


Salophen-Type Schiff Bases Functionalized with Pyridinium Halide Units as Metal-Free Catalysts for Synthesis of Cyclic Carbonates from Carbon Dioxide and Terminal Epoxides


Abstract

1. Introduction
2. Results and Discussion
3. Materials and Methods
Catalytic Examination—General Procedure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Dziejarski, B.; Krzyżyńska, R.; Andersson, K. Current Status of Carbon Capture, Utilization, and Storage Technologies in the Global Economy: A Survey of Technical Assessment. Fuel 2023, 342, 127776. [Google Scholar] [CrossRef]
- Ekemezie, I.O.; Digitemie, W.N. Carbon Capture and Utilization (CCU): A Review of Emerging Applications and Challenges. Eng. Sci. Technol. J. 2024, 5, 949–961. [Google Scholar] [CrossRef]
- Lin, Q.; Zhang, X.; Wang, T.; Zheng, C.; Gao, X. Technical Perspective of Carbon Capture, Utilization, and Storage. Engineering 2022, 14, 27–32. [Google Scholar] [CrossRef]
- Kamkeng, A.D.N.; Wang, M.; Hu, J.; Du, W.; Qian, F. Transformation Technologies for CO2 Utilisation: Current Status, Challenges and Future Prospects. Chem. Eng. J. 2021, 409, 128138. [Google Scholar] [CrossRef]
- Gabrielli, P.; Gazzani, M.; Mazzotti, M. The Role of Carbon Capture and Utilization, Carbon Capture and Storage, and Biomass to Enable a Net-Zero-CO2 Emissions Chemical Industry. Ind. Eng. Chem. Res. 2020, 59, 7033–7045. [Google Scholar] [CrossRef]
- Zhanga, Z.; Pan, S.Y.; Li, H.; Cai, J.; Olabi, A.G.; Anthony, E.J.; Manovic, V. Recent Advances in Carbon Dioxide Utilization. Renew. Sustain. Energy Rev. 2020, 125, 109799. [Google Scholar] [CrossRef]
- Chu, H.; Huang, Z.; Zhang, Z.; Yan, X.; Qiu, B.; Xu, N. Integration of Carbon Emission Reduction Policies and Technologies: Research Progress on Carbon Capture, Utilization and Storage Technologies. Sep. Purif. Technol. 2024, 343, 127153. [Google Scholar] [CrossRef]
- Osorio-Tejada, J.; Escriba-Gelonch, M.; Vertongen, R.; Bogaerts, A.; Hessel, V. CO2 Conversion to CO Via Plasma and Electrolysis: A Techno-Economic and Energy Cost Analysis. Energy Environ. Sci. 2024, 17, 5833–5853. [Google Scholar] [CrossRef]
- Ra, E.C.; Kim, K.Y.; Kim, E.H.; Lee, H.; An, K.; Lee, J.S. Recycling Carbon Dioxide through Catalytic Hydrogenation: Recent Key Developments and Perspectives. ACS Catal. 2020, 10, 11318–11345. [Google Scholar] [CrossRef]
