

Metal-Doped Mesoporous MnO2-CeO2 Catalysts for Low-Temperature Pre-Oxidation of NO to NO2 in Fast SCR Process


Abstract
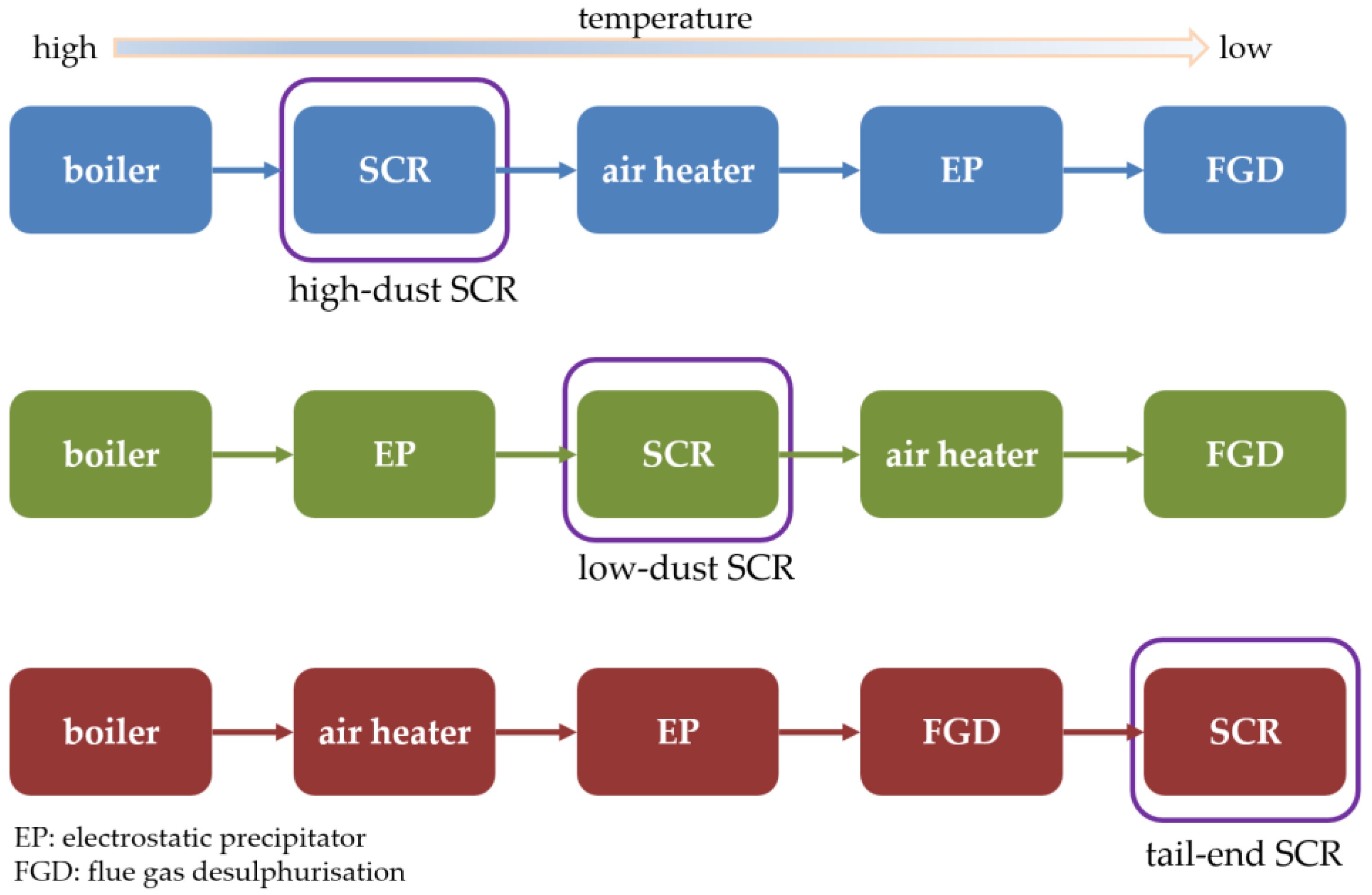
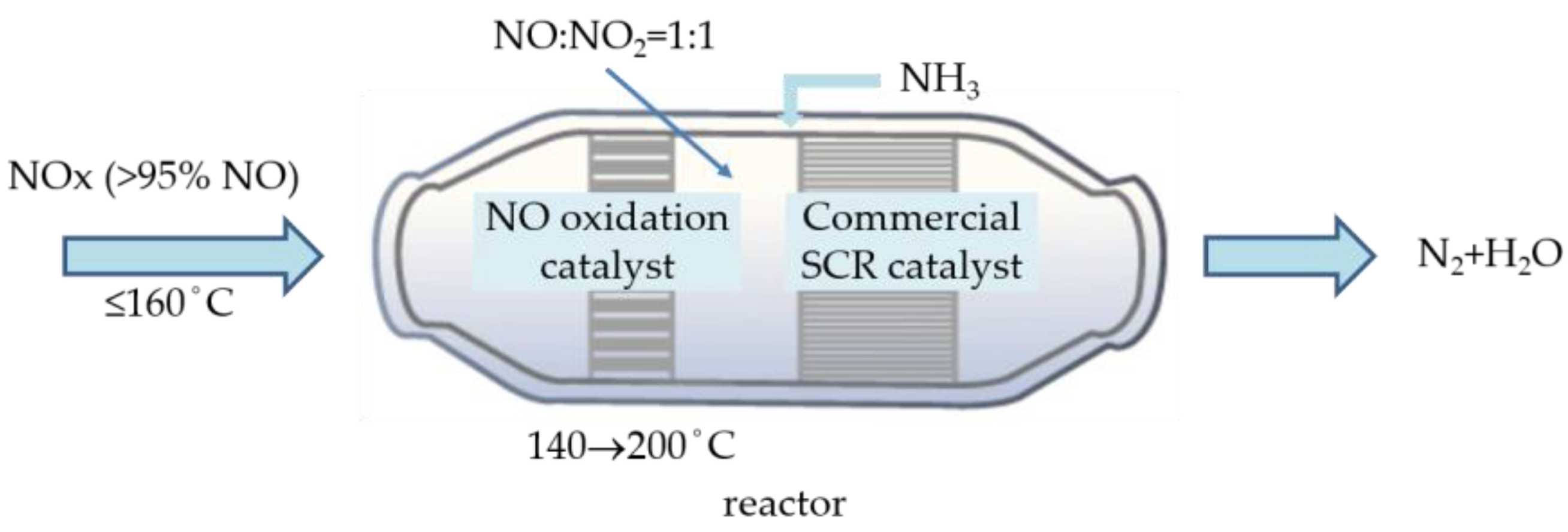
1. Introduction
2. Results and Discussion
2.1. Characterization of NOx Oxidation Catalysts
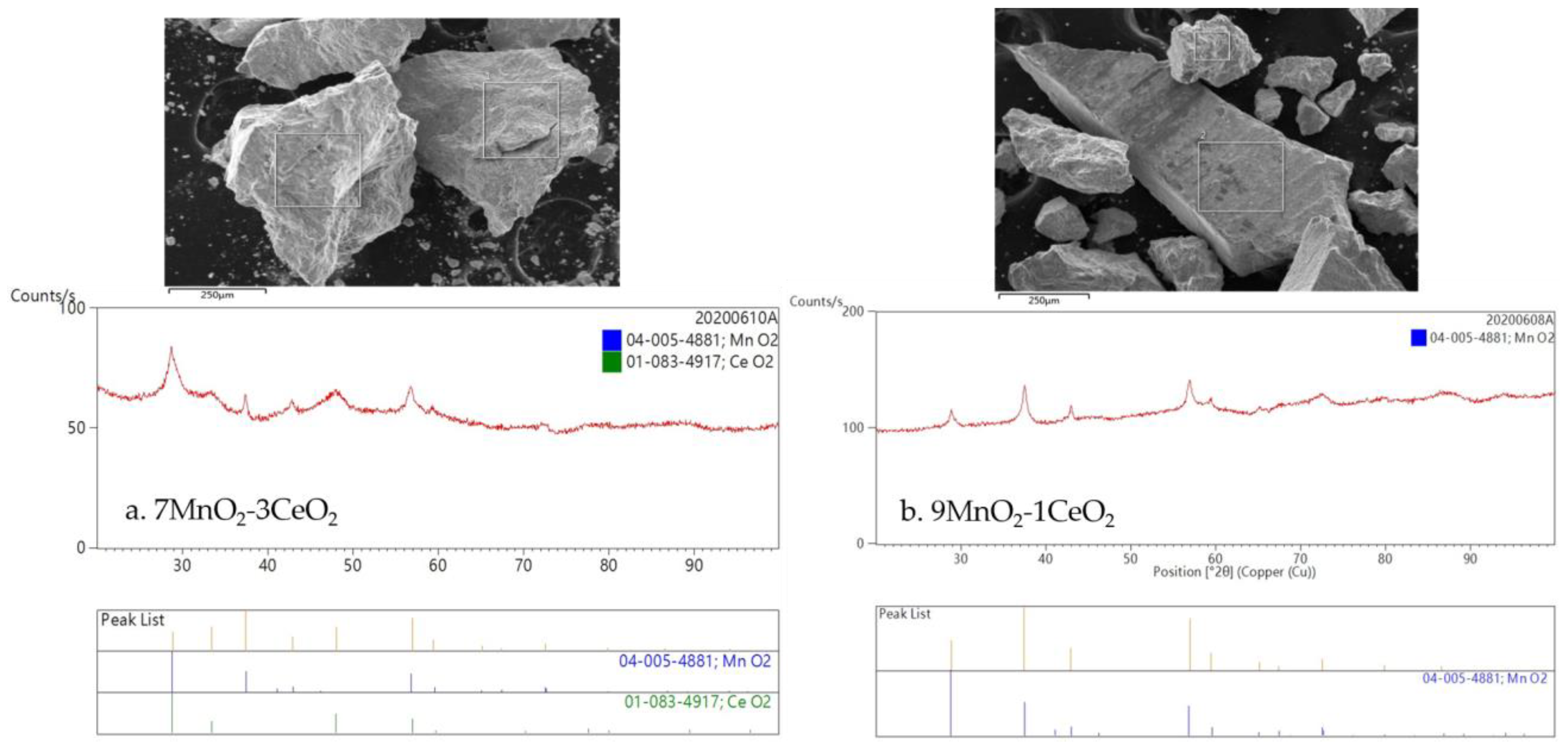
2.1.1. Scanning Electron Microscope (SEM) and X-ray Diffraction (XRD) Analysis
2.1.2. Physical Characteristics Analysis
2.1.3. Transmission Electron Microscopy (TEM) and Energy-Dispersive X-ray Spectroscopy (EDS) Results
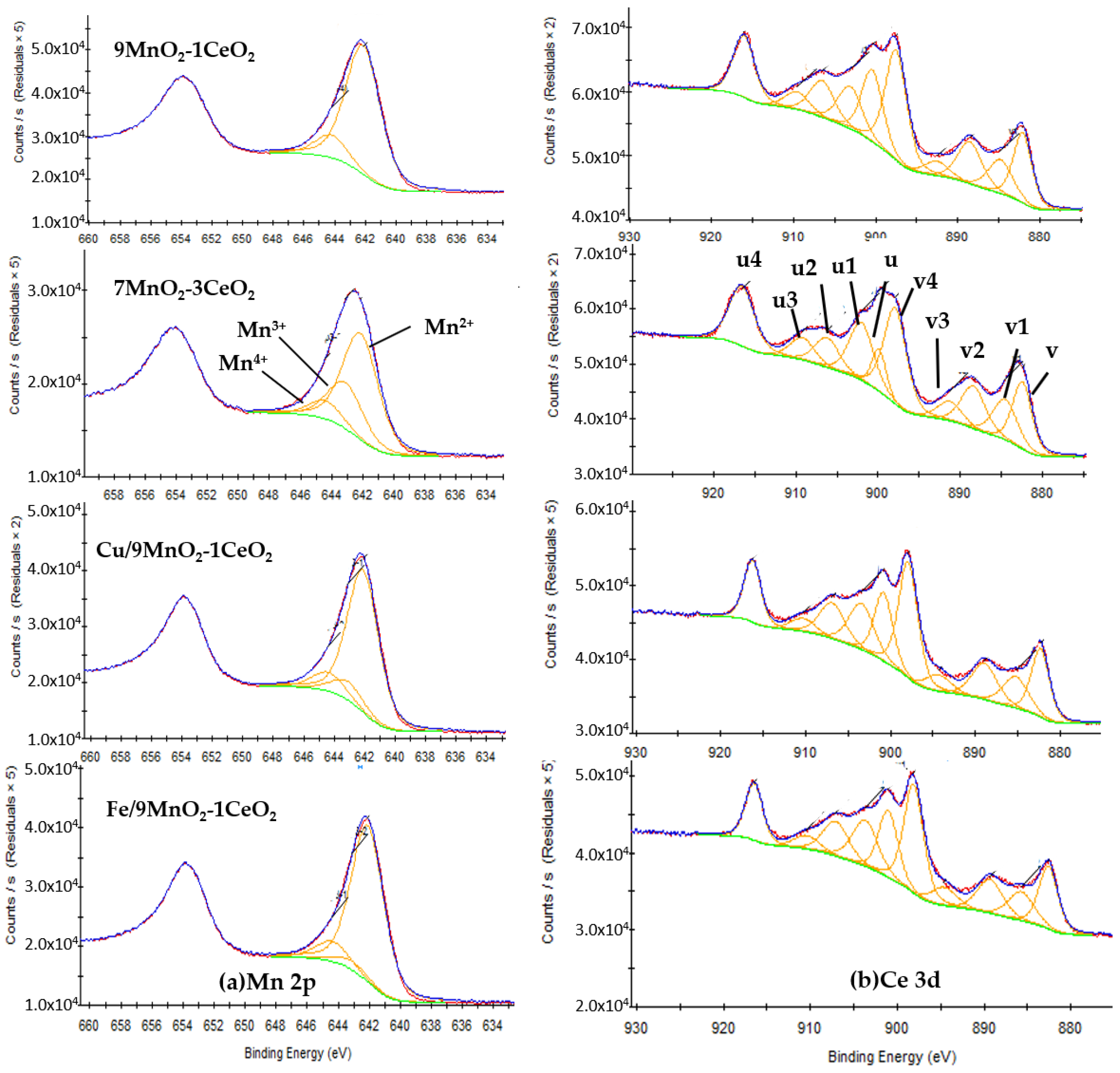
2.1.4. X-ray Photoelectron Spectroscopy (XPS) Analysis
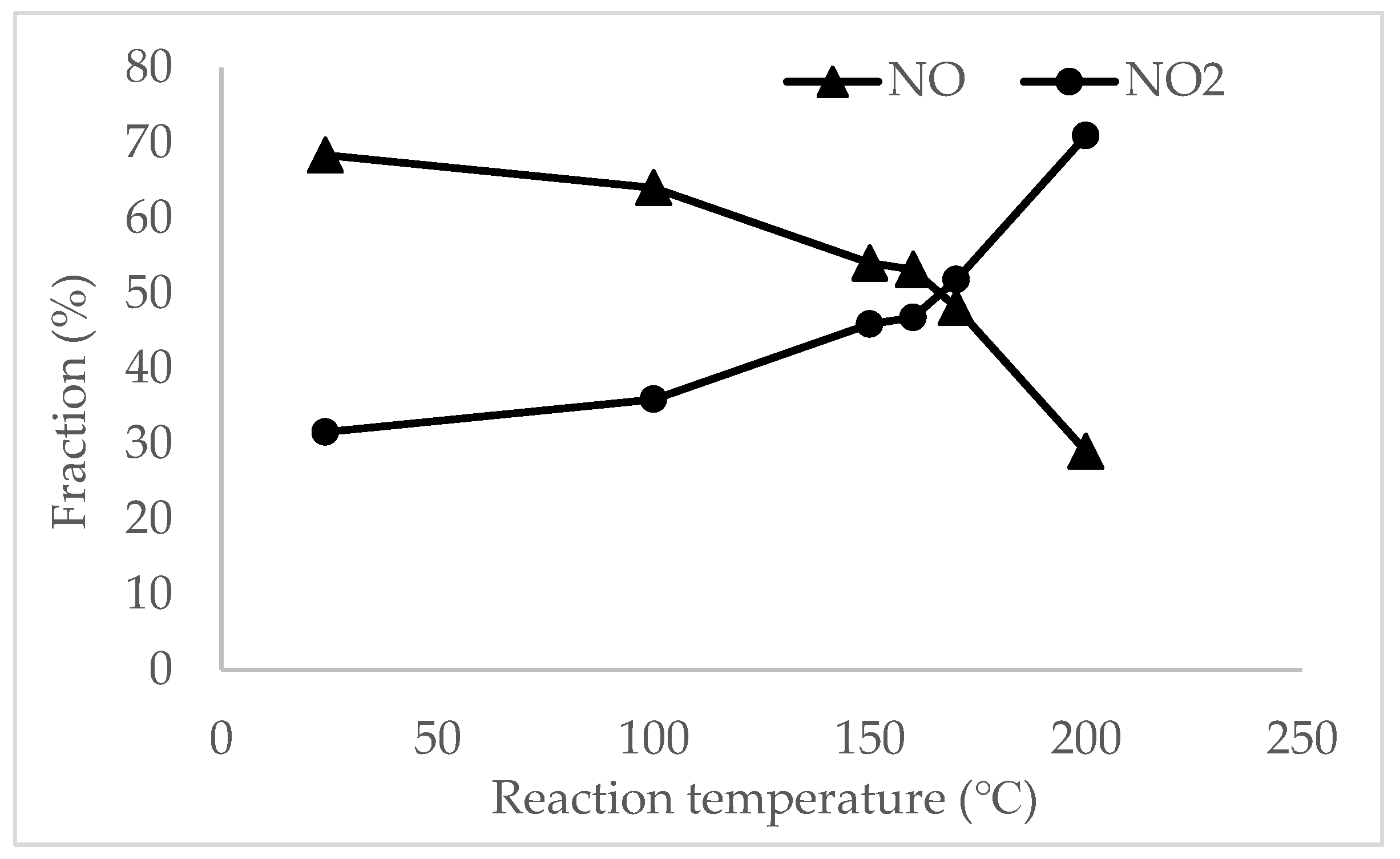
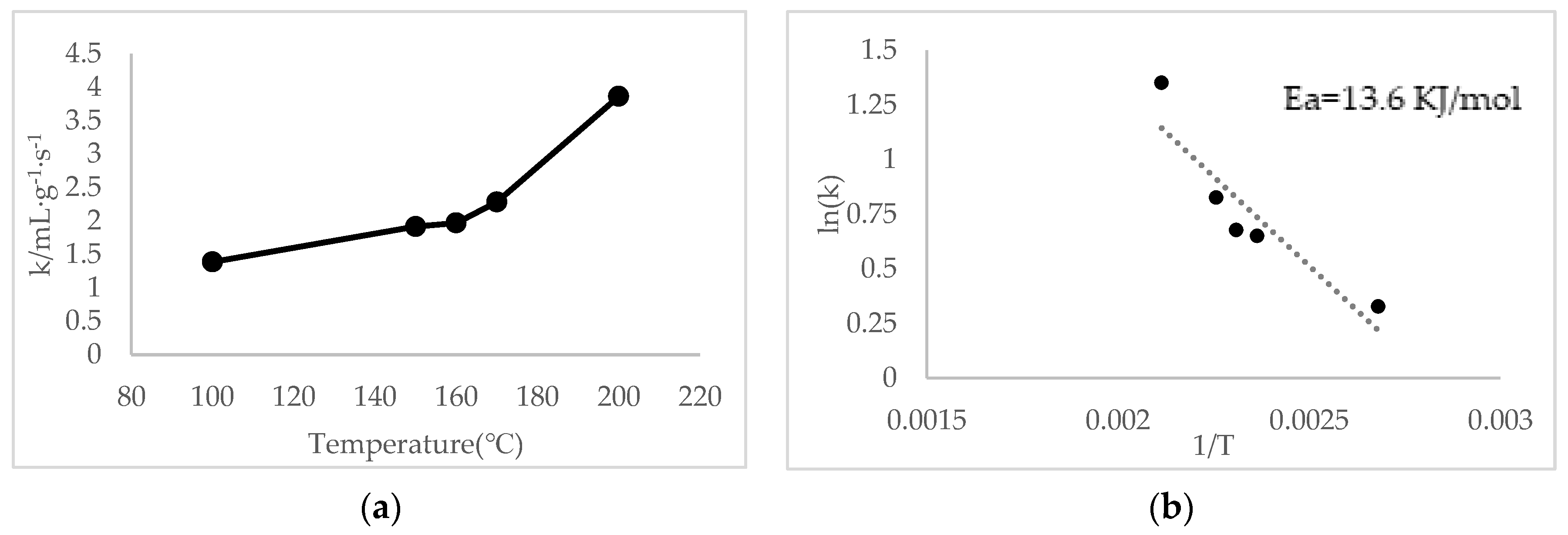
2.2. Catalytic Performance Results
2.3. Mechanisms of NO Oxidation and Fast SCR
3. Materials and Methods
3.1. Materials
