
Recent Advances in the Synthesis of Chiral Tetrahydroisoquinolines via Asymmetric Reduction



Abstract
:1. Introduction
2. Asymmetric Synthesis of Chiral Tetrahydroisoquinolines via Transition-Metal-Catalyzed Hydrogenation
2.1. Asymmetric Synthesis of Chiral Tetrahydroisoquinolines via Titanium-Catalyzed Hydrogenation
2.2. Asymmetric Synthesis of Chiral Tetrahydroisoquinolines via Ruthenium-Catalyzed Hydrogenation
2.3. Asymmetric Synthesis of Chiral Tetrahydroisoquinolines via Rhodium-Catalyzed Hydrogenation
2.3.1. Asymmetric Hydrogenation of DHIQs with Rhodium Catalysts
2.3.2. Asymmetric Hydrogenation of IQs with Rhodium Catalysts
2.4. Asymmetric Synthesis of Chiral Tetrahydroisoquinolines via Iridium-Catalyzed Hydrogenation
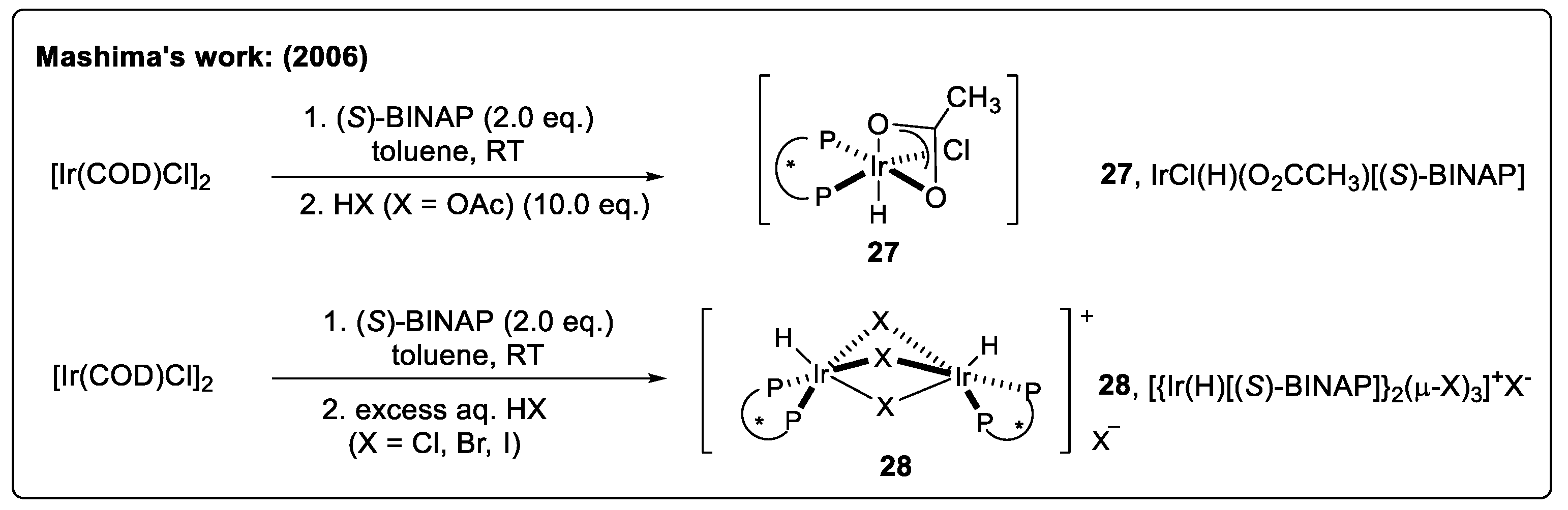
2.4.1. Asymmetric Hydrogenation of DHIQs with Iridium Catalysts
Catalyst Activation Strategy with Imides
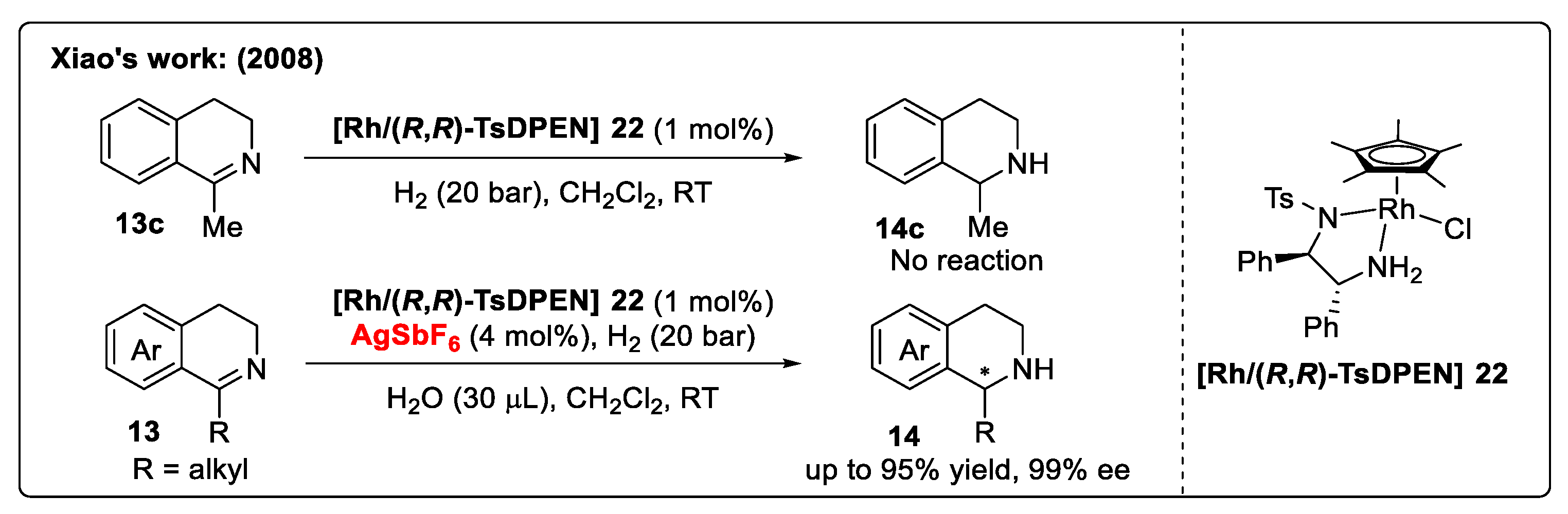
Catalyst Activation Strategy with Brønsted Acids/Oxidizing Halogen Reagents
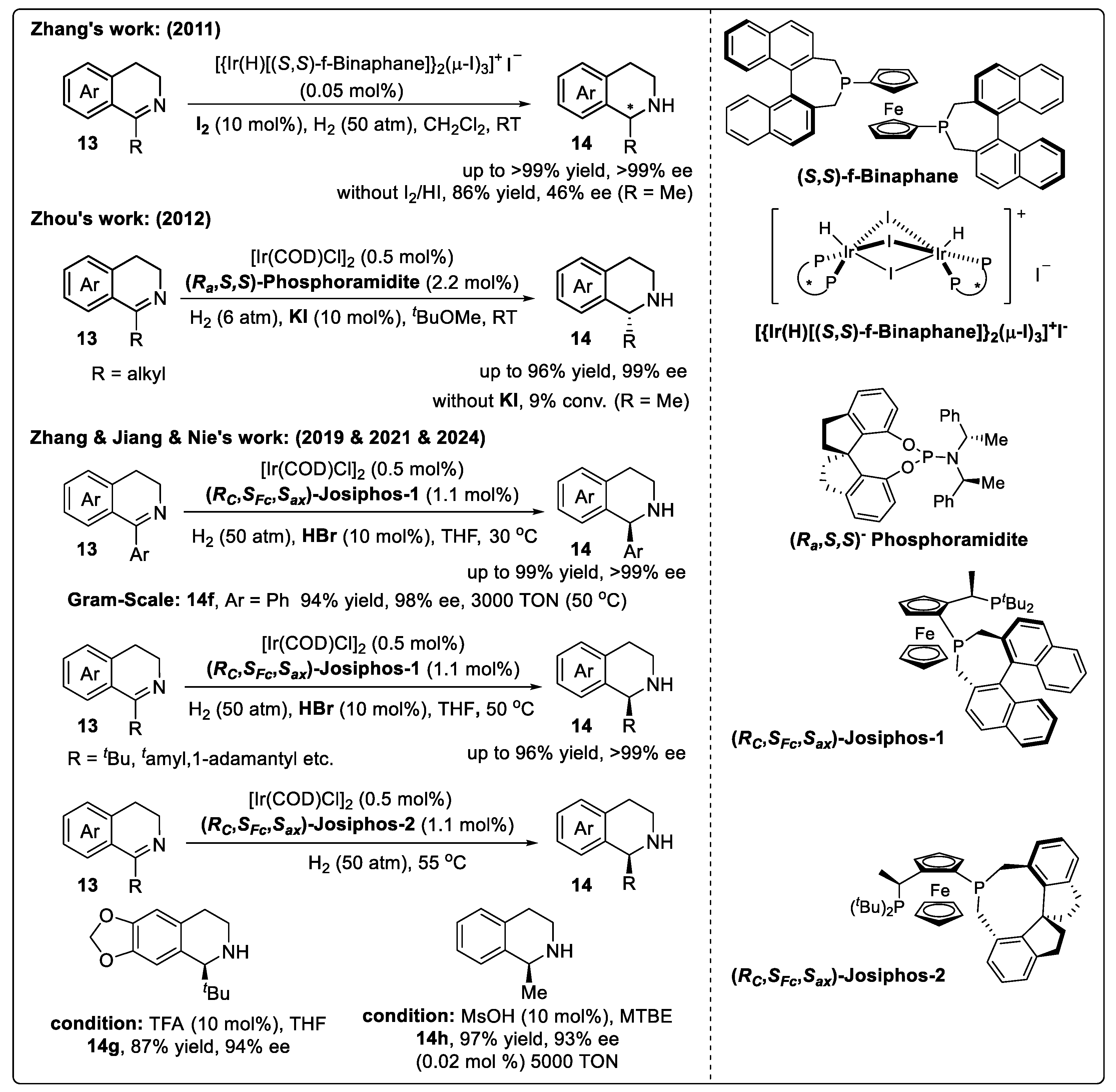
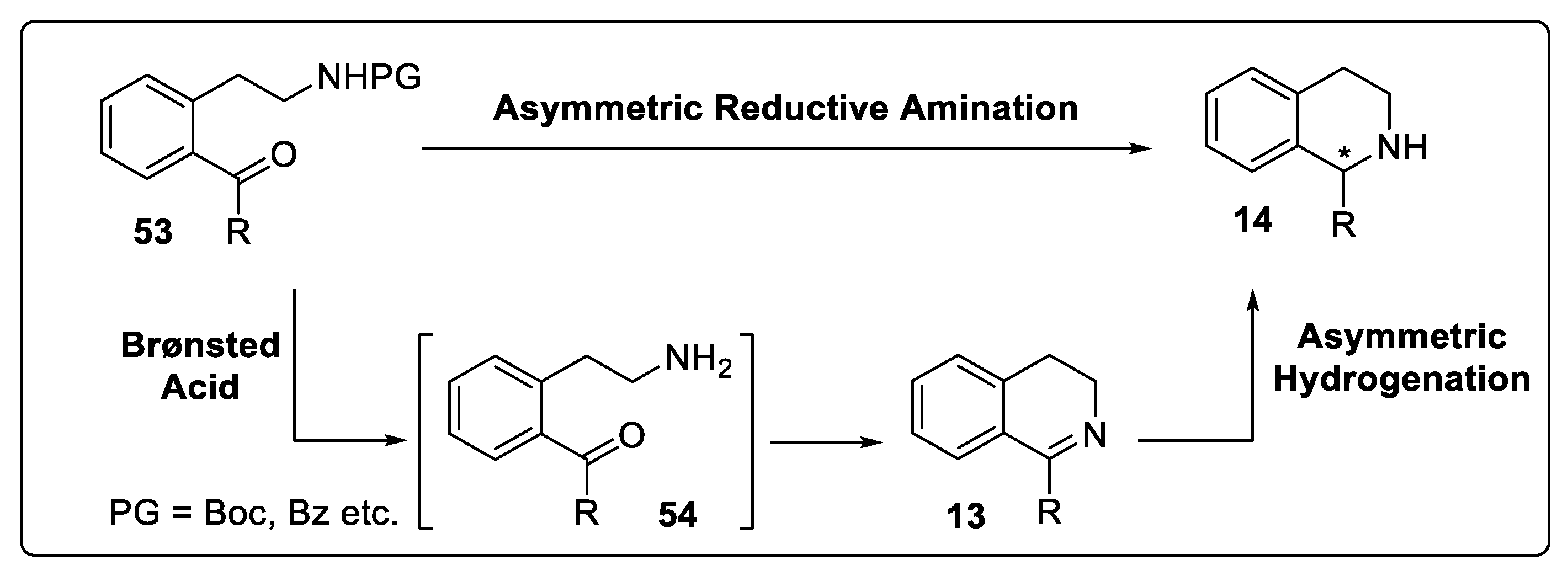
Substrate Activation Strategy of DHIQs with Brønsted Acids
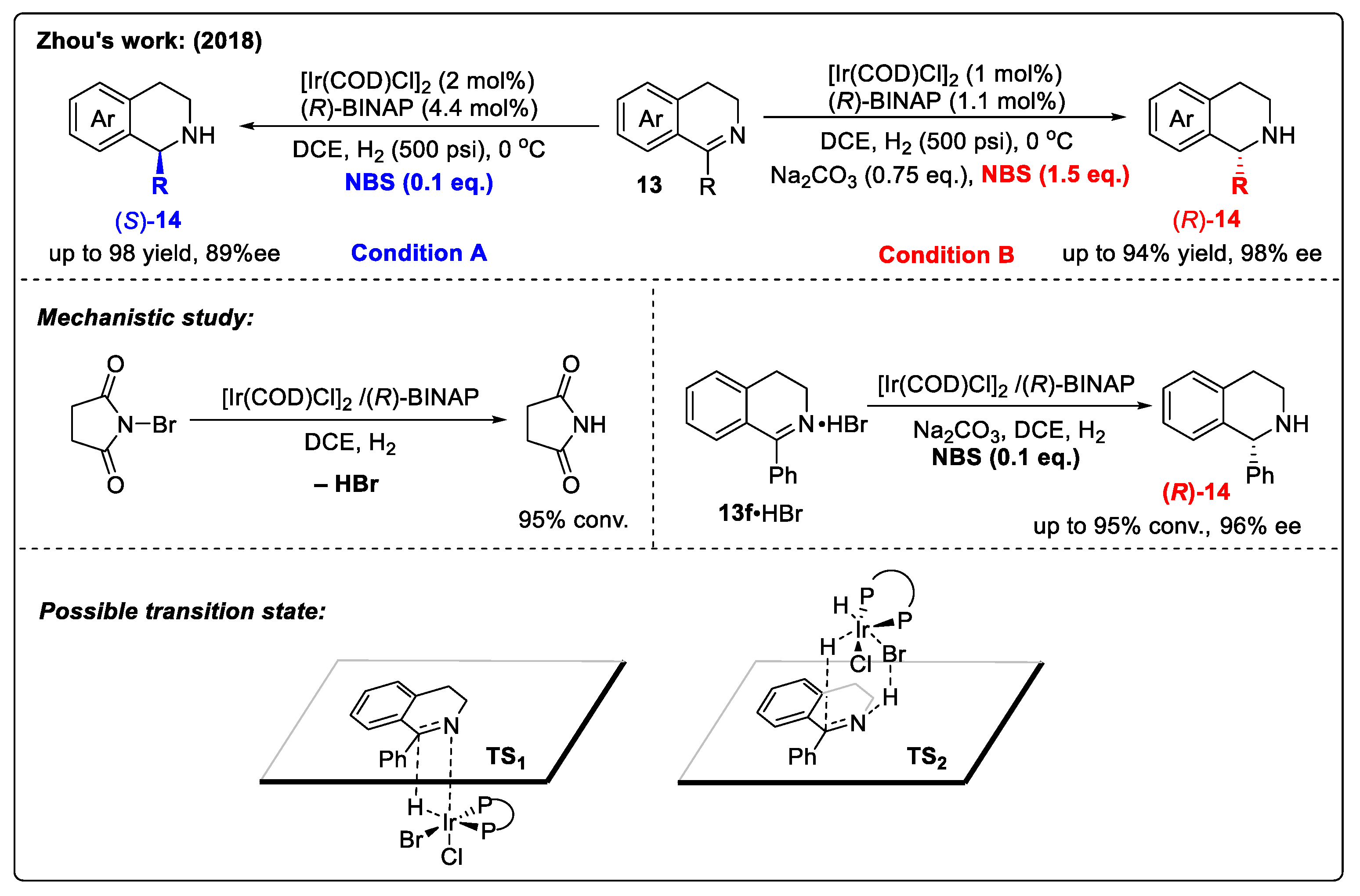
Substrate Activation Strategy of DHIQs with Organic Halides
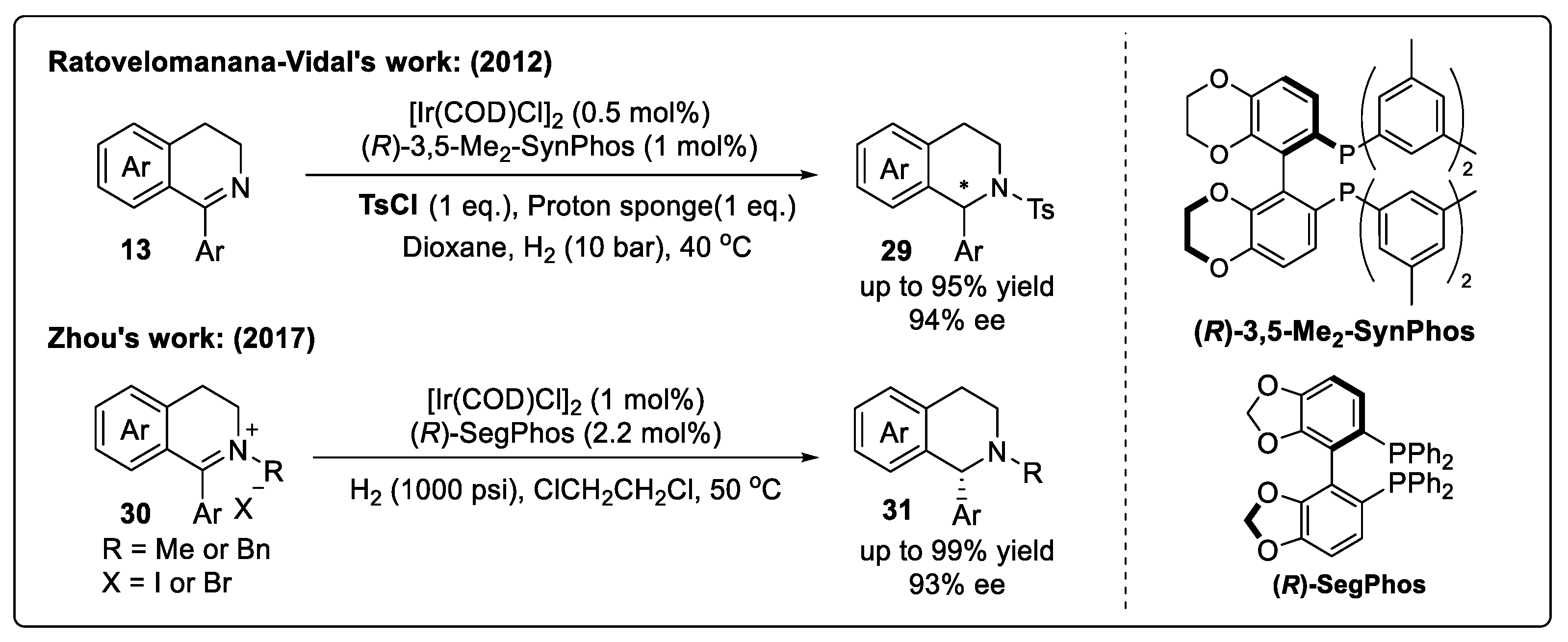
2.4.2. Asymmetric Hydrogenation of IQ-Type Enamines
2.4.3. Asymmetric Hydrogenation of IQs
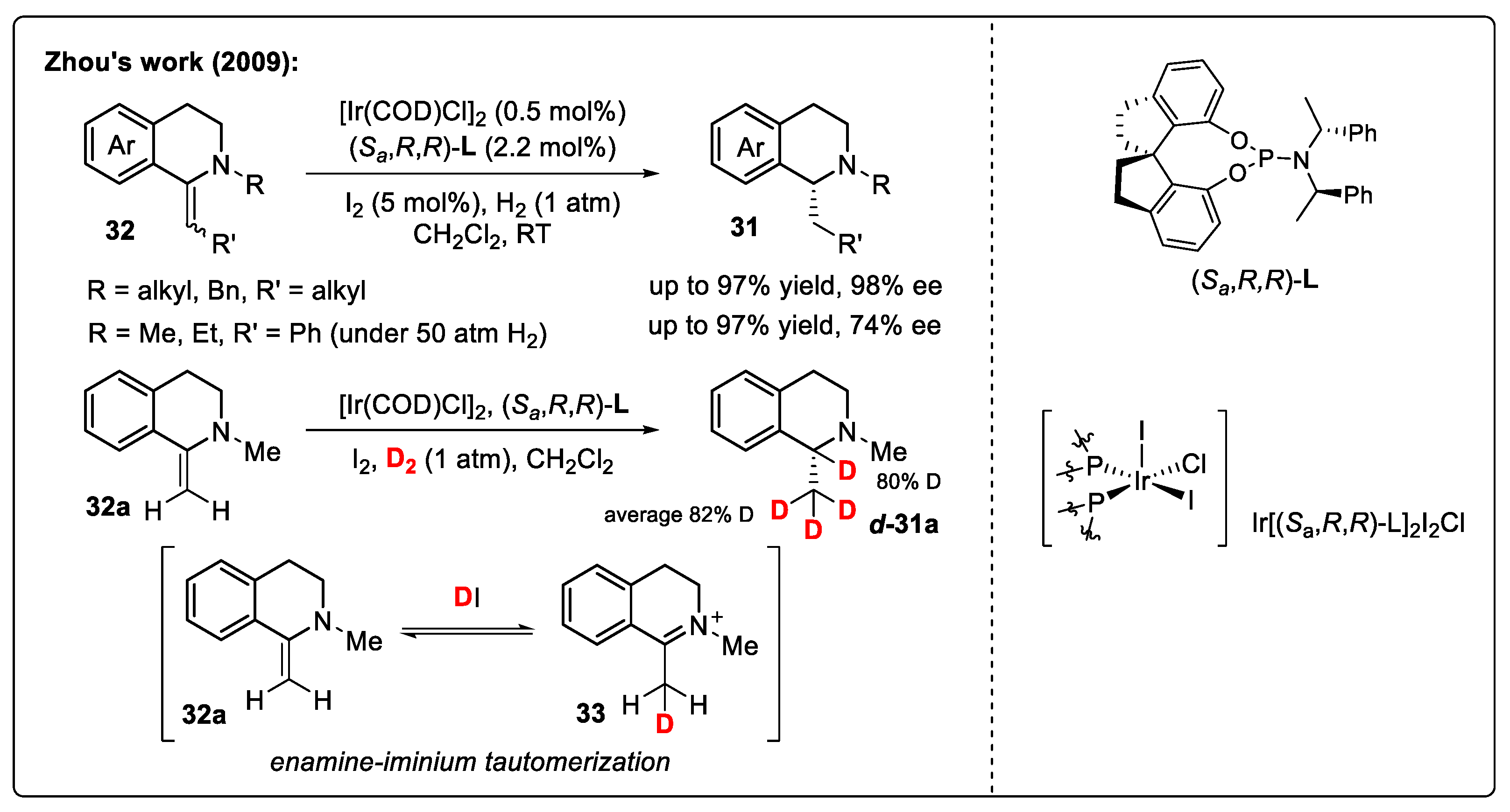
Substrate Activation Strategy of IQs with Organic Halides
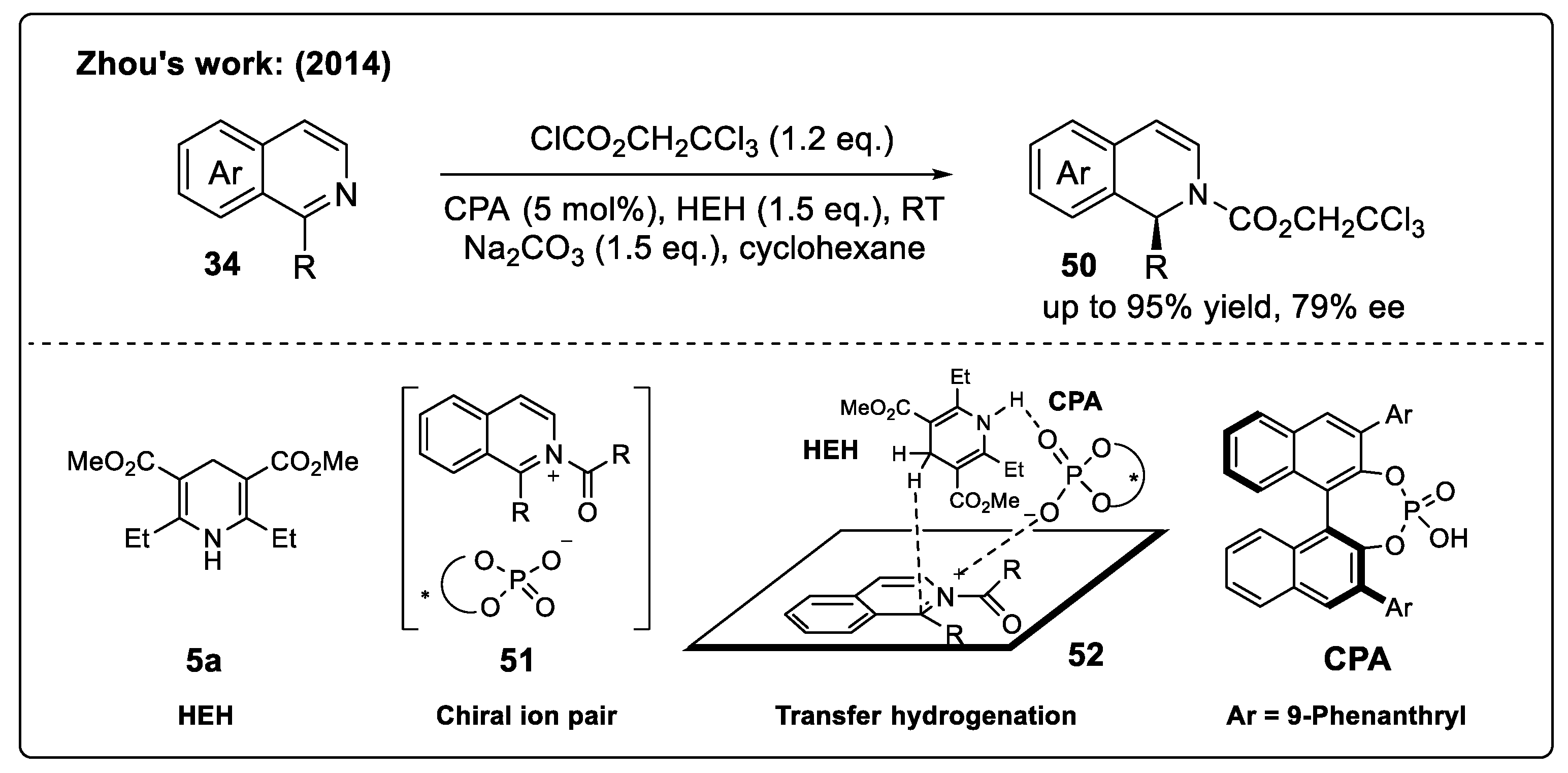
Substrate Activation Strategy of IQs with Brønsted Acids
Substrate Activation Strategy with Directing Groups
Direct Hydrogenation Strategy
3. Asymmetric Synthesis of Chiral THIQs via Catalytic Transfer Hydrogenation
3.1. Asymmetric Synthesis of Chiral Tetrahydroisoquinolines via Ruthenium-Catalyzed Transfer Hydrogenation
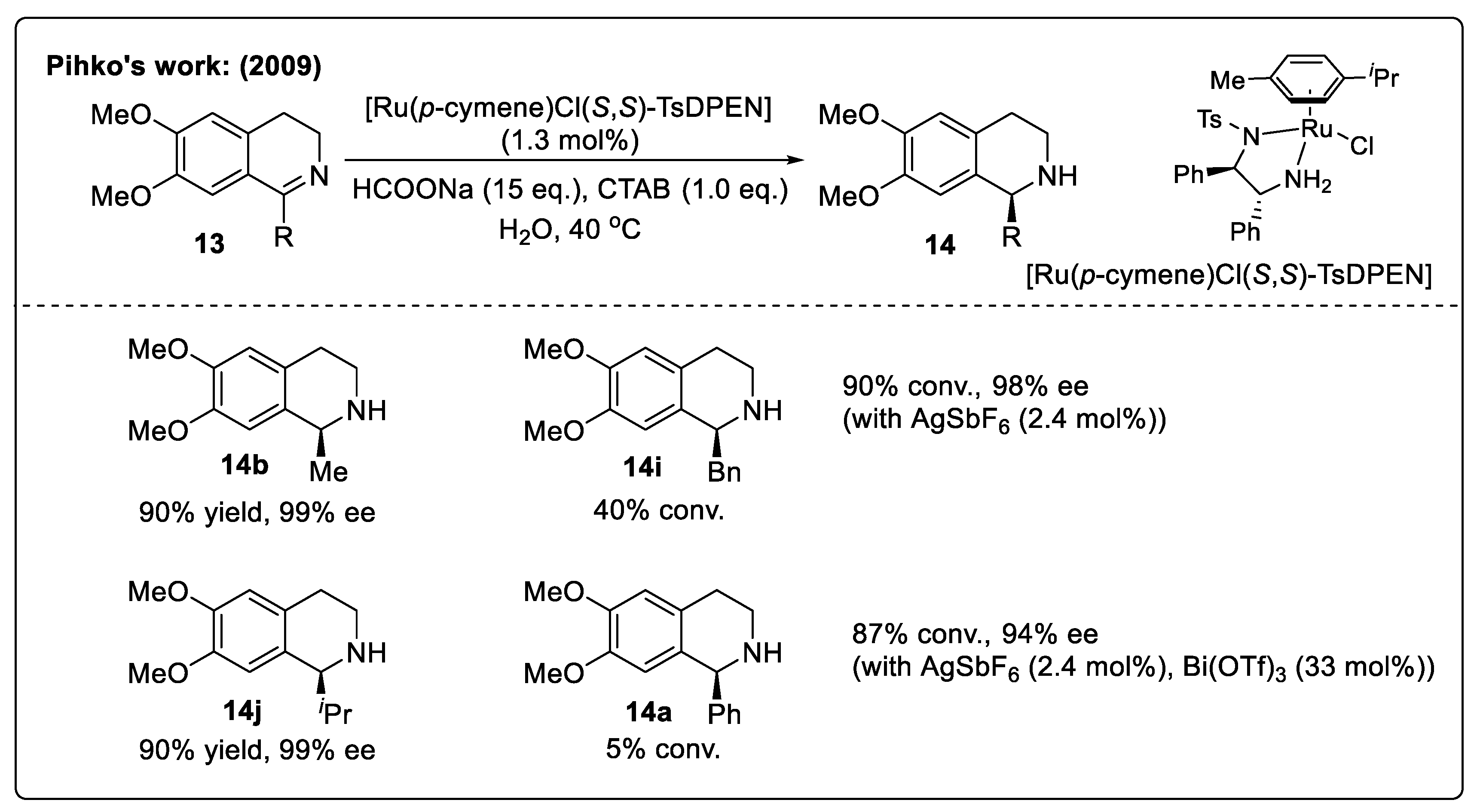
3.1.1. Asymmetric Transfer Hydrogenation of DHIQs

 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | Catalyst | Reaction Conditions | Yield (%) | Ee (%) | Group (Year) | Ref. |
| 1 |  | [Ru/(S,S)-N-N* a] or [Ru/(R,R)-N-N* b] or [Ru/(S,S)-N-N* c] (0.5 mol%) | HCOOH/Et3N (2.5 mL, 5:2) CH3CN or CH2Cl2, 28 °C | 90–97 | 84–95 | Noyori (1996) | [68] |
| 2 |  | [Ru/(R,R)-N-N* e] (1–2 mol%) | HCOOH/Et3N (0.25 mL, 5:2) CH2Cl2, RT | 63–76 | 85–99 | Vedejs (1999) | [69] |
| 3 |  | RuCl2(p-cymene)/ (R,R)-N-N* f (1 mol%)/(2.2 mol%) | HCOONa (5.0 eq.) CTAB (50 mol%) H2O, 28 °C | 68–97 | 90–95 | Zhu and Deng (2006) | [73] |
| 4 |  | [Ru/(S,S)-N-N* a] (0.6 mol%) | HCOONa (5.0 eq.) CTAB (50 mol%) H2O, 40 °C | 87–90 | 99–99.5 | Pihko (2009) | [74] |
| 5 |  | [Ru/(R,R)-N-N* a] or [Ru/(R,R)-N-N* c] or [Ru/(R,R)-N-N* d] (1 mol%) | HCOOH/Et3N (6.3 eq., 5:2) CH3CN or CH2Cl2, 40 °C | 171–261 3.8–45 <1–265 (TOF/h) | 82–94 39–80 63–87 | Kacer (2013) | [75] |
| 6 |  | [Ru/(R,R)-N-N* e] (1 mol%) | HCOOH/Et3N (2 eq., 5:2) iPrOH, 30 °C | 72–97 | 82–98 | Ratovelomanana-Vidal (2013) | [76] |
| 7 |  | [Ru/(R,R)-N-N* e] (1 mol%) | HCOOH/Et3N (2 eq., 5:2) iPrOH, 30 °C | 71–97 | 15–99 | Ratovelomanana-Vidal (2015) | [77] |
| 8 | 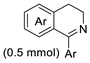 | [Ru/(R,R)-N-N* g] (1 mol%) | HCOOH/Et3N (0.25 mL, 5:2) iPrOH, RT | 25–90 | 88–93 | Wills (2020) | [78] |
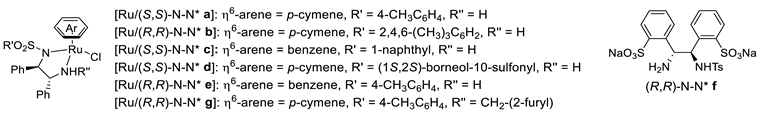 | |||||||
3.1.2. Asymmetric Transfer Hydrogenation of DHIQ-Type Iminium Salts
3.2. Asymmetric Synthesis of Chiral Tetrahydroisoquinolines via Rhodium-Catalyzed Transfer Hydrogenation
3.3. Asymmetric Synthesis of Chiral Tetrahydroisoquinolines via Iridium-Catalyzed Transfer Hydrogenation
3.4. Asymmetric Synthesis of Chiral THIQs via Organocatalytic Transfer Hydrogenation
4. Asymmetric Synthesis of Chiral THIQs via Transition-Metal-Catalyzed Reductive Amination
4.1. Ruthenium-Catalyzed Asymmetric Reductive Amination
4.2. Iridium-Catalyzed Asymmetric Reductive Amination
5. Asymmetric Synthesis of Chiral Tetrahydroisoquinolines via Deracemization
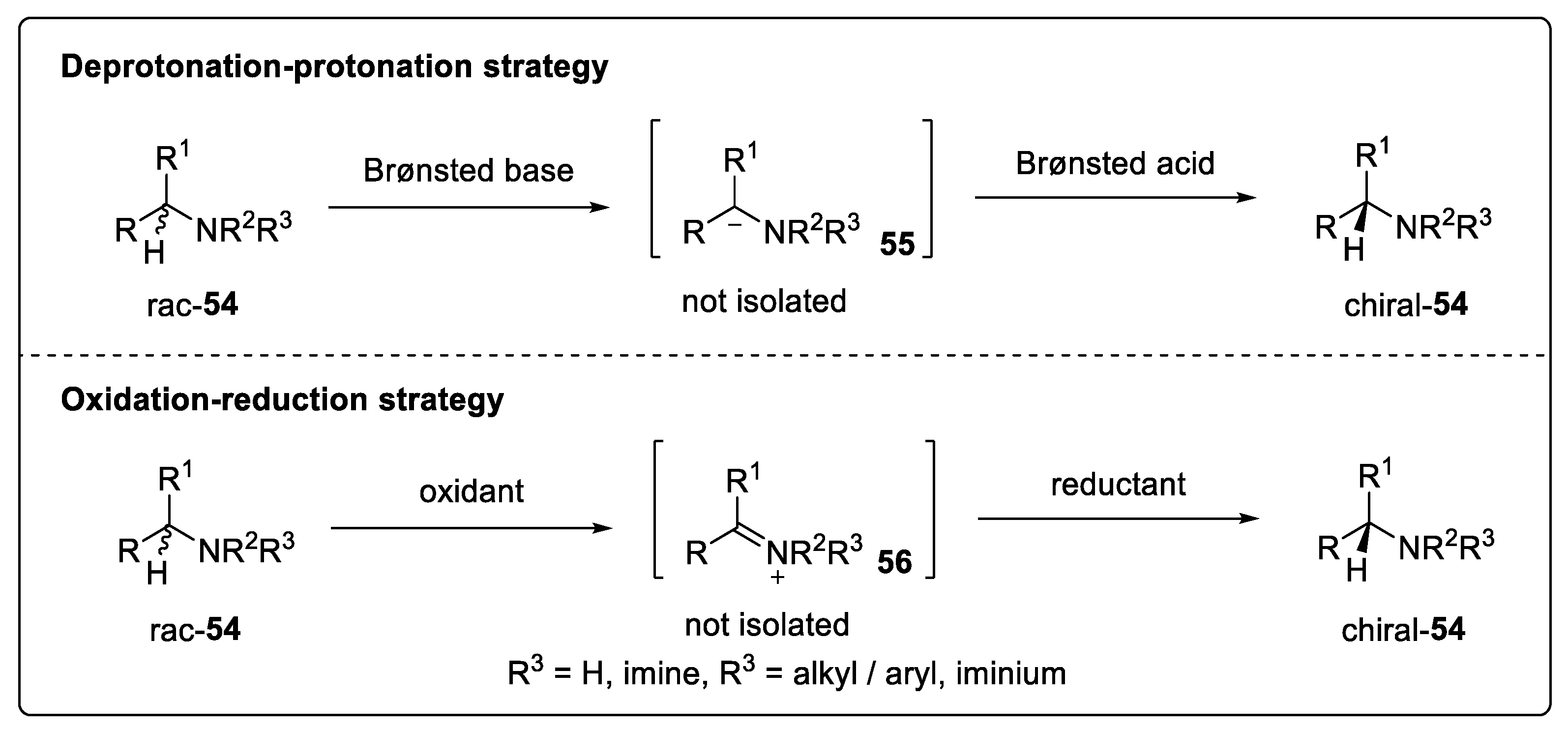
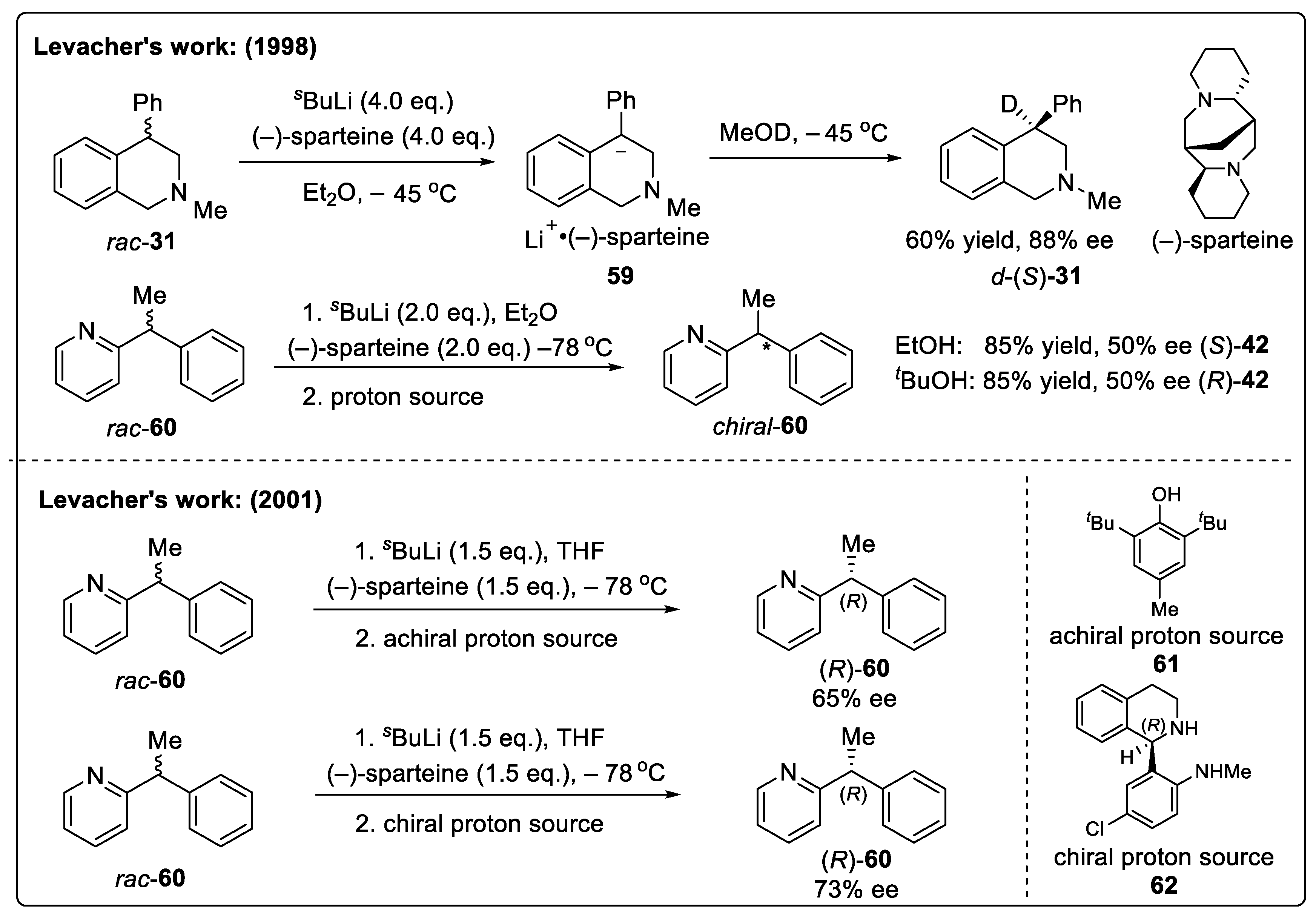
5.1. Deracemization via a Deprotonation-Protonation Strategy
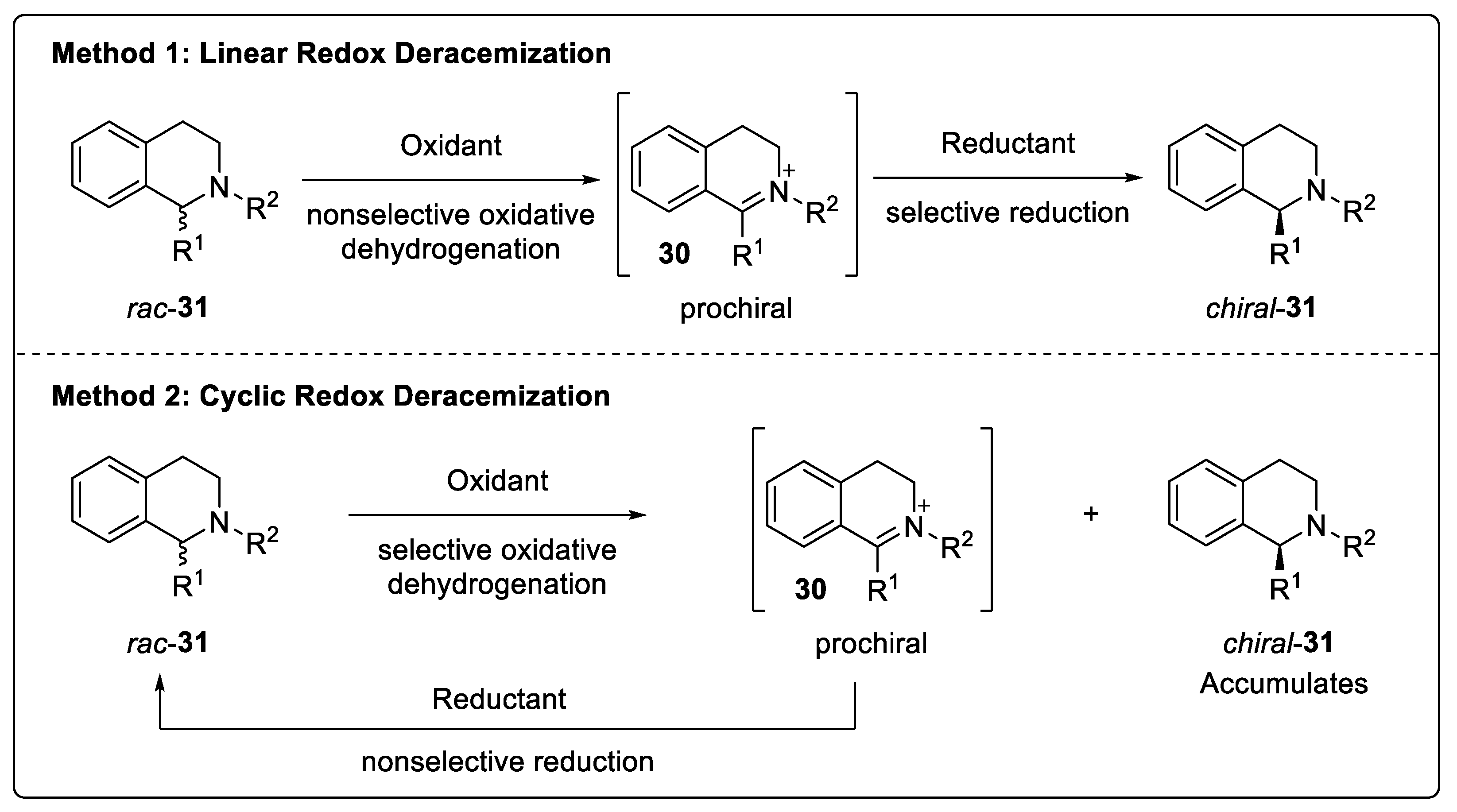
5.2. Deracemization via a Redox Strategy
