1. Introduction
Polyphosphate kinases (PPKs) use inorganic polyphosphate (PolyP) as a phosphate donor. PolyP is a high-energy phosphoanhydride-bound linear polymer consisting of tens or hundreds of orthophosphates (P
i), representing a rich source of energy [
1,
2]. While the linear form is the most common form in living organisms, a cyclic (metaphosphate) or branched (ultraphosphate) form can also occur [
3]. PolyP is found in all living organisms and has many biological functions, such as the generation of adenosine triphosphate (ATP) in kinase reactions, the storage of phosphate (P
i), the chelation of metal ions like Mg
2+, molecular chaperone activity on bacterial virulence, etc. [
2,
4,
5]. However, it also has industrial applications, for example as an antibacterial agent in processed meat, poultry or fish products or for the regeneration of the expensive substrate ATP using PPK in enzymatically catalysed reactions [
2,
6].
Due to the moderate price of PolyP (63 EUR∙kg
−1, sodium polyphosphate, Merck KGaA, retrieved: January 2024) as a substrate, the use of PPKs is particularly promising for the regeneration of expensive nucleotide triphosphates (NTPs) such as cytidine triphosphate (CTP) (12,130 EUR∙kg
−1, cytidine 5′-triphosphate disodium salt, Biosynth, retrieved: January 2024). Other enzymes for CTP regeneration, such as pyruvate kinase with phosphoenolpyruvate [
7], acetate kinase with acetyl phosphate [
8] or creatine kinase with creatine phosphate [
9], have also been described [
10]. However, all these donors are much more expensive than PolyP.
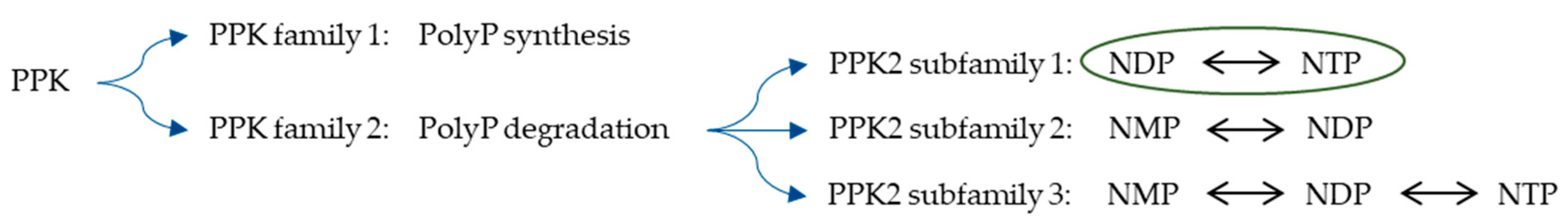
According to their functionality, PPKs can be divided into two families: PPK1 and PPK2 (
Figure 1) [
5,
11]. PPK1s preferentially synthesise long PolyP chains by the reversible transfer of the terminal P
i from ATP, but they also accept all other NTPs [
1,
4,
12]. PPK2s preferentially catalyse the degradation of PolyP to phosphorylate nucleotide monophosphate (NMP) or nucleotide diphosphate (NDP) and are classified into three subfamilies [
12,
13,
14]. Many genomes encode two or three paralogs of PPK2, most of which are one-domain PPK2s of approximately 230 residues or two-domain PPK2s of 496–544 residues [
15]. The one-domain PPK2s generate NTP from NDP and PolyP (class I subfamily), whereas the two-domain PPK2s generate NDP from NMP and PolyP (class II subfamily) [
15,
16]. In addition, a PPK2 class III subfamily with one-domain PPK2s catalyses both NMP and NDP phosphorylation with PolyP [
15]. In this study, an NDP polyphosphate phosphotransferase from the bacterium
Ruegeria pomeroyi (EC 2.7.4.1, gene: SPO1727, UniProt KB: Q5LSN8) is examined. This enzyme was first characterised by Achbergová and Nahálka in 2014 and named
RpPPK2-3 [
17]. The abbreviation
RpPPK2-3 erroneously suggests that it is a PPK2 subfamily III. However, no NMP phosphorylation was detected.
