Abstract
The effect of strontium substitution in the structure of the complex oxide Gd2SrFe2O7 on the production of light olefins by CO hydrogenation was investigated. Perovskite-type oxides Gd2−xSr1+xFe2O7 (x = 0; 0.1; 0.2; 0.3; 0.4) were synthesized by sol–gel technology and characterized by XRD, Mössbauer spectroscopy, BET specific area, acidity testing, and SEM. The experimental data revealed a correlation between the state of iron atoms, acidity, and catalytic performance. It was found that with an increase in the content of Sr2+ in the perovskite phase, the basicity of the surface and the oxygen diffusion rate increased. This contributed to the CO dissociative adsorption, formation of active carbon, and its further interaction with atomic hydrogen.
1. Introduction
The development of new active and selectively acting catalytic systems remains one of the most important trends in modern chemistry. Oxygen-deficient complex oxides with a perovskite-type structure have attracted the attention of a huge number of researchers around the world as promising materials for creating electrochemical, catalytic, and magnetic devices [1,2,3,4,5]. Complex oxides with a layered perovskite-type structure have mixed oxygen, ionic and electronic conductivity and high stability. They are also used as catalysts for high-temperature processes [6,7,8,9,10].
Varying the composition of perovskite, including partial substitution or doping of the cation in the A-site and/or B-site, is an effective way to increase the stability and the oxygen and ionic conductivity, i.e., to improve the physical and chemical properties of perovskites for their further use in catalytic processes. Replacing the cation in the A-site leads to the changing of the size of the perovskite unit cell, and, consequently, the length of the B-O covalent bond in the structure. Cationic substitution in perovskite not only has a significant effect on catalytic activity due to the stabilization of unusual oxidation states, but it also promotes the simultaneous formation of structural defects responsible for oxygen mobility in the crystal lattice [1,3,11]. Doping the A-site of perovskite with alkali and alkaline earth metals leads to an increase in the mobility of lattice oxygen ions, reducing the process temperature in oxidation reactions [1]. It was shown in [12] that a better ability of the catalyst to adsorb CO2 was realized due to the increase in basicity upon the introduction of Ca or Sr. This increased the rate of carbon removal, and thus the catalytic activity. The promoting effect of alkali and alkaline earth metals was also noted by the authors of [13]. It was found [14] that the doping of La3+ with low-valent strontium ions resulted is the appearance of an induced negative charge in the A-site. This was compensated by the formation of oxygen vacancies and/or oxidation of the metal in the B-position to higher oxidation states, thereby leading to the doped perovskite having a high electronic and ionic conductivity.
The catalytic properties of complex perovskite-type oxides, La1−xSrxCoO3, in the selective Fischer–Tropsch synthesis of higher alcohols were studied in [15]. It was found that Sr substitution improved the catalyst’s reducibility and increasing strontium substitution beyond a threshold of 10 mol% Sr had an inverse effect on the catalytic performance in the syngas conversion. For x ≤ 0.1, the catalysts exhibited a stable rhombohedral structure, which led to good dispersion of active center Co0 during the reduction process and displayed a high catalytic activity. For x ≥ 0.2 the catalyst structure was gradually changed, from a rhombohedral to a less stable cubic structure, leading to Co0 sintering. This led to a decrease in the number of active centers and an improvement in the reducibility of samples due to the presence of strontium in the perovskite lattice. In the study of strontium-doped lanthanum cobaltites in the Fischer–Tropsch synthesis, it was found that SrCO3 [16] formed on the surface of the samples after the reaction, with the amount of SrCO3 increasing with an increase in the amount of strontium. It was also reported that this compound was inactive in the Fischer–Tropsch synthesis and coated the catalyst surface [17], blocking active sites, which adversely affected the catalytic activity and partially inhibited the process.
Since the discovery of Fischer–Tropsch (FT) synthesis, iron-based catalysts have been used extensively in industrial FT plants [18,19]. The catalytic performance of iron catalysts depends on their active phase, transition metal promoter, alkali promoter, and support [20,21,22,23,24]. It has been shown that FT synthesis over iron catalysts is a structure-sensitive reaction [1,25,26,27], with the intrinsic reaction rate and product selectivity being functions of iron particle size. The promoted iron catalysts have, so far, shown one of the best performances in direct olefin synthesis from syngas. The particularly attractive features of iron catalysts for light olefin synthesis from syngas are summarized as follows:
- -
- Flexibility in terms of activity, selectivity, and reaction conditions;
- -
- High selectivity to olefins within the Anderson–Schulz–Flory hydrocarbon distribution;
- -
- Sensitivity of reaction rate and selectivity to promotion and support;
- -
- Activity in the water–gas shift (WGS) reaction and the possibility of using syngas with variable H2/CO ratios;
- -
- Presence of different phases that are potentially active in FT synthesis;
- -
- Relative stability in the presence of syngas impurities compared to cobalt catalysts;
- -
- Lower cost compared to other metal catalysts.
Iron-based catalysts are very versatile with rich chemistry during their preparation, activation, and reaction. They can be adapted for various operating conditions of CO/CO2 hydrogenation reactions [28,29]. Also, the structure of iron catalysts can significantly evolve as a function of the operating conditions [1,30]. Thus, during FT synthesis, iron species may be distributed among several phases, e.g., carbides, oxides, and metallic iron. The iron carbides may transform from one to another as a function of the operating conditions [1,2,6,13].
We have previously investigated GdB’xFe1−xO3 (B’ = Co, Mn) and An+1FenO3n+1 (A = Gd, Sr, n = 1, 2,…,∞—number of perovskite layers) in the processes of carbon oxide adsorption, carbon oxide hydrogenation, and dry reforming of methane [31,32,33,34]. It was shown that the sol–gel method makes it possible to obtain samples with better catalytic characteristics compared to ceramic systems. A correlation between the number of perovskite layers (n = 1, 2,…,∞) in the layered oxide structure and its activity was observed. Comparison of the physicochemical and catalytic results of studies in the dry reforming of methane (DRM) and hydrogenation of carbon oxides made it possible to establish that the chemisorption processes for CO, CO2, H2, and CH4 proceed on different types of centers. It has been established that the formation of Gd2O2CO3 carbonate complexes occurs on Gd3+ ions (A-site), and transition metal ions in the B-site are responsible for the formation of atomic hydrogen. The nature of the 3d metal in GdBO3 influences the ratio of molecular and atomic forms of hydrogen and its spillover on the surface of the catalyst. It was also found that GdCoO3, after treatment in a reducing atmosphere at 900 °C and after 100 h of operation, was reduced to Gd2Co2O5 and valence states of cobalt changed from Co3+ to Co2+, while for GdMnO3 and GdFeO3 there was little change in the structure and valence state [19,35].
There have been numerous studies of the acid–base properties of interacting with small carbon molecules [36,37], but not as many studies involving Fischer–Tropsch synthesis. In addition, the possibility of targeted variation of the cationic composition, their unique physicochemical properties, and their high thermal stability make perovskites promising for heterogeneous catalysis. In this regard, the main objectives of this work were to study the catalytic properties of complex layered oxides Gd2−xSr1+xFe2O7 (x = 0.1; 0.2; 0.3; 0.4) in CO hydrogenation and to establish a correlation between the composition of the complex oxide and its physicochemical and catalytic properties.
2. Results and Discussion
2.1. Characterization
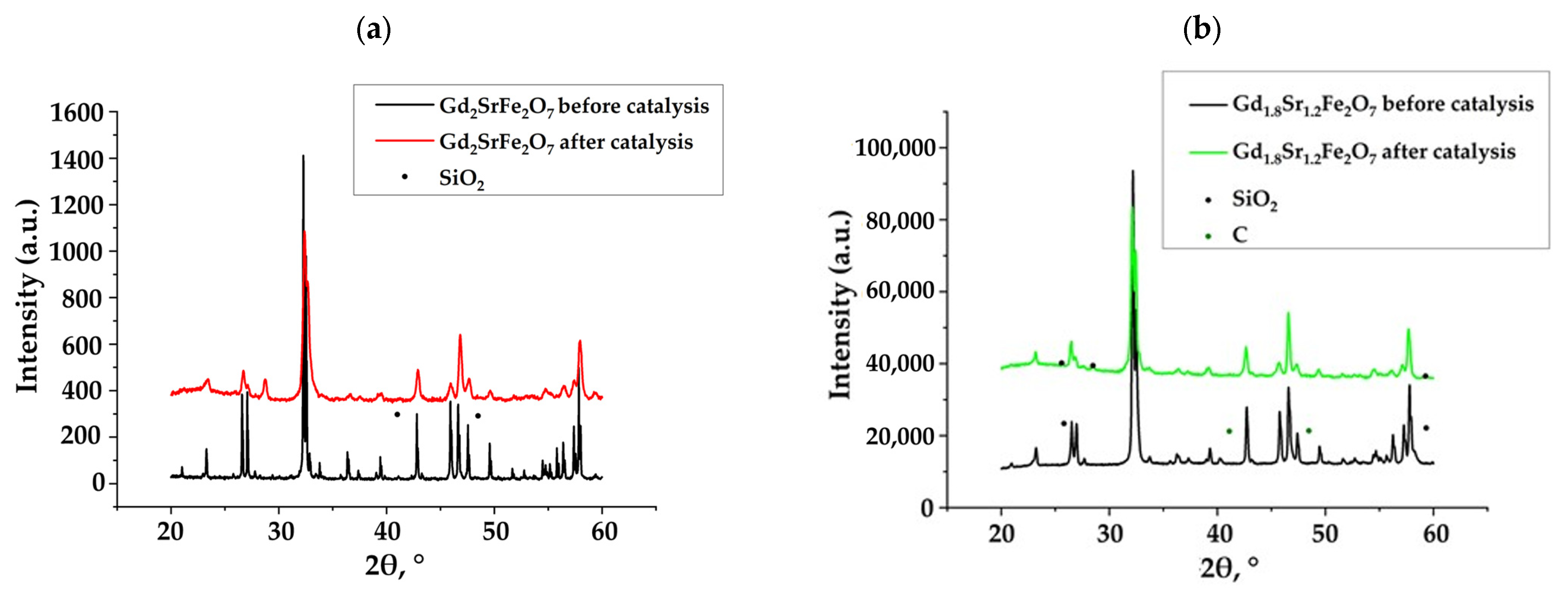
Figure 1 shows X-ray diffraction patterns of synthesized complex oxides Gd2−xSr1+xFe2O7 (x = 0; 0.2) obtained by sol–gel technology (citrate–nitrate method) before and after catalysis. For the initial samples, it was found that the introduction of strontium, i.e., the transition from perovskite to perovskite-like layered structure in complex oxides Gd2−xSr1+xFe2O7, did not lead to a change in the type of crystal lattice—it remained as a tetragonal crystal lattice with the space group I4mmm [38].
