
Properties, Industrial Applications and Future Perspectives of Catalytic Materials Based on Nickel and Alumina: A Critical Review



Abstract
:1. Introduction
2. Solid-State Chemistry of Nickel and Aluminum-Based Catalytic Materials
2.1. Oxide and Hydroxide Phases
2.1.1. Ni Oxides and Hydroxides
2.1.2. Aluminum Oxides and Hydroxides
2.1.3. Mixed Ni-Al Hydroxide and Oxide Phases
2.1.4. Deposition of NiO onto Transitional Aluminas
2.1.5. Deposition of NiO over α-Al2O3 and Alkali Earth Aluminates
2.2. Metallic Phases in the Ni-Al System
2.3. On the Reactivity of the Metallic Ni-Based Phases
2.3.1. Oxidation of Metallic Nickel
2.3.2. Sulfidation of Metallic Nickel
2.3.3. Reactivity with Carbon: Carburization and Formation of Carbon Nanotubes and Ni(CO)4
2.3.4. Nickel Reactivity with Hydrogen
2.3.5. Nickel Reactivity with HCl
3. Surface Properties of Materials Based on Nickel and Alumina
3.1. Characterization of Unreduced Systems
3.1.1. Surface Properties of Aluminas
3.1.2. Surface Properties of Nickel Oxide NiOx (x ≥ 1)
3.1.3. Surface Properties of NiAl2O4 and NiAl2O4-Al2O3 Solid Solutions
3.1.4. Surface Properties of Impregnated NiO/Al2O3 Materials
3.2. Characterization of Reduced Ni/Al2O3 Materials
4. Mechanistic Aspects in Catalysis by Ni and Alumina Materials
4.1. The Activation of Hydrogen on Ni/Al2O3 Catalysts
4.2. The Activation of Carbon Monoxide
4.3. The Activation of Unsaturated Hydrocarbons
4.4. The Activation of CO2
4.5. The Activation of Water
4.6. The Activation of Oxygen
4.7. The Activation of Saturated Hydrocarbons on Reduced and Oxidized Nickel
5. Deactivation and Reactivation of Ni/Al2O3 Metal Catalysts
6. Applications of Catalysts Based on Nickel and Alumina or Aluminates
6.1. Commercial Ni-Al2O3 Catalysts for Hydrogenation Reactions
6.1.1. Hydrogenation of Acetylenics and Dienes
6.1.2. Dearomatization Reactions
6.1.3. Low-Temperature Methanation of COx for Hydrogen Purification
6.1.4. High-Temperature Methanation of COX for the Production of Substitute Natural Gas
6.1.5. Methanation of CO2
6.1.6. Reverse Water–Gas Shift
6.2. Ni/Al2O3-Based Catalysts for Other Hydrogenation Reactions
6.2.1. Application in Amine Syntheses
6.2.2. Hydrogenation of Oxygenates
6.2.3. Hydrogenations and Hydrodeoxygenation of Renewables
6.2.4. Hydrogenolysis and Pyrolysis for the Conversion of Plastic Wastes to Useful Products
6.3. Catalysts for Hydrogen and Syngas Production
6.3.1. Industrial Steam Reforming of Methane, Natural Gas and Higher Hydrocarbons
6.3.2. Industrial Pre-Reforming of Natural Gas
6.3.3. Methane Dry Reforming and Biogas Reforming Reaction
6.3.4. Methane Partial Oxidation, Secondary Reforming and Autothermal Reforming
6.3.5. Methane and Hydrocarbon Decomposition Catalysts for the Production of Hydrogen and Carbon Nanomaterials
6.3.6. Ammonia Cracking Catalysts
6.3.7. Steam Reforming of Biomass Tar for Purification of Biomass Gasification-Derived Syngas
6.3.8. Steam Reforming, Partial Oxidation and Autothermal and Dry Reforming of Renewables for the Production of Renewable Hydrogen
6.3.9. Ni/Al2O3 for Chemical Looping Processes
6.3.10. Ni/Alumina Catalysts in Dehydrogenation and Aromatization Reactions
6.4. Applications of Unreduced NiO-Al2O3 Catalysts
6.4.1. NiO and NiO-Al2O3 as Combustion Catalysts
6.4.2. The Oxidation of Ammonia and Hydrogen
6.4.3. Oxidation of Carbon Monoxide
6.4.4. Oxidative Dehydrogenation of Alkanes
6.4.5. Ozone Decomposition and Use
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Busca, G. Heterogeneous Catalytic Materials; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Busca, G. Critical aspects of energetic transition technologies and the roles of materials chemistry and engineering. Energies 2024, 17, 3565. [Google Scholar] [CrossRef]
- Hughes, A.E.; Haque, N.; Northey, S.A.; Giddey, S. Platinum Group Metals: A Review of Resources, Production and Usage with a Focus on Catalysts. Resources 2021, 10, 93. [Google Scholar] [CrossRef]
- Ananikov, V.P. Nickel: The “Spirited Horse” of Transition Metal Catalysis. ACS Catal. 2015, 5, 1964–1971. [Google Scholar] [CrossRef]
- Serov, A. Nickel catalysts for affordable fuel cells. Nat. Catal. 2022, 5, 971–972. [Google Scholar] [CrossRef]
- Bartholomew, C.H.; Farrauto, R.J. Chemistry of nickel-alumina catalysts. J. Catal. 1976, 45, 41–53. [Google Scholar] [CrossRef]
- Perry, R.H.; Green, D.W. Perry’s Chemical Engineers’ Handbook, 7th ed.; Mc Graw Hill Pub.: New York, NY, USA, 1997; Chapter 4. [Google Scholar]
- Hall, D.S.; Lockwood, D.J.; Bock, C.; MacDougall, B.R. Nickel hydroxides and related materials: A review of their structures, synthesis and properties. Proc. R. Soc. A 2015, 471, 20140792. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.F.; Li, J.L.; Liu, Z.P. Structure and Catalysis of NiOOH: Recent Advances on Atomic Simulation. J. Phys. Chem. C 2021, 125, 27033–27045. [Google Scholar] [CrossRef]
- Kannan, K.; Radhika, D.; Kumar Sadasivuni, K.; Raghava Reddy, K.; Raghu, A.V. Nanostructured metal oxides and its hybrids for photocatalytic and biomedical applications. Adv. Colloid Interface Sci. 2020, 281, 102178. [Google Scholar] [CrossRef]
- Hussein, N.A.; Ali, N.A. Ni2O3 nanomaterial: Synthesis and characterization by simple chemical process. AIP Conf. Proc. 2023, 2834, 030021. [Google Scholar]
- Fingerle, M.; Tengeler, S.; Calvetz, W.; Jaegermann, W.; Mayer, T. Sputtered Nickel Oxide Thin Films on n-Si(100)/SiO2 Surfaces for Photo-Electrochemical Oxygen Evolution Reaction (OER): Impact of Deposition Temperature on OER Performance and on Composition before and after OER. J. Electrochem. Soc. 2020, 167, 136514. [Google Scholar] [CrossRef]
- Pereñíguez, R.; González-DelaCruz, V.M.; Holgado, J.P.; Caballero, A. Synthesis and characterization of a LaNiO3 perovskite as precursor for methane reforming reactions catalysts. Appl. Catal. B Environ. 2010, 93, 346–353. [Google Scholar] [CrossRef]
- Nagle-Cocco, L.A.V.; Bull, C.L.; Ridley, C.J.; Dutton, S.E. Pressure Tuning the Jahn−Teller Transition Temperature in NaNiO2. Inorg. Chem. 2022, 61, 4312–4321. [Google Scholar] [CrossRef]
- Li, Y.; Tan, Z.; Liu, Y.; Lei, C.; He, P.; Li, J.; He, Z.; Cheng, Y.; Wu, F.; Li, Y. Past, present and future of high-nickel materials. Nano Energy 2024, 119, 109070. [Google Scholar] [CrossRef]
- Genreith-Schriever, A.R.; Alexiu, A.; Phillips, G.S.; Coates, C.S.; Nagle-Cocco, L.A.V.; Bocarsly, J.D.; Sayed, F.N.; Dutton, S.E.; Grey, C.P. Jahn−Teller Distortions and Phase Transitions in LiNiO2: Insights from Ab Initio Molecular Dynamics and Variable-Temperature X-ray Diffraction. Chem. Mater. 2024, 36, 2289–2303. [Google Scholar] [CrossRef]
- Busca, G. The surface of transition aluminas. A critical Review. Catal. Today 2014, 226, 2–13. [Google Scholar] [CrossRef]
- Busca, G. Structural, surface and catalytic properties of aluminas. Adv. Catal. 2014, 57, 319–404. [Google Scholar]
- Yang, Y.; Miao, C.; Wang, R.; Zhang, R.; Li, X.; Wang, J.; Wang, X.; Yao, J. Advances in morphology-controlled alumina and its supported Pd catalysts: Synthesis and applications. Chem. Soc. Rev. 2024, 53, 5014–5053. [Google Scholar] [CrossRef]
- Violante, A.; Huang, P.M. Influence of inorganic and organic ligands on the formation of aluminum hydroxides and oxyhydroxides. Clays Clay Min. 1985, 33, 181–192. [Google Scholar] [CrossRef]
- Pereira Antunes, M.L.; de Souza Santos, H.; de Souza Santos, P. Characterization of the aluminum hydroxide microcrystals formed in some alcohol−water solutions. Mater. Chem. Phys. 2002, 76, 243–249. [Google Scholar] [CrossRef]
- Wefers, K.; Mitra, C. Oxides and Hydroxides of Aluminum; Alcoa Laboratories: New Kensington, PA, USA, 1987. [Google Scholar]
- Tsuchida, T. Preparation and reactivity of acicular α-Al2O3 from synthetic diaspore, β-Al2O3·H2O. Solid State Ion. 1993, 63–65, 464–470. [Google Scholar] [CrossRef]
- Marturano, M.; Aglietti, E.F.; Ferretti, O. α-Al2O3 catalyst supports for synthesis gas production: Influence of different alumina bonding agents on support and catalyst properties. Mater. Chem. Phys. 1997, 47, 252–256. [Google Scholar] [CrossRef]
- Deng, B.; Advincula, P.A.; Luong, D.X.; Zhou, J.; Zhang, B.; Wang, Z.; McHugh, E.A.; Chen, J.; Carter, R.A.; Kittrell, C.; et al. High-surface-area corundum nanoparticles by resistive hotspot-induced phase transformation. Nat. Commun. 2022, 13, 5027. [Google Scholar] [CrossRef]
- Levin, I.; Brandon, D. Metastable Alumina Polymorphs: Crystal Structures and Transition Sequences. J. Am. Ceram. Soc. 1998, 81, 1995–2012. [Google Scholar] [CrossRef]
- Dai, J.; Li, S.F.Y.; Xiao, T.D.; Wang, D.M.; Reisner, D.E. Structural stability of aluminum stabilized alpha nickel hydroxide as a positive electrode material for alkaline secondary batteries. J. Power Sources 2000, 89, 40–45. [Google Scholar] [CrossRef]
- Ogata, F.; Nakamura, T.; Toda, M.; Otani, M.; Kawasaki, N. Evaluation of Nickel–Aluminium Complex Hydroxide for Adsorption of Chromium(VI) Ion. Chem. Pharm. Bull. 2020, 68, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Cavani, F.; Trifirò, F.; Vaccari, A. Hydrotalcite-type anionic clays: Preparation, properties and applications. Catal. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- Debecker, D.P.; Gaigneaux, E.M.; Busca, G. Exploring, tuning and exploiting the basicity of hydrotalcites for applications in heterogeneous catalysis. Chem. Eur. J. 2009, 15, 3920–3935. [Google Scholar] [CrossRef] [PubMed]
- Kruissink, E.C.; Van Reijen, L.L.; Ross, J.R.H. Coprecipitated nickel-alumina catalysts for methanation at high temperature. Part 1.—Chemical composition and structure of the precipitates. J. Chem. Soc. Faraday Trans. 1981, 1, 649–663. [Google Scholar] [CrossRef]
- Clause, O.; Rebours, B.; Merlen, E.; Trifiró, F.; Vaccari, A. Preparation and characterization of nickel-aluminum mixed oxides obtained by thermal decomposition of hydrotalcite-type precursors. J. Catal. 1992, 133, 231–246. [Google Scholar] [CrossRef]
- Busca, G.; Lorenzelli, V.; Sanchez Escribano, V. Preparation, solid-state characterization, and surface chemistry of high surface area NixAl2-2xO3-2x mixed oxides. Chem. Mater. 1992, 4, 595–605. [Google Scholar] [CrossRef]
- Busca, G.; Lorenzelli, V.; Sanchez Escribano, V.; Guidetti, R. FT-IR study of the surface properties of the spinels NiAl2O4 and CoAl2O4 in relation to those of transitional aluminas. J. Catal. 1991, 131, 167. [Google Scholar] [CrossRef]
- Morales-Marína, A.; Ayastuya, J.L.; Iriarte-Velasco, U.; Gutiérrez-Ortiz, M.A. Nickel aluminate spinel-derived catalysts for the aqueous phase reforming of glycerol: Effect of reduction temperature. Appl. Catal. B Environ. 2019, 244, 931–945. [Google Scholar] [CrossRef]
- Raghavan, V. Al-Fe-Ni-O (Aluminum-Iron-Nickel-Oxygen). J. Phase Equilib. Diffus. 2010, 31, 377–378. [Google Scholar] [CrossRef]
- O’Neill, H.S.C.; Dollase, W.A.; Ross, C.R. Temperature dependence of the cation distribution in nickel aluminate (NiAl2O4) spinel: A powder XRD study. Phys. Chem. Miner. 1991, 18, 302–319. [Google Scholar] [CrossRef]
- Gil-Calvo, M.; Jiménez-González, C.; de Rivas, B.; Gutiérrez-Ortiz, J.I.; López-Fonseca, R. Effect of Ni/Al molar ratio on the performance of substoichiometric NiAl2O4 spinel-based catalysts for partial oxidation of methane. Appl. Catal. B Environ. 2017, 209, 128–138. [Google Scholar] [CrossRef]
- Rogers, J.L.; Mangarella, M.C.; D’Amico, A.D.; Gallagher, J.R.; Dutzer, M.R.; Stavitski, E.; Miller, J.T.; Sievers, C. Differences in the Nature of Active Sites for Methane Dry Reformingand Methane Steam Reforming over Nickel Aluminate Catalysts. ACS Catal. 2016, 6, 5873–5886. [Google Scholar] [CrossRef]
- Bassoul, P.; Gilles, J.C. Structure and microstructure of the metastable B phase (NiAl10O16): I. Preparation and structural study by X-ray diffraction. J. Solid State Chem. 1985, 58, 383–388. [Google Scholar] [CrossRef]
