Abstract
The search for active, inexpensive, and stable heterogeneous catalysts to produce desired imines in fine chemistry presents an ongoing challenge for both academia and industry. This work reports the utilization of Co nanoparticles supported on TiO2 derived from the H2-assisted reduction of the perovskite-type mixed oxide CoTiO3. The entire preparation process is operationally simple and straightforward, enabling scalability for practical applications. The resulting catalyst comprises metallic cobalt nanoparticles responsible for the hydrogenation process, whereas the TiOx thin layer surrounding the cobalt promotes the adsorption of C=O, thereby enhancing the formation of desired products. Notably, at lower temperatures, the reaction yields the target imine product. Our study demonstrates a synergistic effect between nitrobenzene and benzaldehyde in the presence of a Co-TiOx interface, which reduces the apparent activation energy for the hydrogenation of the-NO2 group. Furthermore, under moderate reaction conditions, the catalytic system offers applicability to various nitrobenzene compounds substituted at the 4-position and benzaldehyde, resulting in high yields of the corresponding imines with electron-density-donating substituent groups. Finally, the catalyst exhibits facile separation for subsequent reuse, displaying moderate stability with minimal selectivity for the desired product.
1. Introduction
Aromatic imines are widely employed as organic intermediates in the production of pharmaceuticals, agrochemicals, and other industrial chemical agents [1,2,3]. Traditionally, these compounds are synthesized through various methods, such as amine condensation with aldehydes, oxidative dehydrogenation of secondary amines using O2, amine self-condensation, and alcohol and or amine condensation [4,5,6]. However, these processes of-ten require environmentally incompatible reaction conditions and result in imine products with low selectivity. An emerging synthetic strategy involves the use of nitroarenes as starting materials, employing heterogeneous catalytic hydrogenation to obtain the respective amines, followed by in situ condensation with an aldehyde or ketone, which has garnered interest in fine chemistry.
The reductive imination of nitroarene–carbonyl compounds is considered a sustainable and cost-effective synthetic protocol due to its high atomic economy and the ready availability of substrates, which combined, minimize toxic waste. The reductive imination of carbonyl compounds with nitroarenes has been studied using Au/TiO2 [7], Pd/Al2O3 [8], Ni/SiO2 [9], and γ-NiSn3/TiO2 [5]. However, these catalysts often exhibit low chemoselectivity, leading to competitive hydrogenation of the C=O group, and overhydrogenation of the C=N bond, yielding N-alkylated amines. In pursuit of economic and sustainable alternatives, non-noble metal-based heterogeneous catalysts have attracted considerable attention because of their high abundance, low cost, low toxicity, high stability, and ease of separation. Schwob et al. demonstrated that Co-based heterogeneous catalysts supported on N-doped carbon (Co@CN) exhibit notable selectivity in the mediation of reductive imination by employing nitroarenes and several aldehydes/ketones [10]. Chen et al. tested NiCo alloys supported on La2O3 for the reductive imination of nitrobenzene and benzaldehyde, demonstrating a clear dependence of the metal content of Co in the alloy and operating conditions on the selectivity toward imine formation [11]. Another interesting study regarding the use of Co in formulations targeted at reductive imination was reported by Zhang et al., who demonstrated that the electronic nature of PtCo alloys on the surface of a CoBOx support controls the selectivity in the production of the respective imine via the preferential adsorption of the -NO2 group versus C=O and C=N during catalytic performance [12]. In addition, Gong et al. demonstrated that a monometallic Co-based catalyst facilitated the one-pot reductive imination of aldehydes with nitroarenes to produce the desired product under mild conditions; concurrently, the Co active phase prevents the hydrogenation of C=O in the production of the corresponding alcohol [13].
The use of Co-supported carriers, responsible for the strong metal support interaction (SMSI) effect, offers another alternative for enhancing the selectivity of the catalyst to ensure an efficient discrimination reaction with relatively strongly bound aldehydes and target imines on the catalyst surface, and preventing their hydrogenation [14,15,16]. The SMSI effect is observed in metals interacting with partially reducible oxides that, when prepared in a reducing atmosphere at temperatures above 300 °C, partially cover the metallic phase, promoting C=O adsorption [17]. In this scenario, the Co active phase displays a low hydrogenation capacity under moderate reaction conditions for aromatic aldehydes, ultimately reducing their side-hydrogenation into aromatic alcohols and avoiding the over-hydrogenation of the imine to amine.
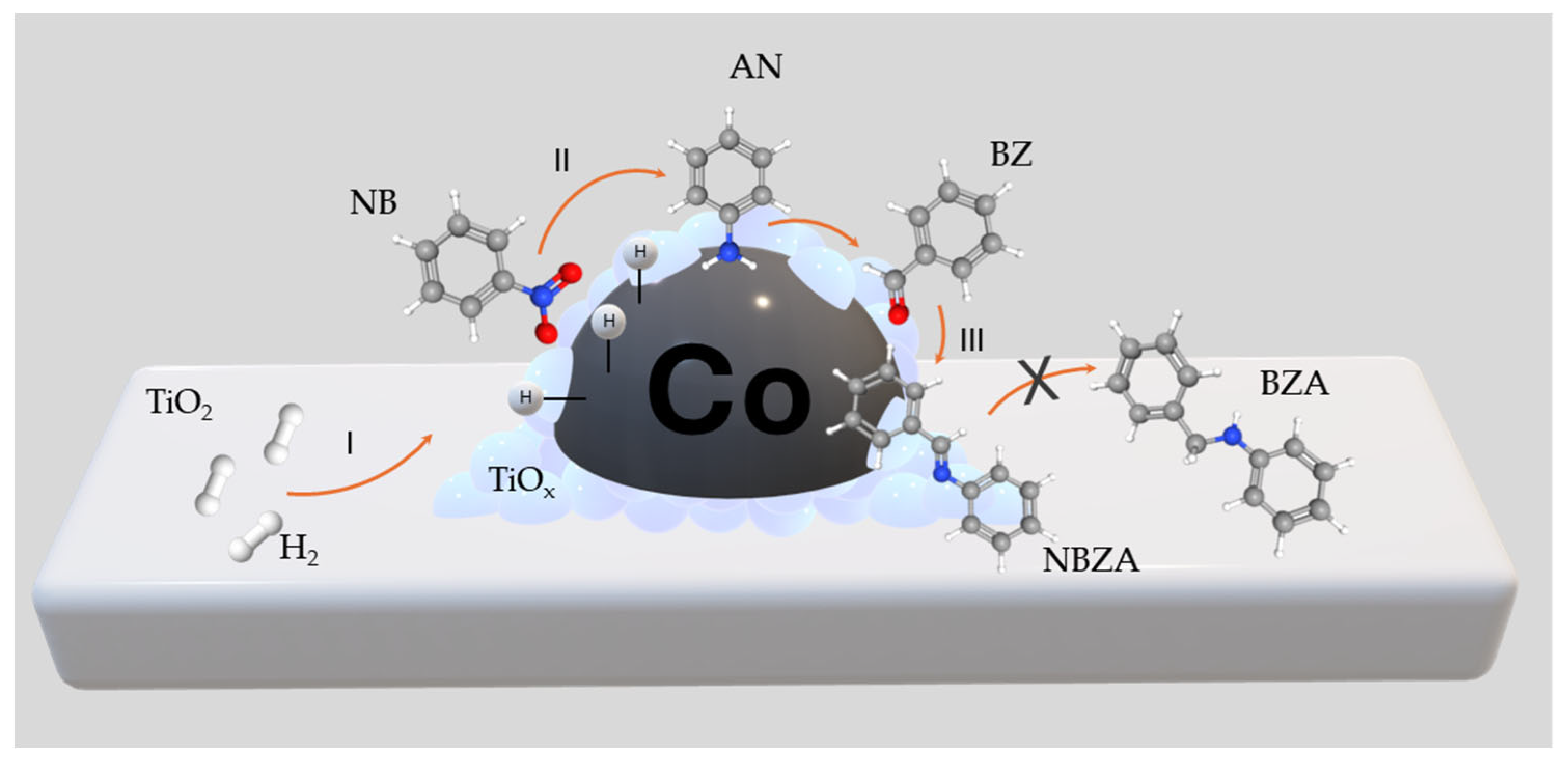
Building upon our previous endeavors to design efficient catalytic formulations for hydrogenation reactions in organic synthesis, we recently developed a Co-based catalyst through the H2-assisted controlled reduction of CoTiO3-type ilmenite. This catalyst demonstrated noteworthy activity and selectivity toward the hydrogenation of nitroarenes with diverse chemical compositions [18]. Encouraged by these findings, we sought to broaden the application of this catalyst to the reductive imination of nitroarenes with aromatic aldehydes leveraging the thermal H2-assisted reduction process and we generated Co supported on TiO2, which serves as a partially reducible support. In this study, we present a heterogeneous Co/TiO2 catalyst exhibiting high catalytic performance for the direct synthesis of imines. Our process involves the selective reduction of the –NO2 group to –NH2 employing nitrobenzene (NB) as the substrate, followed by in situ condensation with the C=O group of benzaldehyde (BZ) to yield N-benzylidenaniline (NBZA), as illustrated in Scheme 1. Various substituted nitrobenzenes bearing electron-density-subtracting or electron-density-giving groups at the 4-position efficiently coupled with benzaldehyde to produce the corresponding imines. Notably, the selectivity towards imines was found to depend on the electronic nature of the aniline employed. Finally, the recyclabilities of the catalysts were evaluated over seven operational cycles.
