellent catalytic activities for three-component coupling of aldehyde, alkyne and amine. J. Mater. Chem. A 2013, 1, 651–656. [Google Scholar] [CrossRef]
- Samai, S.; Nandi, G.C.; Singh, M.S. An efficient and facile one-pot synthesis of propargylamines by three-component coupling of aldehydes, amines, and alkynes via C-H activation catalyzed by NiCl2. Tetrahedron Lett. 2010, 51, 5555–5558. [Google Scholar] [CrossRef]
- Shi, X.-L.; Sun, B.; Chen, Y.; Hu, Q.; Li, P.; Meng, Y.; Duan, P. Tuning anion species and chain length of ligands grafted on the fiber for an efficient polymer-supported Ni(II) complex catalyst in one-pot multicomponent A3-coupling. J. Catal. 2019, 372, 321–329. [Google Scholar] [CrossRef]
- Manikandan, R.; Anitha, P.; Viswanathamurthi, P.; Malecki, J.G. Palladium(II) pyridoxal thiosemicarbazone complexes as efficient and recyclable catalyst for the synthesis of propargylamines by a three-component coupling reactions in ionic liquids. Polyhedron 2016, 119, 300–306. [Google Scholar] [CrossRef]
- Nouruzi, N.; Dinari, M.; Mokhtari, N.; Gholipour, B.; Rostamnia, S.; Khaksar, S.; Boluki, R. Porous triazine polymer: A novel catalyst for the three-component reaction. Appl. Organomet. Chem. 2020, 34, e5677. [Google Scholar] [CrossRef]
- Li, J.; Xu, Y.; Hu, X.; Zhu, S.; Liu, L. Easy access to 2,4-disubstituted cyclopentenones by a gold(III)-catalyzed A3-coupling/cyclization cascade. Org. Lett. 2020, 22, 9478–9483. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Feng, H.; Van der Eycken, E.V. Microwave-assisted Cu(I)-catalyzed synthesis of unsymmetrical 1,4-diamino-2-butynes via cross-A3-coupling/decarboxylative A3-coupling. J. Org. Chem. 2021, 86, 14036–14043. [Google Scholar] [CrossRef]
- Cao, S.; Zou, B.; Yang, J.; Wang, J.; Feng, H. Hollow CuO-CeO2 nanospheres for an effectively catalytic annulation/A3-coupling reaction sequence. ACS Appl. Nano Mater. 2022, 5, 11689–11698. [Google Scholar] [CrossRef]
- Kumar, G.; Pandey, S.; Gupta, R. Ag-based coordination polymers based on metalloligands and their catalytic performance in multicomponent A3-coupling reactions. Cryst. Growth Des. 2022, 18, 5501–5511. [Google Scholar] [CrossRef]
- Costabile, C.; Mariconda, A.; Sirignano, M.; Crispini, A.; Scarpelli, F.; Longo, P. A green approach for A3-coupling reactions: An experimental and theoretical study on NHC silver and gold catalysts. New J. Chem. 2021, 45, 18509–18517. [Google Scholar] [CrossRef]
- Cao, J.; Xu, G.; Li, P.; Tao, M.; Zhang, W. Polyacrylonitrile fiber supported N-heterocyclic carbene Ag(I) as efficient catalysts for three-component coupling and intramolecular 1,3-dipolar cycloaddition reactions under flow conditions. ACS Sustain. Chem. Eng. 2017, 5, 3438–3447. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Q.; Pu, M.; Yang, Z.; Lei, M. Theoretical study on nitrogenous heterocyclic assisted aldimine condensation. Acta Chim. Sin. 2020, 78, 437–443. [Google Scholar] [CrossRef]
- Ciaccia, M.; Di Stefano, S. Mechanisms of imine exchange reactions in organic solvents. Org. Biomol. Chem. 2015, 13, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Boz, E.; Tüzün, N.Ş. Ag-catalyzed azide alkyne cycloaddition: A DFT approach. Dalton Trans. 2016, 45, 5752–5764. [Google Scholar] [CrossRef] [PubMed]
