
Supported Inverse MnOx/Pt Catalysts Facilitate Reverse Water Gas Shift Reaction


Abstract

1. Introduction
2. Results and Discussion
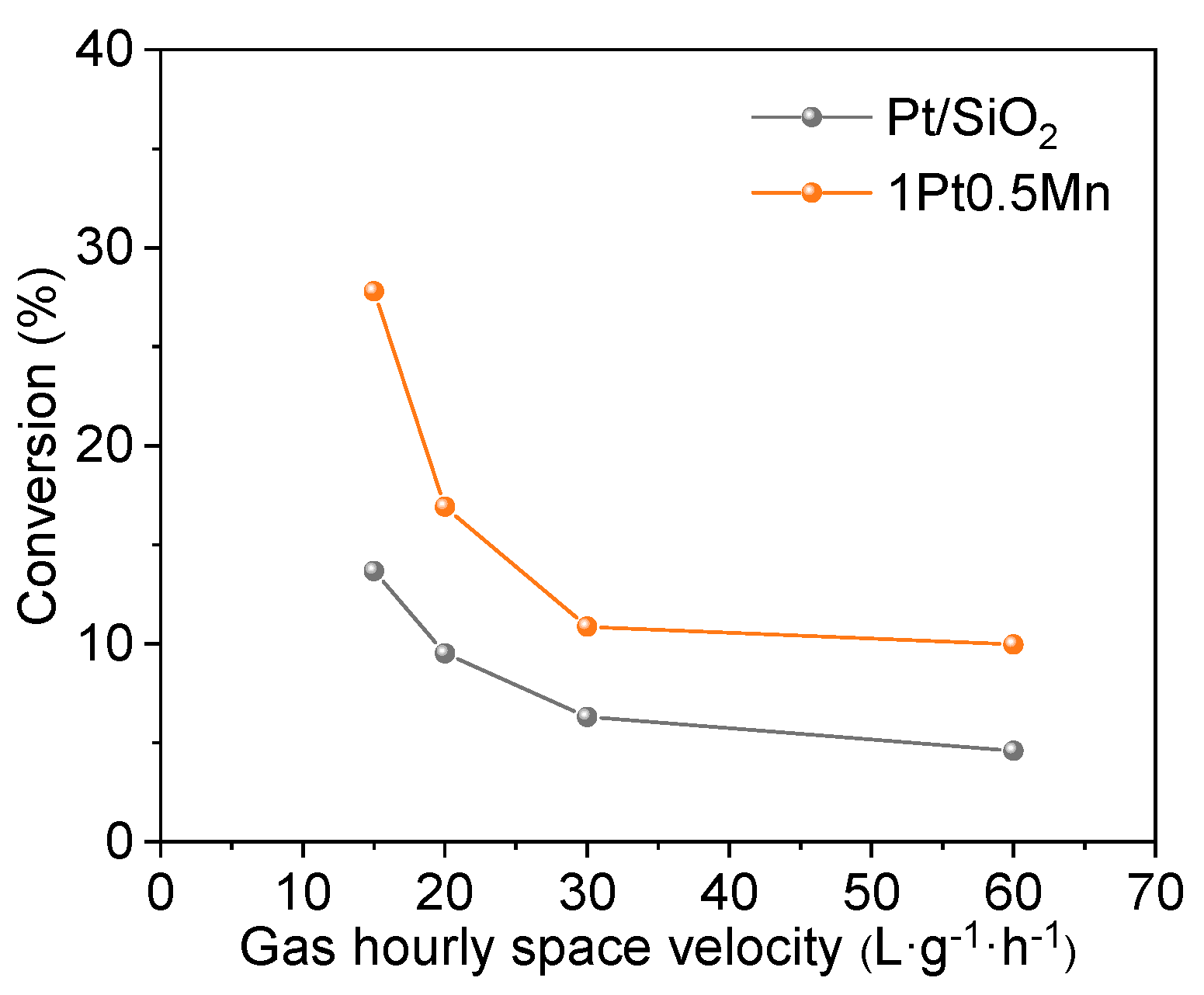
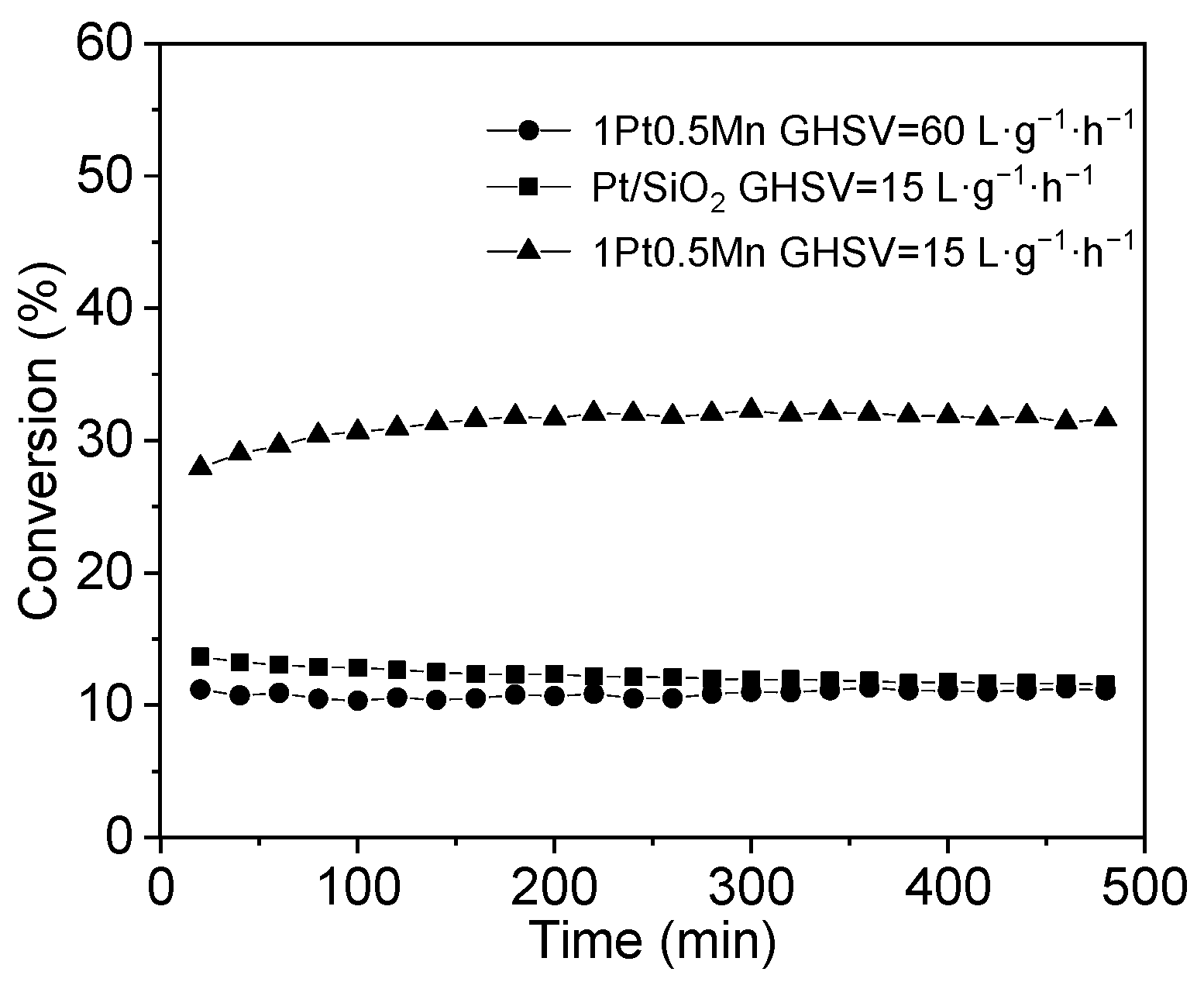
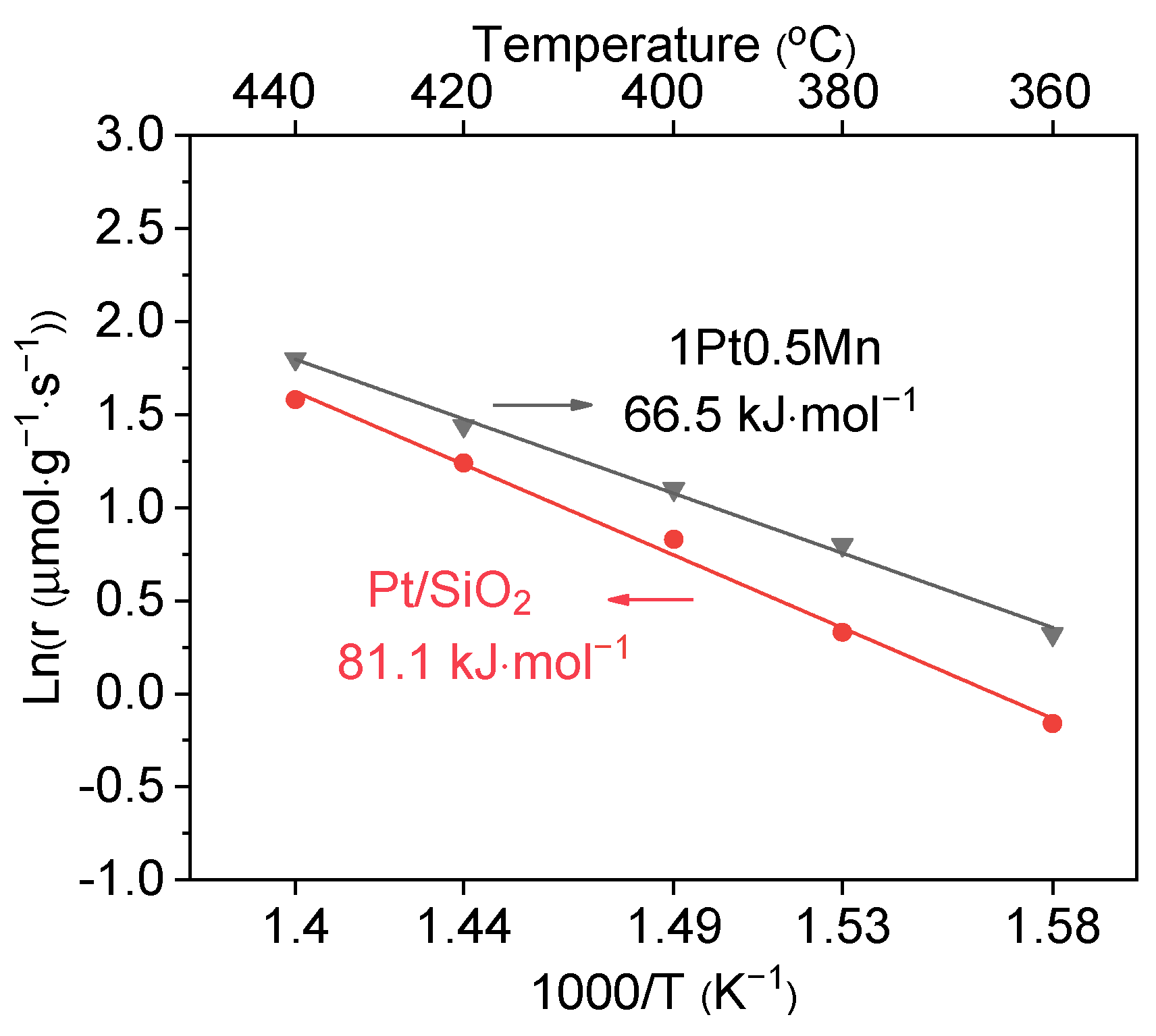
2.1. Catalytic Performance
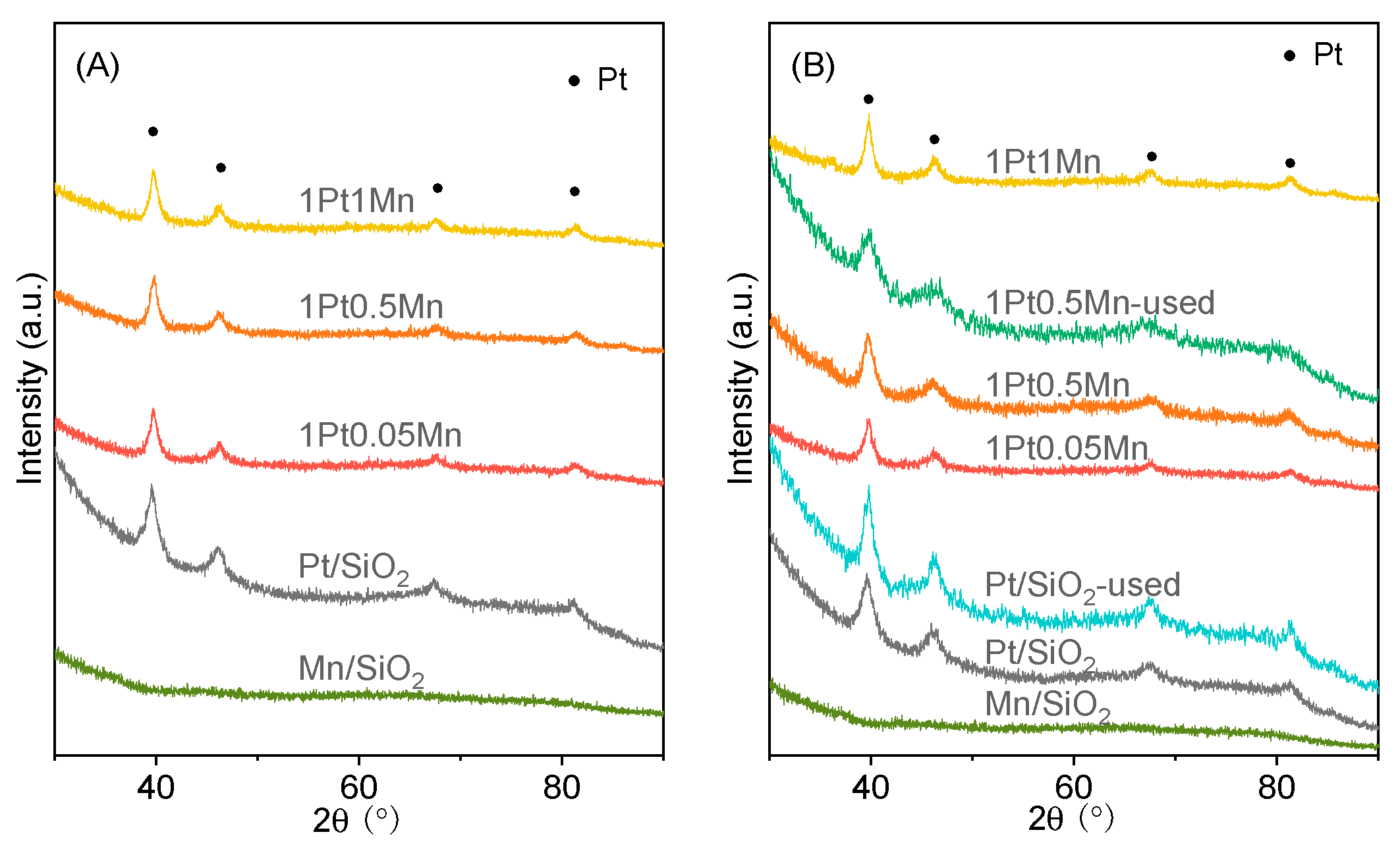
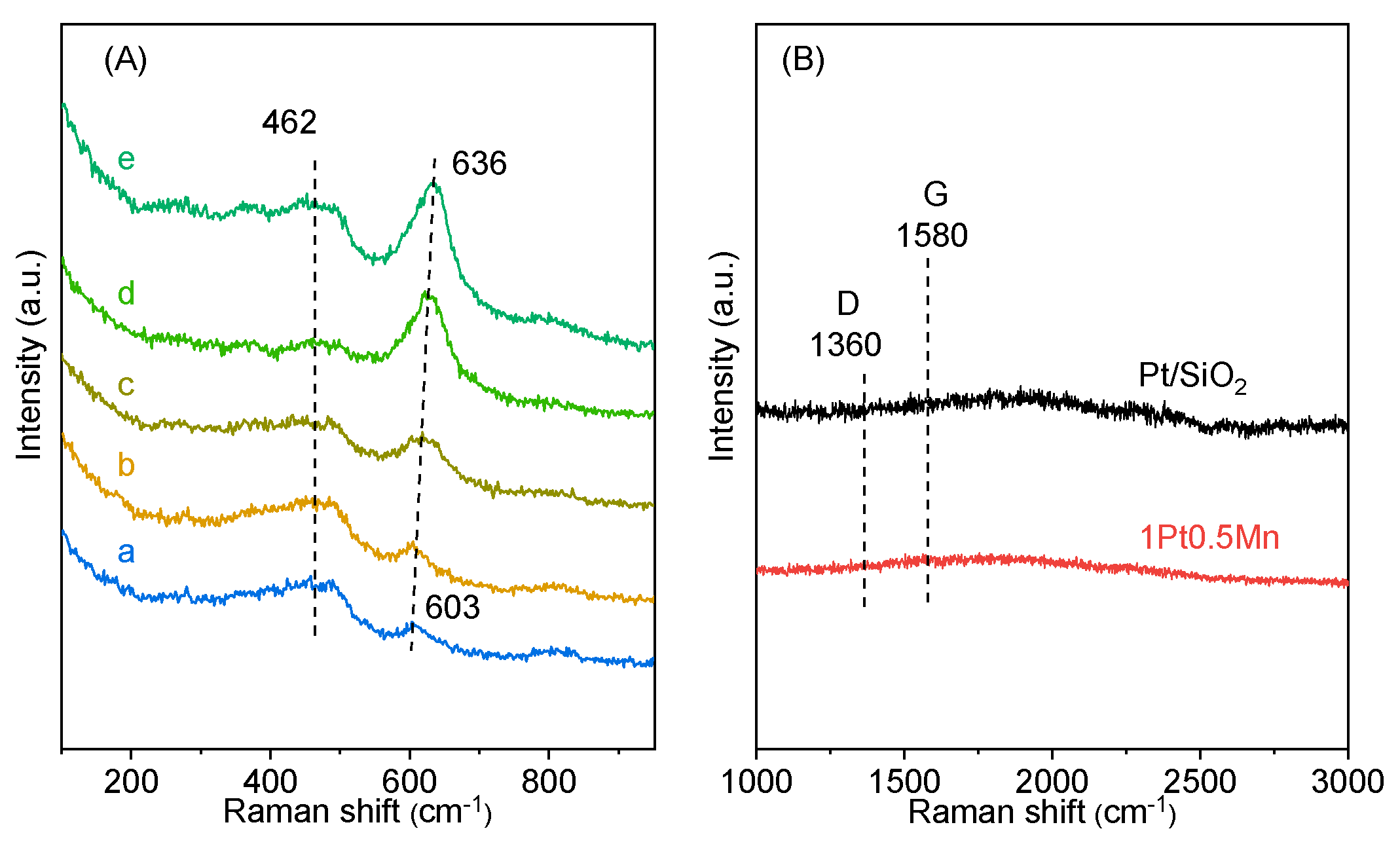
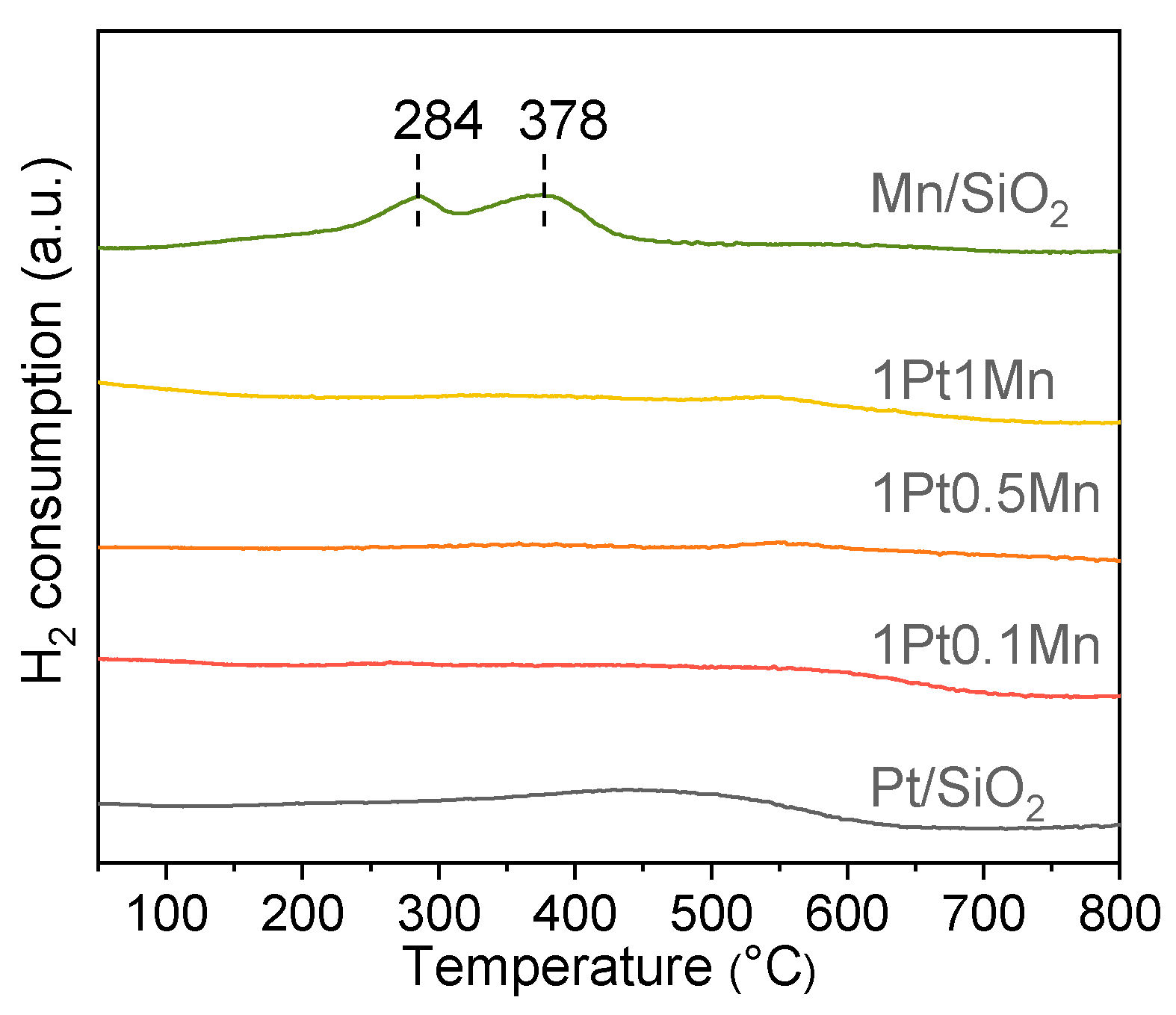
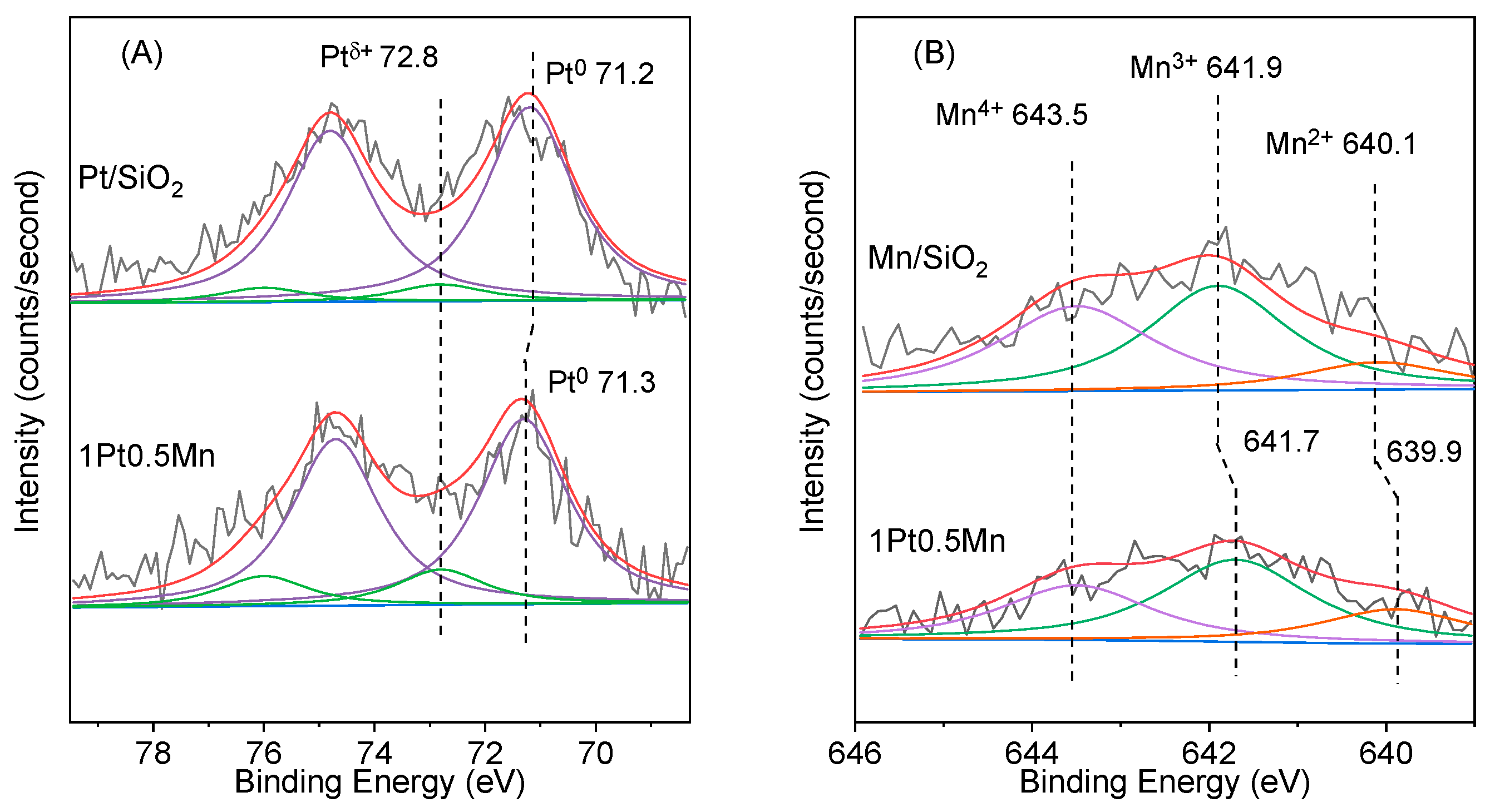
2.2. Catalytic Characterization
2.3. Discussion
3. Experimental
3.1. Catalyst Preparation
3.2. Catalyst Characterization
3.3. Catalytic Activity
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Jiang, L.-Q.; Dunne, J.; Carter, B.R.; Tjiputra, J.F.; Terhaar, J.; Sharp, J.D.; Olsen, A.; Alin, S.; Bakker, D.C.E.; Feely, R.A.; et al. Global surface ocean acidification indicators from 1750 to 2100. J. Adv. Model. Earth Syst. 2023, 15, e2022MS003563. [Google Scholar] [CrossRef]
- Schakel, W.; Oreggioni, G.; Singh, B.; Strømman, A.; Ramírez, A. Assessing the techno-environmental performance of CO2 utilization via dry reforming of methane for the production of dimethyl ether. J. CO2 Util. 2016, 16, 138–149. [Google Scholar] [CrossRef]
- Hu, B.; Guild, C.; Suib, S.L. Thermal, electrochemical, and photochemical conversion of CO2 to fuels and value-added products. J. CO2 Util. 2013, 1, 18–27. [Google Scholar] [CrossRef]
- Galadima, A.; Muraza, O. Catalytic thermal conversion of CO2 into fuels: Perspective and challenges. Renew. Sustain. Energy Rev. 2019, 115, 109333. [Google Scholar] [CrossRef]
- Su, X.; Yang, X.; Zhao, B.; Huang, Y. Designing of highly selective and high-temperature endurable RWGS heterogeneous catalysts: Recent advances and the future directions. J. Energy Chem. 2017, 26, 854–867. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, R.; Liu, Y.; Wu, X.; Wang, H.; Ge, Q.; Zhu, X. Blocking methanation during reverse water gas shift reaction on Ni/SiO2 catalysts by surface Ag. ChemCatChem 2023, 15, e202201284. [Google Scholar] [CrossRef]
- Chen, X.; Su, X.; Liang, B.; Yang, X.; Ren, X.; Duan, H.; Huang, Y.; Zhang, T. Identification of relevant active sites and a mechanism study for reverse water gas shift reaction over Pt/CeO2 catalysts. J. Energy Chem. 2016, 25, 1051–1057. [Google Scholar] [CrossRef]