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, W.; Liu, S.; Jin, R.; Guo, H.; Zhao, J. Novel strategies for catalytic asymmetric synthesis of C1-chiral 1,2,3,4-tetrahydroisoquinolines and 3,4-dihydrotetrahydroisoquinolines. Org. Chem. Front. 2015, 2, 288–299. [Google Scholar] [CrossRef]
- Yet, L. Tetrahydroisoquinolines. In Privileged Structures in Drug Discovery; Wiley: Hoboken, NJ, USA, 2018; pp. 356–413. [Google Scholar] [CrossRef]
- Stöckigt, J.; Antonchick, A.P.; Wu, F.; Waldmann, H. The Pictet—Spengler reaction in nature and in organic chemistry. Angew. Chem. Int. Ed. 2011, 50, 8538–8564. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.N.; Ngamnithiporn, A.; Du, E.; Stoltz, B.M. Recent advances in the total synthesis of the tetrahydroisoquinoline alkaloids (2002–2020). Chem. Rev. 2023, 123, 9447–9496. [Google Scholar] [CrossRef] [PubMed]
- Bembenek, M.E.; Abell, C.W.; Chrisey, L.A.; Rozwadowska, M.D.; Gessner, W.; Brossi, A. Inhibition of monoamine oxidases A and B by simple isoquinoline alkaloids: Racemic and optically active 1,2,3,4-tetrahydro-, 3,4-dihydro-, and fully aromatic isoquinolines. J. Med. Chem. 1990, 33, 147–152. [Google Scholar] [CrossRef]
- Schrittwieser, J.H.; Resch, V.; Wallner, S.; Lienhart, W.-D.; Sattler, J.H.; Resch, J.; Macheroux, P.; Kroutil, W. Biocatalytic organic synthesis of optically pure (S)-scoulerine and berbine and benzylisoquinoline alkaloids. J. Org. Chem. 2011, 76, 6703–6714. [Google Scholar] [CrossRef]
- Klutchko, S.; Blankley, C.J.; Fleming, R.W.; Hinkley, J.M.; Werner, A.E.; Nordin, I.; Holmes, A.; Hoefle, M.L.; Cohen, D.M. Synthesis of novel angiotensin converting enzyme inhibitor quinapril and related compounds. A divergence of structure-activity relationships for non-sulfhydryl and sulfhydryl types. J. Med. Chem. 1986, 29, 1953–1961. [Google Scholar] [CrossRef]
- Robinson, D.; Cardozo, L. The emerging role of solifenacin in the treatment of overactive bladder. Expert Opin. Inv. Drug 2004, 13, 1339–1348. [Google Scholar] [CrossRef]
- Uchida, T.; Krauwinkel, W.J.; Mulder, H.; Smulders, R.A. Food does not affect the pharmacokinetics of solifenacin, a new muscarinic receptor antagonist: Results of a randomized crossover trial. Br. J. Clin. Pharmacol. 2004, 58, 4–7. [Google Scholar] [CrossRef]
- Barrios-Rivera, J.; Xu, Y.; Wills, M.; Vyas, V.K. A diversity of recently reported methodology for asymmetric imine reduction. Org. Chem. Front. 2020, 7, 3312–3342. [Google Scholar] [CrossRef]
- Wang, D.-S.; Chen, Q.-A.; Lu, S.-M.; Zhou, Y.-G. Asymmetric hydrogenation of heteroarenes and arenes. Chem. Rev. 2012, 112, 2557–2590. [Google Scholar] [CrossRef]
- Xie, J.-H.; Zhu, S.-F.; Zhou, Q.-L. Transition metal-catalyzed enantioselective hydrogenation of enamines and imines. Chem. Rev. 2011, 111, 1713–1760. [Google Scholar] [CrossRef] [PubMed]
- Seo, C.S.G.; Morris, R.H. Catalytic homogeneous asymmetric hydrogenation: Successes and opportunities. Organometallics 2019, 38, 47–65. [Google Scholar] [CrossRef]
- Kim, A.N.; Stoltz, B.M. Recent advances in homogeneous catalysts for the asymmetric hydrogenation of heteroarenes. ACS Catal. 2020, 10, 13834–13851. [Google Scholar] [CrossRef]
- Abdine, R.A.A.; Hedouin, G.; Colobert, F.; Wencel-Delord, J. Metal-catalyzed asymmetric hydrogenation of C=N bonds. ACS Catal. 2021, 11, 215–247. [Google Scholar] [CrossRef]
- Cabré, A.; Verdaguer, X.; Riera, A. Recent advances in the enantioselective synthesis of chiral amines via transition metal-catalyzed asymmetric hydrogenation. Chem. Rev. 2022, 122, 269–339. [Google Scholar] [CrossRef]
- Zhou, Y.-G. Asymmetric hydrogenation of heteroaromatic compounds. Acc. Chem. Res. 2007, 40, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Candish, L.; Paul, D.; Glorius, F. N-heterocyclic carbenes in asymmetric hydrogenation. ACS Catal. 2016, 6, 5978–5988. [Google Scholar] [CrossRef]
- Chen, Z.-P.; Zhou, Y.-G. Asymmetric hydrogenation of heteroarenes with multiple heteroatoms. Synthesis 2016, 48, 1769–1781. [Google Scholar]
- Bartoszewicz, A.; Ahlsten, N.; Martín-Matute, B. Enantioselective synthesis of alcohols and amines by iridium-catalyzed hydrogenation, transfer hydrogenation, and related processes. Chem. Eur. J. 2013, 19, 7274–7302. [Google Scholar] [CrossRef]