- Lu, T. Concvar: A computer program for simulating concentration variation of complex chemical reactions. ChemRxiv 2022, preprint. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate Ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Smith, D.G.; Burns, L.A.; Patkowski, K.; Sherrill, C.D. Revised damping parameters for the D3 dispersion correction to density functional theory. J. Phys. Chem. Lett. 2016, 7, 2197–2203. [Google Scholar] [CrossRef]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions-the IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Shermo: A general code for calculating molecular thermochemistry properties. Comput. Theor. Chem. 2021, 1200, 113249. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density functionals with broad applicability in chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. Benchmark energetic data in a model system for grubbs II metathesis catalysis and their use for the development, assessment, and validation of electronic structure methods. J. Chem. Theory Comput. 2009, 5, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Yang, Y.-F.; Hong, X.; Yu, J.-Q.; Houk, K.N. Experimental–computational synergy for selective Pd(II)-catalyzed C–H activation of aryl and alkyl groups. Acc. Chem. Res. 2017, 50, 2853–2860. [Google Scholar] [CrossRef]
- Deng, L.; Fu, Y.; Lee, S.Y.; Wang, C.; Liu, P.; Dong, G. Kinetic resolution via Rh-catalyzed C-C activation of cyclobutanones at room temperature. J. Am. Chem. Soc. 2019, 141, 16260–16265. [Google Scholar] [CrossRef]
- Li, Y.; Chen, H.; Qu, L.-B.; Houk, K.N.; Lan, Y. Origin of regiochemical control in Rh(III)/Rh(V)-catalyzed reactions of unsaturated oximes and alkenes to form pyrdines. ACS Catal. 2019, 9, 7154–7165. [Google Scholar] [CrossRef]
- Palani, V.; Hugelshofer, C.L.; Kevlishvili, I.; Liu, P.; Sarpong, R. A short synthesis of delavatine a unveils new insights into site-selective cross-coupling of 3,5-dibromo-2-pyrone. J. Am. Chem. Soc. 2019, 141, 2652–2660. [Google Scholar] [CrossRef]
- Qi, X.; Kohler, D.G.; Hull, K.L.; Liu, P. Energy decomposition analyses reveal the origins of catalyst and nucleophile effects on regioselectivity in nucleopalladation of alkenes. J. Am. Chem. Soc. 2019, 141, 11892–11904. [Google Scholar] [CrossRef]
- Zhang, C.; Yu, S.; Wang, F.; Wang, F.; Cao, J.; Zheng, H.; Chen, X.; Ren, A. Density functional theory analysis of the copolymerization of cyclopropenone with ethylene using a palladium catalyst. Polymers 2022, 14, 5273. [Google Scholar] [CrossRef]
- Liu, Z.; Lu, Y.; Guo, J.; Hu, W.; Wang, Z.X. DFT mechanistic account for the site selectivity of electron-rich C(sp3)–H bond in the manganese-catalyzed aminations. Org. Lett. 2020, 22, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Lu, Y.; Lu, G.; Wang, Z.X. Density functional theory mechanistic study of Ni-catalyzed reductive alkyne–alkyne cyclodimerization: Oxidative cyclization versus outer-sphere proton transfer. Org. Lett. 2020, 22, 2454–2459. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-F.; Chen, G.; Hong, X.; Yu, J.-Q.; Houk, K.N. The origins of dramatic differences in five-membered vs six-membered chelation of Pd(II) on efficiency of C(sp3)-H bond activation. J. Am. Chem. Soc. 2017, 139, 8514–8521. [Google Scholar] [CrossRef]
- Yu, S.-Y.; Peng, X.; Wang, F.; Cao, J.; Wang, F.; Zhang, C.-G. Density functional theory study of the regioselectivity in copolymerization of bis-styrenic molecules with propylene using zirconocene catalyst. Catalysts 2022, 12, 1039. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q.X. Realization of conceptual density functional theory and information-theoretic approach in Multiwfn program. In Conceptual Density Functional Theory; WILEY-VCH GmbH: Weinheim, Germany, 2022; pp. 631–647. [Google Scholar]
- Parr, R.G.; Pearson, R.G. Absolute hardness-companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Parr, R.G.; Von Szentpaly, L.; Liu, S.B. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Fu, R.; Lu, T.; Chen, F.-W. Comparing methods for predicting the reactive site of electrophilic substitution. Acta Phys. Chim. Sin. 2014, 30, 628–639. [Google Scholar]
- Lu, T.; Chen, F.-W. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Lu, T. TSTcalculator. Available online: http://sobereva.com/310 (accessed on 10 March 2023).