- Odaka, A.; Yamaguchi, T.; Fujita, T.; Taruta, S.; Kitajima, K. Cation dopant effect on phase transformation and microstructural evolution in M2+-substituted γ-alumina powders. J. Mater. Sci. 2008, 43, 2713–2720. [Google Scholar] [CrossRef]
- Azurdia, J.A.; Marchal, J.; Shea, P.; Sun, H.; Pan, X.Q.; Laine, R.M. Liquid-Feed Flame Spray Pyrolysis as a Method of Producing Mixed-Metal Oxide Nanopowders of Potential Interest as Catalytic Materials. Nanopowders along the NiO-Al2O3 Tie Line Including (NiO)0.22(Al2O3)0.78, a New Inverse Spinel Composition. Chem. Mater. 2006, 18, 731–739. [Google Scholar] [CrossRef]
- Piao, L.; Li, Y.; Chen, J.; Chang, L.; Lin, J.Y.S. Methane decomposition to carbon nanotubes and hydrogen on an alumina supported nickel aerogel catalyst. Catal. Today 2002, 74, 145–155. [Google Scholar] [CrossRef]
- Al-Nakoua, M.A.; El-Naas, M.H.; Abu-Jdayil, B. Characterization and testing of sol–gel catalysts prepared as thin layers in a plate reactor. Fuel Process. Technol. 2011, 92, 1836–1841. [Google Scholar] [CrossRef]
- Komeili, S.; Taeb, A.; Ravanchi, M.T.; Fard, M.R. The properties of nickel aluminate nanoparticles prepared by sol–gel and impregnation methods. Res. Chem. Intermed. 2016, 42, 7909–7921. [Google Scholar] [CrossRef]
- Peña, J.A.; Herguido, J.; Guimon, C.; Monzón, A.; Santamaría, J. Hydrogenation of Acetylene over Ni/NiAl2O4 Catalyst: Characterization, Coking, and Reaction Studies. J. Catal. 1996, 159, 313–322. [Google Scholar] [CrossRef]
- Ghule, A.V.; Ghule, K.; Punde, T.; Liu, J.Y.; Tzing, S.H.; Chang, J.Y.; Chang, H.; Ling, J.C. In situ monitoring of NiO–Al2O3 nanoparticles synthesis by thermo-Raman spectroscopy. Mater. Chem. Phys. 2010, 119, 86–92. [Google Scholar] [CrossRef]
- Li, C.Y.; Zhang, H.J.; Chen, Z.Q. Reaction between NiO and Al2O3 in NiO/γ-Al2O3 catalysts probed by positronium Atom. Appl. Surf. Sci. 2013, 266, 17–21. [Google Scholar] [CrossRef]
- Garbarino, G.; Chitsazan, S.; Phung, T.K.; Riani, P.; Busca, G. Preparation of supported catalysts: A study of the effect of small amounts of silica on Ni/Al2O3 catalysts. Appl. Catal. A Gen. 2015, 505, 86–97. [Google Scholar] [CrossRef]
- Riani, P.; Spennati, E.; Villa Garcia, M.; Sanchez Escribano, V.; Busca, G.; Garbarino, G. Ni/Al2O3 catalysts for CO2 methanation: Effect of silica and nickel loading. Int. J. Hydrogen Energy 2023, 48, 24976–24995. [Google Scholar] [CrossRef]
- Jurczyk, K.; Kania, W. Acid-base properties of modified γ-alumina. Appl. Catal. 1987, 34, 1–12. [Google Scholar]
- Jurczyk, K.; Kania, W. Base Properties of Modified y-Alumina. Appl. Catal. 1989, 56, 253–261. [Google Scholar] [CrossRef]
- Margossian, T.; Larmier, K.; Kim, S.M.; Krumeich, F.; Fedorov, A.; Chen, P.; Muller, C.R.; Coperet, C. Molecularly Tailored Nickel Precursor and Support Yield a Stable Methane Dry Reforming Catalyst with Superior Metal Utilization. J. Am. Chem. Soc. 2017, 139, 6919–6927. [Google Scholar] [CrossRef]
- Busca, G.; Riani, P.; Garbarino, G.; Ziemacki, G.; Gambino, L.; Montanari, E.; Millini, R. The state of nickel in spent Fluid Catalytic Cracking catalysts. Appl. Catal. A Gen. 2014, 486, 176–186. [Google Scholar] [CrossRef]
- Li, M.; Fang, S.; Hu, Y.H. Self-stabilization of Ni/Al2O3 Catalyst with a NiAl2O4 Isolation Layer in Dry Reforming of Methane. Catal. Lett. 2022, 152, 2852–2859. [Google Scholar] [CrossRef]
- Williams, A.; Butler, G.A.; Hammonds, J. Sintering of Nickel-Alumina Catalysts. J. Catal. 1972, 24, 352–355. [Google Scholar] [CrossRef]
- Garbarino, G.; Valsamakis, I.; Riani, P.; Busca, G. On the consistency of results arising from different techniques concerning the nature of supported metal oxide (nano)particles. The case of NiO/Al2O3. Catal. Commun. 2014, 51, 37–41. [Google Scholar] [CrossRef]
- Nikolopoulos, I.; Kogkos, G.; Tsavatopoulou, V.D.; Kordouli, E.; Bourikas, K.; Kordulis, C.; Lycourghiotis, A. Nickel—Alumina Catalysts for the Transformation of Vegetable Oils into Green Diesel: The Role of Preparation Method, Activation Temperature, and Reaction Conditions. Nanomaterials 2023, 13, 616. [Google Scholar] [CrossRef] [PubMed]
- Clariant, Catalysts and Adsorbents for Syngas. Available online: https://www.clariant.com/en/Business-Units/Catalysts/Syngas-Catalysts (accessed on 24 July 2024).
- Molina, R.; Poncelet, G. α-Alumina-Supported Nickel Catalysts Prepared from Nickel Acetylacetonate: A TPR Study. J. Catal. 1998, 173, 257–267. [Google Scholar] [CrossRef]
- Bolt, P.; Habraken, F.H.P.M.; Geus, J.W. On the Role of a NiAl2O4 Intermediate Layer in the Sintering Behavior of Ni/α-Al2O3. J. Catal. 1995, 151, 300–306. [Google Scholar] [CrossRef]
- Azancot, L.; Bobadilla, L.F.; Santos, J.L.; Córdoba, J.M.; Centeno, M.A.; Odriozola, J.A. Influence of the preparation method in the metal-support interaction and reducibility of Ni-Mg-Al based catalysts for methane steam reforming. Int. J. Hydrogen Energy 2019, 44, 19827–19840. [Google Scholar] [CrossRef]
- Yang, J.; Xiao, H. Preparation and Oxidation Behavior of Metallic Nickel Containing MgAlON Composite. High Temp. Mater. Proc. 2018, 37, 563–569. [Google Scholar]
- Jacob, K.T.; Alcock, C.B. Activities and their relation to cation distribution in NiAl2O4-MgAl2O4 spinel solid solutions. J. Solid State Chem. 1977, 20, 79–88. [Google Scholar] [CrossRef]
- Qi, C.; Spagnoli, D.; Fourie, A. Structural, electronic, and mechanical properties of calcium aluminate cements: Insight from first-principles theory. Construct. Build. Mater. 2020, 264, 120259. [Google Scholar] [CrossRef]
- Roussière, T.; Schelkle, K.M.; Titlbach, S.; Wasserschaff, G.; Milanov, A.; Cox, G.; Schwab, E.; Deutschmann, O.; Schulz, L.; Jentys, A.; et al. Structure–Activity Relationships of Nickel–Hexaaluminates in Reforming Reactions Part I: Controlling Nickel Nanoparticle Growth and Phase Formation. ChemCatChem 2014, 6, 1438–1446. [Google Scholar] [CrossRef]
- Johnson Matthey, Methanation Catalysts. Available online: https://matthey.com/documents/161599/440146/JM+Methanation+product+brochure+%28c2020%29.pdf/56eaef18-4782-776f-ed2a-c332f5f16659?t=1653488109779 (accessed on 20 July 2024).
- Rossi, P.F.; Busca, G.; Lorenzelli, V.; Waqif, M.; Saur, O.; Lavalley, J.C. Surface basicity of mixed oxides: Magnesium and zinc aluminates. Langmuir 1991, 7, 2677–2681. [Google Scholar] [CrossRef]
- Busca, G.; Cristiani, C.; Forzatti, P.; Groppi, G. Surface characterization of Ba-β-alumina. Catal. Lett. 1995, 31, 65–74. [Google Scholar] [CrossRef]
- Garbarino, G.; Finocchio, E.; Lagazzo, A.; Valsamakis, I.; Riani, P.; Sanchez Escribano, V.; Busca, G. Steam reforming of ethanol–phenol mixture on Ni/Al2O3: Effect of magnesium and boron on catalytic activity in the presence and absence of sulphur. Appl. Catal. B Environ. 2014, 147, 813–826. [Google Scholar] [CrossRef]
- Van Teijlingen, A.; Davis, S.A.; Hall, S.R. Size-dependent melting point depression of nickel nanoparticles. Nanoscale Adv. 2020, 2, 2347–2351. [Google Scholar] [CrossRef]
- Riani, P.; Garbarino, G.; Lucchini, M.A.; Canepa, F.; Busca, G. Unsupported versus alumina-supported Ni nanoparticles as catalysts for steam/ethanol conversion and CO2 methanation. J. Mol. Catal. A Chem. 2014, 383–384, 10–16. [Google Scholar] [CrossRef]
- Riani, P.; Garbarino, G.; Infantes-Molina, A.; Rodríguez-Castellón, E.; Canepa, F.; Busca, G. Hydrogen from steam reforming of ethanol over cobalt nanoparticles: Effect of boron impurities. Appl. Catal. A Gen. 2016, 518, 67–77. [Google Scholar] [CrossRef]
- Peng, B.; Zhang, X.; Aarts, D.G.A.L.; Dullens, R.P.A. Superparamagnetic nickel colloidal nanocrystal clusters with antibacterial activity and bacteria binding ability. Nat. Nanotechnol. 2018, 13, 478–482. [Google Scholar] [CrossRef]
- Rodríguez-González, V.; Marceau, E.; Beaunier, P.; Che, M.; Train, C. Stabilization of hexagonal close-packed metallic nickel for alumina-supported systems prepared from Ni(II) glycinate. J. Solid State Chem. 2007, 180, 22–30. [Google Scholar] [CrossRef]
- Su, J.; Wang, Q.; Fang, M.; Wang, Y.; Ke, J.; Shao, Q.; Lu, J. Metastable Hexagonal-Phase Nickel with Ultralow Pt Content for anEfficient Alkaline/Seawater Hydrogen Evolution Reaction. ACS Appl. Mater. Interfaces 2023, 15, 51160–51169. [Google Scholar] [CrossRef]
- Li, Z.; Wen, X.; Chen, F.; Zhang, Q.; Zhang, Q.; Gu, L.; Cheng, J.; Wu, B.; Zheng, N. Hexagonal Nickel as a Highly Durable and Active Catalyst for Hydrogen Evolutione. ACS Catal. 2021, 11, 8798–8806. [Google Scholar] [CrossRef]
- Shi, D.; Wen, B.; Melnik, R.; Yao, S.; Li, T. First-principles studies of Al–Ni intermetallic compounds. J. Solid State Chem. 2009, 182, 2664–2669. [Google Scholar] [CrossRef]
- Paul, A.R.; Mukherjee, M.; Singh, D. A Critical Review on the Properties of IntermetallicCompounds and Their Application in the Modern Manufacturing. Cryst. Res. Technol. 2022, 57, 2100159. [Google Scholar] [CrossRef]
- Sampath, S.; Ravi, V.P.; Sundararajan, S. An Overview on Synthesis, Processing and Applications of Nickel Aluminides: From Fundamentals to Current Prospects. Crystals 2023, 13, 435. [Google Scholar] [CrossRef]
- Villarroel-Rocha, J.; Gil, A. Modeling the temperature-programmed reduction of metal oxide catalysts by considering the particle-size distribution effect. Chem. Eng. J. 2024, 487, 150722. [Google Scholar] [CrossRef]
- Manukyan, K.V.; Avetisyan, A.G.; Shuck, C.E.; Chatilyan, H.A.; Rouvimov, S.; Kharatyan, S.L.; Mukasyan, A.S. Nickel Oxide Reduction by Hydrogen: Kinetics and Structural Transformations. J. Phys. Chem. C 2015, 119, 16131–16138. [Google Scholar] [CrossRef]
- Braaten, O.; Kjekshus, A.; Kvande, H. The Possible Reduction of Alumina to Aluminum Using Hydrogen. JOM 2000, 52, 47–53. [Google Scholar] [CrossRef]
- Helali, Z.; Jedidi, A.; Syzgantseva, O.A.; Calatayud, M.; Minot, C. Scaling reducibility of metal oxides. Theor. Chem. Acc. 2017, 136, 100. [Google Scholar] [CrossRef]
- Song, P.; Wen, D.; Guob, Z.X.; Korakianitis, T. Oxidation investigation of nickel nanoparticles. Phys. Chem. Chem. Phys. 2008, 10, 5057–5065. [Google Scholar] [CrossRef]
- Unutulmazsoy, Y.; Merkle, R.; Fischer, D.; Mannhart, J.; Maier, J. The oxidation kinetics of thin nickel films between 250 and 500 °C. Phys. Chem. Chem. Phys. 2017, 19, 9045–9052. [Google Scholar] [CrossRef] [PubMed]
- Payne, B.P.; Grosvenor, A.P.; Biesinger, M.C.; Kobe, B.A.; McIntyre, N.S. Structure and growth of oxides on polycrystalline nickel surfaces. Surf. Interface Anal. 2007, 39, 582–592. [Google Scholar] [CrossRef]
- Payne, B.P.; Biesinger, M.C.; McIntyre, N.S. The study of polycrystalline nickel metal oxidation by water vapour. J. Electron Spectrosc. Relat. Phenom. 2009, 175, 55–65. [Google Scholar] [CrossRef]
- Yavuz, A.; Yilmaz, N.F.; Kalkan, M.F. Enhancement in the Corrosion Resistance of Nickel Metal Via a Straightforward Thermal Oxidation Method. J. Bio-Tribo-Corros. 2023, 9, 22. [Google Scholar] [CrossRef]
- Kumar, S.N.; Appari, S.; Vardhan, B.; Kuncharam, R. Techniques for Overcoming Sulfur Poisoning of Catalyst Employed in Hydrocarbon Reforming. Catal. Surv. Asia 2021, 25, 362–388. [Google Scholar] [CrossRef]
- Argyle, M.D.; Bartholomew, C.H. Heterogeneous Catalyst Deactivation and Regeneration: A Review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef]
- Hernandez, J.M.; Lim, D.H.; Nguyen, H.V.P.; Yoon, S.P.; Han, J.; Nam, S.W.; Yoon, C.W.; Kim, S.K.; Ham, H.C. Decomposition of hydrogen sulfide (H2S) on Ni(100) and Ni3Al(100) surfaces from first-principles. Int. J. Hydrogen Energy 2014, 39, 12251–12258. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, Y.; Li, H.; Li, Q.; Qian, Y. Hydrothermal synthesis of NiS nanobelts and NiS2 microspheres constructed of cuboids architectures. J. Solid State Chem. 2010, 183, 223–227. [Google Scholar] [CrossRef]