RpPPK2-3 belongs to the PPK2 family, a class I subfamily based on the classification that was proposed by Motomura and co-workers as well in 2014 [
15].
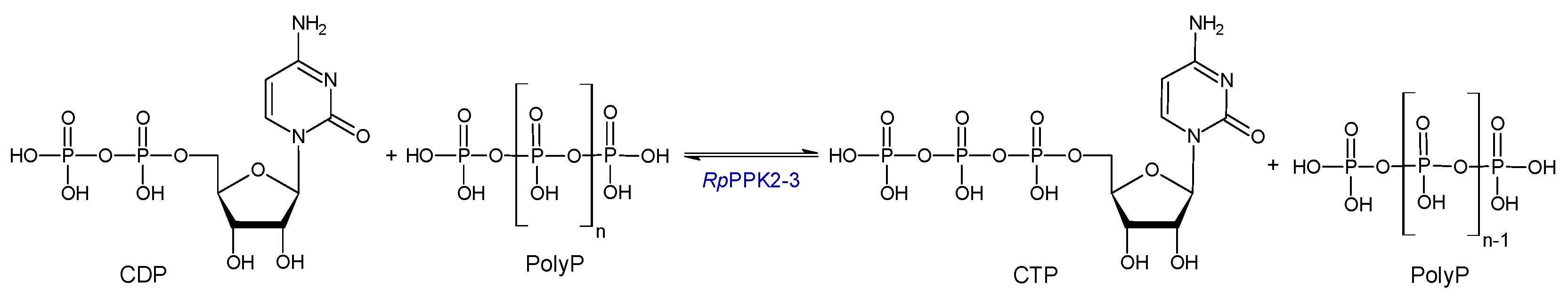
RpPPK2-3 converts NDP to NTP using PolyP as a donor and accepts adenosine diphosphate (ADP), cytidine diphosphate (CDP), uridine diphosphate (UDP), guanosine diphosphate (GDP) and deoxythymidine diphosphate (dTDP) with a preference for UDP, GDP and CDP [
17,
18,
19]. In this study, the conversion of CDP and PolyP to CTP is investigated (
Scheme 1).
The use of
RpPPK2-3 in biocatalytic synthesis has already been described in the literature, and examples are given below. Due to its acceptance of all NDPs, different methods for its utilisation are possible. Nahálka et al. [
18] described the use of
RpPPK2-3 and cytidine 5′-monophosphate kinase (CMP kinase) as an active inclusion body system for CTP regeneration starting from CMP combined with whole cells, co-expressing two enzymes for the synthesis of CMP-sialic-acid. Gottschalk et al. [
19] developed a one-pot synthesis for hyaluronic acid with a combined ATP and UTP regeneration using
RpPPK2-3. The use of
RpPPK2-3 provided a solution for the limitations of ATP, ADP and UDP inhibition [
19]. His-tag-immobilised enzymes on Ni
2+/nitrilotriacetic acid magnetic beads were used, and almost the highest possible loading capacity according to the manufacturer’s specifications for the immobilised
RpPPK2-3 with an increased maximum specific activity was achieved [
20]. Mahour et al. [
21] described the use of
RpPPK2-3 to regenerate ATP and UTP in a five-enzyme cell-free cascade to produce UDP-GlcNAc.