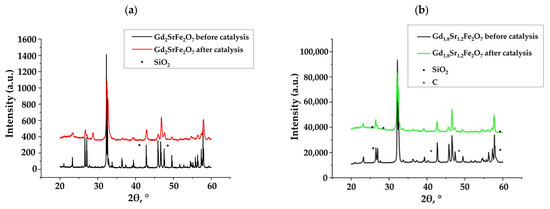
Figure 1.
X-ray diffraction patterns of complex layered ferrites Gd2−xSr1+xFe2O7: (a) x = 0 and (b) x = 0.2.
The unit cell parameters, crystallite sizes, and the specific surface areas of iron-containing oxides are presented in Table 1.

Table 1.
Structural parameters, crystallite sizes, and SBET of Gd2−xSr1+xFe2O7 (x = 0; 0.1; 0.2; 0.3; 0.4) samples.
It was typical for all solid solutions that, with an increase in the content of strontium atoms, the parameter a decreased, while the parameter c increased. These changes in the lattice parameters are explained by the ionic sizes of the substituted atoms (the ionic radius of Sr2+ is 1.31 Å and that of Gd3+ is 1.107 Å), since an atom with a smaller ionic radius is replaced by an atom with a larger ionic radius. According to the data obtained by the method of low-temperature nitrogen adsorption (Table 1), all complex ferrites had a small specific surface of 1.4–4.8 m2/g, so it was quite difficult to determine the porosity. The specific surface area of solid solutions decreased with an increase in the content of strontium atoms; this can be easily explained by the synthesis temperature of these solid solutions. The Gd1.9Sr1.1Fe2O7 solid solution had the maximum surface area and was obtained at a minimum temperature of 1260 °C for 20 min. Solid solutions of Gd1.7Sr1.3Fe2O7 and Gd1.8Sr1.2Fe2O7 were synthesized at the same temperature, but with an annealing time of 1 h, so they had the same surface area. The complex ferrite Gd1.6Sr1.4Fe2O7 was obtained, single-phase only, at 1260 °C after 3 h of calcination, and the Gd1.5Sr1.5Fe2O7 solid solution was obtained single-phase after heating to 1400 °C for 10 min and this was the maximum calcination temperature, and therefore the minimum surface area.
The diffraction patterns of the Gd2−xSr1+xFe2O7 samples after catalysis did not differ from the initial ones. But, in addition to the peaks of complex oxides Gd2−xSr1+xFe2O7, there were peaks of quartz—SiO2, which had been used as an inert additive to increase the volume of the catalyst. Thus, we can conclude that the phase composition of the catalyst after catalysis was unchanged.
Micrographs of Gd2−xSr1+xFe2O7 solid solutions (Figure 2) illustrate that the particle sizes were 150–300 nm. As an example, micrographs of solid solutions with x = 0; 0.2 are presented. It should be noted that the particles were closer to a more regular shape and had approximately the same size.

Figure 2.
Micrographs of the surface of Gd2−xSr1+xFe2O7 (x = 0; 0.2) before and after catalysis (reaction conditions: CO:H2 = 0.5 L h−1:1 L h−1, GHSV = 8700 h−1, 1 atm, time ~100 h).
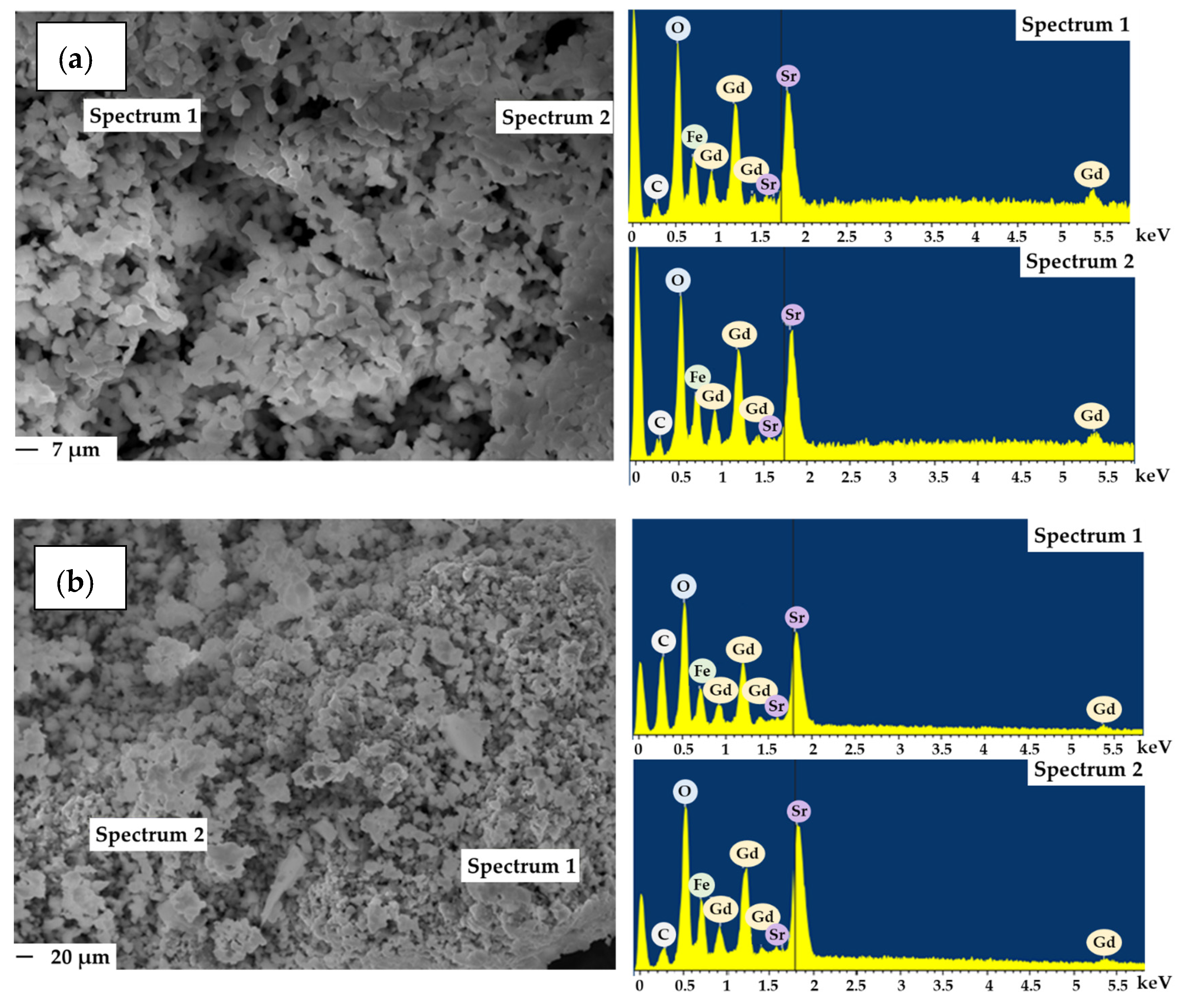
After catalytic tests, the samples showed slight particle agglomeration and the EDX spectra data confirmed the presence of carbon on the surface of spent catalysts (Figure 3).
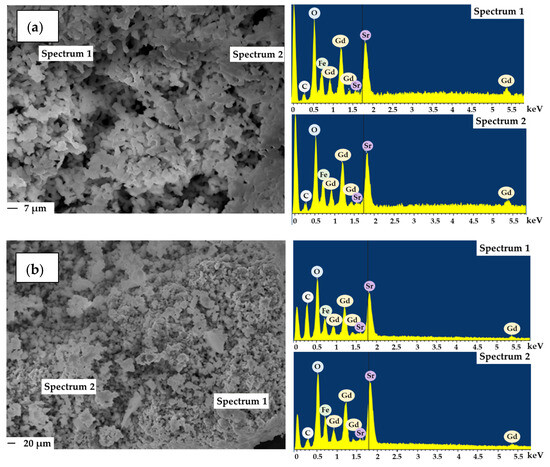
Figure 3.
Micrographs and EDX of complex oxides Gd2SrFe2O7 (a) and Gd1.8Sr1.2Fe2O7 (b) after catalysis (reaction conditions: CO:H2 = 0.5 L h−1:1 L h−1, GHSV = 8700 h−1, 1 atm, time on stream ~ 100 h).
The states of iron atoms in complex perovskite-like ferrites were studied using Mössbauer spectroscopy. All spectra were taken at room temperature.
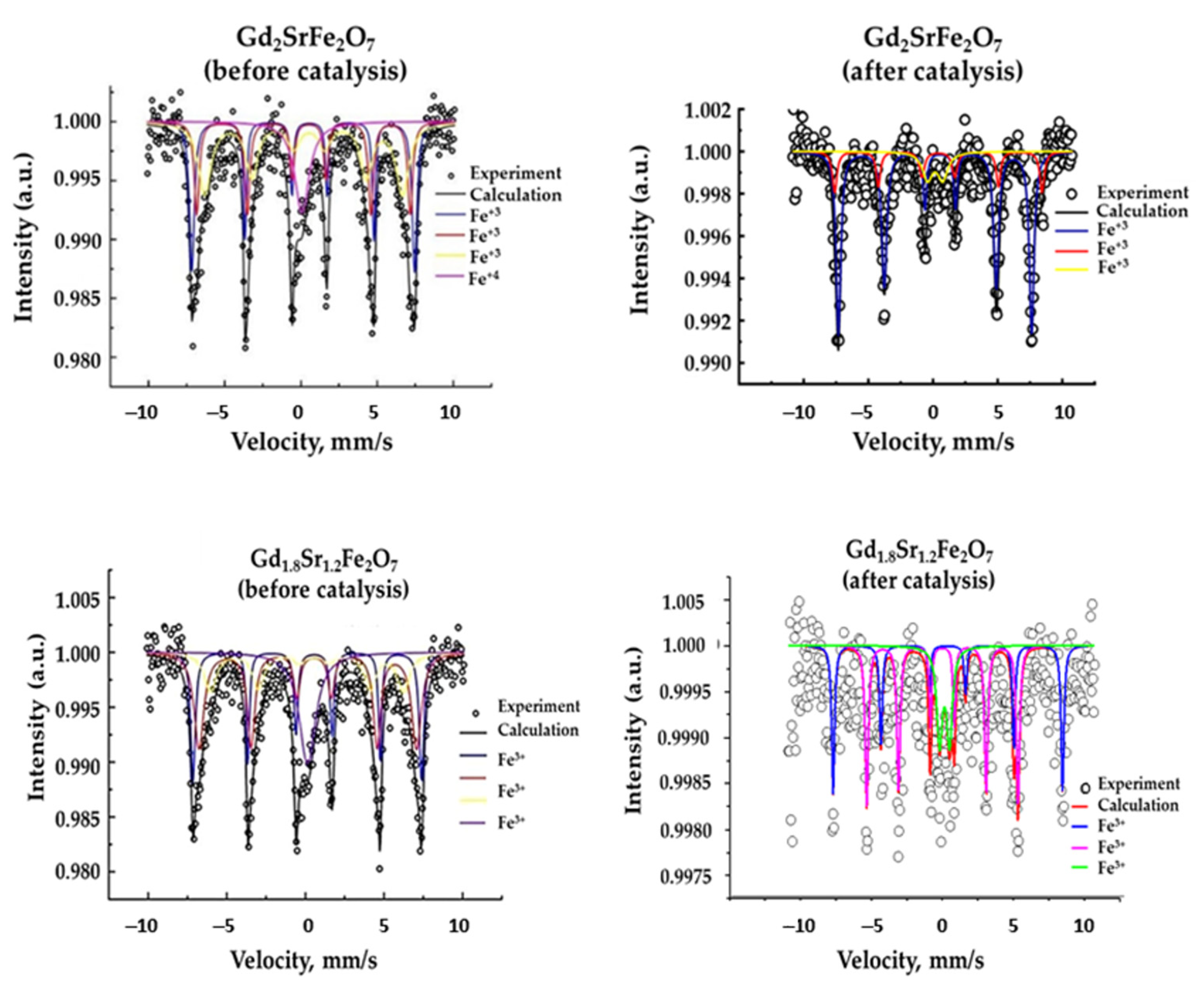
The complete replacement of gadolinium by strontium, as well as the “embedding” of the SrO layer in GdFeO3 (samples of SrFeO3−δ and GdSrFeO4) led to the appearance of an additional form of Fe4+ iron in contrast to gadolinium ferrite (Figure 4, Table 2) [39].