3.2. Preparation
3.2.1. Mn–Ce Catalyst Preparation
9MnO2–1CeO2
7MnO2–3CeO2
3.2.2. DP–Mn–Ce Catalyst Preparation (DP = Fe, Co, Ni, Cu, or Cr)
Co/9MnO2–1CeO2
DP/9MnO2–1CeO2 and DP/7MnO2–3CeO2 (DP = Fe, Ni, Cu, or Cr)
3.2.3. Preparation of Catalysts for Comparison between them in NO Oxidation Performance
Cr2O3, Co3O4, Fe2O3, and MnO2
Pt/Cr2O3, Pt/Fe2O3, Rh/MnO2, and Co/MnO2
3.3. Catalytic Performance: NO Oxidation to NO2
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Directive (EU) 2016/2284 of the European Parliament and of the Council of 14 December 2016 on the Reduction of National Emissions of Certain Atmospheric Pollutants. Available online: http://data.europa.eu/eli/dir/2016/2284/oj (accessed on 31 March 2023).
- Bosch, H.; Janssen, F. Formation and control of nitrogen oxides. Catal. Today 1988, 2, 369–379. [Google Scholar] [CrossRef]
- Janssen, F.; Meijer, R. Quality control of DeNOx catalysts: Performance testing, surface analysis and characterization of DeNOx catalysts. Catal. Today 1993, 16, 157–185. [Google Scholar] [CrossRef]
- Damma, D.; Ettireddy, P.R.; Reddy, B.M.; Smirniotis, P.G. A Review of Low Temperature NH3-SCR for Removal of NOx. Catalysts 2019, 9, 349. [Google Scholar] [CrossRef]
- Zhu, M.; Lai, J.; Wachs, I.E. Formation of N2O greenhouse gas during SCR of NO with NH3 by supported vanadium oxide cata-lysts. Appl. Catal. B Environ. 2018, 224, 836–840. [Google Scholar] [CrossRef]
- Kato, A.; Matsuda, S.; Kamo, T.; Nakajima, F.; Kuroda, H.; Narita, T. Reaction between nitrogen oxide (NOx) and ammonia on iron oxide-titanium oxide catalyst. J. Phys. Chem. 1981, 85, 4099–4102. [Google Scholar] [CrossRef]
- Tuenter, G.; van Leeuwen, W.F.; Snepvangers, L. Kinetics and Mechanism of the NOx Reduction with NH3 on V2O5—WO3-TiO2 Catalyst. Ind. Eng. Chem. Prod. Res. Dev. 1986, 25, 633–636. [Google Scholar] [CrossRef]
- Jiang, B.; Lin, B.; Li, Z.; Zhao, S.; Chen, Z. Mn/TiO2 catalysts prepared by ultrasonic spray pyrolysis method for NOx removal in low-temperature SCR reaction. Colloids Surf. A Physicochem. Eng. Asp. 2020, 586, 124210. [Google Scholar] [CrossRef]
- Koebel, M.; Madia, G.; Elsener, M. Selective catalytic reduction of NO and NO2 at low temperatures. Catal. Today 2002, 73, 239–247. [Google Scholar] [CrossRef]
- Sorrels, J.L. Chapter 2: Selective Catalytic Reduction. Section 4: NOx Controls. In EPA Air Pollution Control Cost Manual. Available online: https://www.epa.gov/economic-and-cost-analysis-air-pollution-regulations/cost-reports-and-guidance-air-pollution (accessed on 31 March 2023).