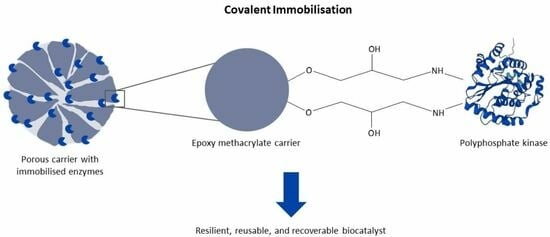
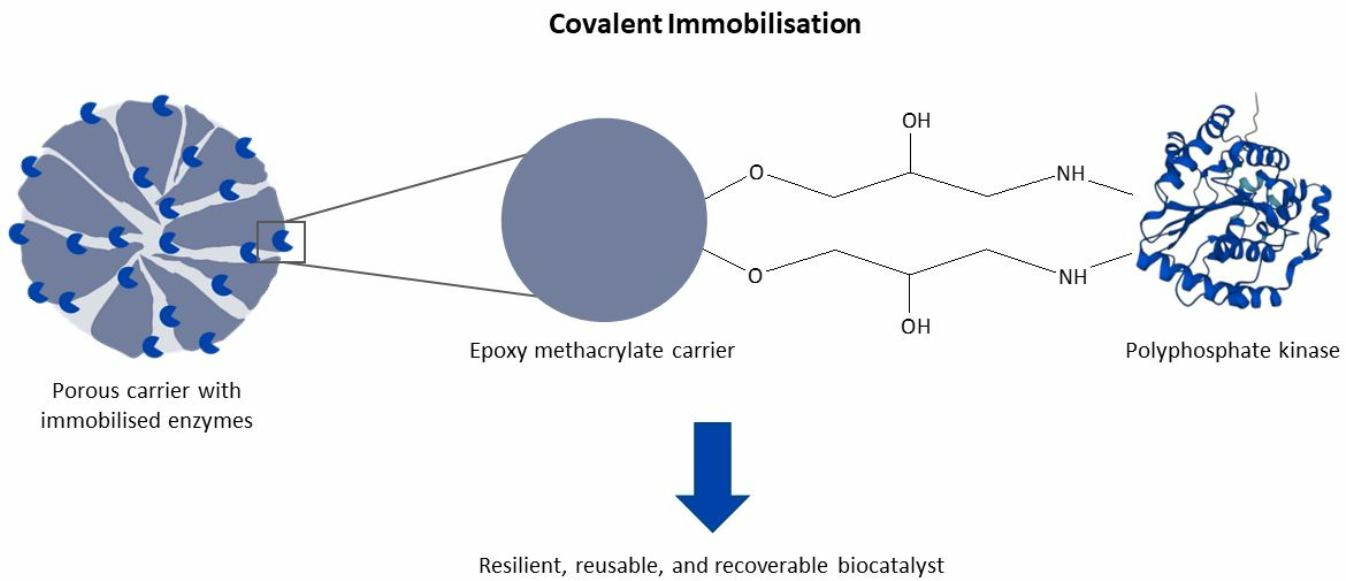
The industrial application of enzymes is often challenged by a lack of long-term stability and the difficulty of enzyme recovery and reuse [
22]. Immobilisation can increase enzyme stability and positively influence their specificity and selectivity and can also potentially reduce inhibition [
23]. The application of immobilised enzymes in different reactor configurations, such as continuous packed-bed reactors (PBR), is another advantage [
23]. The PBR reactor configuration can prevent product contamination by enzymes, allowing for the use of high enzyme concentrations, as there is no risk of enzyme aggregation, and for reducing enzyme costs due to enzyme reusability. Furthermore, it allows for the conversion under high pressures, and the performance of multi-enzyme cascades with co-immobilised enzymes is such that even cofactor regeneration can be implemented [
23,
24]. Thus, immobilisation could offer many advantages for the development of low-cost production processes. Enzyme immobilisation can be achieved by carrier binding, encapsulation or cross-linking [
22]. Covalent carrier binding offers the advantage of stable bonds that prevent leaching of the enzyme as well as its easy recovery and reuse [
23,
25]. For continuous-flow reactors, immobilisation on the reactor wall or on carrier materials such as particles or monoliths has been demonstrated [
25]. In this study, epoxy methacrylate particles with glutaraldehyde-pre-activated amino methacrylate particles as supports were used to covalently immobilise
RpPPK2-3. The covalently immobilised
RpPPK2-3 was then used in a CTP regeneration system in a multi-enzyme cascade performed in a PBR to synthesise the human milk oligosaccharide (HMO) sialyllactose.