- Chen, X.; Su, X.; Duan, H.; Liang, B.; Huang, Y.; Zhang, T. Catalytic performance of the Pt/TiO2 catalysts in reverse water gas shift reaction: Controlled product selectivity and a mechanism study. Catal. Today 2017, 281, 312–318. [Google Scholar] [CrossRef]
- Kim, S.S.; Lee, H.H.; Hong, S.C. A study on the effect of support’s reducibility on the reverse water-gas shift reaction over Pt catalysts. Appl. Catal. A 2012, 423–424, 100–107. [Google Scholar] [CrossRef]
- Chen, Z.; Liang, L.; Yuan, H.; Liu, H.; Wu, P.; Fu, M.; Wu, J.; Chen, P.; Qiu, Y.; Ye, D.; et al. Reciprocal regulation between support defects and strong metal-support interactions for highly efficient reverse water gas shift reaction over Pt/TiO2 nanosheets catalysts. Appl. Catal. B 2021, 298, 120507. [Google Scholar] [CrossRef]
- Wu, X.; Liu, C.-j.; Wang, H.; Ge, Q.; Zhu, X. Origin of strong metal-support interactions between Pt and anatase TiO2 facets for hydrodeoxygenation of m-cresol on Pt/TiO2 catalysts. J. Catal. 2023, 418, 203–215. [Google Scholar] [CrossRef]
- Cui, B.; Wang, H.; Ge, Q.; Zhu, X. Size-dependent strong metal-support interactions of rutile TiO2-supported Ni catalysts for hydrodeoxygenation of m-cresol. Catalysts 2022, 12, 955. [Google Scholar] [CrossRef]
- Cui, B.; Wang, H.; Han, J.; Ge, Q.; Zhu, X. Crystal-phase-depended strong metal-support interactions enhancing hydrodeoxygenation of m-cresol on Ni/TiO2 catalysts. J. Catal. 2022, 413, 880–890. [Google Scholar] [CrossRef]
- Zhao, X.; Wu, X.; Wang, H.; Han, J.; Ge, Q.; Zhu, X. Effect of strong metal-support interaction of Pt/TiO2 on hydrodeoxygenation of m-cresol. ChemistrySelect 2018, 3, 10364–10370. [Google Scholar] [CrossRef]
- Huang, R.; Kwon, O.; Lin, C.; Gorte, R.J. The effects of SMSI on m-cresol hydrodeoxygenation over Pt/Nb2O5 and Pt/TiO2. J. Catal. 2021, 398, 102–108. [Google Scholar] [CrossRef]
- Ro, I.; Resasco, J.; Christopher, P. Approaches for understanding and controlling interfacial effects in oxide-supported metal catalysts. ACS Catal. 2018, 8, 7368–7387. [Google Scholar] [CrossRef]