- Li, P.; Xin, Y.; Li, Q.; Wang, Z.P.; Zhang, Z.L.; Zheng, L. Ce-Ti Amorphous Oxides for Selective Catalytic Reduction of NO with NH3: Confirmation of Ce-O-Ti Active Sites. Environ. Sci. Technol. 2012, 46, 9600–9605. [Google Scholar] [CrossRef]
- Liu, Z.; Yi, Y.; Zhang, S.; Zhu, T.; Zhu, J.; Wang, J. Selective Catalytic Reduction of NOx with NH3 over Mn-Ce Mixed Oxide Catalyst at Low Temperatures. Catal. Today 2013, 216, 76–81. [Google Scholar] [CrossRef]
- Tsukahara, H.; Ishida, T.; Mayumi, M. Gas-Phase Oxidation of Nitric Oxide: Chemical Kinetics and Rate Constant. Nitric Oxide 1999, 3, 191–198. [Google Scholar] [CrossRef]
- Forecasts and Trends—Total Industrial Air Pollution Control Equipment Market. In Global Industrial Air Pollution Control Equipment Market, Forecast to 2025; Frost & Sullivan: San Antonio, TX, USA, 2018.
- Xue, E.; Seshan, K.; Ross, J.R.H. Roles of supports, Pt loading and Pt dispersion in the oxidation of NO to NO2 and of SO2 to SO3. Appl. Catal. B 1996, 11, 65–79. [Google Scholar] [CrossRef]
- Auvray, X.; Olsson, L. Stability and activity of Pd-, Pt- and Pd–Pt catalysts supported on alumina for NO oxidation. Appl. Catal. B 2015, 168–169, 342–352. [Google Scholar] [CrossRef]
- Li, L.; Qu, L.; Cheng, J.; Li, J.; Hao, Z. Oxidation of nitric oxide to nitrogen dioxide over Ru catalysts. Appl. Catal. B 2009, 88, 224–231. [Google Scholar] [CrossRef]
- Shen, M.; Zhao, Z.; Chen, J.; Su, Y.; Wang, J.; Wang, X. Effects of calcium substitute in LaMnO3 perovskites for NO catalytic oxi-dation. J. Rare Earths 2013, 31, 119–123. [Google Scholar] [CrossRef]
- Chen, J.; Shen, M.; Wang, X.; Wang, J.; Su, Y.; Zhao, Z. Catalytic performance of NO oxidation over LaMeO3 (Me=Mn, Fe, Co) perovskite prepared by the sol–gel method. Catal. Commun. 2013, 37, 105–108. [Google Scholar] [CrossRef]
- Ramesh, K.; Chen, L.; Chen, F.; Liu, Y.; Wang, Z.; Han, Y. Re-investigating the CO oxidation mechanism over unsupported MnO, Mn2O3 and MnO2 catalysts. Catal. Today 2008, 131, 477–482. [Google Scholar] [CrossRef]
- Gao, J.; Jia, C.; Zhang, L.; Wang, H.; Yang, Y.; Hung, S.; Hsu, Y.; Liu, B. Tuning chemical bonding of MnO2 through transition-metal doping for enhanced CO oxidation. J. Catal. 2016, 341, 82–90. [Google Scholar] [CrossRef]
- Zhi, B.; Ding, H.; Wang, D.M.; Cao, Y.; Zhang, Y.; Wang, X.; Liu, Y.L.; Huo, Q.S. Ordered mesoporous MnO2 as a synergetic adsorbent for effective arsenic(III) removal. J. Mater. Chem. A 2014, 2, 2374–2382. [Google Scholar] [CrossRef]
- Sa, Y.J.; Kwon, K.; Cheon, J.Y.; Kleitz, F.; Joo, S.H. Ordered mesoporous Co3O4 spinels as stable, bifunctional, noble metal-free oxygen electrocatalysts. J. Mater. Chem. A 2013, 1, 9992–10001. [Google Scholar] [CrossRef]