3. Materials and Methods
3.1. Chemicals
The following chemicals were used in this study with the following manufacturers and order number: acetonitrile (VWR International GmbH, Darmstadt, Germany, 83640.320), albumin from bovine serum (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany, A7906), amino C2 methacrylate (Purolite Ltd., Wales, UK, ECR8309M), amino C6 methacrylate (Purolite Ltd., ECR8409M), Bradford reagent (Sigma-Aldrich, B6916), cytidine 5′-triphosphate disodium salt (Shanghai Tianqi Chemical Limited, Zhengzhou, China), cytidine 5′-diphosphate disodium salt (Biosynth Ltd., Compton, UK, NC09380), dipotassium hydrogen phosphate (Carl Roth GmbH & Co. KG, Karlsruhe, Germany, 6875.1), disodium hydrogen phosphate dihydrate (VWR, 28029.260), epoxy/butyl methacrylate (Purolite Ltd., ECR8285), epoxymethacrylate (Purolite Ltd., ECR8209M/ECR8204M), glutaraldehyde (Carl Roth, 3778.1), 2-propanol (VWR, 20880.320), magnesium chloride hexahydrate (Sigma-Aldrich, M9272), potassium dihydrogen phosphate (Carl Roth, 3904.1), sodium chloride (VWR, 27810.295), sodium phosphate monobasic monohydrate (Sigma-Aldrich, 71507), sodium polyphosphate (Merck KGaA, Darmstadt, Germany, 106529), tetrabutylammonium bromide (Carl Roth, 6633.3) and Tris (AppliChem GmbH, Darmstadt, Germany, A1379). All chemicals were of analytical grade.
3.2. Expression and Purification
The gene for
RpPPK2-3 (SPO1727) cloned into the pET22-b(+) expression vector was kindly provided by the RWTH Aachen University, Laboratory for Biomaterials [
19]. The
E. coli BL21(DE3) was transformed with the plasmid using heat shock. The expression of the C-terminal hexahistidine (His6)-tagged
RpPPK2-3 was performed as a fed-batch cultivation in a bioreactor with a working volume of up to 2 L. Immobilised metal affinity chromatography (IMAC) was used for enzyme purification. The buffer was exchanged to 50 mmol∙L
−1 sodium phosphate buffer at pH 7.4, performed by dialysis or tangential flow filtration, and the enzyme was lyophilised and stored at −20 °C. The purity of the enzyme was qualified by SDS-PAGE. The enzyme concentration was determined by a Bradford assay using albumin from bovine serum (BSA) as a reference.
3.3. Immobilisation
Epoxy- and amino-functionalised carriers were used for the covalent immobilisation of the
RpPPK2-3 (
Table 6). Epoxy-functionalised carriers form stable covalent linkages with the thiol, amino, carboxylic or phenolic groups of the enzyme. The amino-functionalised carriers were pre-activated with glutaraldehyde. The terminal amino groups of the enzyme formed multipoint covalent bonds with the aldehyde group of the resin, resulting in an imino bond [
32]. Immobilisation was performed according to the manufacturer’s instructions [
36] and as described in previous studies [
37]. A sodium phosphate buffer at pH 7.4 with a concentration ranging from 20 to 1000 mmol∙L
−1 was used as the immobilisation buffer for the epoxy-functionalised carriers and a concentration of 20 mmol∙L
−1 for the amino-functionalised carriers. For the mixing during the immobilisation process, a rotary mixer (Sample Mixer MXICI, Dynal) was used. For vacuum filtration, a vacuum pump (Chemistry diaphragm pump ME 2C NT, Vacuubrand GmbH + Co. KG, Wertheim, Germany), a filter attachment (Bottletop-filter, Thermo Fisher Scientific Inc., Waltham, MA, USA) and a membrane filter (Labsolute
® CA membrane filter, Th. Geyer GmbH & Co. KG, Renningen, Germany) was used. The carrier was equilibrated by washing 3 times with immobilisation buffer with a carrier-to-buffer ratio of 1:2 (
w/
v). To pre-activate the amino methacrylate carriers, an additional step was performed with a 1% glutaraldehyde (
v/
v) solution in immobilisation buffer with a ratio of 1:4 (
w/
v). After the addition of the 1% glutaraldehyde solution, mixing was carried out for 1 h at room temperature and at 10 rpm. The pre-activated amino methacrylate carriers were then washed 3 times with immobilisation buffer (ratio of 1:4 (
w/
v)). A 5 mg∙mL
−1 enzyme solution in immobilisation buffer was added to the filtered equilibrated and pre-activated carriers with an enzyme loading of 100 mg
ezyme∙g
carier−1 and was mixed at 20 °C and at 10 rpm for 18 h. The epoxy methacrylate carrier was additionally stored for 20 h at 20 °C without mixing. The immobilisates were washed with immobilisation buffer containing 500 mmol∙L
−1 sodium chloride for the desorption of non-covalently bound proteins and stored in immobilisation buffer at 6 °C. The specific enzyme activity and enzyme concentration (Bradford assay) was determined in the enzyme solution before and after immobilisation. The carrier specific activity was determined with the immobilisate.