- Yu, Z.; Jin, W.; Jiang, Q. Brønsted acid activation strategy in transition-metal catalyzed asymmetric hydrogenation of N-unprotected imines, enamines, and N-heteroaromatic compounds. Angew. Chem. Int. Ed. 2012, 51, 6060–6072. [Google Scholar] [CrossRef]
- Ouellet, S.G.; Walji, A.M.; Macmillan, D.W.C. Enantioselective organocatalytic transfer hydrogenation reactions using Hantzsch esters. Acc. Chem. Res. 2007, 40, 1327–1339. [Google Scholar] [CrossRef] [PubMed]
- Pilar Lamata, M.; Passarelli, V.; Carmona, D. Recent advances in iridium-catalysed transfer hydrogenation reactions. In Iridium Catalysts for Organic Reactions; Oro, L.A., Claver, C., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 67–152. [Google Scholar]
- Rueping, M.; Sugiono, E.; Schoepke, F.R. Thieme chemistry journal awardees—where are they now? Asymmetric brønsted acid catalyzed transfer hydrogenations. Synlett 2010, 2010, 852–865. [Google Scholar] [CrossRef]
- Rueping, M.; Dufour, J.; Schoepke, F.R. Advances in catalytic metal-free reductions: From bio-inspired concepts to applications in the organocatalytic synthesis of pharmaceuticals and natural products. Green Chem. 2011, 13, 1084–1105. [Google Scholar] [CrossRef]
- de Vries, J.G.; Mršić, N. Organocatalytic asymmetric transfer hydrogenation of imines. Catal. Sci. Technol. 2011, 1, 727–735. [Google Scholar] [CrossRef]
- Gladiali, S.; Alberico, E. Asymmetric transfer hydrogenation: Chiral ligands and applications. Chem. Soc. Rev. 2006, 35, 226–236. [Google Scholar] [CrossRef]
- Zheng, C.; You, S.-L. Transfer hydrogenation with Hantzsch esters and related organic hydride donors. Chem. Soc. Rev. 2012, 41, 2498–2518. [Google Scholar] [CrossRef]
- Faísca Phillips, A.M.; Pombeiro, A.J.L. Recent advances in organocatalytic enantioselective transfer hydrogenation. Org. Biomol. Chem. 2017, 15, 2307–2340. [Google Scholar] [CrossRef]
- Zeng, L.; Huang, B.; Shen, Y.; Cui, S. Multicomponent synthesis of tetrahydroisoquinolines. Org. Lett. 2018, 20, 3460–3464. [Google Scholar] [CrossRef]
- Asif, M.M.A.; Lisa, S.R.; Qais, N. Introduction of chirality at C1 position of 1-substituted-3,4-dihydroisoquinoline by its enantioselective reduction: Synthesis of chiral 1-substituted-1,2,3,4-tetrahydroisoquinoline—A review. RSC Adv. 2023, 13, 11010–11036. [Google Scholar] [CrossRef]
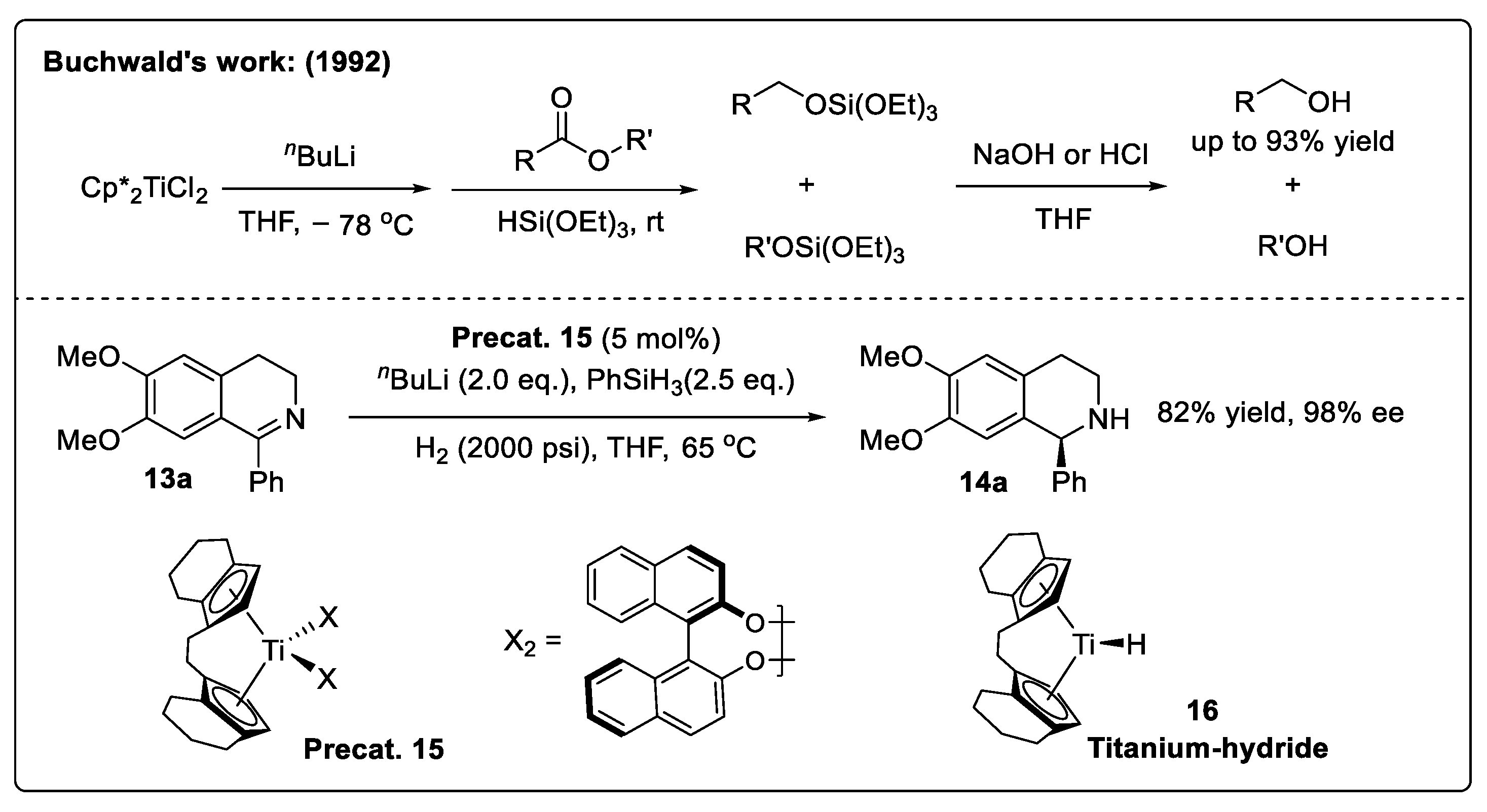
- Berk, S.C.; Kreutzer, K.A.; Buchwald, S.L. A catalytic method for the reduction of esters to alcohols. J. Am. Chem. Soc. 1991, 113, 5093–5095. [Google Scholar] [CrossRef]
- Willoughby, C.A.; Buchwald, S.L. Asymmetric titanocene-catalyzed hydrogenation of imines. J. Am. Chem. Soc. 1992, 114, 7562–7564. [Google Scholar] [CrossRef]
- Balázsik, K.; Szőllősi, G.; Berkesi, O.; Szalontai, G.; Fülöp, F.; Bartók, M. Heterogeneous asymmetric hydrogenation of N-heterocyclic compounds: Hydrogenation of tetrahydroisoquinoline derivatives. Top. Catal. 2012, 55, 880–888. [Google Scholar] [CrossRef]
- Ding, Z.-Y.; Wang, T.; He, Y.-M.; Chen, F.; Zhou, H.-F.; Fan, Q.-H.; Guo, Q.; Chan, A.S.C. Highly enantioselective synthesis of chiral tetrahydroquinolines and tetrahydroisoquinolines by ruthenium-catalyzed asymmetric hydrogenation in ionic liquid. Adv. Synth. Catal. 2013, 355, 3727–3735. [Google Scholar] [CrossRef]
- Li, C.; Xiao, J. Asymmetric hydrogenation of cyclic imines with an ionic cp*Rh(III) catalyst. J. Am. Chem. Soc. 2008, 130, 13208–13209. [Google Scholar] [CrossRef]
- Balakrishna, B.; Núñez-Rico, J.L.; Vidal-Ferran, A. Substrate activation in the catalytic asymmetric hydrogenation of N-heteroarenes. Eur. J. Org. Chem. 2015, 2015, 5293–5303. [Google Scholar] [CrossRef]
- Wen, J.; Tan, R.; Liu, S.; Zhao, Q.; Zhang, X. Strong brønsted acid promoted asymmetric hydrogenation of isoquinolines and quinolines catalyzed by a Rh–thiourea chiral phosphine complex via anion binding. Chem. Sci. 2016, 7, 3047–3051. [Google Scholar] [CrossRef]
- Morimoto, T.; Achiwa, K. An improved diphosphine-iridium(I) catalyst system for the asymmetric hydrogenation of cyclic imines: Phthalimide as an efficient co-catalyst. Tetrahedron Asymmetry 1995, 6, 2661–2664. [Google Scholar] [CrossRef]