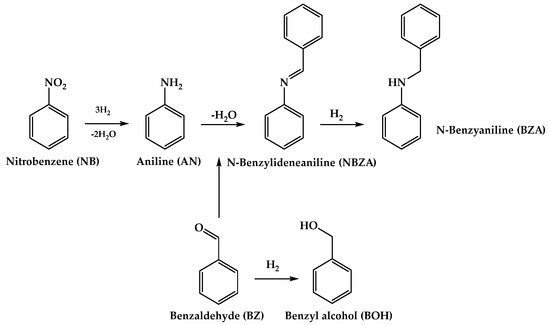
Scheme 1.
General scheme of reaction pathways in reductive imination of NB with BZ.
2. Results and Discussion
2.1. Characterization
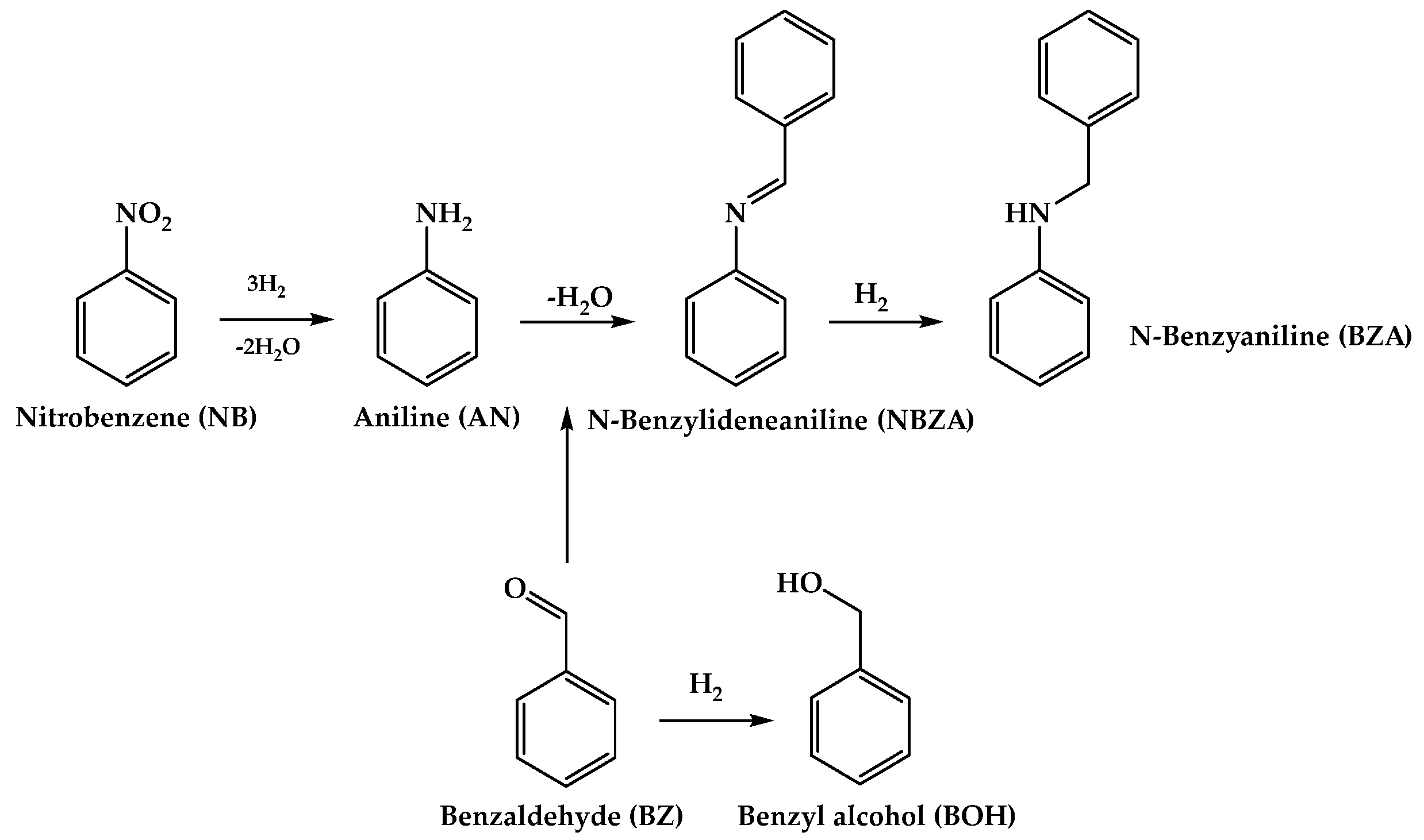
Figure 1a presents the H2 temperature-programmed reduction (TPR) profiles, where the CoTiO3 precursor (TPR1) denotes a single reduction signal centered at 640 °C, which is attributed to the reduction in ionic Co, in the ilmenite structure. Then, a second TPR was performed to determine the reducibility of the system from the in situ reduction of CoTiO3 at 550 °C for 5 h (TPR2). In this analysis, an additional signal of very low intensity is observed at approximately 182 °C, which can be attributed to the CoOx species remaining after the reduction treatment. In comparison with our previous results [18], the CoTiO3 reduction treatment was extended from 3 h at 500 °C to 5 h at 550 °C where a substantial increase in Co reduction was achieved from a 46% to a 94% reduction, respectively.
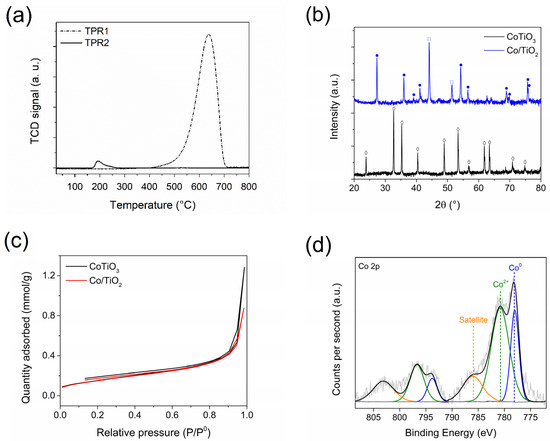
Figure 1.
Characterization of perovskite CoTiO3 precursor and Co/TiO2 catalyst. (a) TPR1 and TPR2, (b) XRD ● CoTiO3 (ICSD 16548), ◊ TiO2-rutile (JCPDS 75-1753), and □ metallic Co (ICSD 52935), (c) N2 adsorption–desorption isotherms at −196 °C and (d) Co 2p XPS spectra for Co/TiO2.
The formation of an ilmenite-type structure in the precursor was confirmed by X-ray diffraction (XRD) (Figure 1b), in which the diffraction patterns of CoTiO3 (ICSD 16548) were determined, showing intense and well-defined signals. Upon reduction, the presence of metallic Co (ICSD 52935) and TiO2-rutile (JCPDS 75-1753) was observed, indicating the collapse of the structure and the formation of metallic Co nanoparticles (NPs) alongside their respective supports. In line with the TPR1-TPR2 characterization, it can be suggested that the materials formed a highly dispersed, partially oxidized phase on the catalyst surface, which however, could not be detected by XRD. For the Co/TiO2 system, the metallic particle size was estimated using a Debye–Sherrer’s equation [19] for the metallic Co phase, revealing a particle size of 25.8 nm. This value exceeds that reported in our previous studies, where a reduction for 3 h at 500 °C yielded a particle size of 19.6 nm. This behavior can be attributed to the duration of the heat treatment time during the reduction in CoTiO3. Increasing the reduction time may enhance the reducibility of the precursor, potentially leading to the coalescence of particles and an increase in the size of metallic crystals during their exsolution from within the precursor oxide crystal. This behavior reflects the results reported by Song et al. in SrFexCo1−xO3 systems [20].
Figure 1c displays the N2 adsorption–desorption isotherms at −196 °C determined for the CoTiO3 precursor and the Co/TiO2 catalyst. Both systems exhibited type-III isotherms according to the IUPAC classification. The specific surface area values of the precursor and catalyst were calculated using the Brunauer–Emmett–Teller (BET) methodology as 6 and 8 m2/g, respectively, which are characteristic of non-porous materials [18]. The characterization results of desorption temperature-programmed desorption (TPD) of ammonia (TPD-NH3) and carbon dioxide (DTP-CO2) are shown in the Supplementary Material (Figure S1). The Co/TiO2 catalyst obtained using the proposed methodology did not exhibit acidic or basic properties.