- Rochet, A.; Ribeiro Passos, A.; Legens, C.; Briois, V. Sulphidation study of a dried Ni/Al2O3 catalyst by time-resolved XAS-MS combined with in situ Raman spectroscopy and multivariate Quick-XAS data analysis. Catal. Struct. React. 2017, 3, 33–42. [Google Scholar] [CrossRef]
- Garbarino, G.; Riani, P.; Magistri, L.; Busca, G. A study of the methanation of carbon dioxide on Ni/Al2O3 catalysts at atmospheric pressure. Int. J. Hydrogen Energy 2014, 39, 11557–11565. [Google Scholar] [CrossRef]
- Enger, B.C.; Holmen, A. Nickel and Fischer-Tropsch Synthesis. Catal. Rev. 2012, 54, 437–488. [Google Scholar] [CrossRef]
- Struis, R.P.W.J.; Bachelin, D.; Ludwig, C.; Wokaun, A. Studying the Formation of Ni3C from CO and Metallic Ni at T = 265 °C in Situ Using Ni K-Edge X-ray Absorption Spectroscopy. J. Phys. Chem. C 2009, 113, 2443–2451. [Google Scholar] [CrossRef]
- Hammer, B.; Nørskov, J.K. Why gold is the noblest of all the metals. Nature 1995, 376, 238. [Google Scholar] [CrossRef]
- Hammer, B.; Nørskov, J.K. Theoretical surface science and catalysis—Calculations and concepts. Adv. Catal. 2000, 45, 71–129. [Google Scholar]
- Nørskov, J.K.; Abild-Pedersen, F.; Studt, F.; Bligaard, T. Density functional theory in surface chemistry and catalysis. Proc. Natl. Acad. Sci. USA 2011, 108, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Sehested, J. Four challenges for nickel steam-reforming catalysts. Catal. Today 2006, 111, 103–110. [Google Scholar] [CrossRef]
- Wen, J.G.; Huang, Z.P.; Wang, D.Z.; Chen, J.H.; Yang, S.X.; Rena, Z.F.; Wang, J.H.; Calvet, L.E.; Chen, J.; Klemic, J.F.; et al. Growth and characterization of aligned carbon nanotubes from patterned nickel nanodots and uniform thin films. J. Mater. Res. 2001, 16, 3246–3253. [Google Scholar] [CrossRef]
- Helveg, S.; Sehested, J.; Rostrup-Nielsen, J.R. Whisker carbon in perspective. Catal. Today 2011, 178, 42–46. [Google Scholar] [CrossRef]
- Ziebro, J.; Skorupińska, S.; Kądziołka, G.; Michalkiewicz, B. Synthesizing Multi-walled Carbon Nanotubes over a Supported-nickel Catalyst, Fullerenes, Nanotubes and Carbon. Nanostructures 2013, 21, 333–345. [Google Scholar]
- Lim, X.X.; Low, S.C.; Oh, W.D. A critical review of heterogeneous catalyst design for carbon nanotubes synthesis: Functionalities, performances, and prospects. Fuel Process. Technol. 2023, 241, 107624. [Google Scholar] [CrossRef]
- Abbas, H.F.; Wan Daud, W.M.A. Hydrogen production by methane decomposition: A review. Int. J. Hydrogen Energy 2010, 35, 1160–1190. [Google Scholar] [CrossRef]
- Goldberger, W.M.; Othmer, D.F. Kinetics of Nickel Carbonyl Formation. Ind. Eng. Chem. Process Des. Dev. 1963, 2, 202–209. [Google Scholar] [CrossRef]
- Baranowski, B.; Filipek, S.M. 45 Years of nickel hydride—History and perspectives. J. Alloys Compd. 2005, 404–406, 2–6. [Google Scholar] [CrossRef]
- Rana, S.; Masli, N.; Monder, D.S.; Chatterjee, A. Hydriding pathway for Ni nanoparticles: Computational characterization provides insights into the nanoparticle size and facet effect on layer-by-layer subsurface hydride formation. Comp. Mat. Sci. 2022, 210, 111482. [Google Scholar] [CrossRef]
- Cecilia, J.A.; Jiménez-Morales, I.; Infantes-Molina, A.; Rodríguez-Castellón, E.; Jiménez-López, A. Influence of the silica support on the activity of Ni and Ni2P based catalysts in the hydrodechlorination of chlorobenzene. Study of factors governing catalyst deactivation. J. Mol. Catal. A Chem. 2013, 368–369, 78–87. [Google Scholar] [CrossRef]
- Hao, Q.; Yang, Z.; Wu, B.; Zhu, J.; Li, Z.; Liu, J.; Ma, L. Study on the deactivation of Ni-based catalyst in the hydrotreating process of waste plastic pyrolysis oil. J. Anal. Appl. Pyrol. 2022, 168, 105789. [Google Scholar] [CrossRef]
- Ohtsuka, Y. Influence of hydrogen chloride treatment on the dispersion of nickel particles supported on carbon. J. Mol. Catal. 1989, 54, 225–235. [Google Scholar] [CrossRef]
- Menini, C.; Park, C.; Brydson, R.; Keane, M.A. Low-Temperature (553 K) Catalytic Growth of Highly Ordered Carbon Filaments during Hydrodechlorination Reactions. J. Phys. Chem. B 2000, 104, 4281–4284. [Google Scholar] [CrossRef]
- Morterra, C.; Magnacca, G. A case study: Surface chemistry and surface structure of catalytic aluminas, as studied by vibrational spectroscopy of adsorbed species. Catal. Today 1996, 27, 497–532. [Google Scholar] [CrossRef]
- Busca, G. Catalytic materials based on silica and alumina: Structural features and generation of surface acidity. Prog. Mater. Sci. 2019, 104, 215–249. [Google Scholar] [CrossRef]
- Escalona Platero, E.; Coluccia, S.; Zecchina, A. CO and NO Adsorption on NiO: A Spectroscopic Investigation. Langmuir 1985, 1, 407–414. [Google Scholar] [CrossRef]
- Escalona Platero, E.; Scarano, D.; Zecchina, A.; Meneghini, G.; De Franceschi, R. Highly sintered nickel oxide: Surface morphology and FTIR investigation of CO adsorbed at low temperature. Surf. Sci. 1996, 350, 113–122. [Google Scholar] [CrossRef]
- Kitakatsu, N.; Maurice, V.; Hinnen, C.; Marcus, P. Surface hydroxylation and local structure of NiO thin films formed on Ni(111). Surf. Sci. 1998, 407, 36–58. [Google Scholar] [CrossRef]
- Zhao, W.; Bajdich, M.; Carey, S.; Vojvodic, A.; Nørskov, J.K.; Campbell, C.T. Water Dissociative Adsorption on NiO(111): Energetics and Structure of the Hydroxylated Surface. ACS Catal. 2016, 6, 7377–7384. [Google Scholar] [CrossRef]
- Tsyganenko, A.A.; Pozdnyakov, D.V.; Filimonov, V.N. Infrared study of surface species arising from ammonia adsorption on oxide surfaces. J. Mol. Struct. 1975, 29, 299–318. [Google Scholar] [CrossRef]
- Matsumoto, T.; Kubota, J.; Kondo, J.N.; Hirose, C.; Domen, K. Adsorption Structures of Carbon Dioxide on NiO(111) and Hydroxylated NiO(111) Studied by Infrared Reflection Adsorption Spectroscopy. Langmuir 1999, 15, 2158–2161. [Google Scholar] [CrossRef]
- Atzori, L.; Cutrufello, M.G.; Meloni, D.; Onida, B.; Gazzoli, D.; Ardu, A.; Monaci, R.; Sini, M.F.; Rombi, E. Characterization and catalytic activity of soft-templated NiO-CeO2 mixed oxides for CO and CO2 co-methanation. Front. Chem. Sci. Eng. 2021, 15, 251–268. [Google Scholar] [CrossRef]
- Bordes-Richard, E.; Courtine, P. Optical basicity: A scale of acidity/basicity of solids and its application to oxidation catalysis. In Metal Oxides: Chemistry and Applications; Fierro, J.L.G., Ed.; CRC Press LLC: Boca Raton, FL, USA, 2006; Volume 108, pp. 319–352. [Google Scholar]
- Busca, G. Bases and basic materials in industrial and environmental chemistry. Liquid versus solid basicity. Chem. Rev. 2010, 110, 2217–2249. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Xing, X.; Xiong, W.; Li, H. Facile synthesis of Ni-based oxides nanocatalyst: Effect of calcination temperature on NRR properties of NiO. Carbon Lett. 2024, 34, 1197–1206. [Google Scholar] [CrossRef]
- Otero Areán, C.; Peñarroya Mentruit, M.; López López, A.J.; Parra, J.B. High surface area nickel aluminate spinels prepared by a sol–gel methoñd. Coll. Surf. A Physicochem. Eng. Asp. 2001, 180, 253–258. [Google Scholar] [CrossRef]
- Garbarino, G.; Campodonico, S.; Romero Perez, A.; Carnasciali, M.M.; Riani, P.; Finocchio, E.; Busca, G. Spectroscopic characterizatin of Ni/Al2O3 catalytic materials for the steam reforming of renewables. Appl. Catal. A Gen. 2013, 452, 163–173. [Google Scholar] [CrossRef]
- Salagre, P.; Fierro, J.L.G.; Medina, F.; Sueiras, J.E. Characterization of nickel species on several γ-alumina supported nickel simples. J. Mol. Catal. A Chem. 1996, 106, 125–134. [Google Scholar] [CrossRef]
- Li, G.; Hu, L.; Hill, J.M. Comparison of reducibility and stability of alumina-supported Ni catalysts prepared by impregnation and co-precipitation. Appl. Catal. A Gen. 2006, 301, 16–24. [Google Scholar] [CrossRef]
- Gericke, S.M.; Rissler, J.; Bermeo, M.; Wallander, H.; Karlsson, H.; Kollberg, L.; Scardamaglia, M.; Temperton, R.; Zhu, S.; Sigfridsson Clauss, K.V.; et al. In situ H2 reduction of Al2O3-supported Ni- and Mo-based catalysts. Catalysts 2022, 12, 755. [Google Scholar] [CrossRef]
- Cho, J.H.; An, S.H.; Chang, T.S.; Shin, C.H. Effect of an Alumina Phase on the Reductive Amination of 2-Propanol to Monoisopropylamine Over Ni/Al2O3. Catal. Lett. 2016, 146, 811–819. [Google Scholar] [CrossRef]
- He, L.; Ren, Y.; Yue, B.; Tsang, S.C.E.; He, H. Tuning Metal–Support Interactions on Ni/Al2O3 Catalysts to Improve Catalytic Activity and Stability for Dry Reforming of Methane. Processes 2021, 9, 706. [Google Scholar] [CrossRef]
- Zieliński, J. Morphology of nickel/alumina catalysts. J. Catal. 1982, 76, 157–163. [Google Scholar] [CrossRef]
- Gupta, S.; Deo, G. Effect of metal amount on the catalytic performance of Ni-Al2O3 catalyst for the Tri-reforming of methane. Int. J. Hydrogen Energy 2023, 48, 5478–5492. [Google Scholar] [CrossRef]
- Li, C.P.; Chen, Y.-W. Temperature-programmed-reduction studies of nickel oxide/alumina catalysts: Effects of the preparation method. Thermochim. Acta 1995, 256, 457–465. [Google Scholar] [CrossRef]
- García-Diéguez, M.; Finocchio, E.; Larrubia, M.A.; Alemany, L.J.; Busca, G. Characterization of alumina-supported Pt, Ni and PtNi alloy catalysts for the dry reforming of methane. J. Catal. 2010, 274, 11–20. [Google Scholar] [CrossRef]
- Amenomiya, Y. Adsorption of Hydrogen and H2-D2 Exchange Reaction on Alumina. J. Catal. 1971, 22, 109–121. [Google Scholar] [CrossRef]
- Weller, S.W.; Montagna, A.A. Studies of Alumina, I. Reaction with Hydrogen at Elevated Temperatures. J. Catal. 1971, 21, 303–311. [Google Scholar] [CrossRef]
- Kramer, R.; Andre, M. Adsorption of Atomic Hydrogen on Alumina by Hydrogen Spillover. J. Catal. 1979, 58, 287–295. [Google Scholar] [CrossRef]
- Cabrejas Manchado, M.; Guil, J.M.; Pérez Masiá, A.; Ruiz Paniego, A.; Trejo Menayo, J.M. Adsorption of H2, O2, CO, and CO2 on a γ-Alumina: Volumetric and Calorimetric Studies. Langmuir 1994, 10, 685–691. [Google Scholar] [CrossRef]
- Digne, M.; Sautet, P.; Raybaud, P.; Euzen, P.; Toulhoat, H. Use of DFT to achieve a rational understanding of acid–basic properties of γ-alumina surfaces. J. Catal. 2004, 226, 54–68. [Google Scholar] [CrossRef]
- Wischert, R.; Laurent, P.; Copéret, C.; Delbecq, F.; Sautet, P. γ-Alumina: The essential and unexpected role of water for the structure, stability, and reactivity of “defect” sites. J. Am. Chem. Soc. 2012, 134, 14430–14449. [Google Scholar] [CrossRef] [PubMed]
- Karim, W.; Spreafico, C.; Kleibert, A.; Gobrecht, J.; VandeVondele, J.; Ekinci, Y.; van Bokhoven, J.A. Catalyst support effects on hydrogen spillover. Nature 2017, 541, 68–71. [Google Scholar] [CrossRef]
- Garbarino, G.; Travi, I.; Pani, M.; Carnasciali, M.M.; Busca, G. Pure vs ultra-pure γ-alumina: A spectroscopic study and catalysis of ethanol conversion. Catal. Commun. 2015, 70, 77–81. [Google Scholar] [CrossRef]
- Shen, H.; Li, H.; Yang, Z.; Li, C. Magic of hydrogen spillover: Understanding and application. Green Energy Environ. 2022, 7, 1161–1198. [Google Scholar] [CrossRef]
- Beck, A.; Rzepka, P.; Marshall, K.P.; Stoian, D.; Willinger, M.G.; van Bokhoven, J.A. Hydrogen Interaction with Oxide Supports in the Presence and Absence of Platinum. J. Phys. Chem. C 2022, 126, 17589–17597. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, M.; Wang, A.; Zhang, T. Selective Hydrogenation over Supported Metal Catalysts: From Nanoparticles to Single Atoms. Chem. Rev. 2020, 120, 683–733. [Google Scholar] [CrossRef] [PubMed]
- Panczyk, T.; Szabelski, P.; Rudzinski, W. Hydrogen Adsorption on Nickel (100) Single-Crystal Face. A Monte Carlo Study of the Equilibrium and Kinetics. J. Phys. Chem. B 2005, 109, 10986–10994. [Google Scholar] [CrossRef]
- Znak, L.; Zieliński, J. Effects of support on hydrogen adsorption/desorption on nickel. Appl. Catal. A Gen. 2008, 334, 268–276. [Google Scholar] [CrossRef]
- Wang, Y.; Winter, L.R.; Chen, J.G.; Yan, B.Y. CO2 hydrogenation over heterogeneous catalysts at atmospheric pressure: From electronic properties to product selectivity. Green Chem. 2021, 23, 249–267. [Google Scholar] [CrossRef]