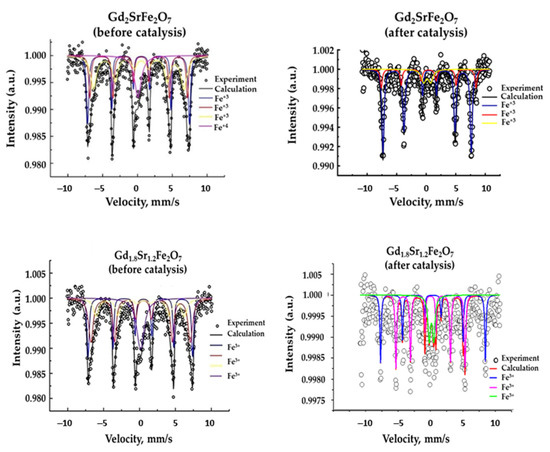
Figure 4.
Mössbauer spectra of complex layered ferrites Gd2−xSr1+xFe2O7 before and after catalysis (reaction conditions: CO:H2 = 0.5 L h−1:1 L h−1, GHSV = 8700 h−1, 1 atm, time on stream ~100 h).
Table 2.
Parameters of the Mössbauer spectra of solid solution Gd2−xSr1+xFe2O7 (x = 0; 0.1; 0.2; 0.3; 0.4) samples.
For Gd2−xSr1+xFe2O7 samples, along with one line corresponding to Fe4+ atoms, there were three sextets characteristic of Fe+3, with Fe+3 atoms in three different environments, and all of them were magnetic, since they had magnetic splitting. The content of Fe4+ was maximum in the complex oxide with x = 0.3 (Table 2). The appearance of such an “exotic” iron oxidation state can be associated with the stabilization of the system due to the redistribution of electron density as a consequence of the introduction of a strontium ion with a lower oxidation state compared to gadolinium into the A-position of the perovskite structure. There was a change in the environment of iron atoms: one Fe atom had more gadolinium atoms, the second had more strontium atoms [39]. In addition to different environments of iron atoms, the crystal structure of the Gd2−xSr1+xFe2O7 solid solution was also stabilized due to the appearance of oxygen vacancies, and the formula of the solid solution should be more accurately written as Gd2−xSr1+xFe2O7−α, where α is close to 0.1.
The Mössbauer spectrum of the complex layered oxide Gd2SrFe2O7 after the catalytic reaction differed from the Mössbauer spectrum of the initial oxide: there was no line corresponding to Fe4+ atoms. From Figure 4 and the data in Table 2, we can conclude that as a result of catalysis under the action of temperature and the reaction medium, changes occurred in the state of iron atoms and their local environment.
Thus, upon non-isovalent substitution of Gd3+→Sr2+ in the completely ordered Gd2SrFe2O7 structure, where strontium atoms are in the perovskite layer and gadolinium atoms are in the rock-salt layer, partial substitution of Gd3+ atoms leads to charge compensation due to the formation of oxygen vacancies in a layer of rock salt [40]. In this case, some of the octahedrons become pyramids, and thus the environment of the Fe+3 atoms changes, which was proved by Mössbauer spectroscopy.
It is known that the properties of perovskite catalysts are affected by the acid–base state of its surface [41,42], so a series of experiments was carried out to test the electron-withdrawing properties of the synthesized ferrites. The results obtained for all the studied samples are presented in Table 3. For some initial samples of gadolinium and strontium ferrites, it was not possible to determine the adsorption rates and the value of Nmax due to the occurrence of a suspension effect, which may have been due to the submicrocrystalline and nanocrystalline state of the ferrites.
Table 3.
Fast and slow adsorption rates and total number of electron-acceptor centers.
The presence of two types of acid sites, differing in strength was established. Moreover, in the initial samples, fast adsorption centers predominated, the number of which increased with an increase in the content of Sr2+ and Fe4+ in the samples. At the same time, the total number of electron-withdrawing centers in the initial samples decreased when Gd was nonstoichiometrically replaced by Sr. After conducting catalytic tests for all samples, an increase in the rates of both slow and fast adsorption, as well as total acidity, was observed. This effect manifested itself most clearly on Gd1.8Sr1.2Fe2O7.
2.2. Catalytic Tests
The hydrogenation of carbon monoxide to various hydrocarbons was carried out to study the catalytic properties of Gd2−xSr1+xFe2O7. Analysis of reactant content in the gas phase near the catalyst surface showed that at room temperature there was intensive adsorption of CO. After adsorption equilibrium was established and up to 573 K, the composition of the gas phase (CO+H2) changed little, while its transition to the catalytic temperature range was accompanied by the formation of CO2, and its amounts formed on complex ferrites Gd2−xSr1+xFe2O7 during the reaction in the CO:H2 = 1:2 ratio several times exceeded the amounts obtained in the hydrogen excess (Figure 5).
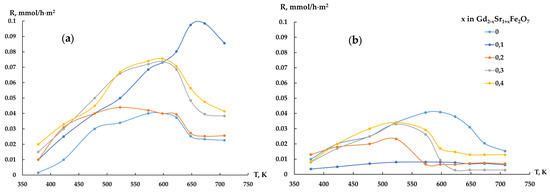
Figure 5.
Temperature dependences of the rate of formation of CO2, Gd1.8Sr1.2Fe2O7 catalyst, CO:H2 = 1:2 mixture (a) and CO:H2 = 1:4 mixture (b).
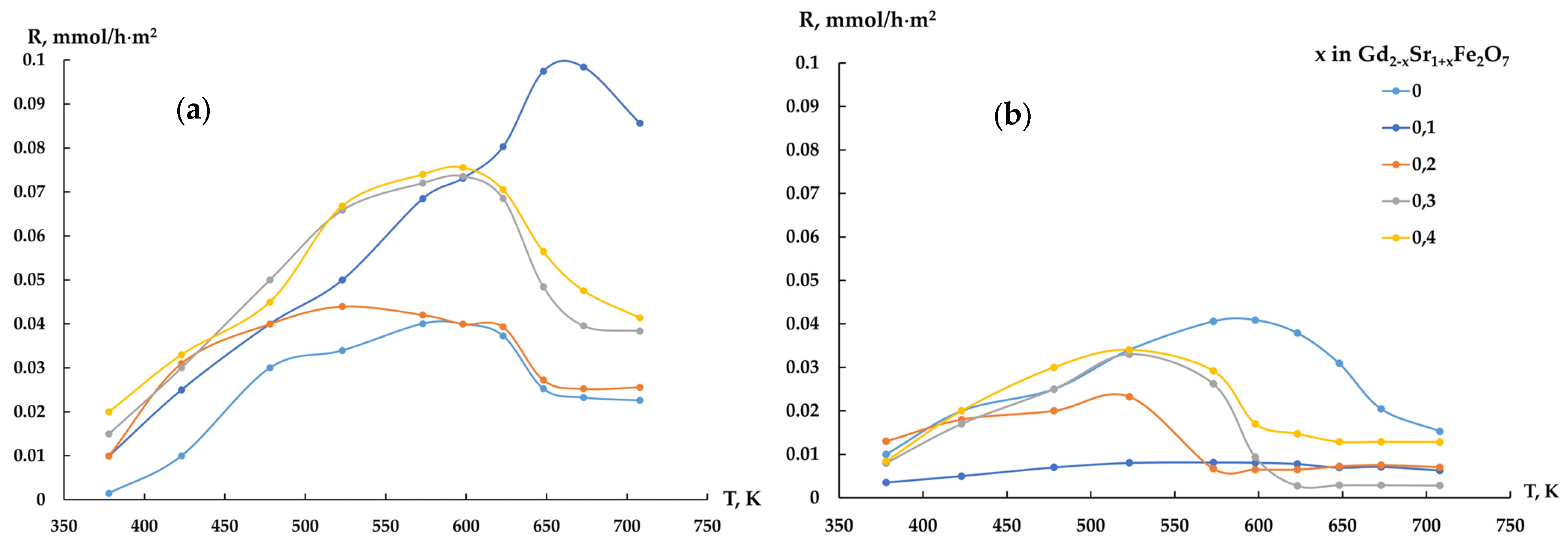
The CO conversions (Figure 6a) on all ferrites were 60–85% at the CO:H2 = 1:2 ratio and varied insignificantly with increasing temperature. Increasing the hydrogen content in the reaction mixture to 1:4 (Figure 6b) led to a decrease in the CO conversion with increasing strontium content in the samples.