- Cao, C.; Zhang, Y.; Liu, D.; Meng, M. Gravity-Driven Multiple Collision-Enhanced Catalytic Soot Combustion over a Space-Open Array Catalyst Consisting of Ultrathin Ceria Nanobelts. Small 2015, 11, 3659–3664. [Google Scholar] [CrossRef]
- Li, C.; Yan, S.; Lai, Y. Catalyst and Method for Manufacturing the Same and Method of Removing VOCs. Taiwan Patent I733200, 11 October 2021. [Google Scholar]
- Xu, H.; Liu, J.; Zhang, Z.; Liu, S.; Lin, Q.; Wang, Y.; Dai, S.; Chen, Y. Design and Synthesis of Highly-Dispersed WO3 Catalyst with Highly Effective NH3–SCR Activity for NOx Abatement. ACS Catal. 2019, 9, 11557–11562. [Google Scholar] [CrossRef]
- Li, H.; Qi, G.; Zhang, X.; Huang, X.; Li, W.; Shen, W. Low-temperature oxidation of ethanol over a Mn0.6Ce0.4O2 mixed oxide. Appl. Catal. B Environ. 2011, 103, 54–61. [Google Scholar] [CrossRef]
- Beche, E.; Charvin, P.; Perarnau, D.; Abanades, S.; Flamant, G. Ce 3d XPS investigation of cerium oxides and mixed cerium oxide (CexTiyOz). Surf. Interface Anal. 2008, 40, 264–267. [Google Scholar] [CrossRef]
- Bai, B.; Li, J.; Hao, J. 1D-MnO2, 2D-MnO2 and 3D-MnO2 for low-temperature oxidation of ethanol. Appl. Catal. B Environ. 2015, 164, 241–250. [Google Scholar] [CrossRef]
- Chen, G.; Zhao, Y.; Fu, G.; Duchesne, P.N.; Gu, L.; Zheng, Y.; Weng, X.; Chen, M.; Zhang, P.; Pao, C.; et al. Interfa-cial Effects in Iron-Nickel Hydroxide–Platinum Nanoparticles Enhance Catalytic Oxidation. Science 2014, 344, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Bunluesin, T.; Cordatos, H.; Gorte, R.J. Study of CO Oxidation Kinetics on Rh/Ceria. J. Catal. 1995, 157, 222–226. [Google Scholar] [CrossRef]
- Amiridis, D.M.; Robert, V.D.; Israel, E.W. The effect of metal oxide additives on the activity of V2O5/TiO2 catalysts for the selective catalytic reduction of nitric oxide by ammonia. Appl. Catal. B Environ. 1999, 20, 111–122. [Google Scholar] [CrossRef]
- Casapu, M.; Krocher, O.; Elsener, M. Screening of doped MnOx–CeO2 catalysts for low-temperature NO-SCR. Appl. Catal. B Environ. 2009, 88, 413–419. [Google Scholar] [CrossRef]
- Tian, W.; Yang, H.; Fan, X.; Zhang, X. Catalytic reduction of NOx with NH3 over different-shaped MnO2 at low temperature. J. Hazard. Mater. 2011, 188, 105–109. [Google Scholar] [CrossRef]
- Joshi, S.; Kumar, A.; Kamasamudram, K.; Currier, N.W.; Yezerets, A. Diagnostic of zeolite-based SCR catalyst deactivation modes using NO oxidation as a probe reaction. In Proceedings of the 23rd North American Catalysts Society Meeting, Louis-ville, KY, USA, 2–7 June 2013. [Google Scholar]
- Mulla, S.; Chen, N.; Delgass, W.; Epling, W.S.; Ribeiro, F.H. NO2 inhibits the catalytic reaction of NO and O2 over Pt. Catal. Lett. 2005, 100, 267–270. [Google Scholar] [CrossRef]