- Blomqvist, J.; Lehman, L.; Salo, P. CO adsorption on metal-oxide surfaces doped with transition-metal adatoms. Phys. Status Solid. 2012, 249, 1046–1057. [Google Scholar] [CrossRef]
- Blyholder, G. Molecular orbital view of chemisorbed carbon monoxide. J. Phys. Chem. 1964, 68, 2772–2777. [Google Scholar] [CrossRef]
- Lupinetti, A.J.; Fau, S.; Frenking, G.; Strauss, S.H. Theoretical Analysis of the Bonding between CO and Positively Charged Atoms. J. Phys. Chem. A 1997, 101, 9551–9559. [Google Scholar] [CrossRef]
- Gopal, P.G.; Schneider, R.L.; Watters, K.L. Evidence for Production of Surface Formate upon Direct Reaction of CO with Alumina and Magnesia. J. Catal. 1987, 105, 366–372. [Google Scholar] [CrossRef]
- Vannice, M.A. Activation of Carbon Monoxide on Metal Surfaces. In Catalysis Science and Technology; Andesron, J.R., Boudart, M., Eds.; Springer: Berlin/Heidelberg, Germany, 1982; Volume 3, p. 139. [Google Scholar]
- Abild-Pedersen, F.; Andersson, M.P. CO adsorption energies on metals with correction for high coordination adsorption sites—A density functional study. Surf. Sci. 2007, 601, 1747–1753. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Sheppard, N. Advances in Infrared and Raman Spectroscopy; Hester, R.E., Clark, R.H.J., Eds.; Heyden: London, UK, 1982; Volume 5, pp. 86–95. [Google Scholar]
- Fielicke, A.; Gruene, P.; Meijer, G.; Rayner, D.M. The adsorption of CO on transition metal clusters: A case study of cluster surface chemistry. Surf. Sci. 2009, 603, 1427–1433. [Google Scholar] [CrossRef]
- Vázquez-Parga, D.; Jurado, A.; Roldan, A.; Viñes, F. A computational map of the probe CO molecule adsorption and dissociation on transition metal low Miller indices surfaces. Appl. Surf. Sci. 2023, 618, 156581. [Google Scholar] [CrossRef]
- Tøttrup, P.B. Kinetics of Decomposition of Carbon Monoxide on a Supported Nickel Catalyst. J. Catal. 1976, 42, 29–36. [Google Scholar] [CrossRef]
- Gałuszka, J.; Chang, J.R.; Amenomiya, Y. Disproportionation of carbon monoxide on supported nickel catalysts. J. Catal. 1981, 68, 172–181. [Google Scholar] [CrossRef]
- Liu, Z.P.; Hu, P. General trends in CO dissociation on transition metal surfaces. J. Chem. Phys. 2001, 114, 8244–8247. [Google Scholar] [CrossRef]
- Schmider, D.; Maier, L.; Deutschmann, O. Reaction Kinetics of CO and CO2 Methanation over Nickel. Ind. Eng. Chem. Res. 2021, 60, 5792–5805. [Google Scholar] [CrossRef]
- Trombetta, M.; Busca, G.; Rossini, S.A.; Piccoli, V.; Cornaro, U. FT-IR studies on light olefin skeletal isomerization catalysis. Part I: The interaction of C4 olefins with pure γ-alumina. J. Catal. 1997, 168, 334–348. [Google Scholar] [CrossRef]
- Vang, R.T.; Honkala, H.; Dahl, S.; Vestergaard, E.K.; Schnadt, J.; Lægsgaard, E.; Clausen, B.S.; Nørskov, J.K.; Besenbacher, F. Ethylene dissociation on flat and stepped Ni(1 1 1): A combined STM and DFT study. Surf. Sci. 2006, 600, 66–77. [Google Scholar] [CrossRef]
- Yilmazer, N.D.; Fellaha, M.F.; Onal, I. A density functional theory study of ethylene adsorption on Ni10(111), Ni13(100) and Ni10(110) surface cluster models and Ni13 nanocluster. Appl. Surf. Sci. 2010, 256, 5088–5093. [Google Scholar] [CrossRef]
- Medlin, J.W.; Allendorf, M.D. Theoretical Study of the Adsorption of Acetylene on the (111) Surfaces of Pd, Pt, Ni, and Rh. J. Phys. Chem. B 2003, 107, 217–223. [Google Scholar] [CrossRef]
- Jenkins, S.J. Aromatic adsorption on metals via first-principles density functional theory. Proc. R. Soc. A 2009, 465, 2949–2976. [Google Scholar] [CrossRef]
- Lobo, L.S.; Trimm, D.L. Carbon formation from light hydrocarbons on nickel. J. Catal. 1973, 29, 15–19. [Google Scholar] [CrossRef]
- Kodde, A.J.; Padin, J.; van der Meer, P.J.; Mittelmeijer-Hazeleger, M.C.; Bliek, A.; Yang, R.T. NiCl2 on γ-Alumina as Selective Adsorbents for Acetylene over Ethylene. Ind. Eng. Chem. Res. 2000, 39, 3108–3111. [Google Scholar] [CrossRef]
- Shimura, K.; Yoshida, S.; Oikawa, H.; Fujitani, T. Impacts of Ni-Loading Method on the Structure and the Catalytic Activity of NiO/SiO2-Al2O3 for Ethylene Oligomerization. Catalysts 2023, 13, 1303. [Google Scholar] [CrossRef]
- Koninckx, E.; Mendes, P.S.F.; Thybaut, J.W.; Broadbelt, L.J. Ethylene oligomerization on nickel catalysts on a solid acid support: From New mechanistic insights to tunable bifunctionality. Appl. Catal. A Gen. 2021, 624, 118296. [Google Scholar] [CrossRef]
- Horiuchi, T.; Hidaka, H.; Fukui, T.; Kubo, Y.; Horio, M.; Suzuki, K.; Mori, T. Effect of added basic metal oxides on CO2 adsorption on alumina at elevated temperatures. Appl. Catal. A Gen. 1998, 167, 195–202. [Google Scholar] [CrossRef]
- Garbarino, G.; Wang, C.; Valsamakis, I.; Chitsazan, S.; Riani, P.; Finocchio, E.; Flytzani-Stephanopoulos, M.; Busca, G. Acido-basicity of lanthana/alumina catalysts and their activity in ethanol conversion. Appl. Catal. B Environ. 2017, 200, 458–468. [Google Scholar] [CrossRef]
- Garbarino, G.; Riani, P.; García, V.; Finocchio, E.; Sánchez Escribano, V.; Busca, G. A study of ethanol conversion over zinc aluminate catalyst. Reac. Kinet. Mech. Cat. 2018, 124, 503–522. [Google Scholar] [CrossRef]
- Nobakht, A.R.; Rezaei, M.; Alavi, S.M.; Akbari, E.; Varbar, M.; Hafezi-Bakhtiari, J. CO2 methanation over NiO catalysts supported on CaO–Al2O3: Effect of CaO:Al2O3 molar ratio and nickel loading. Int. J. Hydrogen Energy 2023, 48, 38664–38675. [Google Scholar] [CrossRef]
- Solymosi, F. The bonding, structure and reactions of CO2 adsorbed on clean and promoted metal surfaces. J. Mol. Catal. 1991, 65, 337–358. [Google Scholar] [CrossRef]
- Freund, H.-J.; Roberts, M.W. Surface chemistry of carbon dioxide. Surf. Sci. Rep. 1996, 25, 225–273. [Google Scholar] [CrossRef]
- Taifan, W.; Boily, J.F.; Baltrusaitis, J. Surface chemistry of carbon dioxide revisited. Surf. Sci. Rep. 2016, 71, 595–671. [Google Scholar] [CrossRef]
- Liu, C.; Cundari, T.R.; Wilson, A.K. CO2 Reduction on Transition Metal (Fe, Co, Ni, and Cu) Surfaces: In Comparison with Homogeneous Catalysis. J. Phys. Chem. C 2012, 116, 5681–5688. [Google Scholar] [CrossRef]
- Liu, X.; Sun, L.; Deng, W.Q. Theoretical Investigation of CO2 Adsorption and Dissociation on Low Index Surfaces of Transition Metals. J. Phys. Chem. C 2018, 122, 8306–8314. [Google Scholar] [CrossRef]
- Jin, W.; Wang, Y.; Liu, T.; Ding, C.; Guo, H. CO2 chemisorption and dissociation on flat and stepped transition metal surfaces. Appl. Surf. Sci. 2022, 599, 154024. [Google Scholar] [CrossRef]
- Cai, J.; Han, Y.; Chen, S.; Crumlin, E.J.; Yang, B.; Li, Y.; Liu, Z. CO2 Activation on Ni(111) and Ni(100) Surfaces in the Presence of H2O: An Ambient-Pressure X-ray Photoelectron Spectroscopy Study. J. Phys. Chem. C 2019, 123, 12176–12182. [Google Scholar] [CrossRef]
- Schreiter, N.; Kirchner, J.; Kureti, S. A DRIFTS and TPD study on the methanation of CO2 on Ni/Al2O3 catalyst. Catal. Commun. 2020, 140, 105988. [Google Scholar] [CrossRef]
- Busca, G.; Spennati, E.; Riani, P.; Garbarino, G. Looking for an Optimal Composition of Nickel-Based Catalysts for CO2 Methanation. Energies 2023, 16, 5304. [Google Scholar] [CrossRef]
- Antill, J.E.; Warburton, J.B. Oxidation of Nickel by Carbon Dioxide. J. Electrochem. Soc. 1967, 114, 1215–1221. [Google Scholar] [CrossRef]
- Napari, M.; Huq, T.N.; Hoye, R.L.Z.; MacManus-Driscoll, J.L. Nickel oxide thin films grown by chemical depositiontechniques: Potential and challenges in next-generationrigid and flexible device applications. InfoMat 2021, 3, 536–576. [Google Scholar] [CrossRef]
- Horn, R.; Schlögl, R. Methane Activation by Heterogeneous Catalysis. Catal. Lett. 2015, 145, 23–39. [Google Scholar] [CrossRef]
- Liu, F.; Sang, Y.; Ma, H.; Li, Z.; Gao, Z. Nickel oxide as an effective catalyst for catalytic combustion of methane. J. Nat. Gas Sci. Eng. 2017, 41, 1–6. [Google Scholar] [CrossRef]
- Lee, M.B.; Yang, Q.Y.; Ceyer, S.T. Dynamics of the Activated Dissociative Chemisorption of CH4 and Implication for the Pressure Gap in Catalysis—A Molecular-Beam High-Resolution Electron-Energy Loss Study. J. Chem. Phys. 1987, 87, 2724–2741. [Google Scholar] [CrossRef]
- Kubokawa, M. The activated adsorption of methane on reduced nickel. Proc. Imp. Acad. 1938, 14, 61–66. [Google Scholar] [CrossRef]
- García-Sancho, C.; Guil-Lopez, R.; Sebastian-Lopez, A.; Navarro, R.M.; Fierro, J.L.G. Hydrogen production by methane decomposition: A comparative study of supported and bulk ex-hydrotalcite mixed oxide catalysts with Ni, Mg and Al. Int. J. Hydrogen Energy 2018, 43, 9607–9621. [Google Scholar] [CrossRef]
- Gubanov, M.A.; Ivantsov, M.I.; Kulikova, M.V.; Kryuchkov, V.A.; Nikitchenko, N.V.; Knyazeva, M.I.; Kulikova, A.B.; Pimenov, A.A.; Maksimova, A.L. Methane Decomposition Nickel Catalysts Based on Structured Supports. Petrol. Chem. 2020, 60, 1043–1051. [Google Scholar] [CrossRef]
- Resasco, D.E.; Marcus, B.K.; Huang, C.S.; Durante, V.A. Isobutane dehydrogenation over sulfided nickle catalysts. J. Catal. 1994, 146, 40–55. [Google Scholar] [CrossRef]
- Tsai, M.C.; Friend, C.M.; Muetterties, E.L. Dehydrogenation processes on nickel and platinum surfaces. Conversion of cyclohexane, cyclohexene, and cyclohexadiene to benzene. J. Am. Chem. Soc. 1982, 104, 2539–2543. [Google Scholar] [CrossRef]
- Ma, R.; Gao, J.; Kou, J.; Dean, D.P.; Breckner, C.J.; Liang, K.; Zhou, B.; Miller, J.T.; Zou, G. Insights into the Nature of Selective Nickel Sites on Ni/Al2O3 Catalysts for Propane Dehydrogenation. ACS Catal. 2022, 12, 12607–12616. [Google Scholar] [CrossRef]
- Diskin, A.M.; Cunningham, R.H.; Ormerod, R.M. The oxidative chemistry of methane over supported nickel catalyst. Catal. Today 1998, 46, 147–154. [Google Scholar] [CrossRef]
- Sehested, J.; Gelten, J.A.P.; Remediakis, J.N.; Bengaard, H.; Nørskov, J.K. Sintering of nickel steam-reforming catalysts: Effects of temperature and steam and hydrogen pressures. J. Catal. 2004, 223, 432–443. [Google Scholar] [CrossRef]
- Clavenna, L.R.; Davis, S.M.; Beasley, B.E. Process for the Reactivation of Nickel-Alumina Catalysts. U.S. Patent 5356845, 18 October 1994. [Google Scholar]
- Hashemnejad, S.M.; Parvari, M. Deactivation and Regeneration of Nickel-Based Catalysts for Steam-Methane Reforming. Chin. J. Catal. 2011, 32, 273–279. [Google Scholar] [CrossRef]
- Dega, F.B.; Abatzoglou, N. H2S Poisoning and Regeneration of a Nickel Spinellized Catalyst Prepared from Waste Metallurgical Residues, During Dry Autothermal Methane Reforming. Catal. Lett. 2019, 149, 1730–1742. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J.R. Some principles relating to the regeneration of sulfur-poisoned nickel catalyst. J. Catal. 1971, 21, 171–178. [Google Scholar] [CrossRef]
- Li, L.; Howard, C.J.; King, D.L.; Gerber, M.A.; Dagle, R.A.; Stevens, B.J. Regeneration of Sulfur Deactivated Ni-Based Biomass Syngas Cleaning Catalysts. Ind. Eng. Chem. Res. 2010, 49, 10144–10148. [Google Scholar] [CrossRef]
- Wachter, P.; Gaber, C.; Raic, J.; Demuth, M.; Hochenauer, C. Experimental investigation on H2S and SO2 sulphur poisoning and regeneration of a commercially available Ni-catalyst during methane tri-reforming. Int. J. Hydrogen Energy 2021, 46, 3437–3452. [Google Scholar] [CrossRef]
- Vogt, E.T.C.; Fu, D.; Weckhuysen, B.M. Carbon Deposit Analysis in Catalyst Deactivation, Regeneration, and Rejuvenation. Angew. Chem. Int. Ed. 2023, 62, e202300319. [Google Scholar] [CrossRef]
- Uemura, Y.; Hatate, Y. Effect of Nickel Concentration Profile on Selectivity of Acetylene Hydrogenation. J. Chem. Eng. Jpn. 1989, 22, 287–291. [Google Scholar] [CrossRef]
- Ryu, J.Y. Ni Catalyst, Process for Making Catalysts and Selective Hydrogenation Process. EP1786747A2, 14 February 2005. [Google Scholar]
- Available online: https://www.clariant.com/en/Solutions/Products/2014/06/16/09/13/OleMax-100-Series (accessed on 26 July 2024).