- Tauster, S.J.; Fung, S.C.; Garten, R.L. Strong metal-support interactions. Group 8 noble metals supported on titanium dioxide. J. Am. Chem. Soc. 1978, 100, 170–175. [Google Scholar] [CrossRef]
- Pesty, F.; Steinrück, H.-P.; Madey, T.E. Thermal stability of Pt films on TiO2(110): Evidence for encapsulation. Surf. Sci. 1995, 339, 83–95. [Google Scholar] [CrossRef]
- Zhu Chen, J.; Talpade, A.; Canning, G.A.; Probus, P.R.; Ribeiro, F.H.; Datye, A.K.; Miller, J.T. Strong metal-support interaction (SMSI) of Pt/CeO2 and its effect on propane dehydrogenation. Catal. Today 2021, 371, 4–10. [Google Scholar] [CrossRef]
- Han, B.; Guo, Y.; Huang, Y.; Xi, W.; Xu, J.; Luo, J.; Qi, H.; Ren, Y.; Liu, X.; Qiao, B.; et al. Strong metal-support interactions between Pt single atoms and TiO2. Angew. Chem. Int. Ed. 2020, 59, 11824–11829. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, F.; Wang, L.; Qi, X.; Shi, F.; Deng, Y. Low-temperature CO oxidation over supported Pt, Pd catalysts: Particular role of FeOx support for oxygen supply during reactions. J. Catal. 2010, 274, 1–10. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, L.; Bao, Y.; Zhang, Y.; Wang, J.; Fu, M.; Wu, J.; Ye, D. The applications of morphology controlled ZnO in catalysis. Catalysts 2016, 6, 188. [Google Scholar] [CrossRef]
- Rieboldt, F.; Helveg, S.; Bechstein, R.; Lammich, L.; Besenbacher, F.; Lauritsen, J.V.; Wendt, S. Formation and sintering of Pt nanoparticles on vicinal rutile TiO2 surfaces. Phys. Chem. Chem. Phys. 2014, 16, 21289–21299. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Zhu, X. Tuning strong metal-support interactions for enhancing direct deoxygenation of biomass-lignin derived phenolics. ChemCatChem 2024, 16, e202301552. [Google Scholar] [CrossRef]
- Rodríguez, J.A.; Hrbek, J. Inverse oxide/metal catalysts: A versatile approach for activity tests and mechanistic studies. Surf. Sci. 2010, 604, 241–244. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Graciani, J.; Evans, J.; Park, J.B.; Yang, F.; Stacchiola, D.; Senanayake, S.D.; Ma, S.; Pérez, M.; Liu, P.; et al. Water-gas shift reaction on a highly active inverse CeOx/Cu(111) catalyst: Unique role of ceria nanoparticles. Angew. Chem. Int. Ed. 2009, 48, 8047–8050. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.A.; Ma, S.; Liu, P.; Hrbek, J.; Evans, J.; Pérez, M. Activity of CeOx and TiOx nanoparticles grown on Au(111) in the water-gas shift reaction. Science 2007, 318, 1757–1760. [Google Scholar] [CrossRef] [PubMed]
- Kapiamba, K.F.; Otor, H.O.; Viamajala, S.; Alba-Rubio, A.C. Inverse oxide/metal catalysts for CO2 hydrogenation to methanol. Energy Fuels 2022, 36, 11691–11711. [Google Scholar] [CrossRef]
- Marlowe, J.; Deshpande, S.; Vlachos, D.G.; Abu-Omar, M.M.; Christopher, P. Effect of dynamic and preferential decoration of Pt catalyst surfaces by WOx on hydrodeoxygenation reactions. J. Am. Chem. Soc. 2024, 146, 13862–13874. [Google Scholar] [CrossRef]
- Barrio, L.; Estrella, M.; Zhou, G.; Wen, W.; Hanson, J.C.; Hungría, A.B.; Hornés, A.; Fernández-García, M.; Martínez-Arias, A.; Rodriguez, J.A. Unraveling the active site in copper-ceria systems for the water-gas shift reaction: In situ characterization of an inverse powder CeO2−x/CuO-Cu catalyst. J. Phys. Chem. C 2010, 114, 3580–3587. [Google Scholar] [CrossRef]
- Singh Negi, S.; Kim, H.-M.; Cheon, B.-S.; Jeong, D.-W. Active nanointerfaces sites for low-temperature water-gas shift reaction over inverse configuration Nb-CeO2/Cu catalysts. Fuel 2024, 362, 130908. [Google Scholar] [CrossRef]
- Shen, Y.; Xiao, Z.; Liu, J.; Wang, Z. Facile preparation of inverse nanoporous Cr2O3/Cu catalysts for reverse water-gas shift reaction. ChemCatChem 2019, 11, 5439–5443. [Google Scholar] [CrossRef]
- Xiong, W.; Wu, Z.; Chen, X.; Ding, J.; Ye, A.; Zhang, W.; Huang, W. Active copper structures in ZnO-Cu interfacial catalysis: CO2 hydrogenation to methanol and reverse water-gas shift reactions. Sci. China Chem. 2024, 67, 715–723. [Google Scholar] [CrossRef]
- Li, Z.-X.; Xu, K.; Wang, W.-W.; Fu, X.-P.; Jia, C.-j. Stabilized inverse Y2O3/Cu interfaces boost the performance of the reverse water-gas shift reaction. Catal. Sci. Technol. 2024, 14, 3483–3492. [Google Scholar] [CrossRef]