To determine the surface composition of the catalysts, X-ray photoelectron spectros-copy (XPS) characterization was performed. The contributions of Co, Ti, and O elements were detected (Figure S2a), with the corresponding proportions of each element on the surface being 10.1%, 19.7%, and 70.2%, respectively. Figure 1d shows the Co 2p spectrum for the in situ-reduced passivated Co/TiO2 catalyst, showing that the signal comprised three Co 2p3/2 binding energies (BE) of 777.8, 780.2, and 786.2 eV, respectively attributed to the Co0, Co2+, and the characteristic satellite signal for this system type [18]. The estimated contributions of the Co0 and Co2+ species were 37% and 63%, respectively. To confirm the reduction of the passivation layer thickness by thermal treatment, TPR analysis was performed (Figure S3). Meanwhile, the total amount of catalytically active Co on the surface was determined by atomic absorption spectroscopy (AAS) considering the amount of total metal. Accordingly, the reducibility was determined by TPR1-TPR2 analyses. The fraction of Co0 was confirmed to be 4.7% via XPS, whereas that for the same system reduced for 3 h at 500 °C was 3.7% [18]. In the case of Ti 2p3/2 (Figure S2b), a BE peak was observed at 458.6 eV, attributable to the Ti–O–Ti bonds in the TiO2 network. This result aligns with the TPR1-TPR2 and XRD results, confirming the transition from CoTiO3 to Co–CoOx/TiO2 under the applicable reduction conditions [21,22]. In conjunction with the TPR1-TPR2 and XRD results, this finding confirms that the reduction process achieved a higher exsolution and reduction of Co deposited on the TiO2 surface than when the catalyst was reduced to a lower temperature and for a shorter time. This correlates with previous studies reported by Bustamante et al. regarding the Co3O4@SiO2 type systems reducibility with respect to the exsolution of metallic Co to form the respective Co@SiO2 systems [23].
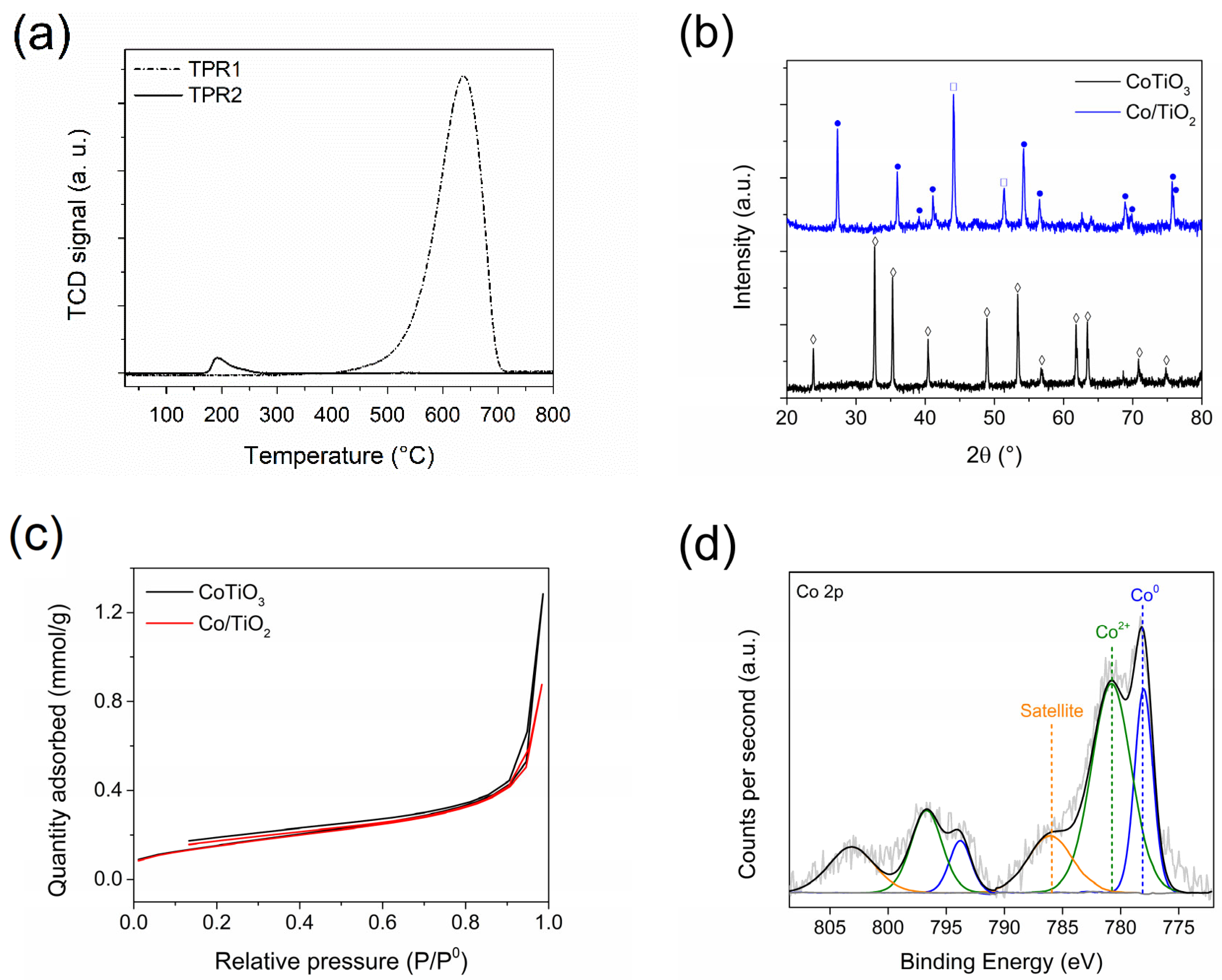
Figure 2a depicts the transmission electron microscopy (TEM) results, wherein a micro-rod structure for the Co/TiO2 catalyst that remained unchanged after the reduction treatment was observed. The average metallic particle size of Co was revealed to be 15.3 ± 1.8 nm, smaller than the corresponding size determined by XRD. This was attributed to the characteristics of the XRD technique, in which the greatest contribution to diffraction stems from larger metallic Co crystals. High resolution TEM (HR-TEM) was performed to evaluate the surface nature of the Co/TiO2 catalyst (see Figure 2b). The analysis of lattice spacing revealed the presence of TiO2-rutile, characterized by a lattice spacing of 0.32 nm corresponding to the (1 1 0) plane. Regarding the Co0 particles, a lattice spacing of 0.20 nm was observed, indicative of the (1 1 1) plane of hcp-Co. These particles Co0 appeared to be covered by a 2.7 nm thick layer, exhibiting a lattice spacing of 0.21 nm, which is characteristic of the (2 0 0) crystallographic plane of CoO. This layer formation is attributed to the passivation process, as well as the presence of residual CoO from the reduction process (see Figure 1a). Additionally, the Co-NPs were enveloped by a thin layer with blurred fringes, suggesting the formation of a TiOx layer around the Co phase. This overcoating phenomenon could be attributed to the reduction at 550 °C under H2 of the CoTiO3 precursor, resulting in an in-situ phase-change from perovskite to the corresponding metallic Co and TiO2-rutile [24].
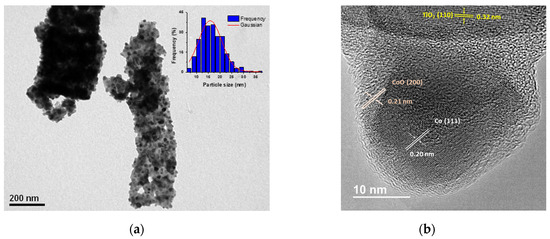
Figure 2.
Microscopy characterization of Co/TiO2 catalyst. (a) TEM image and Co-NPs size distribution (inset) and (b) HR-TEM image.
2.2. Catalytic Activity
2.2.1. Reductive Imination of NB and BZ
Catalytic activity studies were conducted under non-diffusional control conditions, utilizing an H2 pressure of 20 bar. To validate the catalytic performance of the Co/TiO2 catalysts, control experiments were carried out as non-catalyzed reactions, as well as the use of other Co-based or TiO2 materials, as shown in the Supplementary Material (Figure S4a). The non-catalyzed reaction exhibited negligible conversion, remaining below 2% even after 24 h of reaction time. This clearly demonstrates that the reaction proceeds only in the presence of a catalyst. Various materials, including CoTiO3, Co3O4, commercial TiO2-rutile and a partially reduced CoO–CoTiO3 material (CoTiO3 reduced for 1.5 h) were tested as catalysts for this reaction. All systems, except for the CoO–CoTiO3 material, displayed results similar to the non-catalyzed reaction, with minimal conversion observed. However, the CoO–CoTiO3 material exhibited nitrobenzene conversion levels of 15% and 28% at 6 h and 24 h of reaction, respectively. In addition, the metallic Co/TiO2 and Co/SiO2 catalysts prepared via wet impregnation demonstrated excellent performance, reaching maximum conversion levels after 24 h of reaction.