- Krupa, S.; Foley, T.; McColl, S. UOP KLP 1.3-butadiene from acetylene process. In Handbook of Petrochemicals Production Processes; Meyers, R.A., Ed.; McGraw-Hill: New York, NY, USA, 2005; pp. 3.11–3.14. [Google Scholar]
- Bosman, H.J.M.; Grootendorst, E.J.; Postma, L.H.; Smeets, T.M. Process for the Hydrogenation of Phenyl Acetylene in a Styrene-containing Medium with the Aid of a Catalyst. U.S. Patent 6747181B1, 8 June 2004. [Google Scholar]
- Rajeshwer, D.; Sreenivasa Rao, G.; Krishnamurthy, K.R.; Padmavathi, G.; Subrahmanyam, N.; Rachh, J.D. Kinetics of Liquid—Phase Hydrogenation of Straight Chain C10 to C13 Di-Olefins Over Ni/Al2O3 Catalyst. Int. J. Chem. React. Eng. 2006, 4, A17. [Google Scholar] [CrossRef]
- Kravtsov, A.V.; Zuev, V.A.; Kozlov, I.A.; Milishnikov, A.V.; Ivanchina, E.D.; Uriev, E.M.; Ivashkina, E.N.; Fetisova, V.A.; Shnidorova, I.O. Development of control system for nickel-containing catalyst in dienes hydrogenation. Pet. Coal 2009, 51, 248–254. [Google Scholar]
- Hoffer, W.; Bonné, R.L.C.; van Langeveld, A.D.; Griffiths, C.; Lok, C.M.; Moulijn, J.A. Enhancing the start-up of pyrolysis gasoline hydrogenation reactors by applying tailored ex situ presulfided Ni/Al2O3 catalysts. Fuel 2004, 83, 1–8. [Google Scholar] [CrossRef]
- Available online: https://matthey.com/products-and-markets/chemicals/pyrolysis-gasoline-catalysts (accessed on 26 July 2024).
- Available online: https://www.topsoe.com/our-resources/knowledge/our-products/catalysts/sh-501 (accessed on 26 July 2024).
- Available online: https://www.axens.net/solutions/catalysts-adsorbents-grading-supply/selective-hydrogenation-catalysts (accessed on 26 July 2024).
- Fremerey, P.; Jess, A.; Moos, R. Why does the Conductivity of a Nickel Catalyst Increase during Sulfidation? An Exemplary Study Using an In Operando Sensor Device. Sensors 2015, 15, 27021–27034. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://matthey.com/products-and-markets/chemicals/de-aromatisation-catalysts (accessed on 26 July 2024).
- Available online: https://matthey.com/products-and-markets/chemicals/solvent-purification (accessed on 26 July 2024).
- Watson, M.J.; Partridge, M.G.; Appleton, P.; Jackson, S. Chemical Process and Catalyst. WO2010018405A1, 18 February 2010. [Google Scholar]
- Aasberg-Petersen, K.; Dybkjær, I.; Ovesen, C.V.; Schjødt, N.C.; Sehested, J.; Thomsen, S.G. Natural gas to synthesis gas—Catalysts and catalytic processes. J. Nat. Gas Sci. Eng. 2011, 3, 423–459. [Google Scholar] [CrossRef]
- Pearce, B.B.; Twigg, M.V.; Woodward, C. Catalyst Handbook, 2nd ed.; Twigg, M.V., Ed.; Wolfe Pub.: London, UK, 1989; pp. 340–383. [Google Scholar]
- Golosman, E.Z.; Efremov, V.N. Industrial Catalysts for the Hydrogenation of Carbon Oxides. Catal. Ind. 2012, 4, 267–283. [Google Scholar] [CrossRef]
- Miguel, C.V.; Mendes, A.; Madeira, L.M. Intrinsic kinetics of CO2 methanation over an industrial nickel-based catalyst. J. CO₂ Util. 2018, 25, 128–136. [Google Scholar] [CrossRef]
- Available online: https://www.topsoe.com/our-resources/knowledge/our-products/catalysts/pk-7r (accessed on 27 July 2024).
- Available online: https://matthey.com/en/products-and-markets/chemicals/methanation-catalysts (accessed on 27 July 2024).
- Kopyscinski, J.; Schildhauer, T.J.; Biollaz, S.M.A. Production of synthetic natural gas (SNG) from coal and dry biomass e a technology review from 1950 to 2009. Fuel 2010, 89, 1763–1783. [Google Scholar] [CrossRef]
- Røstrup-Nielsen, J.R.; Pedersen, K.; Sehested, J. High temperature methanation. Sintering and structure sensitivity. Appl Catal. A Gen. 2007, 330, 134–138. [Google Scholar] [CrossRef]
- Li, J.; Tian, Y.; Yan, X.; Yang, J.; Wang, Y.; Xu, W.; Xie, K. Approach and potential of replacing oil and natural gas with coal in China. Front. Energy 2020, 14, 419–431. [Google Scholar] [CrossRef]
- Available online: https://www.topsoe.com/our-resources/knowledge/our-products/catalysts/mcr-2 (accessed on 27 July 2024).
- Torcida, M.F.; Curto, D.; Martín, M. Design and optimization of CO2 hydrogenation multibed reactors. Chem. Eng. Res. Des. 2022, 181, 89–100. [Google Scholar] [CrossRef]
- Dagle, R.A.; Wang, Y.; Xia, G.G.; Strohm, J.J.; Holladay, J.; Palo, D.R. Selective CO methanation catalysts for fuel processing applications. Appl. Catal. A Gen. 2007, 326, 213–218. [Google Scholar] [CrossRef]
- Nguyen, T.T.M.; Wissing, L.; Skjøth-Rasmussen, M.S. High temperature methanation: Catalyst considerations. Catal. Today 2013, 215, 233–238. [Google Scholar] [CrossRef]
- Ross, J.R.H. Nickel Catalysts for C1 Reactions: Recollections from a Career in Heterogeneous Catalysis. Top. Catal. 2021, 64, 896–906. [Google Scholar] [CrossRef]
- Available online: https://www.clariant.com/en/Corporate/News/2016/07/VESTA-Oncethrough-Methanation-New-Technology-with-Wide-H2CO-Flexibility-Successfully-Passes-Pilot-Te (accessed on 27 July 2024).
- Romano, L.; Ruggeri, F. Methane from syngas—Status of Amec Foster Wheeler VESTA technology development. Energy Procedia 2015, 81, 249–254. [Google Scholar] [CrossRef]
- Götz, M.; Lefebvre, J.; Mörs, F.; Koch, A.M.; Graf, F.; Bajohr, S.; Reimert, R.; Kolb, R. Renewable Power-to-Gas: A Technological and Economic Review. Renew. Energy 2016, 85, 1371–1390. [Google Scholar] [CrossRef]
- Vega Puga, E.; Moumin, G.; Neumann, N.C.; Roeb, M.; Ardone, A.; Sattler, C. Holistic View on Synthetic Natural Gas Production: A Technical, Economic and Environmental Analysis. Energies 2022, 15, 1608. [Google Scholar] [CrossRef]
- Thema, M.; Bauer, F.; Sterner, M. Sterner Power-to-Gas: Electrolysis and methanation status review. Renew. Sustain. Energy Rev. 2019, 112, 775–778. [Google Scholar] [CrossRef]
- Pintér, G. The development of global power-to-methane potentials between 2000 and 2020: A comparative overview of international projects. Appl. Energy 2024, 353, 122094. [Google Scholar] [CrossRef]
- Tripodi, A.; Conte, F.; Rossetti, I. Carbon Dioxide Methanation: Design of a Fully Integrated Plant. Energy Fuels 2020, 34, 7242–7256. [Google Scholar] [CrossRef]
- Uddin, Z.; Yu, B.Y.; Lee, H.Y. Evaluation of alternative processes of CO2 methanation: Design, optimization, control, techno-economic and environmental analysis. J. CO2 Util. 2022, 60, 101974. [Google Scholar] [CrossRef]
- Available online: https://www.clariant.com/en/Business-Units/Catalysts/Energy-Transition/Carbon-Capture-and-Utilization (accessed on 27 July 2024).
- Mohd Ridzuan, N.D.; Shaharun, M.S.; Anawar, M.A.; Ud-Din, I. Ni-Based Catalyst for Carbon Dioxide Methanation: A Review on Performance and Progress. Catalysts 2022, 12, 469. [Google Scholar] [CrossRef]
- Hanh My Bui, H.M.; Großmann, P.F.; Gros, T.; Blum, M.; Berger, A.; Fischer, R.; Szesni, N.; Tonigold, M.; Hinrichsen, O. 3D printed co-precipitated Ni-Al CO2 methanation catalysts by Binder Jetting: Fabrication, characterization and test in a single pellet string reactor. Appl. Catal. A Gen. 2022, 643, 118760. [Google Scholar]
- Bown, R.M.; Joyce, M.; Zhang, Q.; Ramirez Reina, T.; Duya, M.S. Identifying Commercial Opportunities for the Reverse Water Gas Shift Reaction. Energy Technol. 2021, 9, 2100554. [Google Scholar] [CrossRef]
- Thor Wismann, S.; Larsen, K.E.; Mølgaard Mortensen, P. Electrical Reverse Shift: Sustainable CO2 Valorization for Industrial Scale. Angew. Chem. Int. Ed. 2022, 61, e202109696. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.hydrocarbonengineering.com/clean-fuels/26052022/ineratec-and-clariant-join-forces-for-a-cleaner-future/ (accessed on 30 October 2023).
- Available online: https://matthey.com/en/news/2022/hycogen (accessed on 30 October 2023).
- Available online: https://www.hydrocarbonprocessing.com/news/2023/08/e-fuels-axens-paul-wurth-ifpen-agree-to-co-develop-reverse-water-gas-shift-tech/ (accessed on 30 October 2023).