- Liu, Y.; Li, L.; Zhang, R.; Guo, Y.; Wang, H.; Ge, Q.; Zhu, X. Synergetic enhancement of activity and selectivity for reverse water gas shift reaction on Pt-Re/SiO2 catalysts. J. CO2 Util. 2022, 63, 102128. [Google Scholar] [CrossRef]
- Kattel, S.; Yu, W.; Yang, X.; Yan, B.; Huang, Y.; Wan, W.; Liu, P.; Chen, J.G. CO2 hydrogenation over oxide-supported PtCo catalysts: The role of the oxide support in determining the product selectivity. Angew. Chem. Int. Ed. 2016, 55, 7968–7973. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, D.; Kobayashi, H.; Kusada, K.; Yamamoto, T.; Toriyama, T.; Matsumura, S.; Kawaguchi, S.; Kubota, Y.; Haneda, M.; Aspera, S.M.; et al. Boosting reverse water-gas shift reaction activity of Pt nanoparticles through light doping of W. J. Mater. Chem. A 2021, 9, 15613–15617. [Google Scholar] [CrossRef]
- Kattel, S.; Yan, B.; Chen, J.G.; Liu, P. CO2 hydrogenation on Pt, Pt/SiO2 and Pt/TiO2: Importance of synergy between Pt and oxide support. J. Catal. 2016, 343, 115–126. [Google Scholar] [CrossRef]
- Dalebout, R.; Barberis, L.; Visser, N.L.; van der Hoeven, J.E.S.; van der Eerden, A.M.J.; Stewart, J.A.; Meirer, F.; de Jong, K.P.; de Jongh, P.E. Manganese oxide as a promoter for copper catalysts in CO2 and CO hydrogenation. ChemCatChem 2022, 14, e202200451. [Google Scholar] [CrossRef]
- Dorner, R.W.; Hardy, D.R.; Williams, F.W.; Willauer, H.D. K and Mn doped iron-based CO2 hydrogenation catalysts: Detection of KAlH4 as part of the catalyst’s active phase. Appl. Catal. A 2010, 373, 112–121. [Google Scholar] [CrossRef]
- González-Arias, J.; González-Castaño, M.; Sánchez, M.E.; Cara-Jiménez, J.; Arellano-García, H. Valorization of biomass-derived CO2 residues with Cu-MnOx catalysts for RWGS reaction. Renew. Energy 2022, 182, 443–451. [Google Scholar] [CrossRef]
- Shadravan, V.; Bukas, V.J.; Gunasooriya, G.T.K.K.; Waleson, J.; Drewery, M.; Karibika, J.; Jones, J.; Kennedy, E.; Adesina, A.; Nørskov, J.K.; et al. Effect of manganese on the selective catalytic hydrogenation of COx in the presence of light hydrocarbons over Ni/Al2O3: An experimental and computational study. ACS Catal. 2019, 10, 1535–1547. [Google Scholar] [CrossRef]