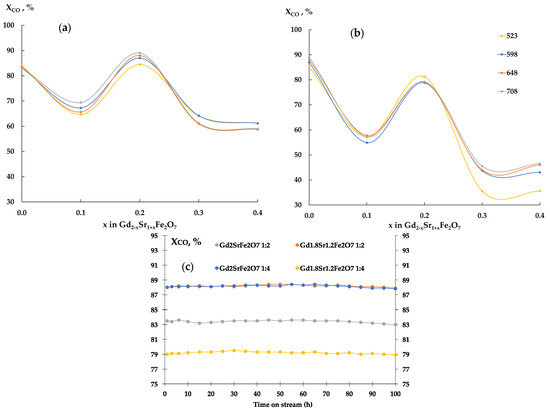
Figure 6.
CO conversion, Gd2−xSr1+xFe2O7 catalyst, CO:H2 = 1:2 mixture (a), CO:H2 = 1:4 mixture (b), and evolution of CO conversion with time on stream (c). Reaction conditions: CO:H2 = 0.5 L h−1:1 L h−1, GHSV = 8700 h−1, 1 atm, time on stream ~100 h.
It should be noted that on the second day of the experiment, the same curve for all gadolinium ferrites was displayed as a line. A similar picture was clearly seen on the third day of testing. In the following days (days 4–6 of the experiment), no changes in catalytic activity were detected (Figure 6c).
The value of CO adsorption depends on the gadolinium–oxygen bond energy in Gd-O-Me, which will change as a result of a third distortion of the perovskite lattice upon non-isovalent substitution of Gd for Sr and a change in the oxygen–metal bond energy. Previously, it was shown that, on complex GdFeO3 oxides, CO adsorption proceeded mainly on the A centers of perovskite [33] with the formation of Gd2O2CO3 carbonate complexes, and the formation of CO2 occurred as a result of the decomposition of these complexes with increasing temperature. The Boudouard mechanism, in which the surface O* species formed by CO* dissociation reacts with another CO* to form CO2, plays a predominant role in CO2 formation on the active χ-Fe5C2 phase [43]. Since the presence of iron carbides on the investigated catalysts was not confirmed, the formation of CO2 by the Boudouard mechanism was unlikely. The formation of CO2 is also possible in the water–gas shift (WGS) reaction (WGS): H2O + CO→CO2 +H2 and in the interaction of the adsorbed COads molecule with (OS) perovskite oxygen [16].
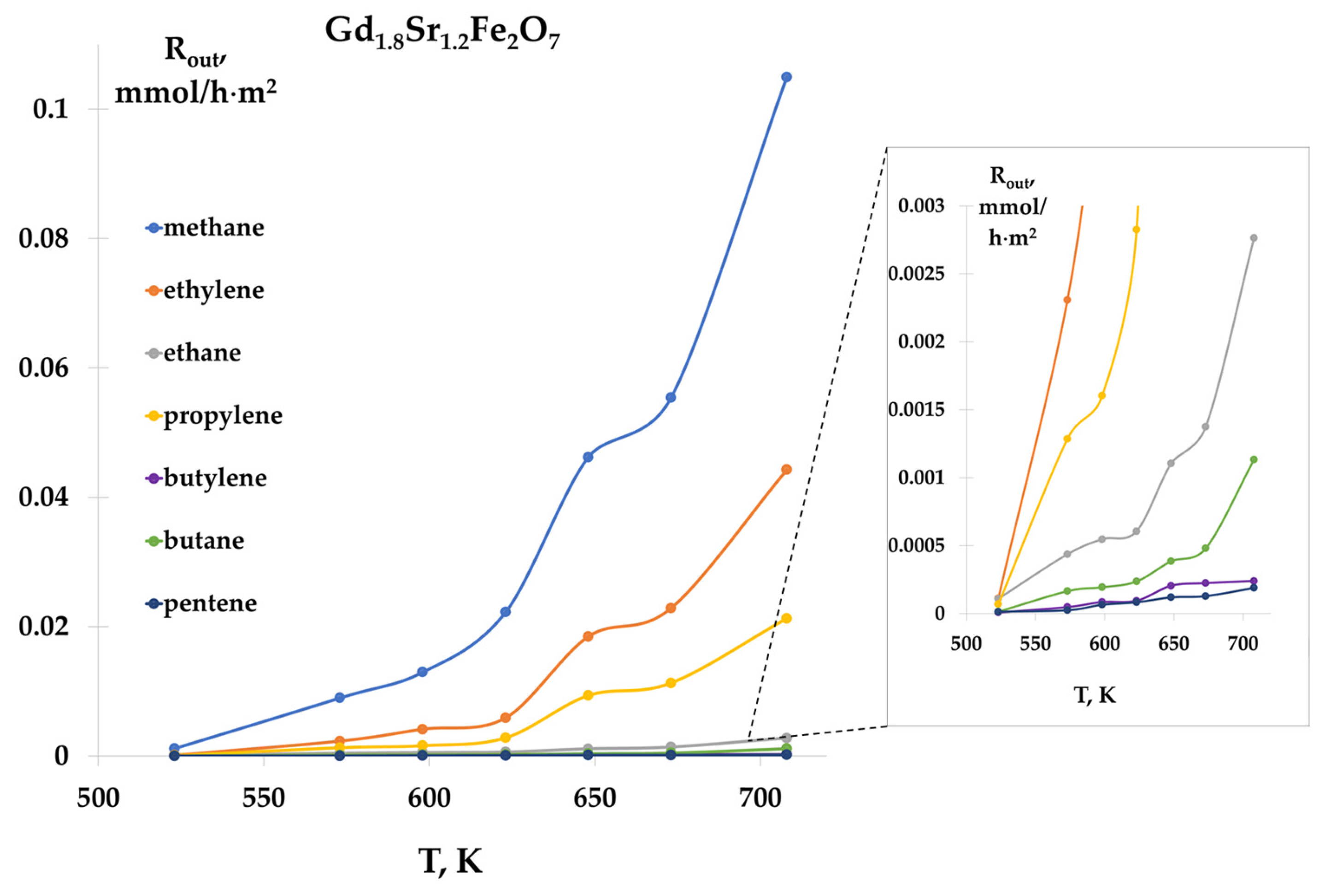
Carrying out the reaction at ratios CO:H2 = 1:2 and 1:4 showed that the products of the reaction were hydrocarbons C1–C5. The formation of all products began at 523 K and increased with increasing temperature. As an example, Figure 7 shows the temperature dependences of the rates of formation of C2–C5 hydrocarbons on a Gd1.8Sr1.2Fe2O7 sample, at a ratio of CO:H2 = 1:2. For all samples, the highest formation rates were observed for methane, ethylene, and propylene. Comparison of the rates of product formation on the studied samples showed that the specific catalytic activity increased in the series:
Gd1.6Sr1.4Fe2O7 < Gd1.9Sr1.1Fe2O7 < Gd2SrFe2O7 ≈ Gd1.8Sr1.2Fe2O7 ≈ Gd1.7Sr1.3Fe2O7
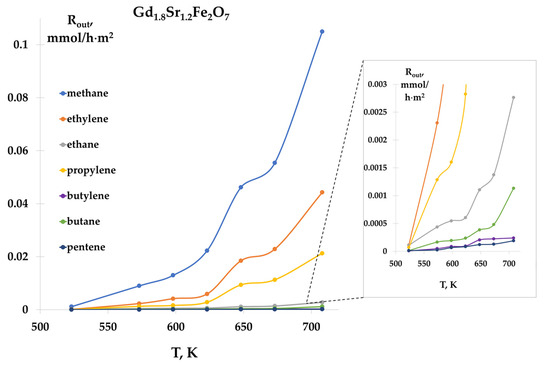
Figure 7.
Temperature dependences of hydrocarbon formation rates, Gd1.8Sr1.2Fe2O7 catalyst, CO:H2 = 1:2 mixture.
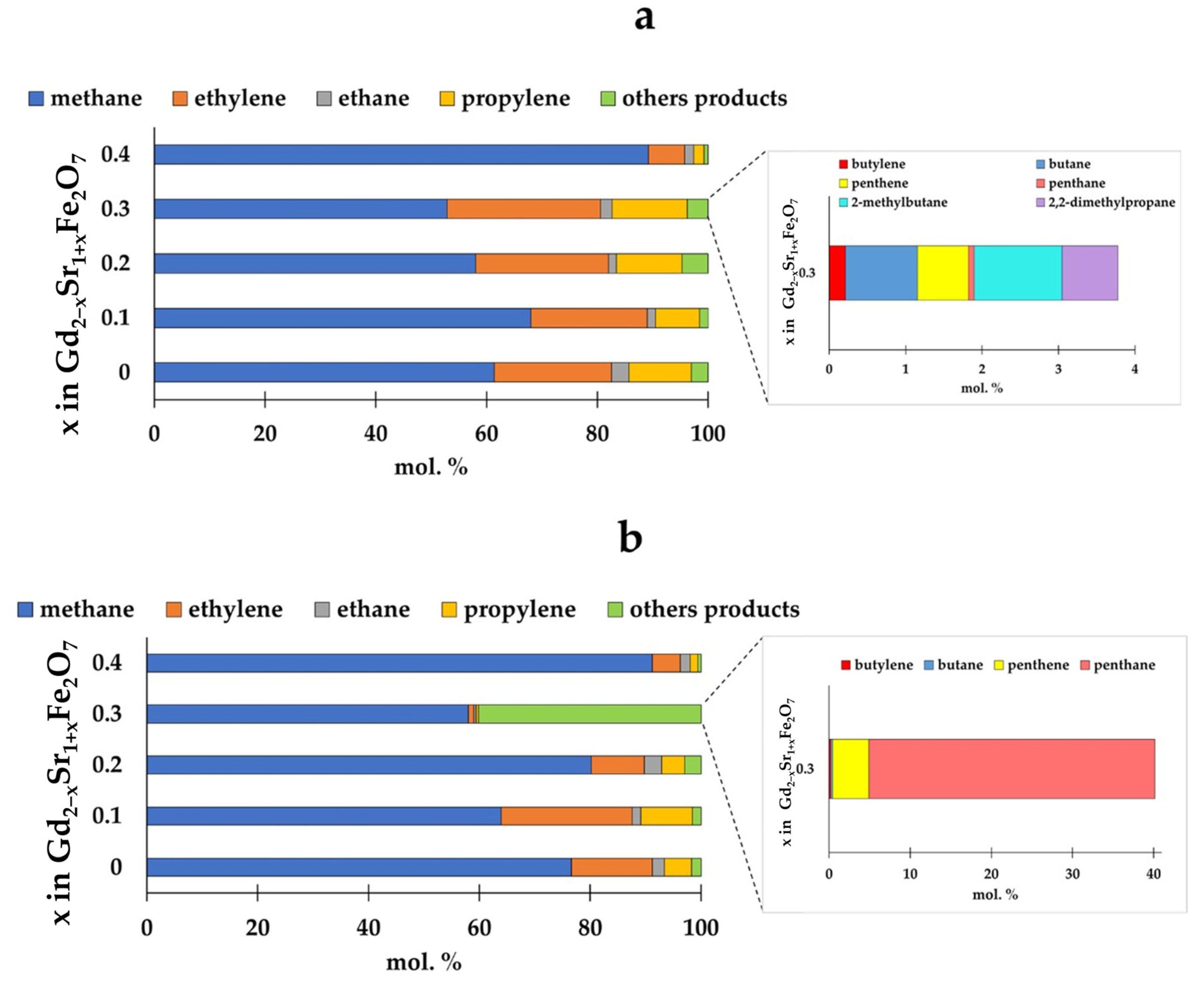
The distribution of hydrogenation products varied depending on the proportion of strontium in the catalyst structure and the composition of the reaction mixture (Figure 8).