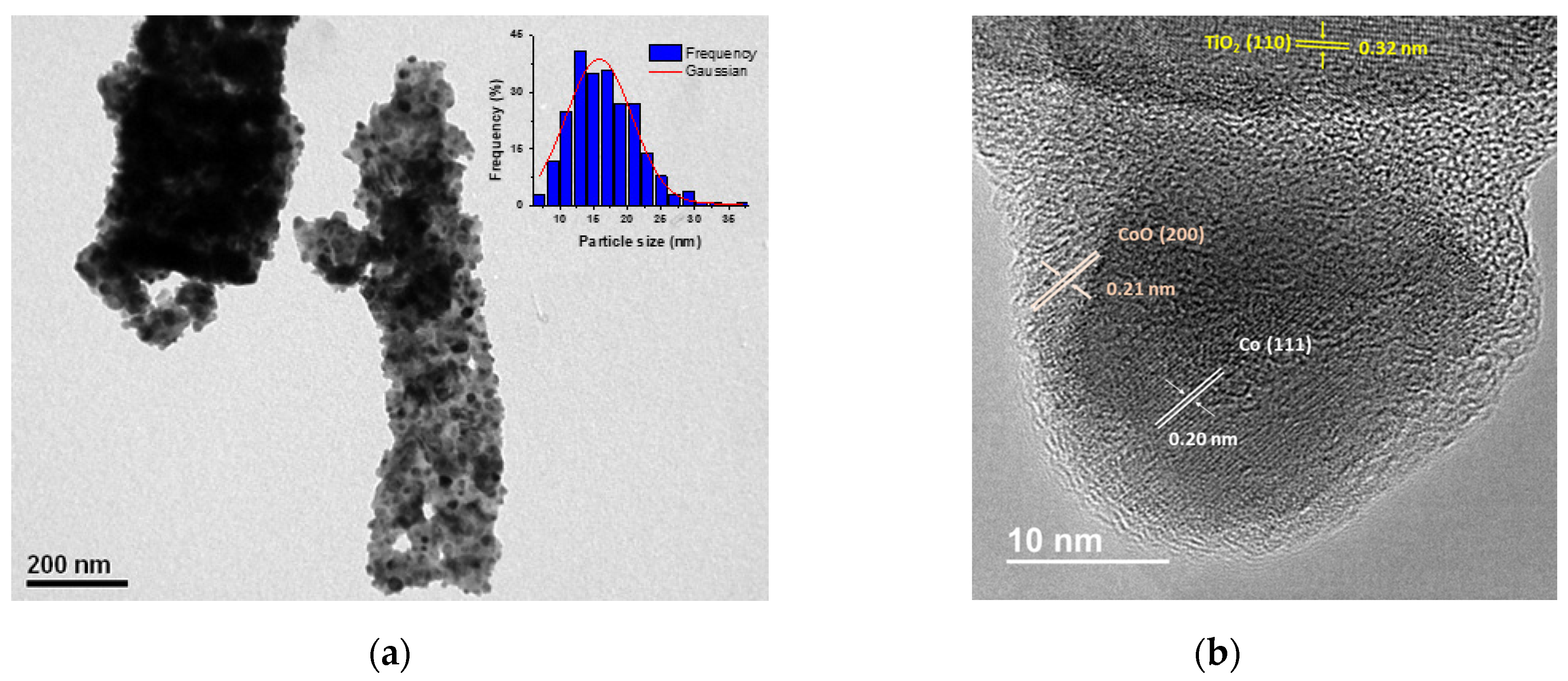
Figure 3a illustrates the conversion of NB consumption, product selectivity, and product distribution over time for reductive imination. NB consumption during the reaction followed pseudo-first-order kinetics (k0), reaching maximum conversion after 360 min. This kinetic model was corroborated by H2 pressure studies, as shown in the Supplementary Material (Figure S4b). In this context, hydrogen pressure ranging from 20 to 50 bar influences NB conversion, suggesting a first-order reaction with respect to hydrogen. According to the characterization results, the layers of CoO and TiOx on the surface of the Co-NPs did not interfere with the catalytic activity under the reaction condition, as evidenced by a non-induction period observed in the conversion of NB. The selectivity shows that only the AN production was detected as a product of NB hydrogenation, which is consumed to lead to the NBZA formation with 96.8%. Only a low amount (<1%) is detected for the NBZA hydrogenation to lead to BZA at 360 min of reaction.
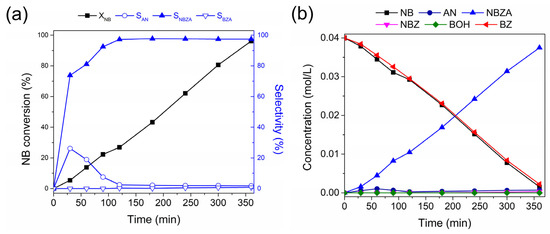
Figure 3.
Catalytic performance of Co/TiO2 for reductive imination of NB and BZ. (a) Conversion and selectivity. (a) NB conversion and main selectivity for the N-based products and (b) product distribution during the reductive amination of NB and BZ. Reaction conditions: NB (2 mmol), BZ (2 mmol), catalyst (20 mg), ethyl acetate (50 mL), H2 (20 bar), 120 °C. The data were obtained based on nitrobenzene.
Figure 3b shows the distribution curves of NB and BZ and their respective reaction products as functions of time. BZ consumption is proportional to NB consumption, corre-lating with continuous NBZA formation over time. However, after 360 min, trace amounts of BOH were detected, identified as the hydrogenation product of BZ (<1%). In agreement with the characterization results, the presence of Co-NPs at 120 °C facilitated –NO2 reduction while avoiding the hydrogenation of C=O bonds. Accordingly, the presence of the TiOx thin layer promoted BZ adsorption adjacent to the metal site. The combined effect of the Co active phase and the TiOx layer promoted the formation of NBZA, while suppressing the overhydrogenation-induced production of BZA (see Scheme 2). On the surface of the Co NPs, H2 underwent dissociation to generate activated hydrogen species (H*) (I). Subsequently, these H* species facilitated the direct reduction of adsorbed NB on the surface of the Co-NPs (II). BZ was adsorbed on both the TiOx layer surrounding the Co-NPs and the TiO2 support. In terms of catalytic kinetics, NB preferentially hydrogenated into AN rather than BZ. Furthermore, the produced AN immediately condensed with the BZ adsorbed on the TiOx thin layer, decreasing the possibility of BZ hydrogenation. The final step involved the desorption of the generated imines from the catalyst surface (III). Due to the strong interaction between BZ and TiOx, the generated NBZA can hydrogenate from the surface of the Co-NPs via the H* species. Nevertheless, overhydrogenation of NBZA was effectively suppressed under these reaction conditions.
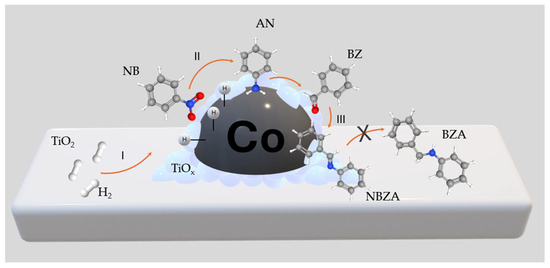
Scheme 2.
Proposed catalytic process to produce NBZA from the H2-assited reductive imination of NB with BZ.
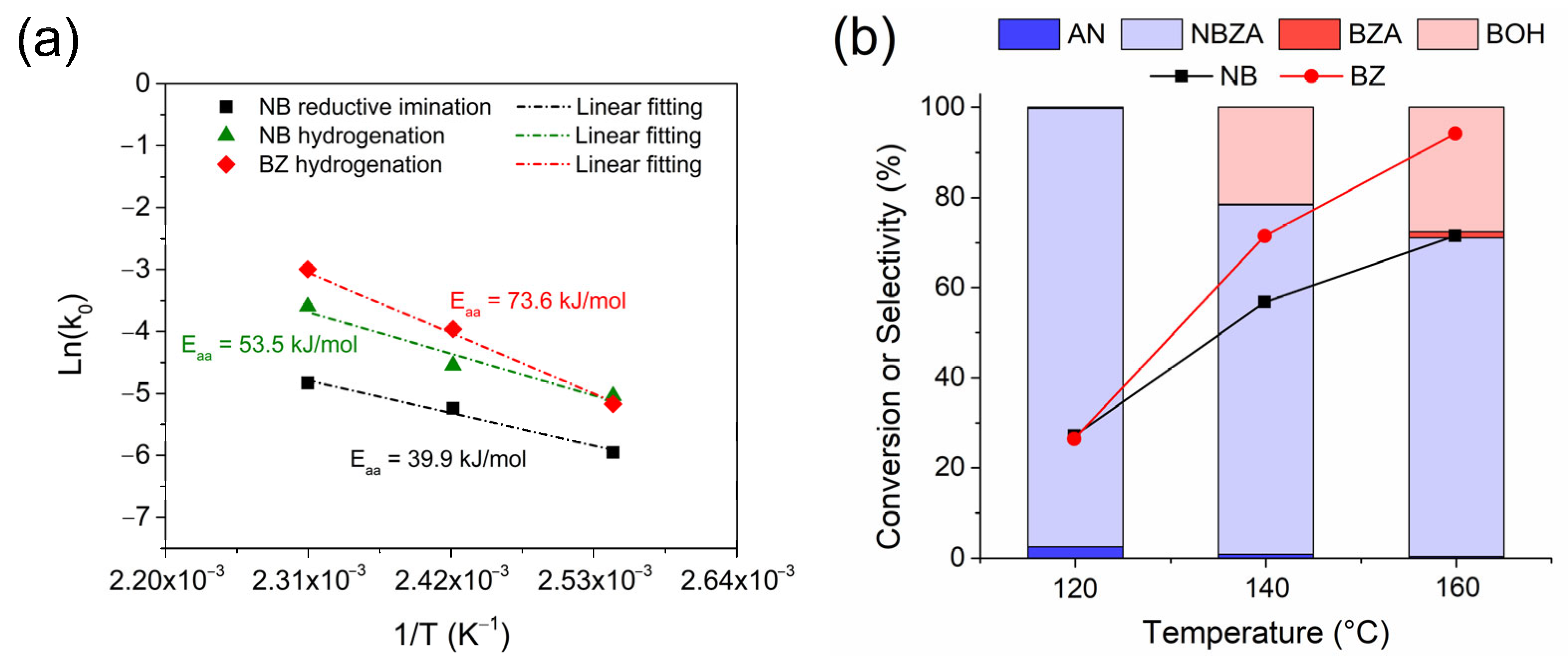
To demonstrate the catalyst selectivity, temperature effect studies were carried out to determine the product distribution and apparent activation energy (Eaa) for the reductive imination reactions of NB and BZ, as well as for the hydrogenation of each substrate separately (Figure 4a). In all cases, hydrogenations were carried out at 20 bar H2 pressure, which assumed a pseudo-first-order kinetic fit for the consumption of each substrate studied. The conversion curves and product distributions for each temperature are shown in the Supplementary Material (Figure S5). The Eaa for NB hydrogenation in the reductive imination reaction was determined to be 39.9 kJ/mol. However, for the hydrogenation of each substrate separately, NB exhibited an Eaa of 53.5 kJ/mol, whereas BZ showed a higher value of Eaa = 73.6 kJ/mol. This suggests that NB hydrogenation in the presence of BZ undergoes a synergistic effect that decreases the activation barrier for the hydrogenation of the –NO2 group compared to hydrogenation in the absence of BZ.