- Guo, Z.; Liang, Q.H.; Yang, Z.; Liu, S.; Huang, Z.H.; Kang, F. Modifying porous carbon nanofibers with MnOx–CeO2–Al2O3 mixed oxides for NO catalytic oxidation at room temperature. Catal. Sci. Technol. 2016, 6, 422–425. [Google Scholar] [CrossRef]
- Ziaei-Azad, H.; Khodadadi, A.; Esmaeilnejad-Ahranjani, P.; Mortazavi, Y. Effects of Pd on enhancement of oxidation activity of LaBO3 (B=Mn, Fe, Co and Ni) pervoskite catalysts for pollution abatement from natural gas fueled vehicles. Appl. Catal. B Environ. 2011, 102, 62–70. [Google Scholar] [CrossRef]
- Esmaeilnejad-Ahranjani, P.; Khodadadi, A.; Ziaei-Azad, H.; Mortazavi, Y. Effects of excess manganese in lanthanum manganite perovskite on lowering oxidation light-off temperature for automotive exhaust gas pollutants. Chem. Eng. J. 2011, 169, 282–289. [Google Scholar] [CrossRef]
- Salman, A.R.; Enger, B.C.; Auvray, X.; Lødeng, R.; Menon, M.; Waller, D.; Rønning, M. Catalytic oxidation of NO to NO2 for nitric acid production over a Pt/Al2O3 catalyst. Appl. Catal. A Gen. 2018, 564, 142–146. [Google Scholar] [CrossRef]
- Tang, X.; Hao, J.; Yi, H.; Li, J. Low-temperature SCR of NO with NH3 over AC/C supported manganese-based monolithic cata-lysts. Catal. Today 2007, 126, 400–405. [Google Scholar] [CrossRef]
- Irani, K.; Epling, W.S.; Blint, R. Effect of hydrocarbon species on no oxidation over diesel oxidation catalysts. Appl. Catal. B Environ. 2009, 92, 422–428. [Google Scholar] [CrossRef]
- Wang, P.; Yin, J.; Lei, L. Evaluation of NO Oxidation Properties over a Mn-Ce/γ-Al2O3 Catalyst. J. Nanomater. 2016, 2016, 2103647. [Google Scholar] [CrossRef]
- Megarajan, S.K.; Rayalu, S.; Teraoka, Y.; Labhsetwar, N. High NO oxidation catalytic activity on non-noble metal based co-balt-ceria catalyst for diesel soot oxidation. J. Mol. Catal. A Chem. 2014, 385, 112–118. [Google Scholar] [CrossRef]
- Tang, N.; Liu, Y.; Wang, H.; Wu, Z. Mechanism Study of NO Catalytic Oxidation over MnOx/TiO2 Catalysts. J. Phys. Chem. C 2011, 115, 8214–8220. [Google Scholar] [CrossRef]
- Zhong, L.; Cai, W.; Zhong, Q. Evaluation of cerium modification over Cr/Ti-PILC for NO catalytic oxidation and their mechanism study. RSC Adv. 2014, 4, 43529–43537. [Google Scholar] [CrossRef]
- Deng, X.; Chen, K.; Tüysüz, H. Protocol for the Nanocasting Method: Preparation of Ordered Mesoporous Metal Oxides. Chem. Mater. 2017, 29, 40–52. [Google Scholar] [CrossRef]
- Yang, K.; Liu, Y.; Deng, J.; Zhao, X.; Yang, J.; Han, Z.; Hou, Z.; Dai, H. Three-dimensionally ordered mesoporous iron oxide-supported single-atom platinum: Highly active catalysts for benzene combustion. Appl. Catal. B Environ. 2019, 244, 650–659. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | SBET (m2/g) | Dpore (nm) | Vpore (cm3/g) | Refs. |
|---|---|---|---|---|
| MnO2 | 61.5 | 8.7 | 0.13 | [25] |
| 9MnO2-1CeO2 | 87.3 | 8.2 | 0.18 | this work |