- Zhukova, A.; Chuklina, S.; Fionov, Y.; Vakhrushev, N.; Sazonova, A.; Mikhalenko, I.; Zhukov, D.; Isaikina, O.; Fionov, A.; Il’Icheva, A. Enhanced ethanol dehydrogenation over Ni-containing zirconia-alumina catalysts with microwave-assisted synthesis. Res. Chem. Intermed. 2024, 50, 1331–1354. [Google Scholar] [CrossRef]
- Janampelli, S.; Darbha, S. Promotional effect of WOx in Pt-WOx/AlPO4-5 catalyzed deoxygenation of fatty acids. ChemistrySelect 2017, 2, 1895–1901. [Google Scholar] [CrossRef]
- Wu, Y.; Sourav, S.; Worrad, A.; Zhou, J.; Caratzoulas, S.; Tsilomelekis, G.; Zheng, W.; Vlachos, D.G. Dynamic formation of brønsted acid sites over supported WOx/Pt on SiO2 inverse catalysts-spectroscopy, probe chemistry, and calculations. ACS Catal. 2023, 13, 7371–7382. [Google Scholar] [CrossRef]
- Patcas, F.; Buciuman, F.C. Investigations on formation and structure of MnOx/SiO2 catalysts. Z. Phys. Chem. 2001, 215, 29–36. [Google Scholar] [CrossRef]
- Chen, Z.; Yang, Q.; Li, H.; Li, X.; Wang, L.; Chi Tsang, S. Cr-MnOx mixed-oxide catalysts for selective catalytic reduction of NO with NH3 at low temperature. J. Catal. 2010, 276, 56–65. [Google Scholar] [CrossRef]
- Zhang, X.; Bi, F.; Zhu, Z.; Yang, Y.; Zhao, S.; Chen, J.; Lv, X.; Wang, Y.; Xu, J.; Liu, N. The promoting effect of H2O on rod-like MnCeOx derived from MOFs for toluene oxidation: A combined experimental and theoretical investigation. Appl. Catal. B 2021, 297, 120393. [Google Scholar] [CrossRef]
- Delimaris, D.; Ioannides, T. VOC oxidation over MnOx-CeO2 catalysts prepared by a combustion method. Appl. Catal. B 2008, 84, 303–312. [Google Scholar] [CrossRef]
- Chen, J.; Chen, X.; Xu, W.; Xu, Z.; Chen, J.; Jia, H.; Chen, J. Hydrolysis driving redox reaction to synthesize Mn-Fe binary oxides as highly active catalysts for the removal of toluene. Chem. Eng. J. 2017, 330, 281–293. [Google Scholar] [CrossRef]
- Ru, X.; Li, W.; Wang, X.; Shi, Z.; Wen, X.; Mo, S.; Zhang, Q.; Mo, D. Regulating the surface local environment of MnO2 materials via metal-support interaction in Pt/MnO2 hetero-catalysts for boosting methanol oxidation. Chem. Eng. Sci. 2023, 281, 119079. [Google Scholar] [CrossRef]