- Meunier, N.; Chauvy, R.; Mouhoubi, S.; Thomas, D.; De Weireld, G. Alternative Production of Methanol from Industrial CO2. Renew. Energy 2020, 146, 1192–1203. [Google Scholar] [CrossRef]
- Mei, Z.; Zhou, Y.; Lv, W.; Tong, S.; Yang, X.; Chen, L.; Zhang, N. Recent Progress in Electrocatalytic Urea Synthesis under Ambient Conditions. ACS Sustain. Chem. Eng. 2022, 10, 12477–12496. [Google Scholar] [CrossRef]
- Kohli, K.; Sharma, B.K.; Panchal, C.B. Dimethyl Carbonate: Review of Synthesis Routes and Catalysts Used. Energies 2022, 15, 5133. [Google Scholar] [CrossRef]
- Lopes, E.J.C.; Ribeiro, A.P.C.; Martins, L.M.D.R.S. New Trends in the conversion of CO2 to cyclic carbonates. Catalysts 2020, 10, 479. [Google Scholar] [CrossRef]
- Pescarmona, P.P. Cyclic Carbonates Synthesised from CO2: Applications, Challenges and Recent Research Trends. Curr. Opin. Green Sustain. Chem. 2021, 29, 100457. [Google Scholar] [CrossRef]
- North, M.; Pasquale, R.; Young, C. Synthesis ff Cyclic Carbonates from Epoxides and CO2. Green Chem. 2010, 12, 1514–1539. [Google Scholar] [CrossRef]
- Alves, M.; Grignard, B.; Mereau, R.; Jerome, C.; Tassaing, T.; Detrembleur, C. Organocatalyzed Coupling of Carbon Dioxide with Epoxides for the Synthesis of Cyclic Carbonates: Catalyst Design and Mechanistic Studies. Catal. Sci. Technol. 2017, 7, 2651–2684. [Google Scholar] [CrossRef]
- Ogasawara, T.; Débart, A.; Holzapfel, M.; Novák, P.; Bruce, P.G. Rechargeable Li2O2 Electrode for Lithium Batteries. J. Am. Chem. Soc. 2006, 128, 1390–1393. [Google Scholar] [CrossRef]
- Kamphuis, A.J.; Picchioni, F.; Pescarmona, P.P. CO2-Fixation into Cyclic and Polymeric Carbonates: Principles and Applications. Green Chem. 2019, 21, 406–448. [Google Scholar] [CrossRef]
- Lanzia, M.; Kleij, A.W. Recent Advances in the Synthesis and Polymerization of New CO2-Based Cyclic Carbonates. Chin. J. Chem. 2024, 42, 430–443. [Google Scholar] [CrossRef]
- Li, H.; Wang, W.; Liu, S.; Xue, D.; Wang, J.; Liu, Y.; Huang, Q. Design and Syntheses of Functional Carbon Dioxide-Based Polycarbonates via Ternary Copolymerization. J. CO2 Util. 2024, 80, 102689. [Google Scholar] [CrossRef]
- Büttner, H.; Longwitz, L.; Steinbauer, J.; Wulf, C.; Werner, T. Recent Developments in the Synthesis of Cyclic Carbonates from Epoxides and CO2. In Chemical Transformations of Carbon Dioxide. Topics in Current Chemistry Collections; Springer: Cham, Switzerland, 2017; Volume 375, pp. 89–144. [Google Scholar]