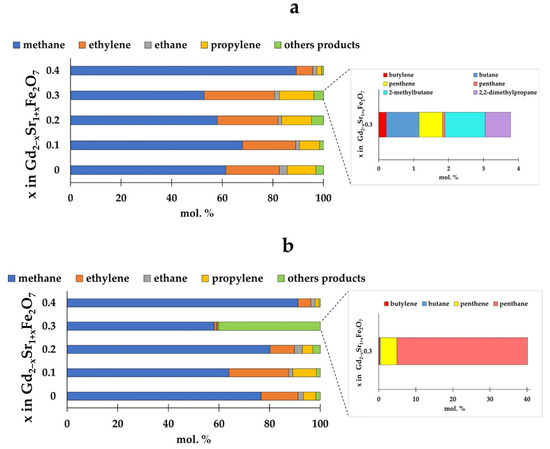
Figure 8.
Distribution of hydrogenation products at CO:H2 = 1:2 (a) and CO:H2 = 1:4 (b) depending on the composition of the catalyst at T = 673 K.
For all catalysts, the proportion of methane among the reaction products was the largest. Moreover, the amount of methane formed changed slightly with an increase in the proportion of strontium in the samples up to x = 0.3. However, on the Gd1.6Sr1.4Fe2O7 sample, both at the ratio CO:H2 = 1:2 and at CO:H2 = 1:4, the proportion of methane exceeded 90%. The content of other C2–C5 paraffins for all samples, except for Gd1.7Sr1.3Fe2O7, was small (1–4%) and changed little depending on the initial composition of the reaction mixture. For Gd1.6Sr1.4Fe2O7 and Gd2SrFe2O7 samples, varying the ratio of CO and H2 had almost no effect on the distribution of reaction products. As for C2–C4 olefins, when the reaction was carried out at a ratio of CO:H2 = 1:2, the introduction of strontium up to x = 0.3 in the amount of ethylene, propylene, and butylene changed little. The anomalous behavior of the Gd1.7Sr1.3Fe2O7 sample should be noted: at the ratio CO:H2 = 1:2, the amounts of olefins were maximum, and when the reaction was carried out in an excess of hydrogen (CO:H2 = 1:4), a significant amount of pentane was observed, comparable to the amount of methane.
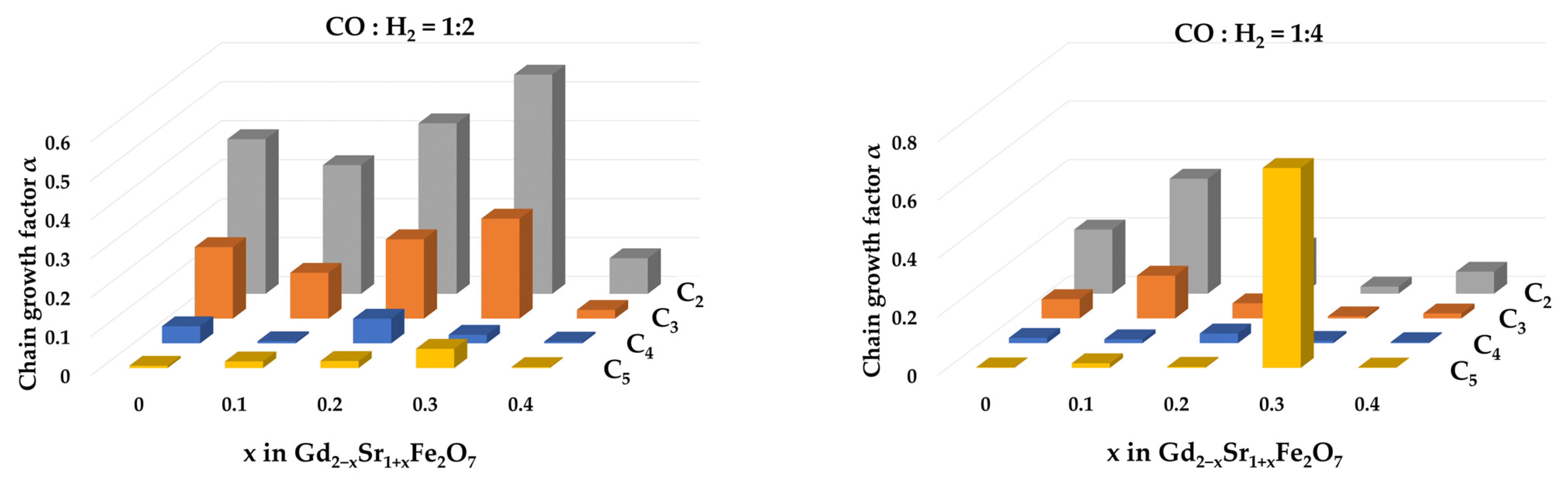
The dependence of the hydrocarbon chain growth factor αi on the number of carbon atoms (i) in the chain of intermediate products is shown in Figure 9.
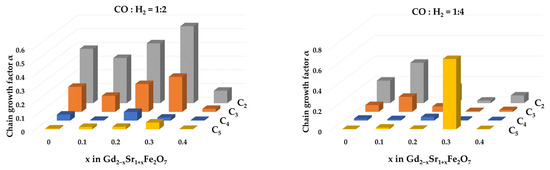
Figure 9.
Chain growth factors αi at 673 K for i = 2, 3, 4, 5 over Gd2−xSr1+xFe2O7 catalysts.
Regardless of the catalyst composition, the highest values were for α1, i.e., the formation of hydrocarbons with two carbon atoms was most likely on the studied systems at CO:H2 = 1:2, and on the Gd1.7Sr1.3Fe2O7 catalyst, this probability was maximum and reached 50%. An increase in the hydrogen content in the reaction mixture to CO:H2 = 1:4 led to the fact that for the same sample the probability of formation of C5 hydrocarbons became the maximum. The calculated values of the chain growth coefficients agreed with the values of selectivity for olefins.
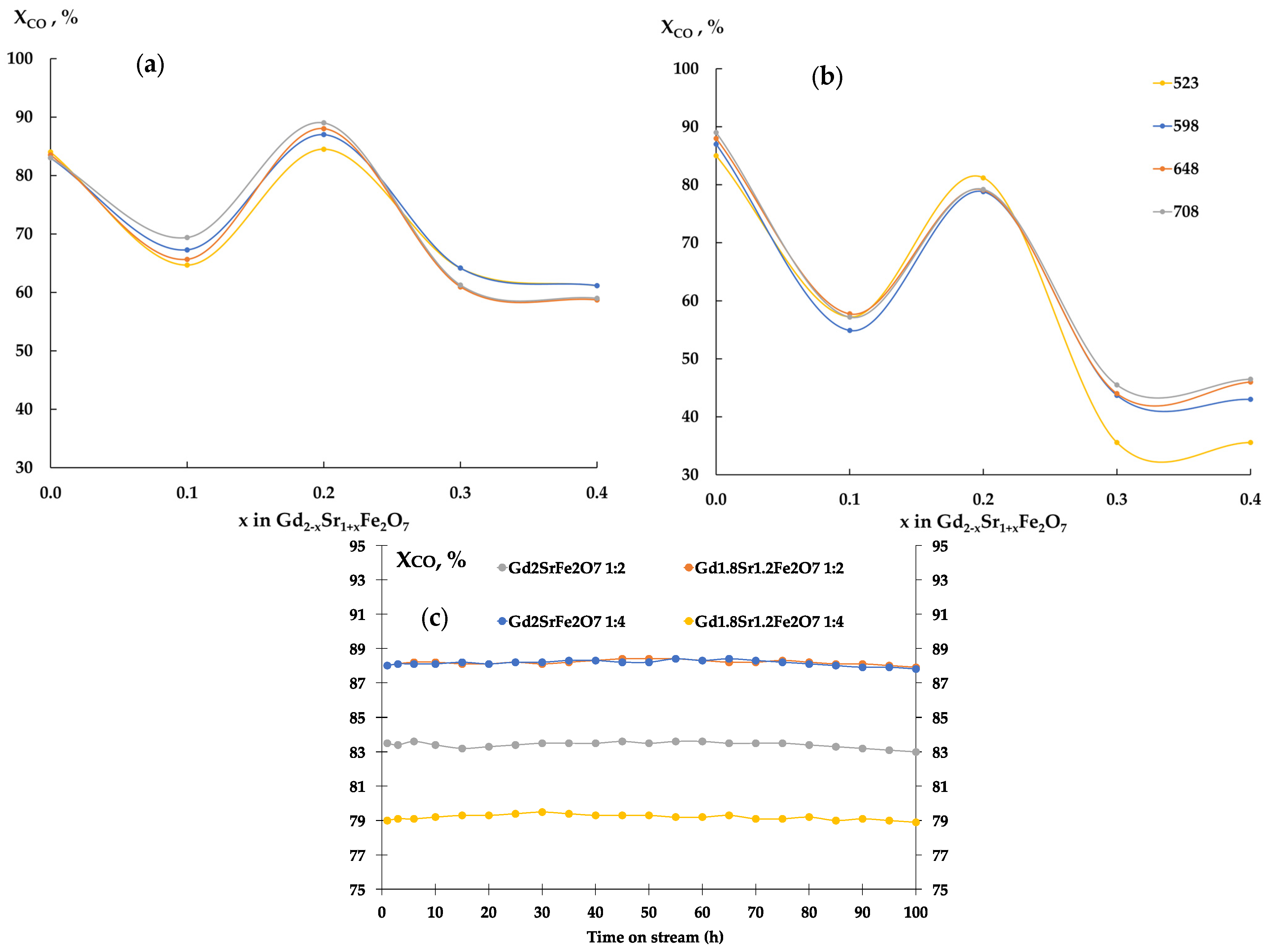
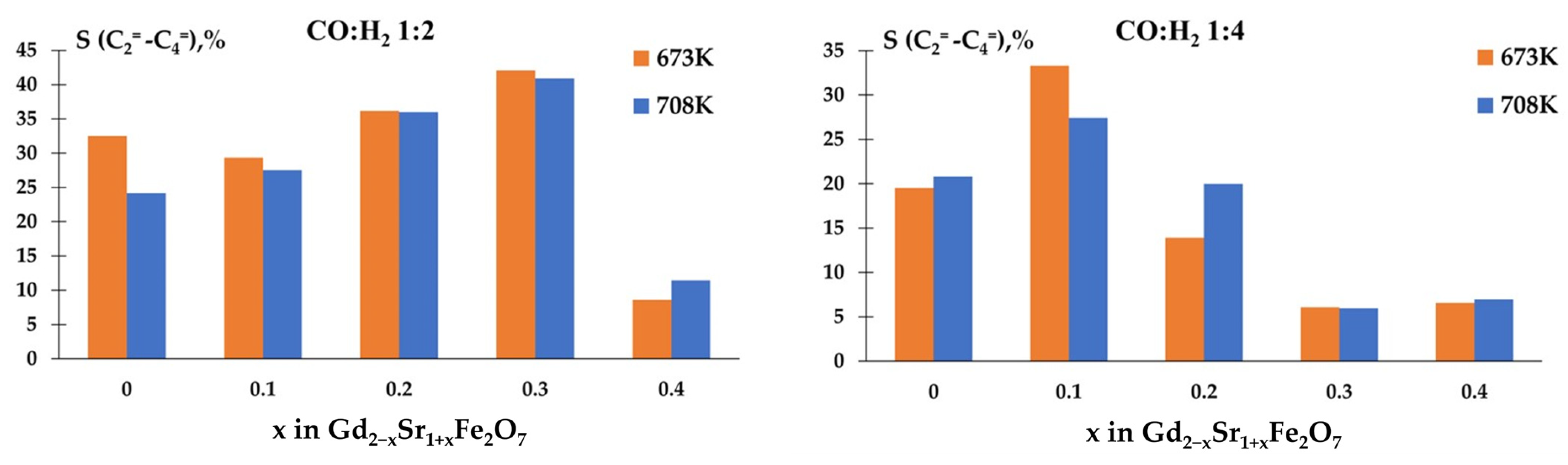
The determination of selectivity for olefins showed that the studied samples had high values of S(CnH2n) when carrying out the reaction in the ratio CO:H2 = 1:2 (Figure 10). Moreover, the non-isovalent substitution of gadolinium for strontium up to x = 0.3 contributed to the growth of S And when the reaction was carried out in an excess of hydrogen, on samples with an amount of strontium with x > 0.2, the selectivity for olefins significantly decreased.