- Gomez, S.; Peters, J.A.; Maschmeyer, T. The Reductive Amination of Aldehydes and Ketones and the Hydrogenation of Nitriles: Mechanistic Aspects and Selectivity Control. Adv. Synth. Catal. 2002, 344, 1037–1057. [Google Scholar] [CrossRef]
- Medina, F.; Dutartre, R.; Tichit, D.; Coq, B.; Dung, N.T.; Salagre, P.; Sueiras, J.E. Characterization and activity of hydrotalcite-type catalysts for acetonitrile hydrogenation. J. Mol. Catal. A Chem. 1997, 119, 201–212. [Google Scholar] [CrossRef]
- Hahn, G.; Kunnas, P.; de Jonge, N.; Kempe, R. General synthesis of primary amines via reductive amination employing a reusable nickel catalyst. Nat. Catal. 2019, 2, 71–77. [Google Scholar] [CrossRef]
- McCullagh, A.M.; Brennan, C.; Davidson, A.L.; Lennon, D.; Ballas, C.E.; How, C. The application of an alumina-supported Ni catalyst for the hydrogenation of nitrobenzene to aniline. Catal. Today 2024, 442, 114933. [Google Scholar] [CrossRef]
- Chen, B.; Dingerdissen, U.; Krauter, J.G.E.; Lansink Rotgerink, H.G.J.; Möbus, K.; Ostgard, D.J.; Panster, P.; Riermeier, T.H.; Seebald, S.; Tack, T.; et al. New developments in hydrogenation catalysis particularly in synthesis of fine and intermediate chemicals. Appl. Catal. A Gen. 2005, 280, 17–46. [Google Scholar] [CrossRef]
- Farahani, M.D.; Valand, J.; Mahomed, A.S.; Friedrich, H.B. A Comparative Study of NiO/Al2O3 Catalysts Prepared by Different Combustion Techniques for Octanal Hydrogenation. Catal. Lett. 2016, 146, 2441–2449. [Google Scholar] [CrossRef]
- Perret, N.; Grigoropoulos, A.; Zanella, M.; Manning, T.D.; Claridge, J.B.; Rosseinsky, M.J. Catalytic Response and Stability of Nickel/Alumina for the Hydrogenation of 5-Hydroxymethylfurfural in Water. ChemSusChem 2016, 9, 521–531. [Google Scholar] [CrossRef]
- Li, J.; Qian, L.P.; Hu, L.Y.; Yue, B.; He, H.Y. Low-temperature hydrogenation of maleic anhydride to succinic anhydride and γ-butyrolactone over pseudo-boehmite derived alumina supported metal (metal = Cu, Co and Ni) catalysts. Chin. Chem. Lett. 2016, 27, 1004–1008. [Google Scholar] [CrossRef]
- Yang, R.; Li, X.; Wu, J.; Zhang, X.; Zhang, Z.; Cheng, Y.; Guo, J. Hydrotreating of crude 2-ethylhexanol over Ni/Al2O3 catalysts: Surface Ni species-catalytic activity correlation. Appl. Catal. A Gen. 2009, 368, 105–112. [Google Scholar] [CrossRef]
- Smejkal, Q.; Smejkalová, L.; Kubicka, D. Thermodynamic balance in reaction system of total vegetable oil hydrogenation. Chem. Eng. J. 2009, 146, 155–160. [Google Scholar] [CrossRef]
- Gousi, M.; Andriopoulou, C.; Bourikas, K.; Ladas, S.; Sotiriou, M.; Kordulis, C.; Lycourghiotis, A. Green diesel production over nickel-alumina co-precipitated catalysts. Appl. Catal. A Gen. 2017, 536, 45–56. [Google Scholar] [CrossRef]
- Morgan, T.; Santillan-Jimenez, E.; Harman-Ware, A.E.; Ji, Y.; Grubb, D.; Crocker, M. Catalytic deoxygenation of triglycerides to hydrocarbons over supported nickel catalysts. Chem. Eng. J. 2012, 189–190, 346–355. [Google Scholar] [CrossRef]
- Shi, F.; Wang, H.; Chen, Y.; Zheng, Z.; Zheng, Y. Green diesel-like hydrocarbon production by H2-free catalytic deoxygenation of oleic acid via Ni/MgO-Al2O3 catalysts: Effect of the metal loading amount. J. Environ. Chem. Eng. 2023, 11, 110520. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, G.; Xie, H.; Jiao, Z.; Zhang, X. Hydrodeoxygenation of methyl laurate over the sulfur-free Ni/γ-Al2O3 catalysts. Appl. Catal. A Gen. 2019, 569, 35–44. [Google Scholar] [CrossRef]
- Jiang, W.; Cao, J.P.; Huan, Z.X.; Hu, X.; Tang, W.; Chen, C.X.; Wang, H.Y.; He, Z.M.; Zhao, X.Y.; Bai, H.C. Relatively Electron-Rich Ni Nanoparticles Supported on α-Al2O3 for High-Efficiency Hydrogenolysis of Lignin and Its Derivatives under Mild Conditions. ACS Sustain. Chem. Eng. 2023, 11, 17646–17661. [Google Scholar] [CrossRef]
- Hou, M.; Chen, H.; Li, Y.; Wang, H.; Zhang, L.; Bi, Y. Reductive Catalytic Fractionation of Lignocellulose over Ni/Al2O3 Catalyst Prepared by an EDTA-Assisted Impregnation Method. Energy Fuels 2022, 36, 1929–1938. [Google Scholar] [CrossRef]
- Van den Bosch, S.; Renders, T.; Kennis, S.; Koelewijn, S.-F.; Van den Bossche, G.; Vangeel, T.; Deneyer, A.; Depuydt, D.; Courtin, C.M.; Thevelein, J.M.; et al. Integrating lignin valorization and bio-ethanol production: On the role of Ni-Al2O3 catalyst pellets during lignin-first fractionation. Green Chem. 2017, 19, 3313–3326. [Google Scholar] [CrossRef]
- Brandon, C.; Vance, B.C.; Najmi, S.; Kots, P.A.; Wang, C.; Jeon, S.; Stach, E.A.; Zakharov, D.N.; Marinkovic, N.; Ehrlich, S.N.; et al. Structure–Property Relationships for Nickel Aluminate Catalysts in Polyethylene Hydrogenolysis with Low Methane Selectivity. JACS Au 2023, 3, 2156–2165. [Google Scholar]
- Alshareef, R.; Nahil, M.A.; Williams, P.T. Hydrogen Production by Three-Stage (i) Pyrolysis, (ii) Catalytic Steam Reforming, and (iii) Water Gas Shift Processing of Waste Plastic. Energy Fuels 2023, 37, 3894–3907. [Google Scholar] [CrossRef]
- Aminu, I.; Nahil, M.A.; Williams, P.T. High-yield hydrogen from thermal processing of waste plastics. Proc. Inst. Civil Eng.—Waste Res. Manag. 2022, 175, 3–13. [Google Scholar] [CrossRef]
- Røstrup-Nielsen, J.R.; Sehested, J.; Nørskov, J.K. Hydrogen and Synthesis Gas by Steam and CO2 Reforming. Adv. Catal. 2002, 47, 65–139. [Google Scholar] [CrossRef]
- Fowles, M.; Carlsson, M. Steam Reforming of Hydrocarbons for Synthesis Gas Production. Top. Catal. 2021, 64, 856–875. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, Z.; Hu, Y.H. Steam reforming of methane: Current states of catalyst design and process upgrading. Renew. Sustain. Energy Rev. 2021, 149, 111330. [Google Scholar] [CrossRef]
- Garbarino, G.; Pugliese, F.; Cavattoni, T.; Busca, G.; Costamagna, P. A Study on CO2 Methanation and Steam Methane Reforming over Commercial Ni/Calcium Aluminate Catalysts. Energies 2020, 13, 2792. [Google Scholar] [CrossRef]
- Røstrup-Nielsen, J.R. Catalytic Steam Reforming. In Catalysis Science and Technology; Anderson, J.R., Boudart, M., Eds.; Springer: Berlin, Germany, 1984; Volume 5, pp. 1–118. [Google Scholar]
- Carlsson, M. Carbon Formation in Steam Reforming and Effect of Potassium Promotion. Johns. Matthey Technol. Rev. 2015, 59, 313–318. [Google Scholar] [CrossRef]
- Goula, M.A.; Charisiou, N.D.; Papageridis, K.N.; Delimitis, A.; Pachatouridou, E.; Iliopoulou, E.F. Nickel on alumina catalysts for the production of hydrogen rich mixtures via the biogas dry reforming reaction: Influence of the synthesis method. Int. J. Hydrogen Energy 2015, 40, 9183–9200. [Google Scholar] [CrossRef]
- Rashid, M.U.; Wan Daud, W.M.A.; Abbas, H.F. Dry reforming of methane: Influence of process parameters—A review. Renew. Sustain. En. Rev. 2015, 45, 710–744. [Google Scholar]
- Aramouni, N.A.K.; Touma, J.G.; Tarboush, B.A.; Zeaiter, J.; Ahmad, M.N. Catalyst design for dry reforming of methane: Analysis review. Renew. Sustain. Energy Rev. 2018, 82, 2570–2585. [Google Scholar] [CrossRef]
- Torrez-Herrera, J.J.; Korili, S.A.; Gil, A. Recent progress in the application of Ni-based catalysts for the dry reforming of methane. Catal. Rev. 2021, 65, 1300–1357. [Google Scholar] [CrossRef]
- Qin, Z.; Chen, J.; Xie, X.; Luo, X.; Su, T.; Ji, H. CO2 reforming of CH4 to syngas over nickel-based catalysts. Environ. Chem. Lett. 2020, 18, 997–1017. [Google Scholar] [CrossRef]
- Liu, Z.W.; Jun, K.W.; Roh, H.S.; Park, S.E.; Oh, Y.S. Partial Oxidation of Methane over Nickel Catalysts Supported on Various Aluminas. Korean J. Chem. Eng. 2002, 19, 735–741. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, Z.; He, Y.; Tang, Y.; Tan, L. Study of Ni Catalysts with Different Al2O3 Supports in the Partial Oxidation of Methane Reaction. Ind. Eng. Chem. Res. 2023, 62, 12120–12132. [Google Scholar] [CrossRef]
- Raza, J.; Khoja, A.H.; Anwar, M.; Saleem, F.; Naqvi, S.R.; Liaquat, R.; Hassan, M.; Javaid, R.; Qazi, U.Y.; Lumbers, B. Methane decomposition for hydrogen production: A comprehensive review on catalyst selection and reactor systems. Renew. Sustain. Energy Rev. 2022, 168, 112774. [Google Scholar] [CrossRef]
- Biondo, L.D.; Manera, C.; Aguzzoli, C.; Godinho, M. DoE-driven thermodynamic assessment of COX-free hydrogen production from methane decomposition. Catal. Commun. 2024, 187, 106874. [Google Scholar] [CrossRef]
- Salmones, J.; Wang, J.A.; Valenzuela, M.A.; Sánchez, E.; Garcia, A. Pore geometry influence on the deactivation behavior of Ni-based catalysts for simultaneous production of hydrogen and nanocarbon. Catal. Today 2009, 148, 134–139. [Google Scholar] [CrossRef]
- Li, Y.; Chen, J.; Chang, L. Catalytic growth of carbon fibers from methane on a nickel-alumina composite catalyst prepared from Feitknecht compound precursor. Appl. Catal. A Gen. 1997, 163, 45–57. [Google Scholar] [CrossRef]
- Kovalenko, G.A.; Rudina, N.A.; Perminova, L.V.; Skrypnik, O.V. Corundum Impregnation Conditions for Preparing Supported Ni Catalysts for the Synthesis of a Uniform Layer of Carbon Nanofibers. Kinet. Catal. 2010, 51, 762–770. [Google Scholar] [CrossRef]
- Wan, Z.; Tao, Y.; Shao, J.; Zhang, Y.; You, H. Ammonia as an effective hydrogen carrier and a clean fuel for solid oxide fuel cells. Energy Convers. Manag. 2021, 228, 113729. [Google Scholar] [CrossRef]
- Yu, X.; Yin, F.; Li, G.; Zhang, J.; Chen, B. Preparation of nickel aluminate supported Ni nanocatalyst and its catalytic activity for ammonia decomposition to produce hydrogen. Int. J. Hydrogen Energy, 2024; in press. [Google Scholar] [CrossRef]
- Ashcroft, J.; Goddin, H. Centralised and Localised Hydrogen Generation by Ammonia Decomposition. A technical review of the ammonia cracking process. Johns. Matthey Technol. Rev. 2022, 66, 375–385. [Google Scholar] [CrossRef]
- Available online: https://matthey.com/products-and-markets/chemicals/ammonia-cracking-catalysts (accessed on 26 July 2024).
- Angelette, L.M.; Belliveau, R.G.; Coopersmith, K.J.; Cooper, J.J.; Steedley, J.A.; Morrell, B.B. Evaluation of catalysts for decomposition of ammonia in hydrogen isotope purification systems. Fusion Eng. Des. 2021, 173, 112895. [Google Scholar] [CrossRef]
- Sansaniwal, S.K.; Pal, K.; Rosen, M.A.; Tyagi, S.K. Recent advances in the development of biomass gasification technology: A comprehensive review. Renew. Sustain. Energy Rev. 2017, 72, 363–384. [Google Scholar] [CrossRef]
- Cortazar, M.; Santamaria, L.; Lopez, G.; Alvarez, J.; Zhang, L.; Wang, R.; Bi, X.; Olazar, M. A comprehensive review of primary strategies for tar removal in biomass gasification. Energy Conv. Manag. 2023, 276, 116496. [Google Scholar] [CrossRef]
- Lorente, E.; Millan, M.; Brandon, N.P. Use of gasification syngas in SOFC: Impact of real tar on anode materials. Int. J. Hydrogen Energy 2012, 37, 7271–7278. [Google Scholar] [CrossRef]
- Røstrup-Nielsen, J.R.; Bøgild Hansen, J. Steam Reforming for Fuel Cells. In Fuel Cells, Technologies for Fuel Processing; Shekhawat, D., Spivey, J.J., Berry, D.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2011; Chapter 4; pp. 49–71. [Google Scholar]
- Andersson, K.J.; Rasmussen, M.S.S.; Højlund Nielsen, P.E. Industrial-scale gas conditioning including Topsoe tar reforming and purification downstream biomass gasifiers: An overview and recent examples. Fuel 2017, 203, 1026–1030. [Google Scholar] [CrossRef]
- Chitsazanm, S.; Sepehri, S.; Garbarino, G.; Carnasciali, M.M.; Busca, G. Steam reforming of biomass-derived organics: Interactions of different mixture components on Ni/Al2O3 based catalysts. Appl. Catal. B Environ. 2016, 187, 386–398. [Google Scholar] [CrossRef]
- Tang, W.; Cao, J.P.; He, Z.M.; Jiang, W.J.; Wang, Z.H.; Zhao, X.Y. Recent Progress of Catalysts for Reforming of BiomassTar/Tar Models at Low Temperatures—A Short Review. ChemCatChem 2023, 15, e202300581. [Google Scholar] [CrossRef]
- Phung, T.K.; Busca, G. Selective Bioethanol Conversion to Chemicals and Fuels via Advanced Catalytic Approaches. In Biorefinery of Alternative Resources: Targeting Green Fuels and Platform Chemicals; Nanda, S., Vo, D.-V.N., Sarangi, P.K., Eds.; Springer: Berlin/Heidelberg, Germany, 2020; pp. 75–103. [Google Scholar]
- Chen, W.H.; Biswas, P.P.; Hwai Chyuan Ong, H.C.; Hoang, A.T.; Nguyen, T.B.; Dong, C.D. A critical and systematic review of sustainable hydrogen production from ethanol/bioethanol: Steam reforming, partial oxidation, and autothermal reforming. Fuel 2023, 333, 126526. [Google Scholar] [CrossRef]
- Alipour, H.; Alavi, S.M.; Rezaei, M.; Akbari, E.; Varbar, M. The role of various preparation techniques and nickel loadings in ethanol steam reforming over mesoporous nanostructured Ni–Al2O3 catalysts. J. Energy Inst. 2024, 112, 101488. [Google Scholar] [CrossRef]
- Garbarino, G.; Cavattoni, T.; Riani, P.; Brescia, R.; Canepa, F.; Busca, G. On the Role of Support in Metallic Heterogeneous Catalysis: A Study of Unsupported Nickel–Cobalt Alloy Nanoparticles in Ethanol Steam Reforming. Catal. Lett. 2019, 149, 929–941. [Google Scholar] [CrossRef]
- Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/866383/Phase_1_-_Wood_-_Novel_Renewable_Ethanol_Steam_Reformer.pdf (accessed on 14 May 2021).