- Calabrese, C.; Giacalone, F.; Aprile, C. Hybrid Catalysts for CO2 Conversion into Cyclic Carbonates. Catalysts 2019, 9, 325. [Google Scholar] [CrossRef]
- Guo, L.; Lamb, K.J.; North, M. Recent Developments in Organocatalysed Transformations of Epoxides and Carbon Dioxide into Cyclic Carbonates. Green Chem. 2021, 23, 77–118. [Google Scholar] [CrossRef]
- Fonseca-Lopez, D.; Ezenarro-Salcedo, D.; Zapata-Rivera, J.; Rojas, R.S.; Hurtado, J.J. Salophen-Type Organocatalysts for the Cycloaddition of CO2 and Epoxides under Solvent, Halide, and Metal-Free Conditions. ACS Omega 2024, 9, 19385–19394. [Google Scholar] [CrossRef] [PubMed]
- North, M.; Young, C. Bimetallic Aluminium(Acen)Complexes as Catalysts for the Synthesis of Cyclic Carbonates from Carbon Dioxide and Epoxides. Catal. Sci. Technol. 2011, 1, 93–99. [Google Scholar] [CrossRef]
- Wu, X.; North, M. Bimetallic Aluminium(Salphen) Complex for the Synthesis of Cyclic Carbonates from Epoxides and Carbon Dioxide. ChemSusChem 2017, 10, 74–78. [Google Scholar] [CrossRef]
- Rulev, Y.A.; Larionov, V.A.; Lokutova, A.V.; Moskalenko, M.A.; Lependina, O.L.; Maleev, V.I.; North, M.; Belokon, Y.N. Chiral Cobalt(III) Complexes as Bifunctional Brönsted Acid-Lewis Base Catalysts for the Preparation of Cyclic Organic Carbonates. ChemSusChem 2016, 9, 216–222. [Google Scholar] [CrossRef]
- Castro-Osma, J.A.; Lamb, K.J.; North, M. Cr(Salophen) Complex Catalyzed Cyclic Carbonate Synthesis at Ambient Temperature and Pressure. ACS Catal. 2016, 6, 5012–5025. [Google Scholar] [CrossRef]
- Taherimehr, M.; Decortes, A.; Al-Amsyar, S.M.; Lueangchaichaweng, W.; Whiteoak, C.J.; Escudero-Ad’an, E.C.; Kleij, A.W.; Pescarmona, P.P. A Highly Active Zn(Salphen) Catalyst for Production of Organic Carbonates in a Green CO2 Medium. Catal. Sci. Technol. 2012, 2, 2231–2237. [Google Scholar] [CrossRef]
- Hui, Z.; Wen-Zhen, Z.; Cui-Hua, L.; Jing-Ping, Q.; Xiao-Bing, L. CO2 Adducts of N-Heterocyclic Carbenes: Thermal Stability and Catalytic Activity toward the Coupling of CO2 with Epoxides. J. Org. Chem. 2008, 73, 8039–8044. [Google Scholar]
- Souleymanou, M.Y.; El-Ouahabi, F.; Masdeu-Bultó, A.M.; Godard, C. Cooperative NHC-based Catalytic System Immobilised onto Carbon Materials for the Cycloaddition of CO2 to Epoxides. ChemCatChem 2021, 13, 1706–1710. [Google Scholar] [CrossRef]