3.3.1. Screening of Different Carriers
To select a suitable support, a screening with five different supports was performed (
Table 6). To obtain a stable immobilisate, carrier materials with a covalent bond toward the enzyme were selected. The support materials differed regarding their functional group, pore and particle diameter, spacer length and hydrophobicity. The
RpPPK2-3 was immobilised on all the supports and compared in terms of the immobilisation yield, immobilisation efficiency, activity yield, carrier loading and carrier specific activity. In addition, the stability of the immobilised enzyme was determined in a reusability and storage study. The immobilisation buffer used for the epoxy-functionalised carriers was a 1000 mmol∙L
−1 sodium phosphate buffer at pH 7.4, and for the amino-functionalised carriers, a 20 mmol∙L
−1 sodium phosphate buffer at pH 7.4 was used.
3.3.2. Optimisation of the Immobilisation Process
To determine the effect of the buffer concentration on the immobilisation efficiency, the immobilisation was performed with different buffer concentrations. Four immobilisations with sodium phosphate buffer concentrations of 20, 250, 500 and 1000 mmol∙L−1 were performed and compared in terms of the immobilisation yield, immobilisation efficiency, activity yield, carrier loading and carrier specific activity.
3.3.3. Calculation of Characteristic Parameters of the Immobilisation Process
The quality of enzyme immobilisation can be assessed by characteristic parameters describing both the immobilisation process and the successfully immobilised enzyme. To characterise the immobilisation process, the immobilisation yield, immobilisation efficiency and activity yield were calculated as described by Syldatk et al. [
38]. The immobilisation yield (
YImmo) describes the percentage of the theoretical maximum enzymatic activity (
EAtotal) (Equation (1)).
EAfree is the activity remaining in the enzyme solution after immobilisation. The immobilisation efficiency (
EffImmo) is the percentage of the measured immobilised activity (apparent activity,
EAapp) of the theoretical immobilised activity (Equation (2)).
The activity yield (
YEA) describes the percentage of the apparent activity (
EAapp) from the theoretical maximum activity (
EAtotal) and is the product of the immobilisation yield and the immobilisation efficiency (Equation (3)).
The activity of the enzyme solution and the immobilisate were measured using the activity assay described in
Section 3.4. The enzyme concentration was determined by a Bradford assay using bovine serum albumin (BSA) as a reference.