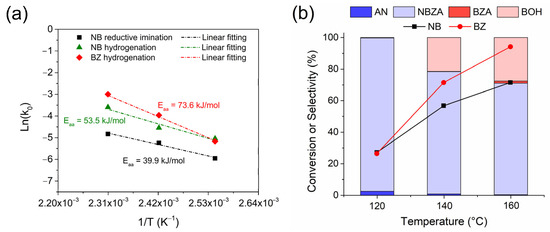
Figure 4.
The effect of reaction temperature on the reductive imination of NB and BZ by the Co/TiO2 catalyst. (a) Apparent energy activation plot and (b) NB and BZ conversion level and selectivity for all the obtained products. Reaction conditions: NB (2 mmol), BZ (2 mmol), catalyst (20 mg), ethyl acetate (50 mL), H2 (20 bar) at 120 min.
This finding is consistent with studies reported by Gong et al., where they demonstrated by density functional theory (DFT) calculations the adsorption energies of NB and BZ on the surface of Co (1 1 1) to be −0.894 eV and −0.368 eV, respectively [13]. According to the above discussion, the selectivity of the reaction changed with an increase in temperature (Figure 3b). The transition from 120 °C to 140 °C resulted in a considerable selectivity toward BOH production, with only 1.8% BZA production. At 160 °C, an increase in BOH production was observed (~28%), accompanied by a higher amount of BZA, achieving a selectivity of 3.9%. This suggests that the hydrogenating capacity of the catalyst follows the trend: –NO2 > C=O >> C=N, respectively. To support this hypothesis, controls for reductive imination in the presence of AN and BZ were performed, employing the catalyst at 20 bar H2 pressure and in an Ar inert gas atmosphere, as shown in the Supplementary Material (Figure S6). In the experiment with H2 and Co/TiO2 catalyst, NBZA production reached a selectivity of 86.0% at 60 min of reaction, with AN and BZ conversion reaching approximately 85%, respectively. After 180 min, BZA accumulation was observed in the reaction medium, with a selectivity of 15% after 360 min. When the reaction was conducted in the presence of the catalyst in an Ar atmosphere, only NBZA was produced reaching a conversion of 96.7% at 360 min of reaction. In addition, controls of the AN imine formation reaction with BZ were performed under the same reaction conditions used for the catalyzed reaction in the presence of 20 bar H2 and Ar; the inert atmosphere is shown in the Supplementary Material (Figure S6). In both experiments, no conversion to BZA or changes in the product distribution were observed compared to the reaction performed in the presence of the catalyst using Ar as an inert gas. These results are consistent with the necessity of a metal site for the hydrogenation of NBZA. According to the proposed reaction pathways (Scheme 2), an increase in temperature enabled the hydrogenation of both BZ and NBZA adsorbed on the TiOx layer around the Co-NPs by H*. From these results, it can be inferred that the presence of NB in the reaction medium inhibited the hydrogenation of BZ to produce BOH at low temperatures. Furthermore, the risk of over-hydrogenation of NBZA and BZ can be effectively minimized, resulting in a significantly enhanced yield and chemoselectivity to produce NBZA at temperatures >120 °C in the presence of the Co/TiO2 catalyst.
2.2.2. Reductive Imination of 4-Substituted Nitrobenzene and Benzaldehyde: Structure–Reactivity Relationship
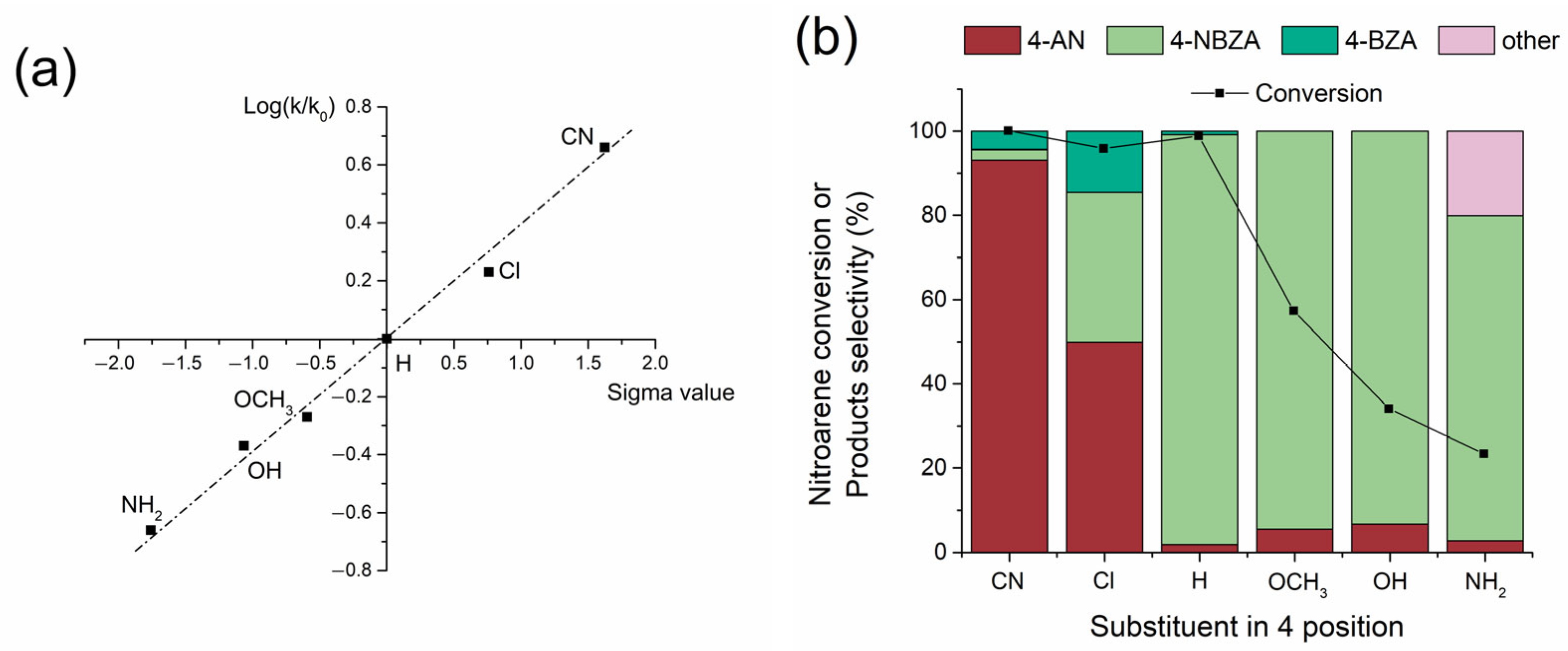
The reductive imination of a series of nitrobenzenes substituted at the 4-position was conducted to elucidate the electronic effects on the reaction rate and yield of the respective imines using the Co/TiO2 system. The discrimination reaction exhibited high selectivity under the Co/TiO2 catalyst, reaching values of approximately 99% toward the hydrogena-tion of the –NO2 group, showing traces of benzyl alcohol as a co-product of the reaction (<1%). The pseudo-first-order kinetic constants (k) for the hydrogenation of the respective nitroarenes and the selectivities for the reaction products were determined (Figure S7), and a Hammett adjustment was applied, as shown in Figure 5a. As demonstrated in previous studies [25], –NO2 group hydrogenation showed a dependence that fits Hammett-type structure–reactivity models (Figure 5a). In this scenario, electron-density-subtracting groups such as (–Cl and –CN) increase the hydrogenation rate of the –NO2 group, whereas electron-density-giving groups (–OCH3, –OH, and –NH2) decrease it. The hydrogenation step favors the formation of 4-substituted anilines at the 4-position. However, in the condensation step, the formation of the respective 4-substituted N-benzylidenaninilines (4-NBZA) and 4-substituted N-benzylanilines (4-BZA) also depended on the reaction selectivity (Figure 5b). Anilines with electron-density substituents, such as 4-cyanoaniline and 4-chloroaniline, showed low selectivity for the product of interest, reaching selectivities of 2.5% and 34%, respectively. Surprisingly, accumulation of the products N-benzyl-4-cyanoaniline and N-benzyl-4-chloroaniline was observed, reaching selectivities of 4.4% and 14.5%, respectively. This can be attributed to the electronic nature of the substituent groups in the respective anilines, which exert an electron density subtracting effect by decreasing the nucleophilicity of the –NH2 group in the formation of the respective imines. Furthermore, N-benzylidene-4-cyanoaniline and N-benzylidene-4-chloroaniline in the reaction medium are affected by the presence of electron-density-subtracting groups, which increase the reactivity of the C=N bond to produce the respective secondary amines.