- Peng, J.; Wang, S.; Yang, H.J.; Ban, B.; Wei, Z.; Wang, L.; Lei, B. Highly Efficient Fixation of Carbon Dioxide to Cyclic Carbonates with New Multi-Hydroxyl Bis-(Quaternary Ammonium) Ionic Liquids as Metal-Free Catalysts under Mild Conditions. Fuel 2018, 224, 481–488. [Google Scholar] [CrossRef]
- Toda, Y.; Komiyama, Y.; Esaki, H.; Fukushima, K.; Suga, H. Methoxy Groups Increase Reactivity of Bifunctional Tetraarylphosphonium Salt Catalysts for Carbon Dioxide Fixation: A Mechanistic Study. J. Org. Chem. 2019, 84, 15578–15589. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zheng, D.; Zhang, J.; Fan, B.; Ma, Y.; Ren, T.; Wang, L.; Zhang, J. Protic Pyrazolium Ionic Liquids: An Efficient Catalyst for Conversion of CO2 in the Absence of Metal and Solvent. ACS Sustain. Chem. Eng. 2018, 6, 2574–2582. [Google Scholar] [CrossRef]
- Rostami, A.; Mahmoodabadi, M.; Hossein Ebrahimi, A.; Khosravi, H.; Al-Harrasi, A. An Electrostatically Enhanced Phenol as a Simple and Efficient Bifunctional Organocatalyst for Carbon Dioxide Fixation. ChemSusChem 2018, 11, 4262–4268. [Google Scholar] [CrossRef]
- Wang, B.; Cao, X.; Wang, L.; Meng, X.; Wang, Y.; Sun, W. Co(II)−N4 Catalysts for the Coupling of CO2 with Epoxides into Cyclic Carbonates: Catalytic Activity, Computational and Kinetic Studies. Inorg. Chem. 2024, 63, 9156–9163. [Google Scholar] [CrossRef] [PubMed]
- Paradiso, V.; Capaccio, V.; Lamparelli, D.H.; Capacchione, C. Metal Complexes Bearing Sulfur-Containing Ligands as Catalysts in the Reaction of CO2 with Epoxides. Catalysts 2020, 10, 825. [Google Scholar] [CrossRef]
- Bester, K.; Bukowska, A.; Kawka, A.; Pytel, M.; Bukowski, W. Salophen Chromium(III) Complexes Functionalized with Pyridinium Salts as Catalysts for Carbon Dioxide Cycloaddition to Epoxides. RSC Adv. 2024, 14, 2466–2480. [Google Scholar] [CrossRef]
- Zhou, Y.; Hu, S.; Ma, X.; Liang, S.; Jiang, T.; Han, B. Synthesis of Cyclic Carbonates from Carbon Dioxide and Epoxides Over Betaine-Based Catalysts. J. Mol. Catal. A Chem. 2008, 284, 52–57. [Google Scholar] [CrossRef]
- Chen, J.-X.; Jin, B.; Dai, W.-L.; Deng, S.-L.; Cao, L.-R.; Cao, Z.-J.; Luo, S.-L.; Luo, X.-B.; Tu, X.-M.; Au, C.-T. Catalytic fixation of CO2 to cyclic carbonates over biopolymer chitosan-grafted quarternary phosphonium ionic liquid as a recylable catalyst. Appl. Catal. A Gen. 2014, 484, 26–32. [Google Scholar]