- Adams, H.; Bailey, N.A.; Briggs, T.N.; McCleverty, J.A.; Colquhoun, H.M.; Williams, D.J. Transition metal complexes with terminal or bridging imidato(1-) ligands. X-ray crystal structures of trans-[Ir(CO)(NCOC2F4CO)(PPh3)2] and [{Pd(o-C6H4CH=NPh)(µ-NCOC2H4CO)}2]·CH2Cl2, spectroscopic studies of [Mn(CO)5(NCOC2H4CO)], and the nature of the metal–nitrogen bond. J. Chem. Soc. Dalton Trans. 1986, 813–819. [Google Scholar] [CrossRef]
- Yamagata, T.; Tadaoka, H.; Nagata, M.; Hirao, T.; Kataoka, Y.; Ratovelomanana-Vidal, V.; Genet, J.P.; Mashima, K. Oxidative addition of RCO2H and hx to chiral diphosphine complexes of iridium(I): Convenient synthesis of mononuclear halo-carboxylate iridium(III) complexes and cationic dinuclear triply halogen-bridged iridium(III) complexes and their catalytic performance in asymmetric hydrogenation of cyclic imines and 2-phenylquinoline. Organometallics 2006, 25, 2505–2513. [Google Scholar]
- Chang, M.; Li, W.; Zhang, X. A highly efficient and enantioselective access to tetrahydroisoquinoline alkaloids: Asymmetric hydrogenation with an iridium catalyst. Angew. Chem. Int. Ed. 2011, 50, 10679–10681. [Google Scholar] [CrossRef]
- Xie, J.-H.; Yan, P.-C.; Zhang, Q.-Q.; Yuan, K.-X.; Zhou, Q.-L. Asymmetric hydrogenation of cyclic imines catalyzed by chiral spiro iridium phosphoramidite complexes for enantioselective synthesis of tetrahydroisoquinolines. ACS Catal. 2012, 2, 561–564. [Google Scholar] [CrossRef]
- Togni, A. Planar-chiral ferrocenes: Synthetic methods and applications. Angew. Chem. Int. Ed. 1996, 35, 1475–1477. [Google Scholar] [CrossRef]
- Nie, H.; Zhu, Y.; Hu, X.; Wei, Z.; Yao, L.; Zhou, G.; Wang, P.; Jiang, R.; Zhang, S. Josiphos-type binaphane ligands for iridium-catalyzed enantioselective hydrogenation of 1-aryl-substituted dihydroisoquinolines. Org. Lett. 2019, 21, 8641–8645. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Liu, R.; Yang, J.; Luo, J.; Yao, L.; Li, M.; Zheng, X.; Jiang, R.; Nie, H.; Zhang, S. Iridium-catalyzed asymmetric hydrogenation of sterically hindered cyclic imines for enantioselective synthesis of tetrahydroisoquinolines. Org. Lett. 2021, 23, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhou, G.; Zhang, D.; Yao, L.; Li, M.; Yang, G.; Zhang, S.; Nie, H. Spiro-Josiphos ligands for the Ir-catalyzed asymmetric synthesis of chiral amines under hydrogenation conditions. Org. Lett. 2024, 26, 2097–2102. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Wang, J.; Chen, M.; Shi, L.; Zhou, Y. Dual stereocontrol for enantioselective hydrogenation of dihydroisoquinolines induced by tuning the amount of N-bromosuccinimide. Chin. J. Chem. 2018, 36, 139–142. [Google Scholar] [CrossRef]
- Facchetti, G.; Christodoulou, M.S.; Binda, E.; Fusè, M.; Rimoldi, I. Asymmetric hydrogenation of 1-aryl substituted-3,4-dihydroisoquinolines with iridium catalysts bearing different phosphorus-based ligands. Catalysts 2020, 10, 914. [Google Scholar] [CrossRef]
- Ružič, M.; Pečavar, A.; Prudič, D.; Kralj, D.; Scriban, C.; Zanotti-Gerosa, A. The development of an asymmetric hydrogenation process for the preparation of solifenacin. Org. Process Res. Dev. 2012, 16, 1293–1300. [Google Scholar] [CrossRef]
- Schwenk, R.; Togni, A. P-trifluoromethyl ligands derived from josiphos in the Ir-catalysed hydrogenation of 3,4-dihydroisoquinoline hydrochlorides. Dalton Trans. 2015, 44, 19566–19575. [Google Scholar] [CrossRef]
- Wang, D.-W.; Wang, X.-B.; Wang, D.-S.; Lu, S.-M.; Zhou, Y.-G.; Li, Y.-X. Highly enantioselective iridium-catalyzed hydrogenation of 2-benzylquinolines and 2-functionalized and 2,3-disubstituted quinolines. J. Org. Chem. 2009, 74, 2780–2787. [Google Scholar] [CrossRef]
- Kita, Y.; Yamaji, K.; Higashida, K.; Sathaiah, K.; Iimuro, A.; Mashima, K. Enhancing effects of salt formation on catalytic activity and enantioselectivity for asymmetric hydrogenation of isoquinolinium salts by dinuclear halide-bridged iridium complexes bearing chiral diphosphine ligands. Chem. Eur. J. 2015, 21, 1915–1927. [Google Scholar] [CrossRef]