- Zhang, H.; Sui, S.; Zheng, X.; Cao, R.; Zhang, P. One-pot synthesis of atomically dispersed Pt on MnO2 for efficient catalytic decomposition of toluene at low temperatures. Appl. Catal. B 2019, 257, 117878. [Google Scholar] [CrossRef]
- Arena, F.; Torre, T.; Raimondo, C.; Parmaliana, A. Structure and redox properties of bulk and supported manganese oxide catalysts. Phys. Chem. Chem. Phys. 2001, 3, 1911–1917. [Google Scholar] [CrossRef]
- Kikkawa, S.; Teramura, K.; Kato, K.; Asakura, H.; Hosokawa, S.; Tanaka, T. Formation of CH4 at the metal-support interface of Pt/Al2O3 during hydrogenation of CO2: Operando XAS-DRIFTS study. ChemCatChem 2022, 14, e202101723. [Google Scholar] [CrossRef]
- Bazin, P.; Saur, O.; Lavalley, J.C.; Daturi, M.; Blanchard, G. FT-IR study of CO adsorption on Pt/CeO2: Characterisation and structural rearrangement of small Pt particles. Phys. Chem. Chem. Phys. 2005, 7, 187. [Google Scholar] [CrossRef]
- Wu, Q.; Li, H.; Yang, G.; Cao, Y.; Wang, H.; Peng, F.; Yu, H. Surface intermediates steer the pathways of CO2 hydrogenation on Pt/γ-Al2O3: Importance of the metal-support interface. J. Catal. 2023, 425, 40–49. [Google Scholar] [CrossRef]
- Liu, D.; Li, Y.; Kottwitz, M.; Yan, B.; Yao, S.; Gamalski, A.; Grolimund, D.; Safonova, O.V.; Nachtegaal, M.; Chen, J.G.; et al. Identifying dynamic structural changes of active sites in Pt-Ni bimetallic catalysts using multimodal approaches. ACS Catal. 2018, 8, 4120–4131. [Google Scholar] [CrossRef]
- Zhang, R.; Wei, A.; Zhu, M.; Wu, X.; Wang, H.; Zhu, X.; Ge, Q. Tuning reverse water gas shift and methanation reactions during CO2 reduction on Ni catalysts via surface modification by MoOx. J. CO2 Util. 2021, 52, 101678. [Google Scholar] [CrossRef]
- Wang, H.; Bootharaju, M.S.; Kim, J.H.; Wang, Y.; Wang, K.; Zhao, M.; Zhang, R.; Xu, J.; Hyeon, T.; Wang, X.; et al. Synergistic interactions of neighboring platinum and iron atoms enhance reverse water-gas shift reaction performance. J. Am. Chem. Soc. 2023, 145, 2264–2270. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Chen, J.G. Trends in the catalytic reduction of CO2 by hydrogen over supported monometallic and bimetallic catalysts. J. Catal. 2013, 301, 30–37. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, M.; Ma, P.; Zheng, Y.; Chen, J.; Li, H.; Zhang, X.; Zheng, K.; Kuang, Q.; Xie, Z.-X. Atomically dispersed Pt/CeO2 catalyst with superior CO selectivity in reverse water gas shift reaction. Appl. Catal. B 2021, 291, 120101. [Google Scholar] [CrossRef]