- Wu, X.; Chen, C.; Guo, Z.; North, M.; Whitwood, A.C. Metal and Halide-Free Catalyst for the Synthesis of Cyclic Carbonates from Epoxides and Carbon Dioxide. ACS. Catal. 2019, 9, 1895–1906. [Google Scholar] [CrossRef]
- Shen, Y.M.; Duan, W.L.; Shi, M. Chemical Fixation of Carbon Dioxide Co-Catalyzed by a Combination of Schiff Bases or Phenols and Organic Bases. Eur. J. Org. Chem. 2004, 2004, 3080–3089. [Google Scholar] [CrossRef]
- Caló, V.; Nacci, A.; Monopoli, A.; Fanizz, A. Cyclic Carbonate Formation from Carbon Dioxide and Oxiranes in Tetrabutylammonium Halides as Solvents and Catalysts. Org. Lett. 2002, 4, 2561–2563. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhang, S.; Cheng, W.; Ren, J. Hydroxyl-Functionalized Ionic Liquid: A Novel Efficient Catalyst for Chemical Fixation of CO2 to Cyclic Carbonate. Tetrahedron Lett. 2008, 49, 3588–3591. [Google Scholar] [CrossRef]
- Wang, J.Q.; Dong, K.; Cheng, W.G.; Sun, J.; Zhang, S.J. Insights into Quaternary Ammonium Salts-Catalyzed Fixation Carbon Dioxide with Epoxides. Catal. Sci. Technol. 2012, 2, 1480–1484. [Google Scholar] [CrossRef]
- Zhao, Y.; Tian, J.S.; Qi, X.H.; Han, Z.N.; Zhuang, Y.Y.; He, L.N. Quaternary Ammonium Salt-Functionalized Chitosan: An Easily Recyclable Catalyst for Efficient Synthesis of Cyclic Carbonates from Epoxides and Carbon Dioxide. J. Mol. Catal. A Chem. 2007, 271, 284–289. [Google Scholar] [CrossRef]
- Clegg, W.; Harrington, R.W.; North, M.; Pasquale, R. Cyclic Carbonate Synthesis Catalysed by Bimetallic Aluminium–Salen Complexes. Chem. Eur. J. 2010, 16, 6828–6843. [Google Scholar] [CrossRef]
- Guo, C.-H.; Liang, M.; Jiao, H. Cycloaddition Mechanisms of CO2 and Epoxide Catalyzed by Salophen—An Organocatalyst Free from Metals and Halides. Catal. Sci. Technol. 2021, 11, 2529–2539. [Google Scholar] [CrossRef]
- Cho, W.; Shin, M.S.; Hwang, S.; Kim, H.; Kim, M.; Gon Kim, J.; Kim, Y. Tertiary Amines: A New Class of Highly Efficient Organocatalysts for CO2 Fixations. J. Ind. Eng. Chem. 2016, 44, 210–215. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, G.; Kodama, K.; Hirose, T. An Efficient Metal- and Solvent-Free Organocatalytic System for Chemical Fixation of CO2 into Cyclic Carbonates under Mild Conditions. Green Chem. 2016, 18, 1229–1233. [Google Scholar] [CrossRef]