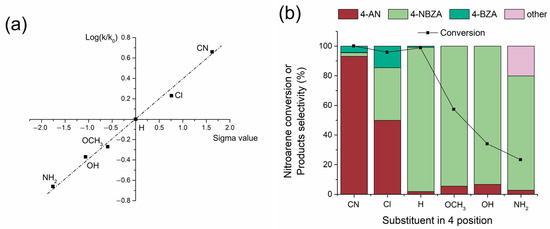
Figure 5.
Catalytic performance Co/TiO2 catalysts for the 4-substituted NB reductive imination with BZ. (a) Hammett plot and (b) Nitroarenes and BZ conversion level and selectivity for all the obtained products. Reaction conditions: nitroarene (2 mmol), BZ (2 mmol), catalyst (20 mg), ethyl acetate (50 mL), H2 (20 bar) at 360 min.
The opposite effect was observed in the case of 4-methoxyaniline, 4-hydroxyaniline, and 4-aminoaniline, wherein a lower production rate of the respective anilines was detected. However, the selectivity of the condensation to the respective imines was substantially increased by the electron-density-giving effect of the substituent group, which increased the nucleophilicity of the –NH2 group. For all these substituents, secondary amine formation was not detected, aligning with the effect of the substituent, which can donate electron density to the C=N bond, thereby decreasing its ability to hydrogenate. Only in the case of the hydrogenation of 4-nitroaniline was the appearance of N-(4-((phenylimino)methyl)benzylidene)aniline detected at 15% due to the fact that the 4-aminoaniline product can form a double imine because it possesses two –NH2 groups.
2.2.3. Recyclability Study
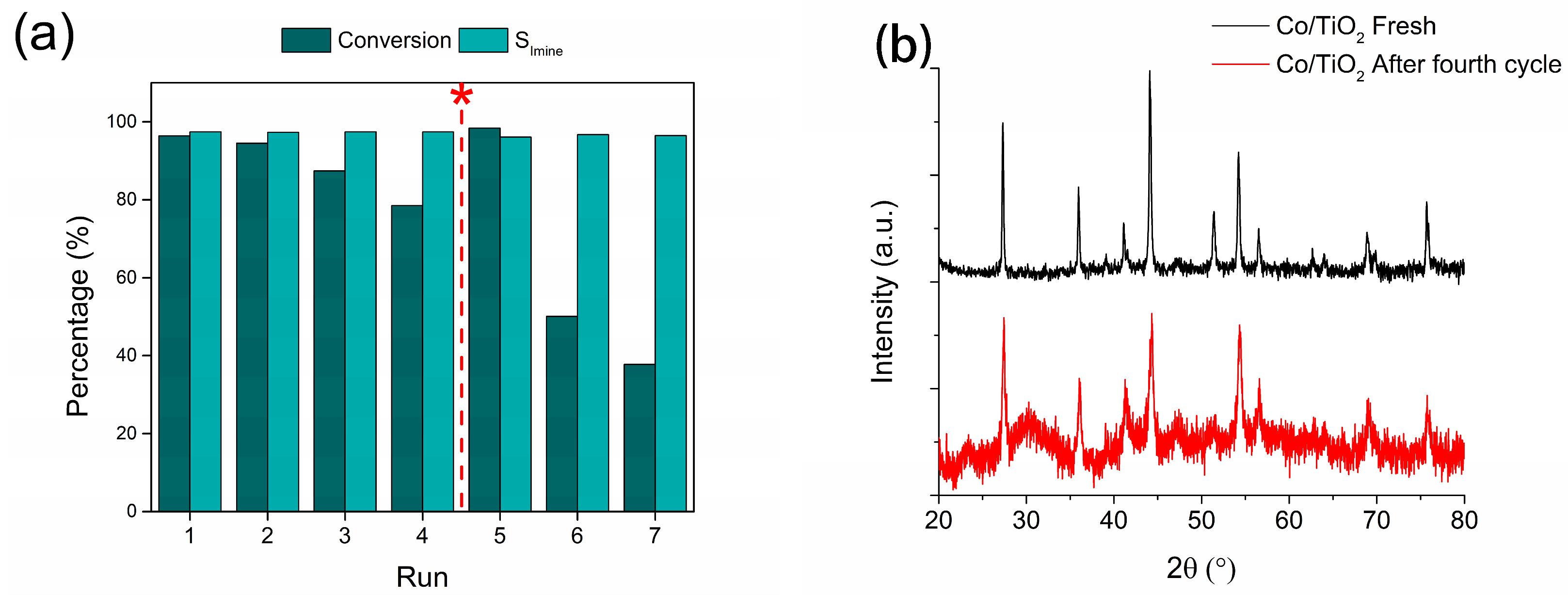
To evaluate the catalyst’s reusability, recycling studies were performed for the reaction of interest (Figure 6a). The catalyst showed moderate stability until the fourth reaction cycle, where a decrease in catalytic activity from a 100% to a 78.5% conversion was observed. However, the selectivity for the imine of interest was maintained at ~97% in all cycles. To evaluate the loss of catalytic activity, a hot filtrate test was performed after the fourth reaction cycle to assess metal leaching during the reaction (Figure S8). The reaction filtrate was subjected to the reaction conditions and was maintained for an additional 6 h under these conditions, with conversion being evaluated afterward. The conversion level for –NO2 and the formation of the respective NBZA remained unchanged. In addition, AAS was performed on the recovered catalyst to determine the post-reaction metal content. Surprisingly, the metal content decreased from 38.4% to 26.9%, indicating leaching of the metal into the liquid phase. However, the hot filtrate test showed no activity, suggesting that the metal leached from the catalyst to the active phase loses its activity by possible oxidation upon contact with the atmosphere during hot filtration, or by interaction with water formed during the hydrogenation of the –NO2 group, or by condensation of AN with BZ to form NBZA. To test this hypothesis, XRD characterization was performed to evaluate the nature of the active phase after the fourth reaction cycle, as shown in Figure 6b. A decrease in the signal intensity attributed to metallic Co diffraction was observed, whereas two contributions attributed to hexagonal CoO species (ICDD 78-0431) appeared at 2θ angles of 36.5° and 42.5°, corresponding to the hkl index diffractions (1 1 1) and (2 0 0), respectively. This suggests that during consecutive cycles, the active phase underwent oxidation either by contact with the atmosphere between operation cycles and/or the presence of water in the reaction medium, which, under the conditions in which reductive imination is performed, can generate hydrothermal reactions that promote the oxidation of Co to CoO in a manner analogous to the results reported in previous works [18].
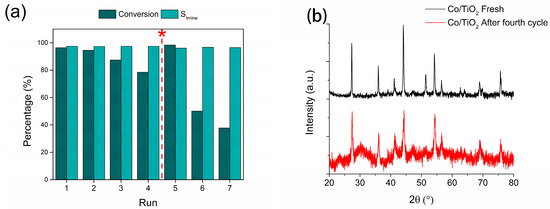
Figure 6.
Reusability of Co/TiO2 catalysts for the NB reductive imination with BZ. (a) Catalytic stability (* reactivation of the used catalyst reducing the recovered solid) and (b) XRD characterization for the fresh and used catalyst. Reaction conditions: nitroarene (2 mmol), BZ (2 mmol), catalyst (20 mg), ethyl acetate (50 mL), H2 (20 bar) at 360 min.
TEM was used to determine the morphology and metallic particle size after the fourth reaction cycle (Figure S9a). The morphology changed from a micro-rod shape to an irregularly shaped nanostructure with an increase in particle size, reaching an average value of 22.5 ± 1.0 nm. In an attempt to restore the catalytic activity of the deactivated Co/TiO2 system after the fourth cycle, a reduction treatment was performed for 5 h at 550 °C and the reaction was repeated for three consecutive cycles (Figure 6a). Although the catalyst regained some activity after the reduction treatment, it experienced a more pronounced loss of activity in the subsequent reaction cycle, reaching a 37.8% conversion in the last cycle of operation for the conversion of NB to AN, as well as a selectivity in the formation of NBZA of 96.5%. A post-reaction TEM analysis was performed for this system (Figure S9b), revealing the loss of the micro-rod morphology and its transition to an irregular shape comprising Co NPs with a mean size of 31.8 ± 1.6 nm, which could be responsible for the deactivation of the catalyst after the regeneration treatment.