- Oakley, J.H.; Hoadley, A.F.A. Industrial scale steam reforming of bioethanol: A conceptual study. Int. J. Hydrogen Energy 2010, 35, 8472–8485. [Google Scholar] [CrossRef]
- Garbarino, G.; Pampararo, G.; Phung, T.K.; Riani, P.; Busca, G. Heterogeneous Catalysis in (Bio)Ethanol Conversion to Chemicals and Fuels: Thermodynamics, Catalysis, Reaction Paths, Mechanisms and Product Selectivities. Energies 2020, 13, 3587. [Google Scholar] [CrossRef]
- Di Nardo, A.; Portarapillo, M.; Russo, D.; Di Benedetto, A. Hydrogen production via steam reforming of different fuels: Thermodynamic comparison. Int. J. Hydrogen Energy 2024, 55, 1143–1160. [Google Scholar] [CrossRef]
- Marquevich, M.; Farriol, X.; Medina, F.; Montané, D. Hydrogen Production by Steam Reforming of Vegetable Oils Using Nickel-Based Catalysts. Ind. Eng. Chem. Res. 2001, 40, 4757–4766. [Google Scholar] [CrossRef]
- Setiabudi, H.D.; Aziz, M.A.A.; Abdullah, S.; The, L.P.; Jusoh, R. Hydrogen production from catalytic steam reforming of biomass pyrolysis oil or bio-oil derivatives: A review. Int. J. Hydrogen Energy 2020, 45, 18376–18397. [Google Scholar] [CrossRef]
- Sepehri, S.; Rezaei, M.; Garbarino, G.; Busca, G. Facile synthesis of a mesoporous alumina and its application as a support of Ni-based autothermal reforming catalysts. Int. J. Hydrogen Energy 2016, 41, 3456–3464. [Google Scholar] [CrossRef]
- Park, S.-W.; Lee, D.; Kim, S.-I.; Kim, Y.J.; Park, J.H.; Heo, I.; Chang, T.S.; Lee, J.H. Effects of Alkali Metals on Nickel/Alumina Catalyzed Ethanol Dry Reforming. Catalysts 2021, 11, 260. [Google Scholar] [CrossRef]
- Nguyen, A.-T.; Ng, K.H.; Kumar, P.S.; Pham, T.P.T.; Setiabudi, H.D.; Yusuf, F.; Pham, L.K.H.; Show, P.L.; Hussain, I.; Vo, D.-V.N. Sustainable hydrogen production and CO2 mitigation from acetic acid dry reforming over Ni/Al2O3 catalyst. Int. J. Hydrogen Energy 2024, 67, 1044–1055. [Google Scholar] [CrossRef]
- Ramezani, R.; Di Felice, L.; Gallucci, F. A review of chemical looping reforming technologies for hydrogen production: Recent advances and future challenges. J. Phys. Energy 2023, 5, 024010. [Google Scholar] [CrossRef]
- Tavanarad, M.; Rezaei, M.; Meshkani, F. Preparation and evaluation of Ni/γ-Al2O3 catalysts promoted by alkaline earth metals in glycerol reforming with carbon dioxide. Int. J. Hydrogen Energy 2021, 46, 24991–25003. [Google Scholar] [CrossRef]
- Song, K.S.; Seo, Y.S.; Yoon, H.K.; Cho, S.J. Characteristics of the NiO/Hexaaluminate for Chemical Looping Combustion Korean. J. Chem. Eng. 2003, 20, 471–475. [Google Scholar]
- Wang, G.; Zhang, S.; Zhu, X.; Li, C. Honghong Shan Dehydrogenation versus hydrogenolysis in the reaction of light alkanes over Ni-based catalysts. J. Ind. Eng. Chem. 2020, 86, 1–12. [Google Scholar] [CrossRef]
- Shuikin, N.I.; Tulupova, E.D. Highly active nickel—Alumina catalyst for the dehydrogenation of the cyclohexane ring. Russ. Chem. Bull. 1960, 9, 668–674. [Google Scholar] [CrossRef]
- Yolcular, S.; Olgun, O. Ni/Al2O3 catalysts and their activity in dehydrogenation of methylcyclohexane for hydrogen production. Catal. Today 2008, 138, 198–202. [Google Scholar] [CrossRef]
- Wang, D.; Lei, Q.; Li, H.; Li, G.; Zhao, Y. Preparation of a Novel NiAlO Composite Oxide Catalyst for the Dehydrogenation of Methylcyclohexane. Catalysts 2022, 12, 958. [Google Scholar] [CrossRef]
- Niermann, M.; Beckendorff, A.; Kaltschmitt, M.; Bonhoff, K. Liquid Organic Hydrogen Carrier (LOHC)—Assessment based on chemical and economic properties. Int. J. Hydrogen Energy 2019, 44, 6631–6654. [Google Scholar] [CrossRef]
- Chu, C.; Wu, K.; Luo, B.; Cao, Q.; Zhang, H. Hydrogen storage by liquid organic hydrogen carriers: Catalyst, renewable carrier, and technology—A review. Carbon Res. Conv. 2023, 6, 334–351. [Google Scholar] [CrossRef]
- Munyentwali, A.; Tan, K.C.; He, T. Advancements in the development of liquid organic hydrogen carrier systems and their applications in the hydrogen economy. Prog. Nat. Sci. Mat. Int. 2024; in press. [Google Scholar] [CrossRef]
- Available online: https://ess.honeywell.com/us/en/solutions/sustainability/hydrogen-solutions/liquid-organic-hydrogen-carrier (accessed on 14 August 2024).
- Kamal, M.S.; Razzak, S.A.; Hossain, M.M. Catalytic oxidation of volatile organic compounds (VOCs). A review. Atmos. Environ. 2016, 140, 117–134. [Google Scholar] [CrossRef]
- Xia, Y.; Lin, M.; Ren, D.; Li, Y.; Hu, F.; Chen, W. Preparation of high surface area mesoporous nickel oxides and catalytic oxidation of toluene and formaldehyde. J. Porous Mater. 2017, 24, 621–629. [Google Scholar] [CrossRef]
- Ozkan, U.S.; Kueller, R.F.; Monteczuma, E. Methanol Oxidation over Nonprecious Transition Metal Oxide Catalysts. Ind. Eng. Chem. Res. 1990, 29, 1136. [Google Scholar] [CrossRef]
- Darvell, L.I.; Heiskanen, K.; Jones, J.M.; Ross, A.B.; Simell, P.; Williams, A. An investigation of alumina-supported catalysts for the selective catalytic oxidation of ammonia in biomass gasification. Catal. Today 2003, 81, 681–692. [Google Scholar] [CrossRef]
- Stoyanova, M.; Konova, P.; Nikolov, P.; Naydenov, A.; Christoskova, S.; Mehandjiev, D. Alumina-supported nickel oxide for ozone decomposition and catalytic ozonation of CO and VOCs. Chem. Eng. J. 2006, 122, 41–46. [Google Scholar] [CrossRef]
- Kozhukhova, A.E.; du Preez, S.P.; Bessarabov, D.G. Catalytic Hydrogen Combustion for Domestic and Safety Applications: A Critical Review of Catalyst Materials and Technologies. Energies 2021, 14, 4897. [Google Scholar] [CrossRef]
- Sadykov, V.A.; Isupova, L.A.; Zolotarskii, I.A.; Bobrova, L.N.; Noskov, A.S.; Parmon, V.N.; Brushtein, E.A.; Telyatnikova, T.V.; Chernyshev, V.I.; Lunin, V.V. Oxide catalysts for ammonia oxidation in nitric acid production: Properties and perspectives. Appl. Catal. A Gen. 2000, 204, 59–87. [Google Scholar] [CrossRef]
- Amblard, M.; Burch, R.; Southward, B.W.L. The selective conversion of ammonia to nitrogen on metal oxide catalysts under strongly oxidising conditions. Appl. Catal. B Environ. 1999, 22, L159–L166. [Google Scholar]