3.4. Activity Assay
The activity assay was performed in duplicate in a 2 mL reaction tube using a thermoshaker with a reaction volume of 1 mL for the soluble enzyme and 1.5 mL for the immobilised enzyme, respectively. The reaction conditions were 20 °C with a rotation speed of 500 rpm for the soluble enzyme and 1000 rpm for the immobilised enzyme, respectively. The reaction contained 5 mmol∙L
−1 CDP, 7.3 g∙L
−1 PolyP, 30 mmol∙L
−1 MgCl
2, 50 mmol∙L
−1 Tris at pH 7.8 and 4 mg∙L
−1 free enzyme or 7 g∙L
−1 immobilised enzyme. The reaction was stopped after 2.5 min with 2-propanol. CDP and CTP were quantified on an Agilent 1100 series HPLC (Agilent Technologies, Santa Clara, USA). A Phenomenex Luna 3u C18(2) 100 Å (150 × 4.6 mm, 3 µm particles) column was used at 40 °C and at a flow rate of 0.8 mL∙min
−1. The isocratic eluent consisted of 46% acetonitrile and 54% 20 mmol∙L
−1 potassium phosphate buffer containing 20 mmol∙L
−1 tetrabutylammonium bromide (TBAB) as an ion-pair reagent at pH 5.9. Cytidine nucleotides were detected at 272 nm (variable wavelength detector (VWD, Agilent Technologies)) with a retention time of 2.1 (CMP), 2.5 (CDP) and 2.9 min (CTP) (
Figure S5).
3.5. Stability Studies
3.5.1. Stabilisation of the Soluble RpPPK2-3 by Additives
To investigate the influence of PolyP, pyrophosphate (PP
i) and Mg
2+ on the stability of the
RpPPK2-3, a storage stability study was performed. To analyse the stability without the addition of additives, the
RpPPK2-3 was stored in 50 mmol∙L