- Berhal, F.; Wu, Z.; Zhang, Z.; Ayad, T.; Ratovelomanana-Vidal, V. Enantioselective synthesis of 1-aryl-tetrahydroisoquinolines through iridium catalyzed asymmetric hydrogenation. Org. Lett. 2012, 14, 3308–3311. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Feng, G.-S.; Chen, M.-W.; Shi, L.; Du, H.; Zhou, Y.-G. Iridium-catalyzed asymmetric hydrogenation of cyclic iminium salts. Org. Chem. Front. 2017, 4, 1125–1129. [Google Scholar] [CrossRef]
- Yan, P.-C.; Xie, J.-H.; Hou, G.-H.; Wang, L.-X.; Zhou, Q.-L. Enantioselective synthesis of chiral tetrahydroisoquinolines by iridium-catalyzed asymmetric hydrogenation of enamines. Adv. Synth. Catal. 2009, 351, 3243–3250. [Google Scholar] [CrossRef]
- Lu, S.-M.; Wang, Y.-Q.; Han, X.-W.; Zhou, Y.-G. Asymmetric hydrogenation of quinolines and isoquinolines activated by chloroformates. Angew. Chem. Int. Ed. 2006, 45, 2260–2263. [Google Scholar] [CrossRef] [PubMed]
- Alexakis, A.; Amiot, F. Enantioselective addition of organolithium reagents on isoquinoline. Tetrahedron Asymmetry 2002, 13, 2117–2122. [Google Scholar] [CrossRef]
- Ye, Z.-S.; Guo, R.-N.; Cai, X.-F.; Chen, M.-W.; Shi, L.; Zhou, Y.-G. Enantioselective iridium-catalyzed hydrogenation of 1- and 3-substituted isoquinolinium salts. Angew. Chem. Int. Ed. 2013, 52, 3685–3689. [Google Scholar] [CrossRef]
- Iimuro, A.; Yamaji, K.; Kandula, S.; Nagano, T.; Kita, Y.; Mashima, K. Asymmetric hydrogenation of isoquinolinium salts catalyzed by chiral iridium complexes: Direct synthesis for optically active 1,2,3,4-tetrahydroisoquinolines. Angew. Chem. Int. Ed. 2013, 52, 2046–2050. [Google Scholar] [CrossRef]
- Li, B.; Wang, J.; Li, X.; Xi, J.; Wang, R.; Zhang, D.; Zheng, X.; Nie, H.; Zhang, S. Iridium-catalyzed enantioselective hydrogenation of 3-substituted isoquinolinium salts. Asian J. Org. Chem. 2021, 10, 2370–2373. [Google Scholar] [CrossRef]
- Guo, R.-N.; Cai, X.-F.; Shi, L.; Ye, Z.-S.; Chen, M.-W.; Zhou, Y.-G. An efficient route to chiral N-heterocycles bearing a C–F stereogenic center via asymmetric hydrogenation of fluorinated isoquinolines. Chem. Commun. 2013, 49, 8537–8539. [Google Scholar] [CrossRef]
- Chen, M.-W.; Ji, Y.; Wang, J.; Chen, Q.-A.; Shi, L.; Zhou, Y.-G. Asymmetric hydrogenation of isoquinolines and pyridines using hydrogen halide generated in situ as activator. Org. Lett. 2017, 19, 4988–4991. [Google Scholar] [CrossRef]
- Kim, A.N.; Ngamnithiporn, A.; Welin, E.R.; Daiger, M.T.; Grünanger, C.U.; Bartberger, M.D.; Virgil, S.C.; Stoltz, B.M. Iridium-catalyzed enantioselective and diastereoselective hydrogenation of 1,3-disubstituted isoquinolines. ACS Catal. 2020, 10, 3241–3248. [Google Scholar] [CrossRef] [PubMed]
- Ngamnithiporn, A.; Welin, E.R.; Pototschnig, G.; Stoltz, B.M. Evolution of a synthetic strategy toward the syntheses of bis-tetrahydroisoquinoline alkaloids. Acc. Chem. Res. 2024, 57, 1870–1884. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Ye, Z.-S.; Cao, L.-L.; Guo, R.-N.; Hu, Y.; Zhou, Y.-G. Enantioselective iridium-catalyzed hydrogenation of 3,4-disubstituted isoquinolines. Angew. Chem. Int. Ed. 2012, 51, 8286–8289. [Google Scholar] [CrossRef] [PubMed]
- Nie, R.; Tao, Y.; Nie, Y.; Lu, T.; Wang, J.; Zhang, Y.; Lu, X.; Xu, C.C. Recent advances in catalytic transfer hydrogenation with formic acid over heterogeneous transition metal catalysts. ACS Catal. 2021, 11, 1071–1095. [Google Scholar] [CrossRef]
- Uematsu, N.; Fujii, A.; Hashiguchi, S.; Ikariya, T.; Noyori, R. Asymmetric transfer hydrogenation of imines. J. Am. Chem. Soc. 1996, 118, 4916–4917. [Google Scholar] [CrossRef]
- Vedejs, E.; Trapencieris, P.; Suna, E. Substituted isoquinolines by Noyori transfer hydrogenation: Enantioselective synthesis of chiral diamines containing an aniline subunit. J. Org. Chem. 1999, 64, 6724–6729. [Google Scholar] [CrossRef]
- Yamashita, Y.; Yasukawa, T.; Yoo, W.-J.; Kitanosono, T.; Kobayashi, S. Catalytic enantioselective aldol reactions. Chem. Soc. Rev. 2018, 47, 4388–4480. [Google Scholar] [CrossRef]
- He, Y.-M.; Fan, Q.-H. Phosphine-free chiral metal catalysts for highly effective asymmetric catalytic hydrogenation. Org. Biomol. Chem. 2010, 8, 2497–2504. [Google Scholar] [CrossRef]
- Sun, H.; Dai, P.; Tian, J.; Xu, Q.; Chen, Q.; Li, L.; Meng, X.; Zhang, L.; Li, C. Cinchona-alkaloid-derived NN ligands for ruthenium catalyzed asymmetric hydrogenation of ketones. Org. Biomol. Chem. 2023, 21, 5537–5541. [Google Scholar] [CrossRef]