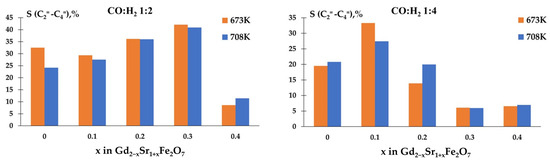
Figure 10.
Temperature dependences of selectivity for olefins.
Probably, with an excess of hydrogen in the reaction mixture, there is almost complete hydrogenation of the surface-active carbon, which arises as a result of dissociative adsorption of CO, occurs [34,44,45,46]. A decrease in the H2 content leads to a decrease in the amount of atomic hydrogen on the catalyst surface and the appearance of CHx radicals and their interaction with the formation of unsaturated hydrocarbons.
As mentioned above, the specific catalytic activity increased in the series:
and the highest selectivity for olefins was obtained on the last three. The low activity of Gd1.6Sr1.4Fe2O7 can be due to a number of factors. This sample had the smallest crystallite size and specific surface area, which was primarily due to prolonged annealing during synthesis. Also, unlike all other samples, the Fe+3 atoms in Gd1.6Sr1.4Fe2O7 are found only in two different environments.
Gd1.6Sr1.4Fe2O7 < Gd1.9Sr1.1Fe2O7 < Gd2SrFe2O7 ≈ Gd1.8Sr1.2Fe2O7 ≈ Gd1.7Sr1.3Fe2O7,
According to Mössbauer spectroscopy data, in Gd2−xSr1+xFe2O7, iron is in the heterovalent state (Fe+3, Fe4+), and the amount of Fe4+ increases with an increase in the proportion of strontium in the samples. The number of electron-withdrawing centers and total acidity decreased in the same sequence. With an increase in the content of Sr2+ in the perovskite phase, the basicity of the surface, and the rate of diffusion of oxygen into the volume, increase due to an increase in the amount of rapidly exchanging oxygen [47,48]. This promotes the dissociative chemisorption of CO [31] and the formation of active carbon (graphite deposited directly on the catalyst surface) and its further interaction with atomic hydrogen to form CHx radicals. Further recombination of CHx into olefins becomes more likely, but polymerization into longer hydrocarbons was not observed. The presence of carbon on the surface was confirmed by EDX data. The preservation of catalytic characteristics for a long time and with an increase in temperature makes it possible to consider this carbon as active. The change in surface acidity and the absence of Fe4+ in the samples after catalysis indicate the formation of active sites under the action of the reaction medium. In accordance with the scheme presented in [11], in the case of a strontium-containing catalyst, the intermediate compound Ln2O2CO3 is formed, which interacts with carbon particles or atomic hydrogen. Surface carbonates are formed on Gd atoms (A-site of the perovskite structure), while atomic hydrogen and CHx radicals are formed on Fe atoms (B-site) [35,49]. Differences in the catalytic activity of ferrite samples can be associated both with the amount of weakly bound (superstoichiometric) oxygen, which is formed in cation-deficient lattices, and with different diffusion rates of weakly bound hydrogen (HI) over the catalyst surface (spillover effect). An increase in the number of perovskite layers in complex oxides led [50] to a decrease in the activation energy for the desorption of weakly bound hydrogen; the Meδ+—H2 bond weakens, and therefore the ability of hydrogen to migrate over the surface and penetrate the volume increases. But, since samples with low acidity had the highest activity and selectivity, it can be assumed that an increase in surface basicity with increasing Sr2+ content in the perovskite phase, on the one hand, is favorable for the dissociative chemisorption of carbon monoxide, and, on the other hand, hinders the formation of atomic hydrogen. As a result, under conditions of hydrogen deficiency, the formation of unsaturated hydrocarbons occurs.
3. Materials and Methods
3.1. Catalyst Preparation
The synthesis of Gd2−xSr1+xFe2O7 (x = 0; 0.1; 0.2; 0.3; 0.4) complex oxides was performed using the citrate–nitrate sol–gel technique [32,51]. Stoichiometric amounts of Sr(NO3)2 (99.5%, Vecton, St. Petersburg, Russia), Gd(NO3)3·6H2O (99.9%, Vecton, St. Petersburg, Russia), Fe(NO3)3·9H2O (98%, Vecton, St. Petersburg, Russia) nitrates were dissolved in a flask with distilled water, then placed on a magnetic stirrer with a thermostat. Citric acid was added to the obtained solution of salts with constant stirring. After complete dissolution of the citric acid, ammonia solution was added to establish pH ~6. Then the solution was evaporated at a temperature of ~120 °C until the self-ignition of the resulting gel and the formation of black powder. Then, the temperature was raised to 450 °C and the resulting powder was calcined for 2 h.
The obtained Gd2−xSr1+xFe2O7 powders were ground in an agate mortar, pressed into tablets, and calcined in an oven at the following temperature regimes:
- -
- Gd2SrFe2O7 and Gd1.9Sr1.1Fe2O7—at 1523 K for 20 min;
- -
- Gd1.7Sr1.3Fe2O7 and Gd1.8Sr1.2Fe2O7—at 1523 K for 1 h;
- -
- Gd1.6Sr1.4Fe2O7—at 1523 K for 3 h.
3.2. Characterization
Powder X-ray diffraction (XRD) was used to determine the phase composition and structural data of the obtained compounds. The analysis was carried out using a Rigaku MiniFlex II diffractometer (Rigaku Corporation, Tokyo, Japan) with CuKα radiation. Diffraction patterns of all investigated compounds were obtained under the same conditions sufficient to detect all characteristic reflections: the range of angles was 2θ = 10–60°, the scanning speed was 5°/min. The obtained diffraction patterns were analyzed by comparing the present reflections with the data of the ICDD-PDF2 database; the data obtained allowed us to determine the phase composition of the studied compounds. Crystallite sizes were estimated using the Scherrer equation:
where λ is the wavelength, CuKα = 1.5406 Å, θ is the Bragg angle, and β is the width of the reflection at half height.
The morphology of the samples was studied using a ZeissMerlin scanning electron microscope (Carl Zeiss AG, Oberkochen, Germany) with an accelerating voltage of 10 kV, which operates in a low vacuum and does not require sputtering for nonconductive samples. A Carl Zeiss Supra 40VP scanning electron microscope (Carl Zeiss AG, Oberkochen, Germany) at 20 kV was also used.
The state of iron atoms was determined by Mössbauer spectroscopy on a WISSEL spectrometer (WissEL—Wissenschaftliche Elektronik GmbH, Starnberg, Germany). Isotope 57Co in the Rh matrix with an activity of 10 mKu was used as a radioactive source and all measurements were performed under standard conditions in the absorption geometry. An α-Fe foil was used to calibrate the velocity scale and the zero position of the chemical shift.
The specific surface area was determined by the method of low-temperature nitrogen adsorption at T = 77 K on a Nova 4200e (Quantachrome) and QuadrasorbSI devices (Quantachrome Instruments, Boynton Beach, FL, USA); the samples were preliminarily degassed at 300 °C for 5 h. The obtained adsorption–desorption isotherms were used to estimate the specific surface area of the samples by the BET method.
The acid–base properties of the catalysts were determined using UV spectroscopy of pyridine adsorption. An SF-103 single-beam scanning spectrophotometer (U-Therm International (H.K.) Limited, Hong Kong) was used to measure concentrations in liquids. The pyridine adsorption spectra in the ultraviolet region of a blank pyridine solution in octane and solutions of adsorption systems with supports and catalyst were recorded at room temperature for 60 min. There were no changes in absorption maxima at 253 nm (analytical absorption band) with pyridine concentration. This was performed using a calibration curve plotted against the optical density of solution D and pyridine concentration. Equation (3) was used to compute Gibbsian adsorption (G, mol/g):
where V (solution volume) is 10 mL, m (sample mass) is 0.1 g, D0 and Dt are equal to the optical density of pyridine at maximum absorption prior to and during the adsorption process, l (cuvette length) is 1 cm, and is the molar absorption coefficient (extinction, ).