3. Conclusions
In conclusion, the Co catalyst supported on TiO2, derived from the reduction of the precursor CoTiO3, proved to be highly efficient for the reductive imination of nitrobenzene with benzaldehyde, yielding the corresponding N-benzylidenaniline under moderate operating conditions and employing ethyl acetate as the solvent. Our study suggests that the active phase, operating at 120 °C and 20 bar pressure, in conjunction with the TiOx layer, can achieve a nitrobenzene conversion of 100% and a selectivity of 97.4% for the desired imine, effectively suppressing both benzaldehyde and imine hydrogenation to yield the secondary amine. Investigation into the influence of the reaction temperature corroborated the results reported in the literature, where reductive imination promoted the hydrogenation of the –NO2 group. The apparent activation energy for the formation of the –NH2 group was 1.3 times lower than that when nitrobenzene was hydrogenated in the absence of benzaldehyde. The catalyst demonstrated high activity in the reductive imination of a series of substituted nitrobenzenes at the 4-position, where the electron-density-giving groups exhibited a lower rate of hydrogenation of the –NO2 group. Furthermore, 100% selectivity was achieved for the formation of the respective imines, contrasting with electron-density-subtracting groups that exhibited higher selectivity for the formation of the respective secondary amines. Although the stability of the catalyst was moderate, it could be operated for four cycles with acceptable conversion and a constant selectivity of 97.8%. Furthermore, our findings present a general approach for obtaining metallic catalysts for chemical reactions with desired activity and selectivity, underscoring the potential for broader applications in organic synthesis.
4. Materials and Methods
4.1. General Information
Cobalt(II) acetate tetrahydrate (Co(CH3CO2)2·4H2O), titanium butoxide (Ti(CH3CH2CH2CH2CH2O)4), nitrobenzene (NB), and aniline (AN) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Ethyl acetate, isopropanol, benzaldehyde (BZ), and ethylene glycol were purchased from Merck (Darmstadt, Germany). All purchased reagents were used without further purification. H2 (99.99%), He (99.99%), synthetic air, and nitrogen gases were provided by Linde Chile (Santiago, Chile).
4.2. Catalyst Preparation
The CoTiO3 precursor was synthesized using the methodology reported in a previous study [18]. Equimolar amounts of Co(CH3COO)2 and Ti(CH3(CH2)3O)4 were dissolved in a 1:1 (v/v) mixture of ethylene glycol and isopropanol. The mixture was stirred for 24 h until a milky purple dispersion was formed. The obtained material was aged for 24 h at 20 °C, oven-heated at 70 °C for 24 h, stirred for 10 min, and finally washed with absolute ethanol (3 × 50 mL) to remove excess ethylene glycol. The recovered product was oven-dried at 100 °C for 12 h and heated in a static air atmosphere from 30 °C to 700 °C at a rate of 5 °C/min until a green solid was obtained. The calcined material was subjected to reduction treatment using H2 as the reducing agent at a heating rate of 10 °C/min from room temperature up to 550 °C, under isothermal conditions for 5 h. Once the reduced material was obtained, a controlled passivation treatment was performed before characterization to avoid violent oxidation of the active phase, according to the procedure reported by Morales et al. [26]. For the catalytic experiments, the CoTiO3 was reduced first to start the reaction.
4.3. Characterization
Chemical analysis was performed via AAS using a Perkin Elmer 3100 instrument (PerkinElmer, Waltham, MA, USA) according to a previously reported protocol [18]. The Co content was 38.1%. TPR was performed using a Micromeritics 2900 TPR/TPD (Micromeritics Instrument Co., Norcross, GA, USA) system equipped with a thermal conductivity detector. Two reduction profiles were recorded. In the first measurement profile (TPR1), the TPR data were recorded for a 0.050 g CoTiO3 sample calcined using a 5% H2/Ar flow rate of 40 mL/min and a heating rate of 10°/min from 30 °C to 800 °C. In the second measurement profile (TPR2), a calcined sample of 0.050 g CoTiO3 was thermally reduced in the TPR apparatus under a pure H2 flow rate of 30 mL/min from room temperature to 550 °C at 10°/min, under isothermal conditions for 5 h. After reductive treatment, the samples were cleaned at 100 °C under Ar flow (30 mL/min) for 2 h; then, a second TPR process was performed using H2-TPR1 conditions. The reducibility of the system was calculated from the TPR data, as follows:
In this expression, the peak areas were normalized by the total experimental Co–Ni content determined by AAS characterization.
TPD-NH3 experiments were conducted with in situ reduced Co/TiO2 systems in a Micromeritics 2900 TPR/TPD (Micromeritics Instrument Co., Norcross, GA, USA), saturating the surface of the materials at 100 °C with ammonia pulses. The samples were then cooled to 30 °C. Once the baseline was re-stored, the temperature was increased linearly at a heating rate of 10°/min up to 500 °C using He as the carrier gas. TPD-CO2 analysis was performed to determine the basic properties of CoTiO3 and Co/TiO2 using Micromeritics 2900 TPR/TPD (Micromeritics Instrument Co., Norcross, GA, USA). In both cases, 0.050 g samples underwent cleaning under a He flow rate of 50 mL/min at 110 °C for 30 min. Subsequently, CO2 adsorption occurred at 80 °C, followed by CO2 desorption using He as the carrier gas at a flow rate of 50 mL/min, heating from 40 to 700 °C at a rate of 10 °C/min. Specific areas were calculated using the Brunauer–Emmett–Teller (BET) method from the N2 adsorption–desorption isotherms obtained at −196 °C using a Tristar II 3020 surface area and porosity analyzer (Micromeritics Instrument Co., Norcross, GA, USA),. Powder XRD patterns of CoTiO3 and Co/TiO2 were acquired using Cu Kα1 radiation (λ = 1.5418 Å) with a Rigaku diffractometer (Rigaku, Tokyo, Japan). A range of angles from 2θ = 20° to 90° was employed. Transmission electron microscopy (TEM) images were obtained using a JEOL JEM-1200 EX II microscope (JEOL, Tokyo, Japan). The passivated catalyst materials were dispersed on a carbon grid. Up to 300 individual metal particles were counted for each catalyst. The mean diameter of Co (dp) weighted by the surface area was calculated using the following equation:
where ni is the number of particles of diameter di.
Average cobalt particle sizes were corrected for a 2.7 nm thick CoO passivation outlayer as measured by high-resolution transmission electron microscopy (HR-TEM).
X-ray photoelectron spectroscopy (XPS) was performed using a SPECS® spectrometer (SPECS, Berlin, Germany) with a PHOIBOS® 150 WAL hemispherical energy analyzer with angular resolution (<0.5°), equipped with an XR 50 X-ray Al-X-ray and μ-FOCUS 500 X-ray monochromator sources (Al excitation line). The passivated Co/TiO2 catalyst was reduced in situ in the XPS prechamber prior to analysis.
4.4. Catalytic Activity
The catalytic activity was performed in a stainless-steel semi-batch reactor, where the reaction conditions used were a H2 pressure of 20 bar and reaction temperature of 120 °C. All reactions were performed in duplicate. The reactor was loaded with 0.0250 g of catalyst with a ratio NB/Co = 100 and NB/BZ = 1 in 50 mL of ethyl acetate at an agitation speed of 800 rpm. The progress of the reaction was followed using a Clarus® 680 Perkin Elmer gas chromatograph (PerkinElmer, Waltham, MA, USA) fitted with a flame ionization detector and an Rtx-5 Amine column. The conversion and yield were calculated using calibration curves and the following equations:
The Co fraction available at the surface obtained from the TPR1-TPR2 measurements, along with the estimate of Co0 in the elemental state extracted from XPS, were considered for calculating the metal content at the surface. Apparent activation energies were determined using the Arrhenius equation.
where k is the pseudo rate constant for the hydrogenation process, A is the frequency factor, and T is the reaction temperature (K).
For the hydrogenation reaction of the 4-position substituted nitroarenes, the operating conditions were the same as that of the NB reaction. The products of all reactions were followed using a GC–MS (Perkin Elmer GCMS-SQ8T, PerkinElmer, Waltham, MA, USA). The kinetic rate constants for the substituted nitroarenes (k) were determined considering pseudo-first order reactions for the hydrogenation of the –NO2 group and related to those obtained with NB according to the equation proposed by Hammett:
The reaction constant ρ is an estimate of the charge development during the reaction and provides a measure of the susceptibility to electronic effects of the substituents. The σ term is the Hammett’s substituent constant and was extracted from values reported in previous studies [25].
Recycling experiments for NB hydrogenation were performed using the recovered catalysts under conditions identical to those mentioned in the kinetic experiments. The catalyst was recovered via centrifugation, washed three times with the reaction solvent, and utilized in subsequent cycles for up to five consecutive reaction cycles. After Cycle 5, the catalyst underwent a regeneration treatment through H2-assisted thermal reduction at a heating ramp of 10 °C/min from 30 °C to 550 °C under isothermal conditions for 5 h. Subsequently, the regenerated catalyst was reused for three consecutive cycles. Hot filtration tests were performed to assess metal leachate, where the reaction was halted after 120 min. The liquid medium was extracted from the reactor, centrifuged at 9000 rpm, and then subjected to the reaction conditions in the absence of the catalyst.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal14040272/s1, Figure S1: Temperature programmed desorption experiments. (a) NH3 and (b) CO2, Figure S2: (a) XPS survey spectrum and (b) XPS high-resolution XPS scan spectra over Ti 2p, Figure S3: TPR characterization of passivated Co/TiO2 catalyst, Figure S4: (a) The effect of non-catalyzed and different catalysts in the reductive imination of NB and BZ. Reaction conditions: Nitrobenzene (2 mmol), benzaldehyde (2 mmol), catalyst mass (20 mg), ethyl acetate (50 mL), H2 (20 bar) at 120 °C; and (b) Conversion vs time profiles for the Co/TiO2 catalysts at different pressures, Figure S5: Kinetic profiles for the Co/TiO2 catalysts at different temperatures, Figure S6: Concentration vs time profile for the control experiments for the imina-formation using AN and BZ, Figure S7: Pseudo-first kinetic adjustment for the conversion profiles of different 4-substituted NB substrates, Figure S8: Reusability and hot filtration after the fourth cycle of Co/TiO2 catalysts for NB reductive imination with BZ, Figure S9: TEM post-reaction for the Co/TiO2 catalyst.