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WBI | QNBO (e) | |||||
|---|---|---|---|---|---|---|
| B (C2–H1) | B (C2–C3) | B (Ag–C1) | B (Ag–C2) | C2 | Ag | |
| CatAg + PAE | 0.934 | 2.825 | 0.482 | 0.000 | −0.201 | 0.701 |
| Ag_PAE | 0.899 | 2.623 | 0.491 | 0.199 | −0.187 | 0.469 |
| WBI | QNBO (e) | ||||||
|---|---|---|---|---|---|---|---|
| B (C2–H1) | B (N1–H1) | B (C2–C3) | B (N1–H2) | H1 | N1 | H2 | |
| Ag_PAE + Amine | 0.899 | 0.000 | 2.623 | 0.845 | 0.174 | −0.703 | 0.362 |
| TS1 | 0.464 | 0.347 | 2.617 | 0.810 | 0.362 | −0.438 | 0.263 |
| Ag_PAI + AmineH | 0.000 | 0.739 | 2.759 | 0.792 | 0.458 | −0.564 | 0.425 |
| WBI | QNBO (e) | ||||||
|---|---|---|---|---|---|---|---|
| B (N1–H1) | B (C4–O1) | B (O1–H1) | B (O1–H2) | O1 | H1 | H2 | |
| AmineH + Ald | 0.739 | 1.886 | −0.515 | 0.458 | 0.425 | ||
| IM1 | 0.693 | 1.734 | 0.071 | −0.639 | 0.459 | 0.423 | |
| TS2 | 0.008 | 1.405 | 0.697 | −0.526 | 0.521 | 0.360 | |
| IM2 | 0.009 | 0.950 | 0.745 | −0.740 | 0.491 | 0.417 | |
| TS3 | 0.917 | 0.750 | 0.004 | −0.766 | 0.482 | 0.484 | |
| IM3 | 0.893 | 0.756 | 0.004 | −0.785 | 0.478 | 0.485 | |
| TS4 | 0.636 | 0.783 | 0.299 | −0.963 | 0.463 | 0.471 | |
| IM4 | 0.002 | 0.795 | 0.670 | −0.984 | 0.456 | 0.493 | |
| Imine + H2O | 0.813 | 0.813 | −0.877 | 0.438 | 0.438 | ||
| TS3A | 0.790 | 0.674 | 0.411 | −0.695 | 0.526 | 0.524 | |
| WBI | QNBO (e) | |||||
|---|---|---|---|---|---|---|
| B (Ag–C2) | B (C4–N1) | B (C2–C4) | C2 | N1 | C4 | |
| Ag_PAI + Imine | 0.627 | 1.669 | 0.000 | −0.386 | −0.313 | 0.316 |
| TS5 | 0.399 | 1.332 | 0.378 | −0.449 | −0.412 | 0.223 |
| IM5 | 0.162 | 0.974 | 0.991 | −0.097 | −0.513 | −0.062 |
| CatAg + PPA | 0.000 | 0.946 | 1.034 | −0.386 | −0.534 | −0.082 |
| WBI | QNBO (e) | ||||||
|---|---|---|---|---|---|---|---|
| B (C2–C3) | B (C2–N3) | B (C3–N5) | B (N3–N4) | B (N4–N5) | C2 | C3 | |
| CatAg + PPA | 2.716 | 0.002 | 0.001 | 1.529 | 2.315 | −0.002 | −0.028 |
| IM6 | 2.550 | 0.004 | 0.003 | 1.504 | 2.334 | −0.084 | −0.013 |
| TS6 | 2.249 | 0.316 | 0.285 | 1.455 | 2.144 | −0.021 | −0.105 |
| PR + CatAg | 1.420 | 1.206 | 1.310 | 1.221 | 1.537 | 0.140 | 0.075 |
| η a | μ b | ω c | NNu d | f +e | f −f | ||
|---|---|---|---|---|---|---|---|
| Ag | C4 | C2 | |||||
| CatAg | 7.424 | −7.623 | 3.795 | −0.330 | 0.693 | ||
| PAE | 9.577 | −3.561 | 0.839 | 2.825 | 0.197 | ||
| PPA | 8.347 | −3.278 | 0.847 | 3.216 | 0.064 | ||
| Imine | 8.598 | −8.503 | 4.172 | −1.172 | 0.212 | ||
| Ag_PAI | 7.073 | −3.077 | 0.818 | 4.215 | 0.172 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Yu, S.; Wang, F.; Cao, J.; Liang, X.; Wang, F.; Zheng, H.; Zhang, Y.; Yang, M.; Zhao, B. Insights into the Three-Component Coupling Reactions of Aldehydes, Alkynes, and Amines Catalyzed by N-heterocyclic Carbene Silver: A DFT Study. Catalysts 2023, 13, 646. https://doi.org/10.3390/catal13040646
Zhang C, Yu S, Wang F, Cao J, Liang X, Wang F, Zheng H, Zhang Y, Yang M, Zhao B. Insights into the Three-Component Coupling Reactions of Aldehydes, Alkynes, and Amines Catalyzed by N-heterocyclic Carbene Silver: A DFT Study. Catalysts. 2023; 13(4):646. https://doi.org/10.3390/catal13040646
Chicago/Turabian StyleZhang, Chenggen, Shuyuan Yu, Fei Wang, Jian Cao, Xinru Liang, Fuping Wang, Huimin Zheng, Yaning Zhang, Mengyao Yang, and Boyu Zhao. 2023. "Insights into the Three-Component Coupling Reactions of Aldehydes, Alkynes, and Amines Catalyzed by N-heterocyclic Carbene Silver: A DFT Study" Catalysts 13, no. 4: 646. https://doi.org/10.3390/catal13040646
APA StyleZhang, C., Yu, S., Wang, F., Cao, J., Liang, X., Wang, F., Zheng, H., Zhang, Y., Yang, M., & Zhao, B. (2023). Insights into the Three-Component Coupling Reactions of Aldehydes, Alkynes, and Amines Catalyzed by N-heterocyclic Carbene Silver: A DFT Study. Catalysts, 13(4), 646. https://doi.org/10.3390/catal13040646