From the kinetic dependences of pyridine adsorption, the rates of this process, which characterize the strength of acid sites, were determined. The rate of fast adsorption W1 (stronger centers of fast adsorption) was determined from the tangent of the slope of the tangent to the initial part of the kinetic dependence. The rate of slow adsorption W2 on centers of the second type (weak centers of slow adsorption) was determined from the slope of the following sections.
3.3. Catalytic Activity Tests
Catalyst performance of the samples in the carbon monoxide hydrogenation reaction was evaluated in a quartz tubular microreactor containing 0.1 g of the sample diluted in 0.5 g of quartz to avoid the formation of hot spots in the catalytic bed alongside increasing the volume. Activity tests were carried out in the temperature range 523–708 K under atmospheric pressure with the feed composition CO:H2 = 1:2 and CO:H2 = 1:4 and total flow rate of 1.5 L·h−1, which corresponds to GHSV of 8700 h−1. Gaseous effluent was analyzed online using a gas chromatograph instrument (Crystal 5000.2 (Chromatec Instruments, Yoshkar-Ola, Mari El, Russia), a column of stainless steel filled with Porapack Q, argon as a carrier gas) fitted with thermal conductivity and flame ionization detectors.
The catalytic characteristics were calculated using the following equations:
where nint и nout is the initial amount of substance and mole yield of products, ∑ni is the mole yield of hydrocarbon reaction products (mole), Ri is the rate of formation of the i-th reaction product per 1 g catalyst (mol/h·g), ΣRi is the sum of yields of the i-th reaction products per 1 g catalyst (mol/h·g), ω is volumetric rate of the reaction mixture (L/h), V is the volume of the chromatograph loop (0.153 × 10−3 L), and m is the mass of the catalyst, g.
The carbon-chain growth factor αi was calculated using Equation (7) [52]:
where is carbon-chain growth factor for the intermediate with the number of carbon atoms I, and Yk is the yield of the component with the number of carbon atoms k.
The coefficient α1 is the probability of the addition of CO to the intermediate containing one carbon atom to form an intermediate with two carbon atoms, α2 is the probability of the next step of the addition of CO to the intermediate with two carbon atoms, etc.
4. Conclusions
In conclusions, complex perovskite-type oxides Gd2−xSr1+xFe2O7 (x = 0; 0.1; 0.2; 0.3; 0.4), prepared by the sol–gel method were studied as catalysts for hydrogenation of carbon monoxide. The effect of gadolinium partial nonisovalent substitution by strontium on the physicochemical and catalytic properties in the process was investigated. It was found that the iron atoms in the samples were in the heterovalent state Fe4+/Fe3++, which was compensated by oxygen vacancies. It has been suggested that Cd3+ and Fe3+ in the composition of Gd-O-Me are active centers, while the presence of Sn2+ somewhat complicates the process and shifts it towards the formation of olefins. The formation of catalytically active centers under the action of the reaction medium was found. It was also found that varying the composition of Gd2−xSr1+xFe2O7 complex oxides led to changes in the oxygen–metal bond energy in Gd-O-Me, in the ratio of metals in different oxidation states, and in the acid–base surface properties, which was reflected in the adsorption and catalytic characteristics of the complex oxides.
Author Contributions
Conceptualization and methodology, I.A.Z., T.F.S. and A.O.T.; investigation, E.M.B., T.A.K., E.B.M. and L.V.Y.; data curation, T.A.K.; writing—original draft, E.M.B., L.V.Y. and T.A.K.; writing—review and editing, T.A.K., T.F.S. and I.A.Z.; supervision, A.G.C. and A.O.T.; project administration, A.G.C. All authors have read and agreed to the published version of the manuscript.
Funding
This publication was supported by the RUDN University Scientific Projects Grant System, project No 021521-2-174 and the scholarship of the President of the Russia (No CП-686.2021.1).
Data Availability Statement
The original data are available from T.F.S., E.M.B. and L.V.Y.
Acknowledgments
The authors are grateful to Saint Petersburg State University Research Park: Research Center for X-ray Diffraction Studies, Center of Thermal Analysis and Calorimetry, Interdisciplinary Centre for Nanotechnology, Centre for Diagnostics of Functional Materials for Medicine, Pharmacology and Nanoelectronics.
Conflicts of Interest
The authors declare no conflict of interest. The funders played no role in the design of the study; in the collection, analysis or interpretation of data; in the writing of the manuscript or in the decision to publish the results.
References
- Peña, M.A.; Fierro, J.L.G. Chemical Structures and Performance of Perovskite Oxides. Chem. Rev. 2001, 101, 1981–2018. [Google Scholar] [CrossRef] [PubMed]
- Royer, S.; Duprez, D.; Can, F.; Courtois, X.; Batiot-Dupeyrat, C.; Laassiri, S.; Alamdari, H. Perovskites as Substitutes of Noble Metals for Heterogeneous Catalysis: Dream or Reality. Chem. Rev. 2014, 114, 10292–10368. [Google Scholar] [CrossRef] [PubMed]
- Giordano, L.; Akkiraju, K.; Jacobs, R.; Vivona, V.; Morgan, D.; Shao-Horn, Y. Electronic structure-based descriptors for oxide properties and functions. Acc. Chem. Res. 2022, 55, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Rao, R.R.; Giordano, L.; Katayama, Y.; Yu, Y.; Shao-Horn, Y. Perovskites in catalysis and electrocatalysis. Science 2017, 358, 751–756. [Google Scholar] [CrossRef]
- Xu, X.; Zhong, Y.; Shao, Z. Double Perovskites in Catalysis, Electrocatalysis, and Photo(electro)catalysis. Trends Chem. 2019, 1, 410–424. [Google Scholar] [CrossRef]
- Zhao, K.; He, F.; Huang, Z.; Wei, G.; Zheng, A.; Li, H.; Zhao, Z. Perovskite-type oxides LaFe1−xCoxO3 for chemical looping steam methane reforming to syngas and hydrogen co-production. Appl. Energy 2016, 168, 193–203. [Google Scholar] [CrossRef]
- Salker, A.V.; Gurav, S.M. Electronic and catalytic studies on Co1−xCuxMn2O4 for CO oxidation. J. Mater. Sci. 2000, 35, 4713–4719. [Google Scholar] [CrossRef]
- de Caprariis, B.; de Filippisa, P.; Palma, V.; Petrullo, A.; Ricca, A.; Ruocco, C.; Scarsella, M. Rh, Ru and Pt ternary perovskites type oxides BaZr(1−x)MexO3 for methane dry reforming. Appl. Catal. A Gen. 2016, 517, 47–55. [Google Scholar] [CrossRef]
- Amin, A.S.; Yaw, T.C. Thermodynamic equilibrium analysis of combined carbon dioxide reforming with partial oxidation of methane to syngas. Int. J. Hydrogen Energy 2007, 32, 1789–1798. [Google Scholar] [CrossRef]
- Song, X.; Dong, X.; Yin, S.; Wang, M.; Li, M.; Wang, H. Effects of Fe partial substitution of La2NiO4/LaNiO3 catalyst precursors prepared by wet impregnation method for the dry reforming of methane. Appl. Catal. A Gen. 2016, 526, 132–138. [Google Scholar] [CrossRef]
- Aksenova, T.V.; Mysik, D.K.; Cherepanov, V.A. Crystal Structure and Properties of Gd1−xSrxCo1−yFeyO3−δ Oxides as Promising Materials for Catalytic and SOFC Application. Catalysts 2022, 12, 1344. [Google Scholar] [CrossRef]
- Yang, E.; Noh, Y.; Ramesh, S.; Lim, S.S.; Moon, D.J. The effect of promoters in La0.9M0.1Ni0.5Fe0.5O3 (M = Sr, Ca) perovskite catalysts on dry reforming of methane. Fuel Process. Technol. 2015, 134, 404–413. [Google Scholar] [CrossRef]
- Zhang, Z.; Ou, Z.; Qin, C.; Ran, J.; Wu, C. Roles of alkali/alkaline earth metals in steam reforming of biomass tar for hydrogen production over perovskite supported Ni catalysts. Fuel 2019, 257, 116032. [Google Scholar] [CrossRef]
- Wei, T.; Jia, L.; Luo, J.; Chi, B.; Pu, J.; Li, J. CO2 dry reforming of CH4 with Sr and Ni co-doped LaCrO3 perovskite catalysts. Appl. Surf. Sci. 2020, 506, 144699. [Google Scholar] [CrossRef]
- Ao, M.; Pham, G.H.; Sage, V.; Pareek, V. Structure and activity of strontium substituted LaCoO3 perovskite catalysts for syngas conversion. J. Mol. Catal. A Chem. 2016, 416, 96–104. [Google Scholar] [CrossRef]
- Escalona, N.; Fuentealba, S.; Pecchi, G. Fischer–Tropsch synthesis over LaFe1−xCoxO3 perovskites from a simulated biosyngas feed. Appl. Catal. A Gen. 2010, 381, 253–260. [Google Scholar] [CrossRef]
- Morales, M.; Segarra, M. Steam reforming and oxidative steam reforming of ethanol over La0.6Sr0.4CoO3− δ perovskite as catalyst precursor for hydrogen production. Appl. Catal. A Gen. 2015, 502, 305–311. [Google Scholar] [CrossRef]
- Gholami, Z.; Gholami, F.; Tišler, Z.; Hubáček, J.; Tomas, M.; Bǎciak, M.; Vakili, M. Production of Light Olefins via Fischer-Tropsch Process Using Iron-Based Catalysts: A Review. Catalysts 2022, 12, 174. [Google Scholar] [CrossRef]