| 7MnO2-3CeO2 | 124.2 | 7.3 | 0.23 | this work |
| CeO2 | 135.0 | 12.8 | 0.43 | [25] |
| Catalyst | Mn/Ce Atom Ratio | (Mn + Ce)/DP Atom Ratio DP = Doped Metal |
|---|---|---|
| 9MnO2-1CeO2 | 8.77 | - |
| Co/9MnO2-1CeO2 | 9.14 | 71.9 |
| Ni/9MnO2-1CeO2 | 9.21 | 66.8 |
| Fe/9MnO2-1CeO2 | 9.11 | 71.4 |
| Cu/9MnO2-1CeO2 | 9.19 | 64.6 |
| Cr/9MnO2-1CeO2 | 9.01 | 99.6 |
| Catalyst | Mn ratio | (Mn2++ Mn3+)/Mn4+ | Ce3+/Ce4+ | ||
|---|---|---|---|---|---|
| Mn2+ | Mn3+ | Mn4+ | |||
| 9MnO2-1CeO2 | 6.32 | 0 | 1 | 6.3 | 0.75 |
| 7MnO2-3CeO2 | 6.17 | 2.67 | 1 | 9.0 | 0.52 |
| Cu/9MnO2-1CeO2 | 7.18 | 0.95 | 1 | 8.1 | 0.72 |
| Fe/9MnO2-1CeO2 | 7.90 | 0.48 | 1 | 8.2 | 0.72 |
| Catalyst | NO Inlet (ppm) | Fraction of NO Converted to NO2@200 °C (%) | Temperature of 50% NO Oxidation to NO2 (°C) |
|---|---|---|---|
| Pt/Cr2O3 | 263 | 29.8 | >200 |
| Co3O4 | 260 | 55.6 | 190 |
| Pt/Fe2O3 | 250 | 35.7 | >200 |
| MnO2 | 310 | 56.8 | 189 |
| Rh/MnO2 | 245 | 59.1 | 185 |
| Co/MnO2 | 260 | 61.5 | 180 |
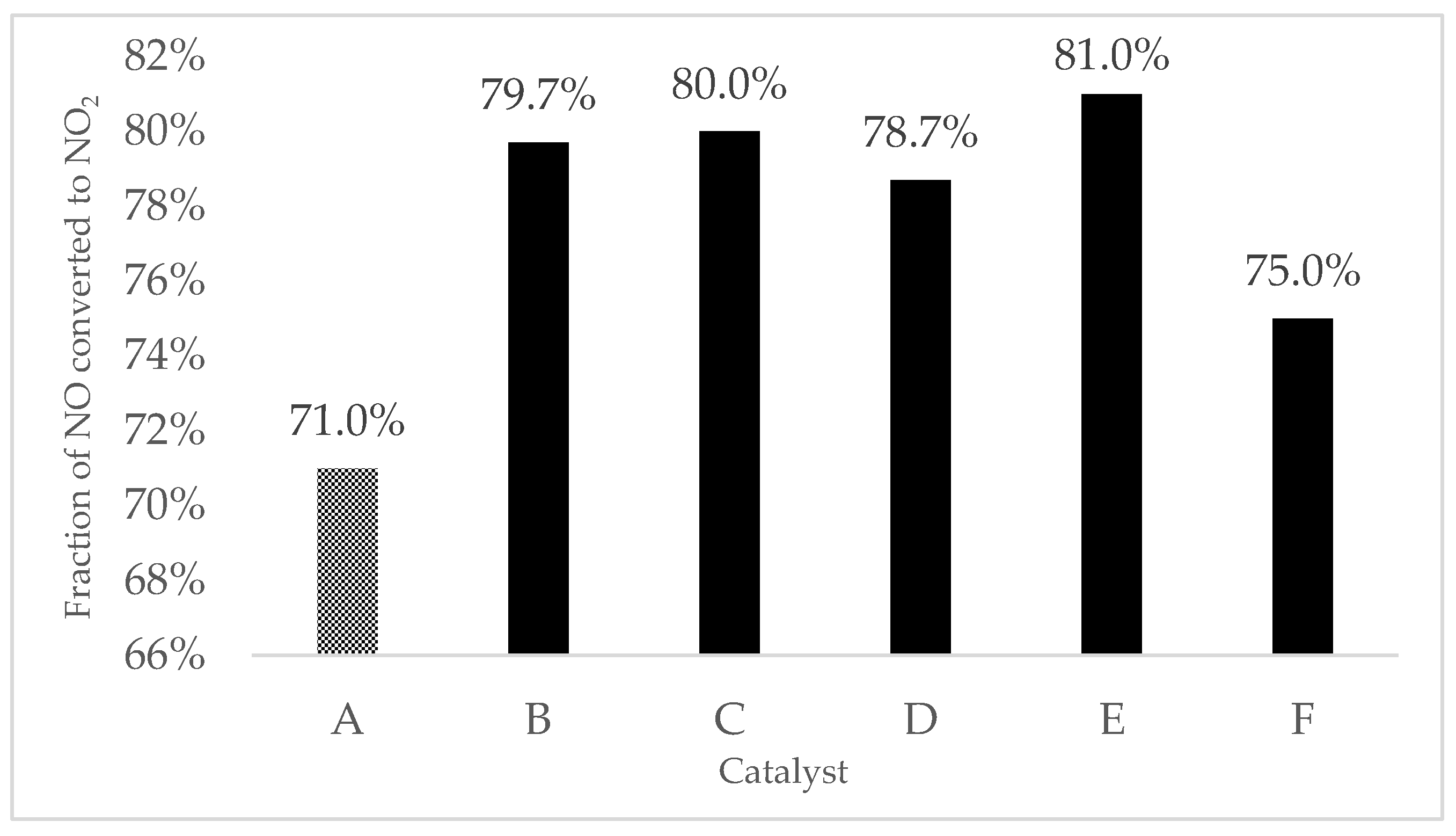
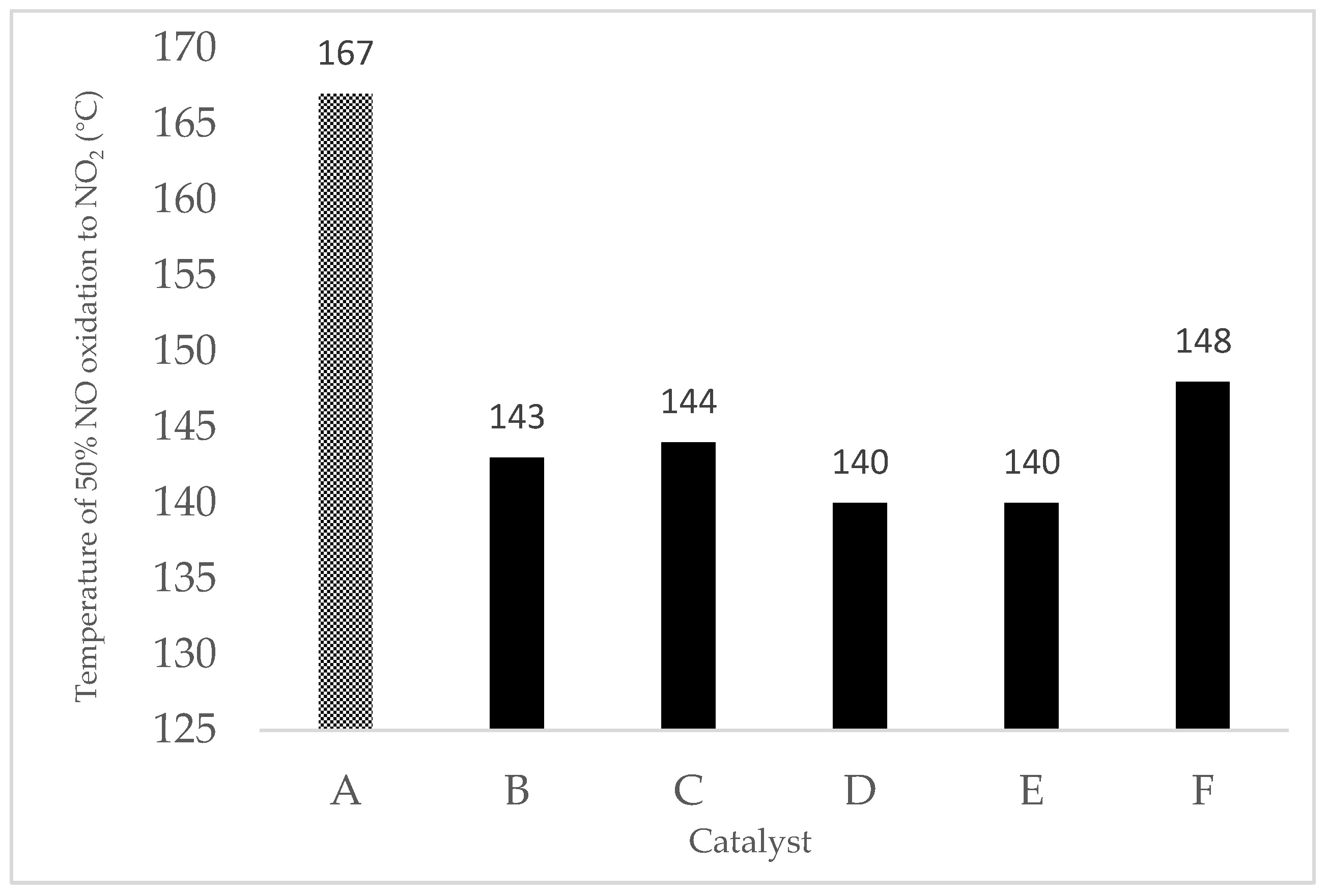
| 9MnO2-1CeO2 | 266 | 71.0 | 167 |
| 7MnO2-3CeO2 | 252 | 65.5 | 172 |
| Catalyst | Fraction of NO Converted to NO2@200 °C (%) | Temperature of 50% NO Oxidation to NO2 (°C) |
|---|---|---|
| 9MnO2-1CeO2 | 71.0 | 167 |
| 7MnO2-3CeO2 | 65.5 | 172 |
| Fe/9MnO2-1CeO2 | 78.7 | 140 |
| Fe/7MnO2-3CeO2 | 82.0 | 140 |
| Cu/9MnO2-1CeO2 | 81.0 | 140 |
| Cu/7MnO2-3CeO2 | 78.0 | 138 |
| Catalyst | Fraction of NO Converted to NO2@200 °C (%) | Temperature of 50% NO Oxidation to NO2 (°C) | Refs. |
|---|---|---|---|
| DP/mMnO2-nCeO2 (m:n = 9:1 and 7:3) | 75~81% | 140~200 | This work |
| Pt/Al2O3 | 40% | 250 | [40] |
| La1−xCexCoO3 | 5% | 270 | [41] |
| Pt-Pd/Al2O3 | 10% | >400 | [42] |
| Mn-Ce/γ-Al2O3 | 40% | 225 | [43] |
| Co3O4-CeO2 | 20% | 230 | [44] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuo, C.-N.; Li, C.-S.; Lai, Y.-L.; Yen, S.-I. Metal-Doped Mesoporous MnO2-CeO2 Catalysts for Low-Temperature Pre-Oxidation of NO to NO2 in Fast SCR Process. Catalysts 2023, 13, 694. https://doi.org/10.3390/catal13040694
Kuo C-N, Li C-S, Lai Y-L, Yen S-I. Metal-Doped Mesoporous MnO2-CeO2 Catalysts for Low-Temperature Pre-Oxidation of NO to NO2 in Fast SCR Process. Catalysts. 2023; 13(4):694. https://doi.org/10.3390/catal13040694
Chicago/Turabian StyleKuo, Chun-Nan, Cheng-Shiuan Li, Yu-Lun Lai, and Shao-I Yen. 2023. "Metal-Doped Mesoporous MnO2-CeO2 Catalysts for Low-Temperature Pre-Oxidation of NO to NO2 in Fast SCR Process" Catalysts 13, no. 4: 694. https://doi.org/10.3390/catal13040694
APA StyleKuo, C.-N., Li, C.-S., Lai, Y.-L., & Yen, S.-I. (2023). Metal-Doped Mesoporous MnO2-CeO2 Catalysts for Low-Temperature Pre-Oxidation of NO to NO2 in Fast SCR Process. Catalysts, 13(4), 694. https://doi.org/10.3390/catal13040694