- Nassos, S.; Svensson, E.E.; Boutonnet, M.; Järås, S.G. The influence of Ni load and support material on catalysts for the selective catalytic oxidation of ammonia in gasified biomass. Appl. Catal. B Environ. 2007, 74, 92–102. [Google Scholar] [CrossRef]
- Chmielarz, L.; Kustrowski, P.; Rafalska-Łasocha, A.; Dziemba, R. Selective oxidation of ammonia to nitrogen on transition metal containing mixed metal oxides. Appl. Catal. B Environ. 2005, 58, 235–244. [Google Scholar] [CrossRef]
- Weissenberger, T.; Zapf, R.; Pennemann, H.; Kolb, G. Effect of the Active Metal on the NOx Formation during Catalytic Combustion of Ammonia SOFC Off-Gas. Catalysts 2022, 12, 1186. [Google Scholar] [CrossRef]
- Dey, S.; Dhal, G.C.; Mohan, D.; Prasad, R. Advances in transition metal oxide catalysts for carbon monoxide oxidation: A review. Advan. Compos. Hybrid Mat. 2019, 2, 626–656. [Google Scholar] [CrossRef]
- Dey, S.; Mehta, N.S. Oxidation of carbon monoxide over various nickel oxide catalysts in different conditions: A review. Chem. Eng. J. Adv. 2020, 1, 100008. [Google Scholar] [CrossRef]
- Han, S.W.; Kim, D.H.; Jeong, M.; Park, K.J.; Kim, Y.D. CO oxidation catalyzed by NiO supported on mesoporous Al2O3 at room temperature. Chem. Eng. J. 2016, 283, 992–998. [Google Scholar] [CrossRef]
- Jiajian, G.; Jia, C.; Li, J.; Zhang, M. Ni/Al2O3 catalysts for CO methanation: Effect of Al2O3 supports calcined at different temperatures. J. Energy Chem. 2013, 22, 919–927. [Google Scholar]
- Maharaj, C.; Lokhat, D.; Rawatlal, R. Oxidation of carbon monoxide over supported nickel oxide catalyst: Kinetic model development and identification. S. Afr. J. Chem. Eng. 2022, 39, 106–116. [Google Scholar] [CrossRef]
- Yousra Abdelbaki, Y.; de Arriba, A.; Solsona, B.; Delgado, D.; García-González, E.; Issaadi, R.; López Nieto, J.M. The nickel-support interaction as determining factor of the selectivity to ethylene in the oxidative dehydrogenation of ethane over nickel oxide/alumina catalysts. Appl. Catal. A Gen. 2021, 623, 118242. [Google Scholar] [CrossRef]
- Zhang, Q.; Jiang, X.; Li, Y.; Tan, Y.; Jiang, Q.; Liu, X.; Qiao, B. Oxidative Dehydrogenation of Propane over Supported Nickel Single-Atom Catalyst. Chin. J. Chem. 2023, 42, 370–376. [Google Scholar] [CrossRef]
- Li, X.; Ma, J.; He, H. Recent advances in catalytic decomposition of ozone. J. Environ. Sci. 2020, 94, 14–31. [Google Scholar] [CrossRef]
- Yu, X.; Wang, S.; Gao, H. Spinel NiAl2O4 Based Catalysts: Past, Present and Future. J. Environ. Sci. Eng. Tech. 2023, 11, 12–27. [Google Scholar] [CrossRef]
- Dilshara, P.; Abeysinghe, B.; Premasiri, R.; Dushyantha, N.; Ratnayake, N.; Senarath, S.; Ratnayake, S.A.; Batapola, N. The role of nickel (Ni) as a critical metal in clean energy transition: Applications, global distribution and occurrences, production-demand and phytomining. J. Asian Earth Sci. 2024, 259, 105912. [Google Scholar] [CrossRef]

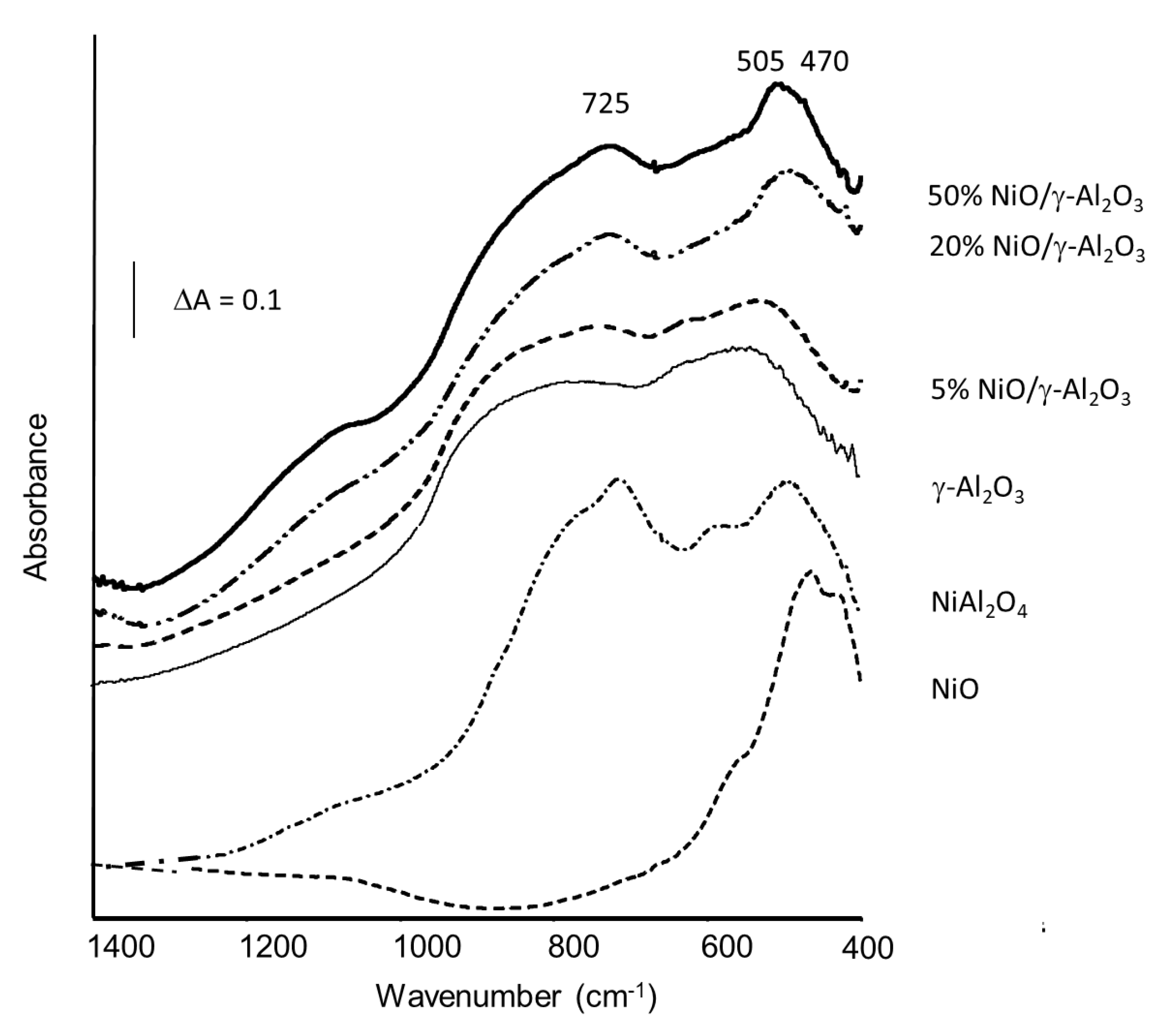


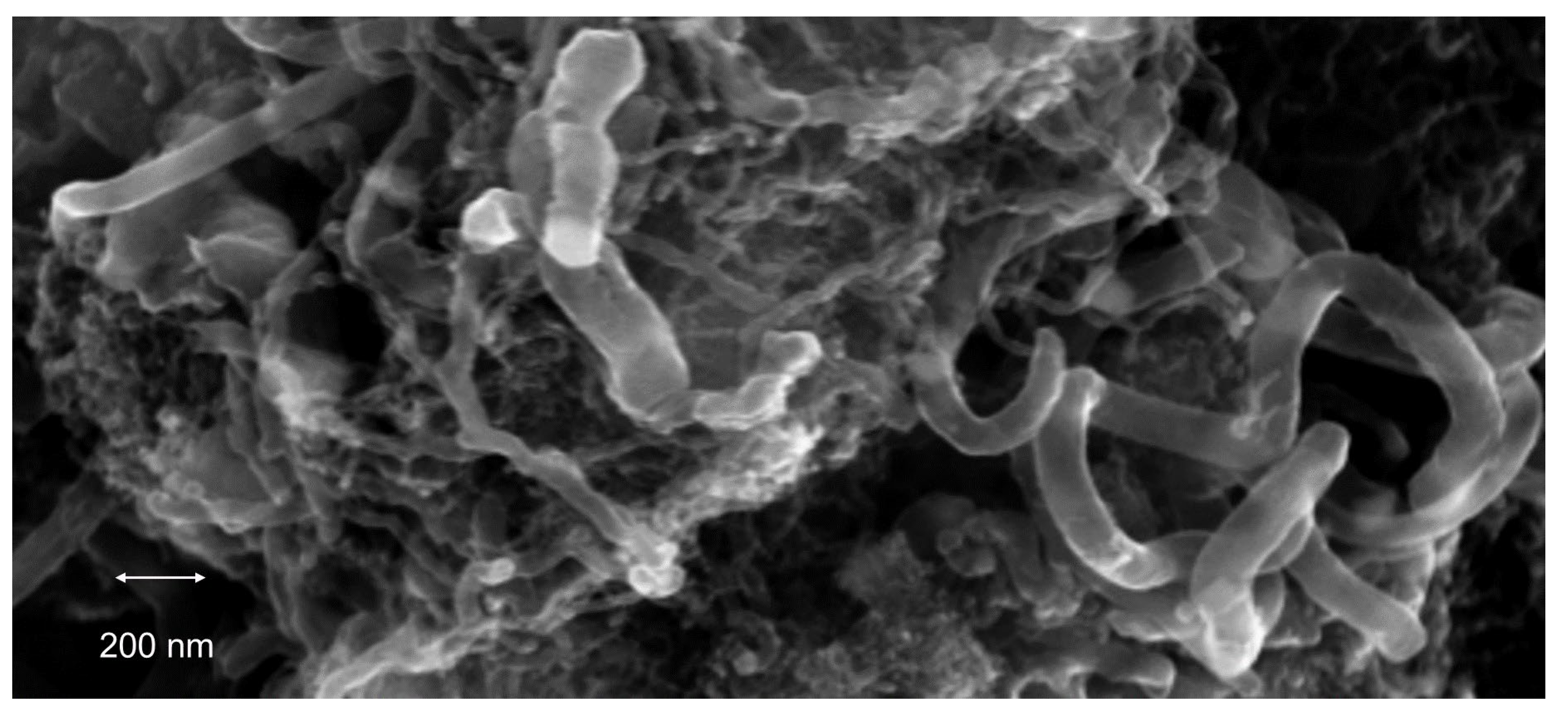

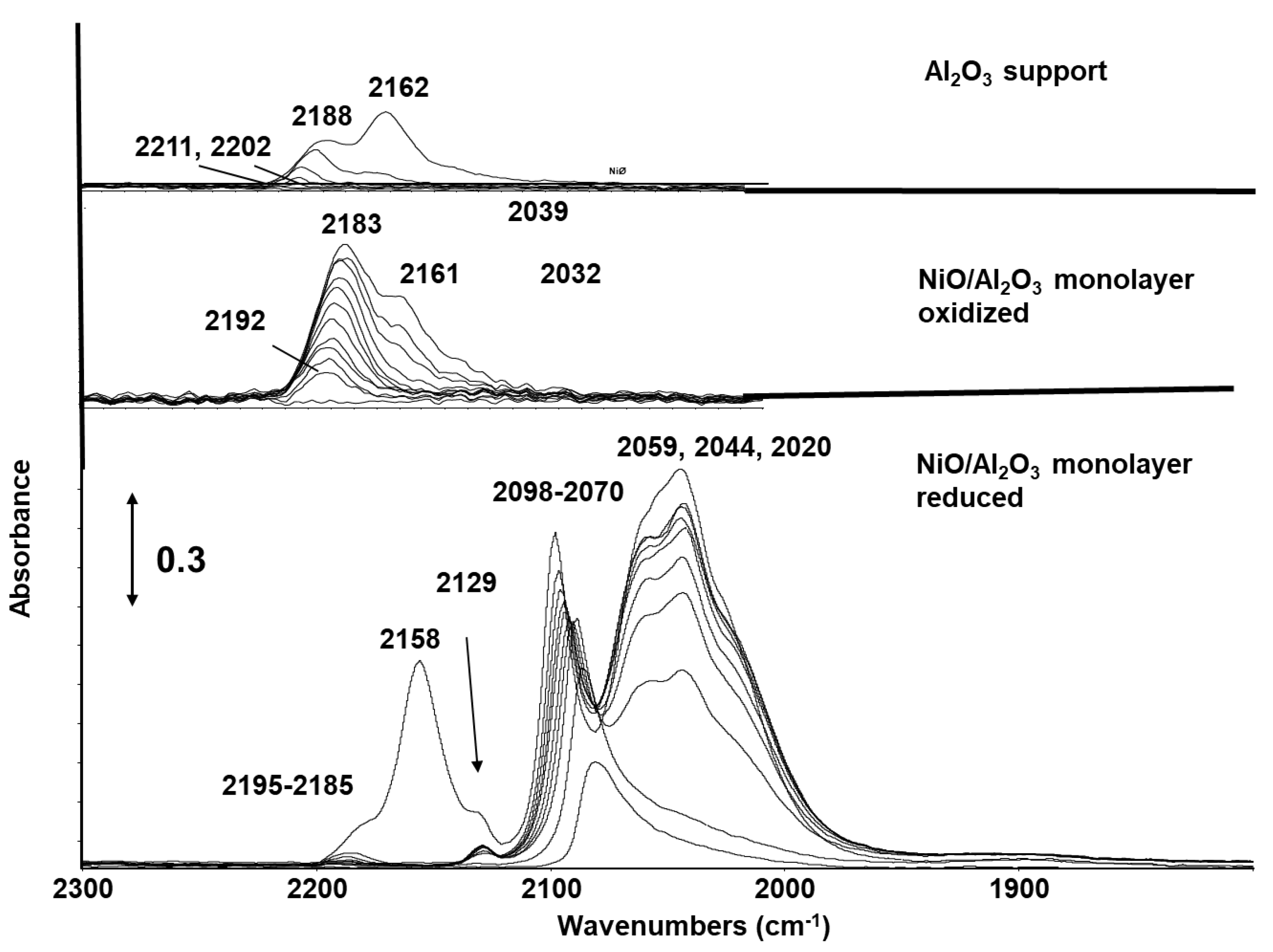


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Name | Stoichiometry | ΔH°298 (Gas) | Typical Reaction Conditions | ||
|---|---|---|---|---|---|
| (kJ/molR) | Phase | T (°C) | P (Bar) | ||
| Acetylene partial hydrogenation in C2 cuts | HC≡CH + H2 → H2C=CH2 | −172 | Gas Liq | 100–250 25–100 | 5 20–35 |
| C3–C5 steam cracking cut purification | Diene and acetylenic hydrogenation | ~−240 | Liq | 10–60 | <100 |
| Pygas hydrotreatment 1 step | Diene and acetylenic hydrogenation | ~−150 | Liq | 70–200 | 20–50 |
| Phenylacetylene partial hydrogenation | C6H5-C≡CH + H2 → C6H5-HC=CH2 | ~−120 | Liq | 15–50 | <50 |
| Linear diene hydrogenation for LAB synthesis | R-CH=CH-CH=CH2 → R-CH2CH2CH2CH3 | −240 | Liq | 170–230 | 10–20 |
| Benzene saturation | C6H6 + 3 H2 ⇆ C6H12 | −208 | Gas Liq | 500 200–350 | 30–50 |
| CO methanation | CO + 3 H2 → CH4 + H2O | −206 | Gas | 250–350 | 20–50 |
| CO2 methanation | CO2 + 4 H2 → CH4 + 2 H2O | −165 | Gas | 300–400 | 20–50 |
| Reverse water–gas shift | CO2 + H2 → CO + H2O | +41 | Gas | >850 | 1–30 |
| Acetonitrile hydrogenation | CH3C ≡ N + H2 → CH3CH2NH2 | −121 | Gas | 100–200 | 1–20 |
| n-octanal hydrogenation | C7H15-CH=O + H2 → C7H15CH2OH | −57 | Gas | 100–200 | 20–50 |
| Methane steam reforming | CH4 + H2O → CO + 3H2 | +206 | Gas | 500–800 | 1–40 |
| Methane dry reforming | CH4 + CO2 → 2CO + 2H2 | +247 | Gas | 500–700 | 1–20 |
| Methane partial oxidation | CH4 + ½ O2 → CO + 2 H2 | −36 | Gas | 500–1000 | 1–40 |
| Methane combustion to CO | CH4 + O2 → CO + 2 H2O | −519 | Gas | >200 | 1–40 |
| Methane decomposition | CH4 → C + 2 H2 | +76 | Gas | >400 | 1–10 |
| Ammonia decomposition | NH3 → ½ N2+ ³/₂ H2 | +46 | Gas | >400 | 1–10 |
| Cyclohexane dehydrogenation | C6H12 ⇆ C6H6 + 3 H2 | +208 | Gas | 400 | 1–3 |
| Ammonia oxidation to NO | NH3 + 5/4 O2 → NO + 3/2 H2O | −226 | Gas | >600 | 1–20 |
| Selective oxidation of ammonia to N2 | NH3 + ¾ O2 → ½ N2 + 3/2 H2O | −317 | Gas | >600 | 1–20 |
| CO oxidation | CO+ ½ O2 → CO2 | −283 | Gas | >0 | 1–20 |
| Hydrogen combustion | H2 + ½ O2 → H2O | −242 | Gas | >100 | 1–20 |
| Ethane oxidative dehydrogenation | H3C-CH3 + ½ O2 → H2=CH2 + H2O | −105 | Gas | 500–700 | 1–10 |
| Ozone decomposition | O3 → 3/2 O2 | −143 | Gas | −50–0 | <1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Busca, G.; Spennati, E.; Riani, P.; Garbarino, G. Properties, Industrial Applications and Future Perspectives of Catalytic Materials Based on Nickel and Alumina: A Critical Review. Catalysts 2024, 14, 552. https://doi.org/10.3390/catal14080552
Busca G, Spennati E, Riani P, Garbarino G. Properties, Industrial Applications and Future Perspectives of Catalytic Materials Based on Nickel and Alumina: A Critical Review. Catalysts. 2024; 14(8):552. https://doi.org/10.3390/catal14080552
Chicago/Turabian StyleBusca, Guido, Elena Spennati, Paola Riani, and Gabriella Garbarino. 2024. "Properties, Industrial Applications and Future Perspectives of Catalytic Materials Based on Nickel and Alumina: A Critical Review" Catalysts 14, no. 8: 552. https://doi.org/10.3390/catal14080552