- Azzouz, R.; Moreno, V.C.; Herasme-Grullon, C.; Levacher, V.; Estel, L.; Ledoux, A.; Derrouiche, S.; Marsais, F.; Bischoff, L. Efficient Conversion of Epoxides into Carbonates with CO2 and a Single Organocatalyst: Laboratory and Kilogram-Scale Experiments. Synlett 2020, 31, 183–188. [Google Scholar] [CrossRef]
- Wang, Z.; Li, D.; Chen, S.; Hu, J.; Gong, Y.; Guo, Y.; Deng, T. Ionic Liquid [DBUH][BO2]: An Excellent Catalyst for Chemical Fixation of CO2 under Mild Conditions. New J. Chem. 2021, 45, 4611–4616. [Google Scholar] [CrossRef]
- Zhang, Z.; Fan, F.; Xing, H.; Yang, Q.; Bao, Z.; Ren, Q. Efficient Synthesis of Cyclic Carbonates from Atmospheric CO2 Using a Positive Charge Delocalized Ionic Liquid Catalyst. ACS Sustain. Chem. Eng. 2017, 5, 2841–2846. [Google Scholar] [CrossRef]
- Yue, Z.; Pudukudy, M.; Chen, S.; Liu, Y.; Zhao, W.; Wang, J.; Shan, S.; Jia, Q. A Non-Metal Acen-H Catalyst for the Chemical Fixation of CO2 into Cyclic Carbonates under Solvent- and Halide-Free Mild Reaction Conditions. Appl. Catal. A Gen. 2020, 601, 117646. [Google Scholar] [CrossRef]
- Larrow, J.F.; Jacobsen, E.N.; Gao, Y.; Hong, Y.; Nie, X.; Zepp, C.M. A Practical Method for the Large-Scale Preparation of [N,N’-Bis(3,5-di-tertbutylsalicylidene)-1,2-cyclohexanediaminato(2-)]manganese(III) chloride, a Highly Enantioselective Epoxidation Catalyst. J. Org. Chem. 1994, 59, 1939–1942. [Google Scholar] [CrossRef]
- Casiraghi, G.; Casnati, G.; Puglia, G.; Sartori, G.; Terenghi, G.J. Selective reactions between phenols and formaldehyde. A novel route to salicylaldehydes. Chem. Soc. Perkin Trans. 1980, 1, 1862–1865. [Google Scholar] [CrossRef]
- Meier, P.; Broghammer, F.; Latendorf, K.; Rauhut, G.; Peters, R. Cooperative Al(Salen)-Pyridinium Catalysts for the Asymmetric Synthesis of trans-Configured β-Lactones by [2+2]-Cyclocondensation of Acylbromides and Aldehydes: Investigation of Pyridinium Substituent Effects. Molecules 2012, 17, 7121–7150. [Google Scholar] [CrossRef]
- Titze, M.; Heitkämper, J.; Junge, T.; Kästner, J.; Peters, R. Highly active cooperative Lewis acid—Ammonium salt catalyst for the enantioselective hydroboration of ketones. Angew. Chem. Int. Ed. 2021, 60, 5544–5553. [Google Scholar] [CrossRef]
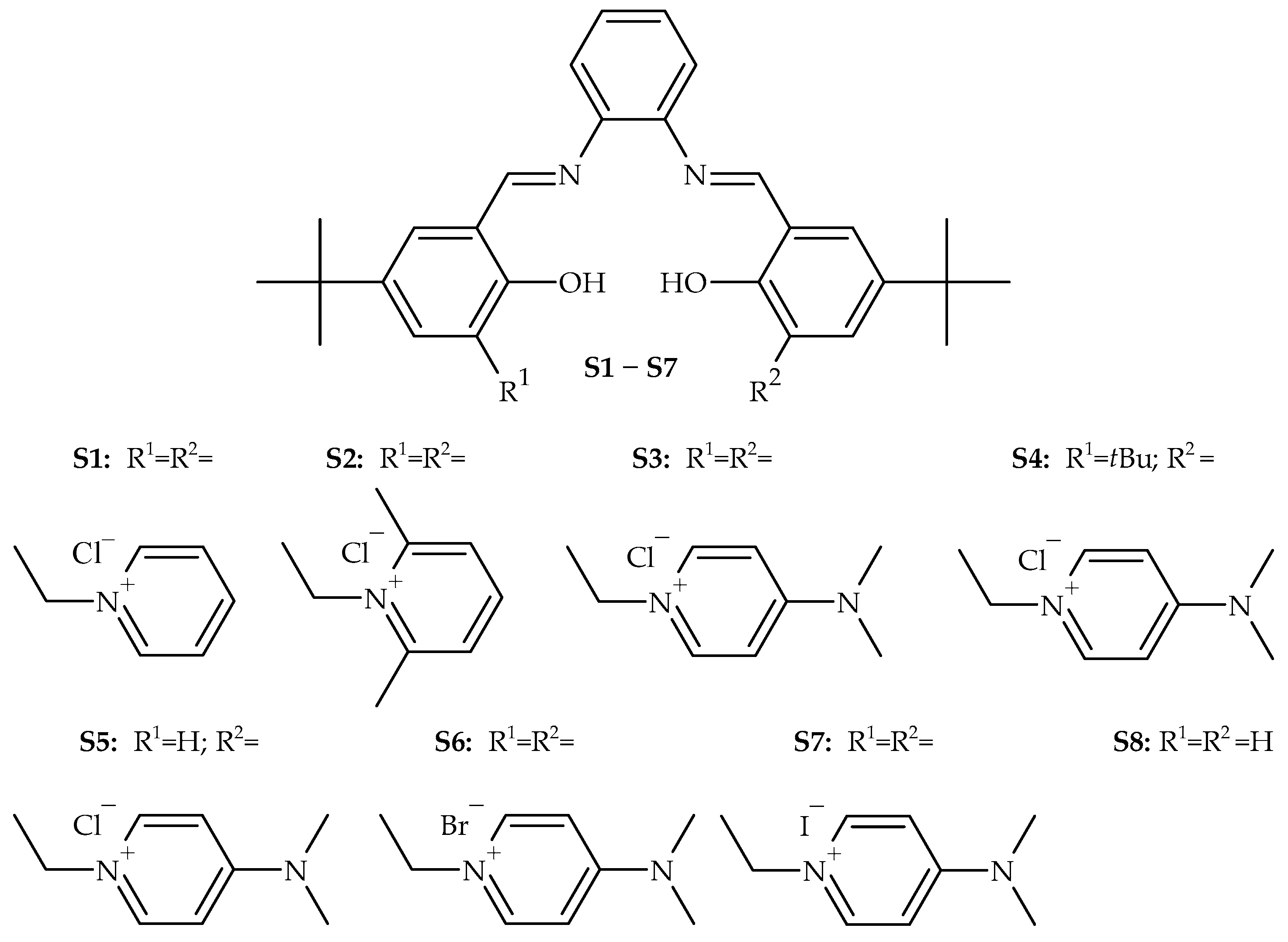