- Lin, Q.; Cheng, M.; Zhang, K.; Li, W.; Wu, P.; Chang, H.; Lv, Y.; Men, Z. Development of an Iron-Based Fischer–Tropsch Catalyst with High Attrition Resistance and Stability for Industrial Application. Catalysts 2021, 11, 908. [Google Scholar] [CrossRef]
- Chang, H.; Lin, Q.; Cheng, M.; Zhang, K.; Feng, B.; Chai, J.; Lv, Y.; Men, Z. Effects of Potassium Loading over Iron–Silica Interaction, Phase Evolution and Catalytic Behavior of Precipitated Iron-Based Catalysts for Fischer-Tropsch Synthesis. Catalysts 2022, 12, 916. [Google Scholar] [CrossRef]
- Zheng, Y.; Xu, C.; Zhang, X.; Wu, Q.; Liu, J. Synergistic Effect of Alkali Na and K Promoter on Fe-Co-Cu-Al Catalysts for CO2 Hydrogenation to Light Hydrocarbons. Catalysts 2021, 11, 735. [Google Scholar] [CrossRef]
- Gong, N.; Wu, Y.; Ma, Q.; Tan, Y. A Simple Strategy Stabilizing for a CuFe/SiO2 Catalyst and Boosting Higher Alcohols’ Synthesis from Syngas. Catalysts 2023, 13, 237. [Google Scholar] [CrossRef]
- Pan, Y.; Ding, X.; Zhang, C.; Zhu, M.; Yang, Z.; Han, Y.-F. Effects of Different Reductive Agents on Zn-Promoted Iron Oxide Phases in the CO2–Fischer–Tropsch to Linear-Olefins. Catalysts 2023, 13, 594. [Google Scholar] [CrossRef]
- Fatih, Y.; Burgun, U.; Sarioglan, A.; Atakül, H. Effect of sodium incorporation into Fe-Zn catalyst for Fischer- Tropsch synthesis to light olefins. Mol. Catal. 2023, 535, 112866. [Google Scholar] [CrossRef]
- Mierczy’nski, P.; Dawid, B.; Mierczynska-Vasilev, A.; Maniukiewicz, W.; Witońska, I.; Vasilev, K.; Szynkowska-Jóźwik, M.I. Novel bimetallic 1%M-Fe/Al2O3-Cr2O3 (2:1) (M = Ru, Au, Pt, Pd) catalysts for Fischer-Tropsch synthesis. Catal. Commun. 2022, 172, 106559. [Google Scholar] [CrossRef]
- Wang, C.; Zhu, H.; Zhang, L.; Huang, Z.; Chen, J. Tuning Fischer-Tropsch synthesis product distribution toward light olefins over nitrided Fe-Mn bimetallic catalysts. Fuel 2023, 343, 127977. [Google Scholar] [CrossRef]
- Hu, R.; Wang, T.; Wang, Y.; Zhu, Y.; Xie, L.; Xing, E.; Wu, Y.; Da, Z. Nano-Hollow Zeolite-Encapsulated Highly Dispersed Ultra-Fine Fe Nanoparticles as Fischer–Tropsch Catalyst for Syngas-to-Olefins. Catalysts 2023, 13, 948. [Google Scholar] [CrossRef]
- Pawelec, B.; Guil-López, R.; Mota, N.; Fierro, J.L.G.; Navarro Yerga, R.M. Catalysts for the Conversion of CO2 to Low Molecular Weight Olefins—A Review. Materials 2021, 14, 6952. [Google Scholar] [CrossRef]
- Li, Y.; He, Y.; Fujihara, K.; Wang, C.; Sun, X.; Gao, W.; Guo, X.; Yasuda, S.; Yang, G.; Tsubaki, N. A Core-Shell Structured Na/Fe@Co Bimetallic Catalyst for Light-Hydrocarbon Synthesis from CO2 Hydrogenation. Catalysts 2023, 13, 1090. [Google Scholar] [CrossRef]
- Chernyak, S.A.; Corda, M.; Dath, J.-P.; Ordomsky, V.V.; Khodakov, A.Y. Light olefin synthesis from a diversity of renewable and fossil feedstocks: State-of the-art and outlook. Chem. Soc. Rev. 2022, 51, 7994–8044. [Google Scholar] [CrossRef]
- Sheshko, T.F.; Markova, E.B.; Sharaeva, A.A.; Kryuchkova, T.A.; Zvereva, I.A.; Chislova, I.V.; Yafarova, L.V. Carbon Monoxide Hydrogenation over Gd(Fe/Mn)O3 Perovskite-Type Catalysts. Pet. Chem. 2019, 59, 1307–1313. [Google Scholar] [CrossRef]
- Yafarova, L.V.; Chislova, I.V.; Zvereva, I.A.; Kryuchkova, T.A.; Kost, V.V.; Sheshko, T.F. Sol–gel synthesis and investigation of catalysts on the basis of perovskite-type oxides GdMO3 (M = Fe, Co). J. Sol-Gel Sci. Technol. 2019, 92, 264–272. [Google Scholar] [CrossRef]
- Sheshko, T.F.; Kryuchkova, T.A.; Serov, Y.M.; Chislova, I.V.; Zvereva, I.A. New Mixed Perovskite-Type Gd2−xSr1+xFe2O7 Catalysts for Dry Reforming of Methane, and Light Olefins Production. Catal. Ind. 2017, 9, 162–167. [Google Scholar] [CrossRef]
- Sheshko, T.F.; Serov, Y.M.; Kryuchkova, T.A.; Khairullina, I.A.; Chislova, I.V.; Zvereva, I.A. Interaction between carbon oxides, hydrogen and Fe2O3 and An+1FenO3n +1 (A = Gd, Sr, n = 1, 2, …, ∞). Russ. J. Phys. Chem. 2016, 90, 1137–1142. [Google Scholar] [CrossRef]
- Ivanov, D.V.; Sadovskaya, E.M.; Pinaeva, L.G.; Isupova, L.A. Influence of Oxygen Mobility on Catalytic Activity of La-Sr-Mn-O Composites in the Reaction of High Temperature N2O Decomposition. J. Catal. 2009, 267, 5–13. [Google Scholar] [CrossRef]
- Sugunan, S.; Meera, V. Acid-base properties and catalytic activity of ABO3 (perovskite-type) oxides consisting of rare earth and 3d transition metals. React. Kinet. Catal. 1997, 62, 327–332. Available online: https://link.springer.com/article/10.1007/BF02475471 (accessed on 1 July 2023). [CrossRef]
- Polo-Garzon, F.; Wu, Z. Acid–base catalysis over perovskites: A review. J. Mater. Chem. A 2018, 6, 2877–2894. Available online: https://pubs.rsc.org/en/content/articlelanding/2018/ta/c7ta10591f (accessed on 1 July 2023). [CrossRef]
- Sharma, I.B.; Singh, D.; Magotra, S.K. Effect of substitution of magnetic rare earths for La on the structure, electric transport and magnetic properties of La2SrFe2O7. J. Alloys Compd. 1998, 269, 13–16. [Google Scholar] [CrossRef]
- Shilova, A.; Chislova, I.; Panchuk, V.; Semenov, V.; Zvereva, I. Evolution of iron electronic state in the solid solutions Gd2−xSr1+xFe2O7−δ. Solid State Phenom. 2013, 194, 116–119. [Google Scholar] [CrossRef]
- Sheshko, T.F.; Serov, Y.M.; Dement’eva, M.V.; Shul’ga, A.; Chislova, I.V.; Zvereva, I.A. Catalytic Hydrogenation of Carbon Monoxide over Nanostructured Perovskite-Like Gadolinium and Strontium Ferrites. Russ. J. Phys. Chem. A 2016, 90, 926–931. [Google Scholar] [CrossRef]
- Pylinina, A.I.; Povarova, E.I.; Mikhalenko, I.I.; Yagodovskaya, T.V. Effect of plasma-chemical and thermal treatment in oxygen on the activity of Na3ZrM(PO4)3 phosphates (M = Zn, Co, Cu) in the transformation of butanol-2. Russ. J. Phys. Chem. 2013, 87, 929–934. [Google Scholar] [CrossRef]
- Liu, B.; Li, W.; Zheng, J.; Lin, Q.; Zhang, X.; Zhang, J.; Jiang, F.; Xu, Y.; Liu, X. CO2 formation mechanism in Fischer–Tropsch synthesis over iron-based catalysts: A combined experimental and theoretical study. Catal. Sci. Technol. 2018, 8, 5288. [Google Scholar] [CrossRef]
- Ma, Z.; Wang, X.; Ma, X.; Tan, M.; Yang, G.; Tan, Y. Catalytic roles of acid property in different morphologies of HZSM-5 zeolites for syngas-to-aromatics conversion over ZnCrOx/H-ZSM-5 catalysts. Microporous Mesoporous Mater. 2022, 349, 112420. [Google Scholar] [CrossRef]
- Dry, M.E. Practical and theoretical aspects of the catalytic Fischer-Tropsch process. Appl. Catal. A Gen. 1996, 138, 319–344. [Google Scholar] [CrossRef]
- Ding, M.; Yang, Y.; Wu, B.; Li, Y.; Wang, T.; Ma, L. Study on reduction and carburization behaviors of iron phases for iron-based Fischer-Tropsch synthesis catalyst. Appl. Energy 2015, 160, 982–989. [Google Scholar] [CrossRef]
- Calderone, V.R.; Shiju, N.R.; Ferr’e, D.C.; Rothenberg, G. Bimetallic catalysts for the Fischer–Tropsch reaction. Green Chem. 2011, 13, 1950–1959. [Google Scholar] [CrossRef]
- Ivanov, D.V.; Pinaeva, L.G.; Isupova, L.A.; Nadeev, A.N.; Prosvirin, I.P.; Dovlitova, L.S. Insights into the Reactivity of La1−xSrxMnO3 (x = 0 ÷ 0.7) in High Temperature N2O Decomposition. Catal. Lett. 2011, 141, 322–331. [Google Scholar] [CrossRef]
- Sheshko, T.F.; Kryuchkova, T.A.; Yafarova, L.V.; Borodina, E.M.; Serov, Y.M.; Zvereva, I.A.; Cherednichenko, A.G. Gd-Co-Fe perovskite mixed oxides as catalysts for dry reforming of methane. Sustain. Chem. Pharm. 2022, 30, 100897. [Google Scholar] [CrossRef]
- Kryuchkova, T.A.; Kost’, V.V.; Sheshko, T.F.; Chislova, I.V.; Yafarova, L.V.; Zvereva, I.A. Effect of Cobalt in GdFeO3 Catalyst Systems on Their Activity in the Dry Reforming of Methane to Synthesis Gas. Pet. Chem. 2020, 60, 609–615. [Google Scholar] [CrossRef]
- Wu, Y.; Li, L.; Chu, B.; Yi, Y.; Qin, Z.; Fan, M.; Qin, Q.; He, H.; Zhang, L.; Dong, L.; et al. Catalytic reduction of NO by CO over B-site partially substituted LaM0.25Co0.75O3 (M = Cu, Mn, Fe) perovskite oxide catalysts: The correlation between physicochemical properties and catalytic performance. Appl. Catal. A Gen. 2018, 568, 43–53. [Google Scholar] [CrossRef]
- Chislova, I.V.; Matveeva, A.A.; Volkova, A.V.; Zvereva, I.A. Sol-gel synthesis of nanostructured perovskite-like gadolinium ferrites. Glass Phys. Chem. 2011, 37, 653–660. [Google Scholar] [CrossRef]
- Osman, M.E.; Maximov, V.V.; Dorokhov, V.S.; Mukhin, V.M.; Sheshko, T.F.; Kooyman, P.J.; Kogan, V.M. Carbon-Supported KCoMoS2 for Alcohol Synthesis from Synthesis Gas. Catalysts 2021, 11, 1321. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).