- Wu, J.; Wang, F.; Ma, Y.; Cui, X.; Cun, L.; Zhu, J.; Deng, J.; Yu, B. Asymmetric transfer hydrogenation of imines and iminiums catalyzed by a water-soluble catalyst in water. Chem. Commun. 2006, 1766–1768. [Google Scholar] [CrossRef]
- Evanno, L.; Ormala, J.; Pihko, P.M. A highly enantioselective access to tetrahydroisoquinoline and β-carboline alkaloids with simple Noyori-type catalysts in aqueous media. Chem. Eur. J. 2009, 15, 12963–12967. [Google Scholar] [CrossRef] [PubMed]
- Přech, J.; Václavík, J.; Šot, P.; Pecháček, J.; Vilhanová, B.; Januščák, J.; Syslová, K.; Pažout, R.; Maixner, J.; Zápal, J.; et al. Asymmetric transfer hydrogenation of 1-phenyl dihydroisoquinolines using Ru(II) diamine catalysts. Catal. Commun. 2013, 36, 67–70. [Google Scholar] [CrossRef]
- Wu, Z.; Perez, M.; Scalone, M.; Ayad, T.; Ratovelomanana-Vidal, V. Ruthenium-catalyzed asymmetric transfer hydrogenation of 1-aryl-substituted dihydroisoquinolines: Access to valuable chiral 1-aryl-tetrahydroisoquinoline scaffolds. Angew. Chem. Int. Ed. 2013, 52, 4925–4928. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.; Wu, Z.; Scalone, M.; Ayad, T.; Ratovelomanana-Vidal, V. Enantioselective synthesis of 1-aryl-substituted tetrahydroisoquinolines through Ru-catalyzed asymmetric transfer hydrogenation. Eur. J. Org. Chem. 2015, 2015, 6503–6514. [Google Scholar] [CrossRef]
- Barrios-Rivera, J.; Xu, Y.; Wills, M. Asymmetric transfer hydrogenation of unhindered and non-electron-rich 1-aryl dihydroisoquinolines with high enantioselectivity. Org. Lett. 2020, 22, 6283–6287. [Google Scholar] [CrossRef]
- Szawkało, J.; Zawadzka, A.; Wojtasiewicz, K.; Leniewski, A.; Drabowicz, J.; Czarnocki, Z. First enantioselective synthesis of the antitumour alkaloid (+)-crispine A and determination of its enantiomeric purity by 1H NMR. Tetrahedron Asymmetry 2005, 16, 3619–3621. [Google Scholar] [CrossRef]
- Szawkało, J.; Czarnocki, S.J.; Zawadzka, A.; Wojtasiewicz, K.; Leniewski, A.; Maurin, J.K.; Czarnocki, Z.; Drabowicz, J. Enantioselective synthesis of some tetrahydroisoquinoline and tetrahydro-β-carboline alkaloids. Tetrahedron Asymmetry 2007, 18, 406–413. [Google Scholar] [CrossRef]
- Roszkowski, P.; Maurin, J.K.; Czarnocki, Z. Synthesis of new mono-N-tosylated diamine ligands based on (R)-(+)-limonene and their application in asymmetric transfer hydrogenation of ketones and imines. Tetrahedron Asymmetry 2013, 24, 643–650. [Google Scholar] [CrossRef]
- Mao, J.; Baker, D.C. A chiral rhodium complex for rapid asymmetric transfer hydrogenation of imines with high enantioselectivity. Org. Lett. 1999, 1, 841–843. [Google Scholar] [CrossRef]
- Facchetti, G.; Neva, F.; Coffetti, G.; Rimoldi, I. Chiral 8-amino-5,6,7,8-tetrahydroquinoline derivatives in metal catalysts for the asymmetric transfer hydrogenation of 1-aryl substituted-3,4-dihydroisoquinolines as alkaloids precursors. Molecules 2023, 28, 1907. [Google Scholar] [CrossRef]
- Blackmond, D.G.; Ropic, M.; Stefinovic, M. Kinetic studies of the asymmetric transfer hydrogenation of imines with formic acid catalyzed by Rh−diamine catalysts. Org. Process Res. Dev. 2006, 10, 457–463. [Google Scholar] [CrossRef]
- Yamakawa, M.; Ito, H.; Noyori, R. The metal−ligand bifunctional catalysis: A theoretical study on the ruthenium(II)-catalyzed hydrogen transfer between alcohols and carbonyl compounds. J. Am. Chem. Soc. 2000, 122, 1466–1478. [Google Scholar] [CrossRef]
- Noyori, R.; Yamakawa, M.; Hashiguchi, S. Metal−ligand bifunctional catalysis: A nonclassical mechanism for asymmetric hydrogen transfer between alcohols and carbonyl compounds. J. Org. Chem. 2001, 66, 7931–7944. [Google Scholar] [CrossRef] [PubMed]
- Václavíková Vilhanová, B.; Budinská, A.; Václavík, J.; Matoušek, V.; Kuzma, M.; Červený, L. Asymmetric transfer hydrogenation of 1-aryl-3,4-dihydroisoquinolines using a cp*Ir(TsDPEN) complex. Eur. J. Org. Chem. 2017, 2017, 5131–5134. [Google Scholar] [CrossRef]
- Shi, L.; Ji, Y.; Huang, W.-X.; Zhou, Y.-G. Application of chiral anion metathesis strategy in asymmetric transfer hydrogenation of isoquinolines. Acta Chim. Sin. 2014, 72, 820–824. [Google Scholar] [CrossRef]