- Huang, Z.; Yuan, Y.; Song, M.; Hao, Z.; Xiao, J.; Cai, D.; Ibrahim, A.-R.; Zhan, G. CO2 hydrogenation over mesoporous Ni-Pt/SiO2 nanorod catalysts: Determining CH4/CO selectivity by surface ratio of Ni/Pt. Chem. Eng. Sci. 2022, 247, 117106. [Google Scholar] [CrossRef]
- Xu, Z.; Mo, S.; Li, Y.; Zhang, Y.; Wu, J.; Fu, M.; Niu, X.; Hu, Y.; Ye, D. Pt/MnOx for toluene mineralization via ozonation catalysis at low temperature: SMSI optimization of surface oxygen species. Chemosphere 2022, 286, 131754. [Google Scholar] [CrossRef]
- Yang, X.; Su, X.; Chen, X.; Duan, H.; Liang, B.; Liu, Q.; Liu, X.; Ren, Y.; Huang, Y.; Zhang, T. Promotion effects of potassium on the activity and selectivity of Pt/zeolite catalysts for reverse water gas shift reaction. Appl. Catal. B 2017, 216, 95–105. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Ptδ+/(Pt0 + Ptδ+) | Mn Distribution (%) | Mn/Pt Atomic Ratio | ||
|---|---|---|---|---|---|
| Mn4+ | Mn3+ | Mn2+ | |||
| Pt/SiO2 | 0.087 | - | - | - | - |
| 1Pt0.5Mn | 0.158 | 32.1 | 48.3 | 19.6 | 3.44 |
| Mn/SiO2 | - | 41.3 | 46.0 | 12.7 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bi, W.; Zhang, R.; Ge, Q.; Zhu, X. Supported Inverse MnOx/Pt Catalysts Facilitate Reverse Water Gas Shift Reaction. Catalysts 2024, 14, 456. https://doi.org/10.3390/catal14070456
Bi W, Zhang R, Ge Q, Zhu X. Supported Inverse MnOx/Pt Catalysts Facilitate Reverse Water Gas Shift Reaction. Catalysts. 2024; 14(7):456. https://doi.org/10.3390/catal14070456
Chicago/Turabian StyleBi, Wenli, Ruoyu Zhang, Qingfeng Ge, and Xinli Zhu. 2024. "Supported Inverse MnOx/Pt Catalysts Facilitate Reverse Water Gas Shift Reaction" Catalysts 14, no. 7: 456. https://doi.org/10.3390/catal14070456
APA StyleBi, W., Zhang, R., Ge, Q., & Zhu, X. (2024). Supported Inverse MnOx/Pt Catalysts Facilitate Reverse Water Gas Shift Reaction. Catalysts, 14(7), 456. https://doi.org/10.3390/catal14070456