−1 Tris at pH 7.8 with an enzyme concentration of 0.04 g∙L
−1 at 22 °C for 3 h in a 1.5 mL reaction tube. To investigate the influence of additives on enzyme stability, additional analyses were performed with added PP
i, PolyP and PolyP with Mg
2+. PP
i was added at a concentration of 10 mmol∙L
−1, PolyP at 8.5 g∙L
−1 and MgCl
2 at 33 mmol∙L
−1. The
RpPPK2-3 was stored in 50 mmol∙L
−1 Tris at pH 8.0 with an enzyme concentration of 0.04 g∙L
−1 at 30 °C for up to 45 d in a 1.5 mL reaction tube on a thermoshaker. The activities were analysed according to
Section 3.4.
3.5.2. Stability of the Soluble RpPPK2-3 in Sodium Phosphate Buffer
To analyse the stability of the
RpPPK2-3 in sodium phosphate buffer, a storage stability study was performed. The enzyme was stored in sodium phosphate buffer with concentrations of 20, 250, 500 or 1000 mmol∙L
−1 at pH 7.8 with an enzyme concentration of 0.04 g∙L
−1 at 20 °C and 300 rpm in a 2 mL reaction tube on a thermoshaker. The activity was measured according to
Section 3.4 at the beginning of the study and after 3 days of storage.
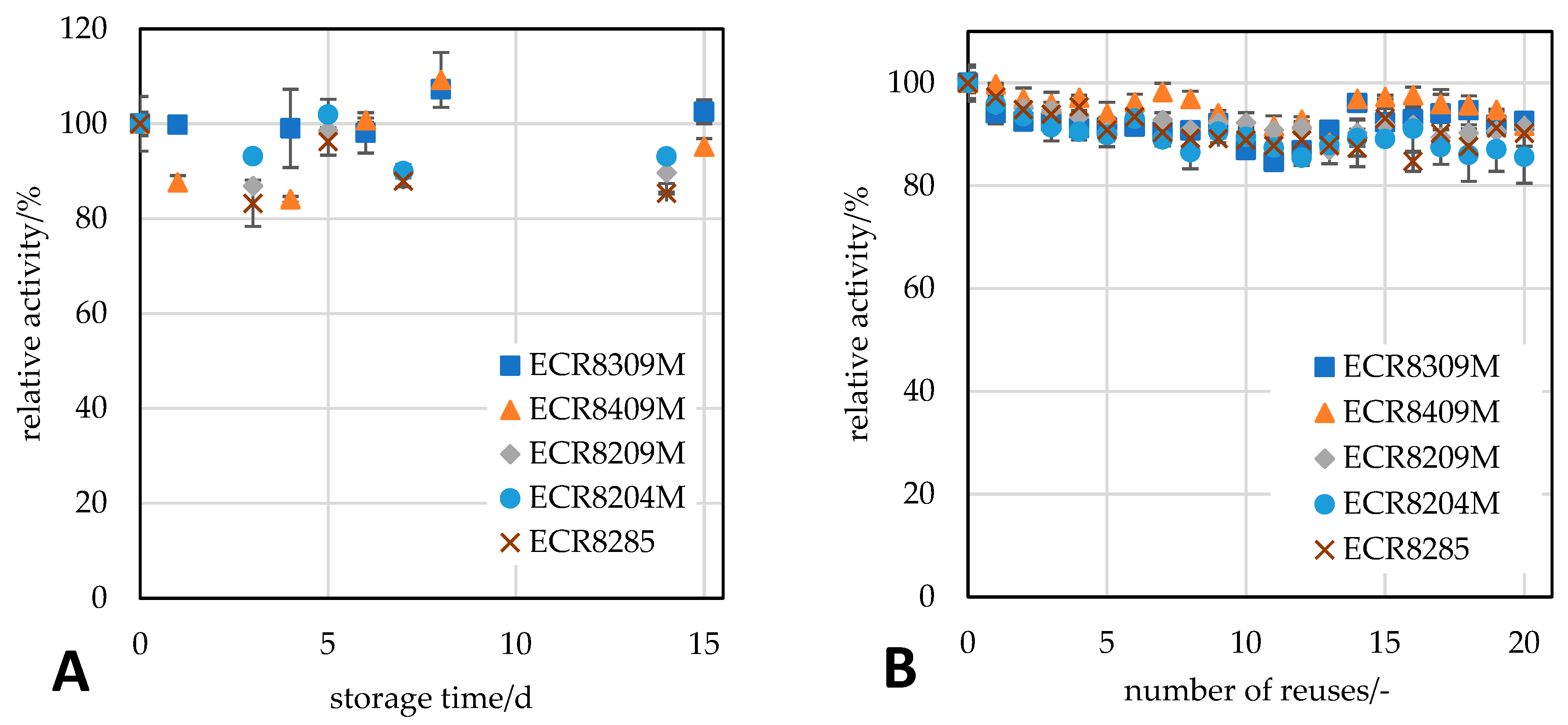
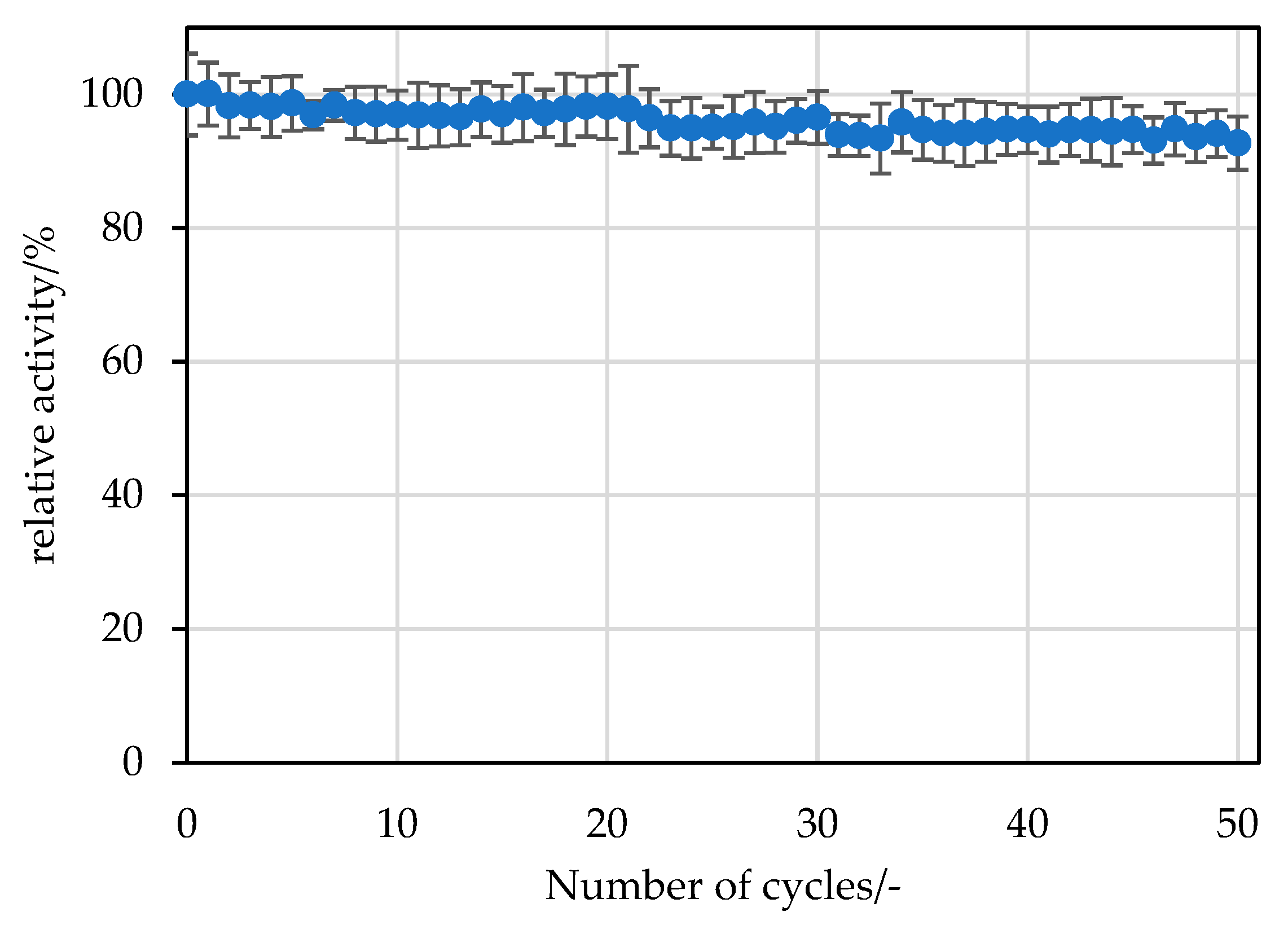
3.5.3. Reusability Studies of the Immobilisate
The reusability was analysed in duplicate with 10 mg of immobilised
RpPPK2-3 in a 2 mL reaction tube using a thermoshaker. The reaction was started by adding 1.5 mL of substrate solution (see
Section 3.4). After 15 min, a sample of the reaction mixture was analysed for product formation at a conversion of 25% at the first use. The remaining substrate was then pipetted off, and the supports were washed twice with 20 mmol∙L
−1 sodium phosphate buffer at pH 7.4 and used for the next cycle or stored at 6 °C until the next experiment. For the next experiment, the wash buffer was removed by pipetting, and the experiment was started by adding the substrate as described above. For the screening experiment with different carriers, 20 replicate batches were performed. For the selected carrier (ECR8209M), 50 replicate batches were performed.
3.5.4. Storage Stability Studies of the Immobilisate
For the stability studies of the immobilisate, aliquots containing 10 mg of immobilised
RpPPK2-3 in 2 mL reaction tubes were stored under six different conditions: in 1 mL of 20 mmol∙L
−1 sodium phosphate buffer at pH 7.4 or water at 6 °C, in 1 mL of 20 mmol∙L
−1 sodium phosphate buffer at pH 7.4 at 20 °C with and without shaking at 300 rpm, and in 1 mL of 20 mmol∙L
−1 sodium phosphate buffer at pH 7.4 at 40 °C. The aliquots were stored for 200 days, and the activity was measured at regular time points over the storage period. At each time point, the initial activity was analysed using the standard activity assay (
Section 3.4). The stability was assessed by the half-life, which is the time taken to halve the initial activity. The deactivation constant k
d was determined by exponential fitting, and the half-life time was calculated according to the following equation [
39]:


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}