- Murugesan, K.; Senthamarai, T.; Chandrashekhar, V.G.; Natte, K.; Kamer, P.C.J.; Beller, M.; Jagadeesh, R.V. Catalytic reductive aminations using molecular hydrogen for synthesis of different kinds of amines. Chem. Soc. Rev. 2020, 49, 6273–6328. [Google Scholar] [CrossRef]
- Irrgang, T.; Kempe, R. Transition-metal-catalyzed reductive amination employing hydrogen. Chem. Rev. 2020, 120, 9583–9674. [Google Scholar] [CrossRef]
- Reshi, N.U.D.; Saptal, V.B.; Beller, M.; Bera, J.K. Recent progress in transition-metal-catalyzed asymmetric reductive amination. ACS Catal. 2021, 11, 13809–13837. [Google Scholar] [CrossRef]
- Tian, Y.; Hu, L.a.; Wang, Y.-Z.; Zhang, X.; Yin, Q. Recent advances on transition-metal-catalysed asymmetric reductive amination. Org. Chem. Front. 2021, 8, 2328–2342. [Google Scholar] [CrossRef]
- Song, B.; Yu, C.-B.; Ji, Y.; Chen, M.-W.; Zhou, Y.-G. Synthesis of chiral sultams via palladium-catalyzed intramolecular asymmetric reductive amination. Chem. Commun. 2017, 53, 1704–1707. [Google Scholar] [CrossRef]
- Song, B.; Ji, Y.; Hu, S.-B.; Yu, C.-B.; Zhou, Y.-G. Synthesis of chiral γ-sultams through intramolecular reductive amination with sulfonylcarbamate as N-source. Tetrahedron Lett. 2017, 58, 1528–1530. [Google Scholar] [CrossRef]
- Chen, Z.-P.; Hu, S.-B.; Zhou, J.; Zhou, Y.-G. Synthesis of chiral trifluoromethyl-substituted hydrazines via Pd-catalyzed asymmetric hydrogenation and reductive amination. ACS Catal. 2015, 5, 6086–6089. [Google Scholar] [CrossRef]
- Chi, Y.; Zhou, Y.-G.; Zhang, X. Highly enantioselective reductive amination of simple aryl ketones catalyzed by Ir−f-binaphane in the presence of titanium(IV) isopropoxide and iodine. J. Org. Chem. 2003, 68, 4120–4122. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.D.; Pike, R.A.; Wade, C.E.; Wills, M. A one-pot process for the enantioselective synthesis of amines via reductive amination under transfer hydrogenation conditions. Org. Lett. 2003, 5, 4227–4230. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, Y.; Yang, S.; Zhou, L.; Chang, M. One-pot N-deprotection and catalytic intramolecular asymmetric reductive amination for the synthesis of tetrahydroisoquinolines. Angew. Chem. Int. Ed. 2017, 56, 2725–2729. [Google Scholar] [CrossRef]
- Yang, T.; Yin, Q.; Gu, G.; Zhang, X. A one-pot process for the enantioselective synthesis of tetrahydroquinolines and tetrahydroisoquinolines via asymmetric reductive amination (ARA). Chem. Commun. 2018, 54, 7247–7250. [Google Scholar] [CrossRef]
- Liu, R.; Han, J.; Li, B.; Liu, X.; Wei, Z.; Wang, J.; Wang, Q.; Jiang, R.; Nie, H.; Zhang, S. Enantioselective synthesis of tetrahydroisoquinolines via catalytic intramolecular asymmetric reductive amination. Org. Chem. Front. 2021, 8, 1930–1934. [Google Scholar] [CrossRef]
- Leithe, W. Über die natürliche drehung des polarisierten lichtes durch optisch aktive basen. Monash. Chem. 1929, 53, 956–962. [Google Scholar] [CrossRef]
- Naito, R.; Yonetoku, Y.; Okamoto, Y.; Toyoshima, A.; Ikeda, K.; Takeuchi, M. Synthesis and antimuscarinic properties of quinuclidin-3-yl 1,2,3,4-tetrahydroisoquinoline-2-carboxylate derivatives as novel muscarinic receptor antagonists. J. Med. Chem. 2005, 48, 6597–6606. [Google Scholar] [CrossRef]
- Zhu, R.; Xu, Z.; Ding, W.; Liu, S.; Shi, X.; Lu, X. Efficient and practical syntheses of enantiomerically pure (S)-(−)-norcryptostyline I, (S)-(−)-norcryptostyline II, (R)-(+)-salsolidine and (S)-(−)-norlaudanosine via a resolution-racemization method. Chin. J. Chem. 2014, 32, 1039–1048. [Google Scholar] [CrossRef]
- Faber, K. Non-sequential processes for the transformation of a racemate into a single stereoisomeric product: Proposal for stereochemical classification. Chem. Eur. J. 2001, 7, 5004–5010. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Zou, Y.; Xiao, W. Recent advances in photocatalytic deracemization. Chin. J. Org. Chem. 2022, 42, 3201–3212. [Google Scholar] [CrossRef]
- Großkopf, J.; Bach, T. Catalytic photochemical deracemization via short-lived intermediates. Angew. Chem. Int. Ed. 2023, 62, e202308241. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Pan, T.; Jiang, X.; Luo, S. Catalytic deracemization reactions. J. Am. Chem. Soc. 2023, 145, 10917–10929. [Google Scholar] [CrossRef]