{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| No. | Cat. | Temp. [°C] | Conversion a [%] | TON [-] | TOF [h−1] |
| 1 | S1 | 80 | 20 | 36 | 18 |
| 2 | S2 | 4 | 8 | 4 | |
| 3 | S3 | 52 | 100 | 50 | |
| 4 | S4 | 36 | 72 | 36 | |
| 5 | S5 | 37 | 72 | 36 | |
| 6 | S1 | 100 | 44 | 84 | 42 |
| 7 | S2 | 15 | 28 | 14 | |
| 8 | S3 | 73 | 140 | 70 | |
| 9 | S4 | 68 | 128 | 64 | |
| 10 | S5 | 69 | 132 | 66 | |
| 11 | S1 | 120 | 62 | 112 | 56 |
| 12 | S2 | 47 | 90 | 45 | |
| 13 | S3 | 83 | 156 | 78 | |
| 14 | S4 | 77 | 142 | 71 | |
| 15 | S5 | 72 | 136 | 68 | |
| 16 | S6 | 86 | 158 | 79 | |
| 17 | S7 | 77 | 138 | 69 | |
| 18 | S8 | 0 | 0 | 0 | |
| 19 | S1 | 140 | 59 | 96 | 48 |
| 20 | S2 | 48 | 78 | 39 | |
| 21 | S3 | 78 | 136 | 68 | |
| 22 | S4 | 67 | 118 | 59 | |
| 23 | S5 | 70 | 122 | 61 | |
| 24 | DMAP | 120 | 49 | 92 | 46 |
| 25 | [BnDMAP]Cl | 52 | 98 | 49 | |
| No. | Cat. | Time [h] | pCO2 [bar] | Temp. [°C] | Conversion a [%] | TON [-] | TOF [h−1] |
|---|---|---|---|---|---|---|---|
| 1 | S1 | 2 | 4 | 100 | 56 | 106 | 53 |
| 2 | S2 | 2 | 4 | 15 | 28 | 14 | |
| 3 | S3 | 2 | 4 | 82 | 156 | 78 | |
| 4 | S4 | 2 | 4 | 68 | 132 | 66 | |
| 5 | S5 | 2 | 4 | 74 | 144 | 72 | |
| 6 | S3 | 2 | 2 | 120 | 83 | 156 | 78 |
| 7 | S3 | 4 | 2 | 91 | 162 | 41 | |
| 8 | S3 | 6 | 2 | 95 | 166 | 28 | |
| 9 | S3 | 2 | 4 | 85 | 156 | 78 | |
| 10 | S3 | 2 | 6 | 87 | 158 | 79 |
| No. | Loading S3, [mol%] | Conversion E3 a [%] | TON [-] | TOF [h−1] |
|---|---|---|---|---|
| 1 | 0.05 | 48 | 893 | 447 |
| 2 | 0.1 | 56 | 513 | 257 |
| 3 | 0.2 | 66 | 300 | 150 |
| 4 | 0.5 | 83 | 156 | 78 |
| No. | Catalyst | Temperature [°C] | Pressure [bar] | Time [h] | Conversion [%] | Yield [%] | TON | TOF [h−1] | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 |  0.1 mol% | 120 | 10 | 10 | 97 | 97 | 967 | 97 | [49] |
| 2 |  8 mol% + 8 mol% nBu4NI | 25 | 1 | 20 | - | 97 | - | - | [50] |
| 3 |  5 mol% | 45 | 12 | 16 | - | 95 | - | - | [51] |
| 4 |  75 mol% | 30 | 1 | 6 | - | 94 | - | - | [52] |
| 5 |  1 mol% | 120 | 1 | 4 | 96 | - | - | - | [53] |
| 6 | 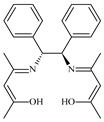 0.2 mol% | 110 | 10 | 4 | 99 | 98.5 | 495.2 | 123.8 | [54] |
| 7 | 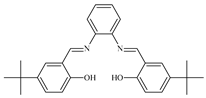 1 mol% | 120 | 10 | 3.5 | 84 | 76 | - | - | [41] |
| 8 | 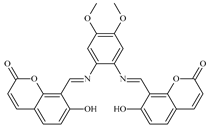 0.5 mol% | 100 | 8 | 9 | 92 | - | 184 | 20.4 | [24] |