Author Contributions
Investigation and data curation, D.G.-V.; Formal analysis and Investigation, T.M.B. and J.N.D.d.L.; Funding acquisition, D.G.-V., C.H.C. and C.C.T.; Resources, J.N.D.d.L. and C.H.C.; Writing—original draft, C.C.T.; Writing—review and editing, C.H.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Agencia Nacional de Investigación y Desarrollo de Chile programa FONECYT grant number 1231395 and grant number 1231352.
Data Availability Statement
The data presented in this study are available in article and Supplementary Materials.
Acknowledgments
D. González-Vera is grateful for her doctoral scholarship ANID 21200167, and to David Domiguez by his expert technical assistance.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kawahara, R.; Fujita, K.-i.; Yamaguchi, R. N-Alkylation of Amines with Alcohols Catalyzed by a Water-Soluble Cp*Iridium Complex: An Efficient Method for the Synthesis of Amines in Aqueous Media. Adv. Synth. Catal. 2011, 353, 1161–1168. [Google Scholar] [CrossRef]
- Sui, D.; Mao, F.; Fan, H.; Qi, Z.; Huang, J. General Reductive Amination of Aldehydes and Ketones with Amines and Nitroaromatics under H2 by Recyclable Iridium Catalysts. Chin. J. Chem. 2017, 35, 1371–1377. [Google Scholar] [CrossRef]
- Kwon, M.S.; Kim, S.; Park, S.; Bosco, W.; Chidrala, R.K.; Park, J. One-Pot Synthesis of Imines and Secondary Amines by Pd-Catalyzed Coupling of Benzyl Alcohols and Primary Amines. J. Org. Chem. 2009, 74, 2877–2879. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Angelici, R.J. Non-nanogold catalyzed aerobic oxidation of secondary amines to imines. Chem. Commun. 2007, 21, 2157–2159. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, N.; Hara, T.; Ichikuni, N.; Shimazu, S. Chemoselective synthesis of imine and secondary amine from nitrobenzene and benzaldehyde by Ni3Sn2 alloy catalyst supported on TiO2. Mol. Catal. 2021, 505, 111503. [Google Scholar] [CrossRef]
- Tang, L.; Sun, H.; Li, Y.; Zha, Z.; Wang, Z. Highly active and selective synthesis of imines from alcohols and amines or nitroarenes catalyzed by Pd/DNA in water with dehydrogenation. Green Chem. 2012, 14, 3423–3428. [Google Scholar] [CrossRef]
- Huang, J.; Yu, L.; He, L.; Liu, Y.-M.; Cao, Y.; Fan, K.-N. Direct one-pot reductive imination of nitroarenes using aldehydes and carbon monoxide by titania supported gold nanoparticles at room temperature. Green Chem. 2011, 13, 2672–2677. [Google Scholar] [CrossRef]
- Xiang, Y.; Meng, Q.; Li, X.; Wang, J. In situ hydrogen from aqueous-methanol for nitroarene reduction and imine formation over an Au–Pd/Al2O3 catalyst. Chem. Commun. 2010, 46, 5918–5920. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ma, K.; Li, H.; Li, J.; He, J.; Sun, X.; Li, R.; Ma, J. One Pot Synthesis of Imines from Aromatic Nitro Compounds with a Novel Ni/SiO2 Magnetic Catalyst. Catal. Lett. 2009, 128, 465–474. [Google Scholar] [CrossRef]
- Schwob, T.; Kempe, R. A Reusable Co Catalyst for the Selective Hydrogenation of Functionalized Nitroarenes and the Direct Synthesis of Imines and Benzimidazoles from Nitroarenes and Aldehydes. Angew. Chem. Int. Ed. 2016, 55, 15175–15179. [Google Scholar] [CrossRef]
- Chen, C.; Fan, R.; Han, M.; Zhu, X.; Zhang, Y.; Zhang, H.; Zhao, H.; Wang, G. Tunable synthesis of imines and secondary-amines from tandem hydrogenation-coupling of aromatic nitro and aldehyde over NiCo5 bi-metallic catalyst. Appl. Catal. B Environ. 2021, 280, 119448. [Google Scholar] [CrossRef]
- Zhang, S.; Xia, Z.; Ma, Y.; Li, J.; Qu, Y. Competitive adsorption on PtCo/CoBOx catalysts enables the selective hydrogen-reductive-imination of nitroarenes with aldehydes into imines. J. Catal. 2019, 374, 72–81. [Google Scholar] [CrossRef]
- Gong, W.; Han, M.; Chen, C.; Lin, Y.; Wang, G.; Zhang, H.; Zhao, H. Rational Design of Cobalt-Platinum Alloy Decorated Cobalt Nanoparticles for One-Pot Synthesis of Imines from Nitroarenes and Aldehydes. ChemCatChem 2020, 12, 5948–5958. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Qian, K.; Huang, W. Metal–Support Interactions in Metal/Oxide Catalysts and Oxide–Metal Interactions in Oxide/Metal Inverse Catalysts. ACS Catal. 2022, 12, 1268–1287. [Google Scholar] [CrossRef]
- van Deelen, T.W.; Hernández Mejía, C.; de Jong, K.P. Control of metal-support interactions in heterogeneous catalysts to enhance activity and selectivity. Nature Catal. 2019, 2, 955–970. [Google Scholar] [CrossRef]
- Kim, J.; Choi, H.; Kim, D.; Park, J.Y. Operando Surface Studies on Metal-Oxide Interfaces of Bimetal and Mixed Catalysts. ACS Catal. 2021, 11, 8645–8677. [Google Scholar] [CrossRef]
- Park, J.Y.; Baker, L.R.; Somorjai, G.A. Role of Hot Electrons and Metal–Oxide Interfaces in Surface Chemistry and Catalytic Reactions. Chem. Rev. 2015, 115, 2781–2817. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, T.M.; Dinamarca, R.; Torres, C.C.; Pecchi, G.; Campos, C.H. Pd-Co catalysts prepared from palladium-doped cobalt titanate precursors for chemoselective hydrogenation of halonitroarenes. Mol. Catal. 2020, 482, 110702. [Google Scholar] [CrossRef]
- Moosavi, S.; Zakaria, S.; Chia, C.H.; Gan, S.; Azahari, N.A.; Kaco, H. Hydrothermal synthesis, magnetic properties and characterization of CoFe2O4 nanocrystals. Ceram. Int. 2017, 43, 7889–7894. [Google Scholar] [CrossRef]
- Song, J.; Zhu, T.; Chen, X.; Ni, W.; Zhong, Q. Cobalt and Titanium substituted SrFeO3 based perovskite as efficient symmetrical electrode for solid oxide fuel cell. J Mater. 2020, 6, 377–384. [Google Scholar] [CrossRef]
- de la Peña O’Shea, V.A.; Consuelo Álvarez Galván, M.; Platero Prats, A.E.; Campos-Martin, J.M.; Fierro, J.L.G. Direct evidence of the SMSI decoration effect: The case of Co/TiO2 catalyst. Chem. Commun. 2011, 47, 7131–7133. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yang, Y.; Suo, H.; Qing, M.; Yan, L.; Wu, B.; Xu, J.; Xiang, H.; Li, Y. Effect of TiO2 promotion on the structure and performance of silica-supported cobalt-based catalysts for Fischer–Tropsch synthesis. J. Mol. Catal. A Chem. 2014, 390, 52–62. [Google Scholar] [CrossRef]
- Bustamante, T.M.; Fraga, M.A.; Fierro, J.L.G.; Campos, C.H.; Pecchi, G. Cobalt SiO2 core-shell catalysts for chemoselective hydrogenation of cinnamaldehyde. Catal. Today. 2020, 356, 330–338. [Google Scholar] [CrossRef]
- Lee, J.; Burt, S.P.; Carrero, C.A.; Alba-Rubio, A.C.; Ro, I.; O’Neill, B.J.; Kim, H.J.; Jackson, D.H.K.; Kuech, T.F.; Hermans, I.; et al. Stabilizing cobalt catalysts for aqueous-phase reactions by strong metal-support interaction. J. Catal. 2015, 330, 19–27. [Google Scholar] [CrossRef]
- Torres, C.C.; Jiménez, V.A.; Campos, C.H.; Alderete, J.B.; Dinamarca, R.; Bustamente, T.M.; Pawelec, B. Gold catalysts supported on TiO2-nanotubes for the selective hydrogenation of p-substituted nitrobenzenes. Mol. Catal. 2018, 447, 21–27. [Google Scholar] [CrossRef]
- Morales, R.; Campos, C.H.; Fierro, J.L.G.; Fraga, M.A.; Pecchi, G. Stable reduced Ni catalysts for xylose hydrogenation in aqueous medium. Catal. Today. 2018, 310, 59–67. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).