- Qiao, K.-K.; Feng, G.-S.; Shi, L. Nonenzymatic catalytic deracemization. J. Catal. 2023, 422, 99–116. [Google Scholar] [CrossRef]
- Wang, J.; Lv, X.; Jiang, Z. Visible-light-mediated photocatalytic deracemization. Chem. Eur. J. 2023, 29, e202204029. [Google Scholar] [CrossRef]
- Wan, Q.; Wang, Z.; Sun, H.; Song, D.; Wang, X.; Liu, L. Enzymatic and chemo–catalytic redox deracemization: Recent progress. ChemCatChem 2024, 16, e202301757. [Google Scholar] [CrossRef]
- Zhu, D. Chemoenzymatic concurrent deracemization. In Chemo-Enzymatic Cascade Reactions; Wiley: Hoboken, NJ, USA, 2021; pp. 217–244. [Google Scholar]
- Prat, L.; Mojovic, L.; Levacher, V.; Dupas, G.; Quéguiner, G.; Bourguignon, J. Deracemization of diarylmethanes via lateral lithiation–protonation sequences by means of sparteine. Tetrahedron Asymmetry 1998, 9, 2509–2516. [Google Scholar] [CrossRef]
- Fehr, C. Enantioselective protonation of enolates and enols. Angew. Chem. Int. Ed. 1996, 35, 2566–2587. [Google Scholar] [CrossRef]
- Prat, L.; Dupas, G.; Duflos, J.; Quéguiner, G.; Bourguignon, J.; Levacher, V. Deracemization of alkyl diarylmethanes using (−)-sparteine or a chiral proton source. Tetrahedron Lett. 2001, 42, 4515–4518. [Google Scholar] [CrossRef]
- Ji, Y.; Shi, L.; Chen, M.-W.; Feng, G.-S.; Zhou, Y.-G. Concise redox deracemization of secondary and tertiary amines with a tetrahydroisoquinoline core via a nonenzymatic process. J. Am. Chem. Soc. 2015, 137, 10496–10499. [Google Scholar] [CrossRef] [PubMed]















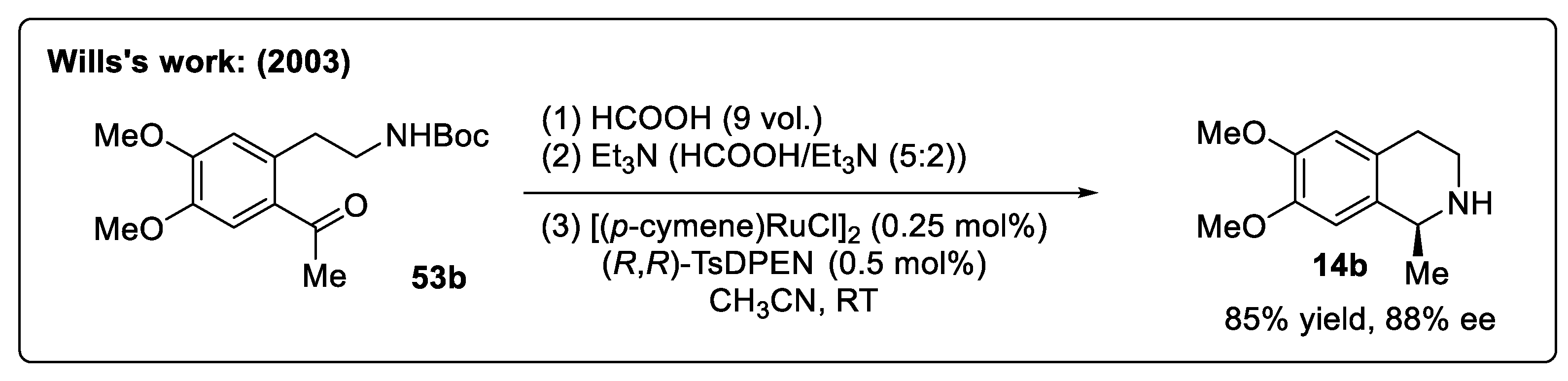

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | Catalyst | Reaction Conditions | Yield (%) | Ee (%) | Group (Year) | Ref. |
| 1 |  | [RuCl(C6H6)(S,S)-N-N* e] (10 mol%) | HCOOH/Et3N (2.5 mL, 5:2) CH3CN, 0 °C | 96 | 92 | Czarnocki (2005) | [79] |
| 2 | 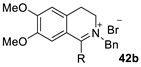 | [RuCl2(p-cymene)]2 /(R,R)-N-N* f (1 mol%/2.2 mol%) | HCOONa (5.0 eq.) CTAB (50 mol%) H2O, 28 °C | 86 (R = Me) 94 (R = Ph) | 90 (R = Me) 95 (R = Ph) | Zhu and Deng (2006) | [73] |
| 3 | 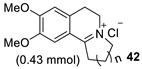 | [RuCl(C6H6)(S,S)-N-N* e] (10 mol%) | HCOOH/Et3N (2.5 mL, 5:2) CH3CN, 0 °C | 91 (n = 1) 92 (n = 2) | 92 (n = 1) 87 (n = 2) | Czarnocki (2007) | [80] |
| 4 | 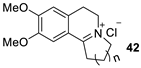 | [RuCl(p-cymene)(S,S)-N-N* a] (1.3 mol%), AgSbF6 (7 mol%) | HCOONa (15.0 eq.) CTAB (1.0 eq.), H2O, 40 °C | 45 (n = 1) 65 (n = 2) | 94 (n = 1) 96 (n = 2) | Pihko (2009) | [74] |
| 5 |  | [RuCl(benzene)((R,R)-N-N* h)] [RuCl(benzene)((R,R)-N-N* i)] (0.2 mol%) | HCOOH/Et3N (2.5 mL, 5:2) CH3CN, 0 °C | 72 (n = 1) 89 (n = 2) | 84 (n = 1) 56 (n = 2) | Czarnocki (2013) | [81] |
 | |||||||
| Entry | Compound | Chiral Acid | ee | Yield a | Entry | Compound | Chiral Acid | ee | Yield a |
|---|---|---|---|---|---|---|---|---|---|
| 1 |  | 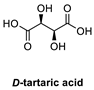 | >98% (S) | 33% | 2 | 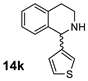 | 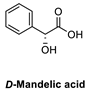 | >98% (S) | 20% |
| 3 | 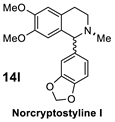 |  | >98% (S) | 45% | 4 | 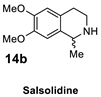 |  | >98% (R) | 41% |
| 5 | 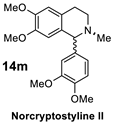 |  | >98% (S) | 40% | 6 |  | 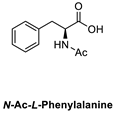 | >98% (S) | 38% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, Y.; Gao, Q.; Han, W.; Fang, B. Recent Advances in the Synthesis of Chiral Tetrahydroisoquinolines via Asymmetric Reduction. Catalysts 2024, 14, 884. https://doi.org/10.3390/catal14120884
Ji Y, Gao Q, Han W, Fang B. Recent Advances in the Synthesis of Chiral Tetrahydroisoquinolines via Asymmetric Reduction. Catalysts. 2024; 14(12):884. https://doi.org/10.3390/catal14120884
Chicago/Turabian StyleJi, Yue, Qiang Gao, Weiwei Han, and Baizeng Fang. 2024. "Recent Advances in the Synthesis of Chiral Tetrahydroisoquinolines via Asymmetric Reduction" Catalysts 14, no. 12: 884. https://doi.org/10.3390/catal14120884
APA StyleJi, Y., Gao, Q., Han, W., & Fang, B. (2024). Recent Advances in the Synthesis of Chiral Tetrahydroisoquinolines via Asymmetric Reduction. Catalysts, 14(12), 884. https://doi.org/10.3390/catal14120884