| 9 | S3 | 100 | 4 | 2 | 82 | - | 156 | 78 | This work |
| 10 | S3 | 120 | 6 | 2 | 87 | 78 | 158 | 79 | This work |
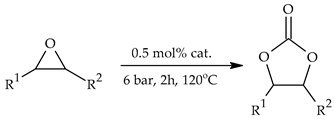 | ||||||||
|---|---|---|---|---|---|---|---|---|
| No. | Epoxide | Epoxide Ab’br. | Conversion a [%] | Cyclic Carbonate | Cyclic Carbonate Abbr. | Yield b [%] | TON | TOF [h−1] |
| 1 c |  | E1 | 53 |  | C1 | 26 | 56 | 28 |
| 2 c |  | E2 | 38 | 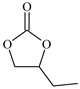 | C2 | 26 | 58 | 29 |
| 3 | 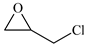 | E3 | 87 | 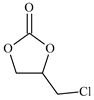 | C3 | 78 | 158 | 79 |
| 4 |  | E4 | 92 | 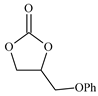 | C4 | 87 | 178 | 89 |
| 5 |  | E5 | 59 | 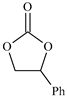 | C5 | 50 | 107 | 54 |
| 6 | 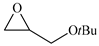 | E6 | 52 | 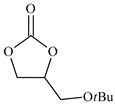 | C6 | 47 | 96 | 48 |
| 7 | 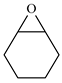 | E7 | 0 |  | C7 | 0 | 0 | 0 |
| 8 | 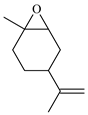 | E8 | 0 | 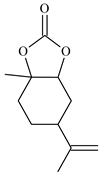 | C8 | 0 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawka, A.; Bester, K.; Bukowska, A.; Bukowski, W. Salophen-Type Schiff Bases Functionalized with Pyridinium Halide Units as Metal-Free Catalysts for Synthesis of Cyclic Carbonates from Carbon Dioxide and Terminal Epoxides. Catalysts 2024, 14, 658. https://doi.org/10.3390/catal14100658
Kawka A, Bester K, Bukowska A, Bukowski W. Salophen-Type Schiff Bases Functionalized with Pyridinium Halide Units as Metal-Free Catalysts for Synthesis of Cyclic Carbonates from Carbon Dioxide and Terminal Epoxides. Catalysts. 2024; 14(10):658. https://doi.org/10.3390/catal14100658
Chicago/Turabian StyleKawka, Aleksandra, Karol Bester, Agnieszka Bukowska, and Wiktor Bukowski. 2024. "Salophen-Type Schiff Bases Functionalized with Pyridinium Halide Units as Metal-Free Catalysts for Synthesis of Cyclic Carbonates from Carbon Dioxide and Terminal Epoxides" Catalysts 14, no. 10: 658. https://doi.org/10.3390/catal14100658
APA StyleKawka, A., Bester, K., Bukowska, A., & Bukowski, W. (2024). Salophen-Type Schiff Bases Functionalized with Pyridinium Halide Units as Metal-Free Catalysts for Synthesis of Cyclic Carbonates from Carbon Dioxide and Terminal Epoxides. Catalysts, 14(10), 658. https://doi.org/10.3390/catal14100658