
Advancements in Cobalt-Based Catalysts for Enhanced CO2 Hydrogenation: Mechanisms, Applications, and Future Directions: A Short Review

Abstract
:1. Introduction
2. Thermodynamic Considerations in CO2 Hydrogenation
(ΔH°298K = 293.0 kJ/mol ΔG°298K = 257.2 kJ/mol)
(ΔH°298K = 41.2 kJ/mol ΔG°298K =28.6 kJ/mol)
(ΔH°298K = −49.5 kJ/mol ΔG°298K = 3.5 kJ/mol)
(ΔH°298K = −86.7 kJ/mol ΔG°298K = −32.4 kJ/mol)
3. Mechanism of Reaction Co-Based Catalysts
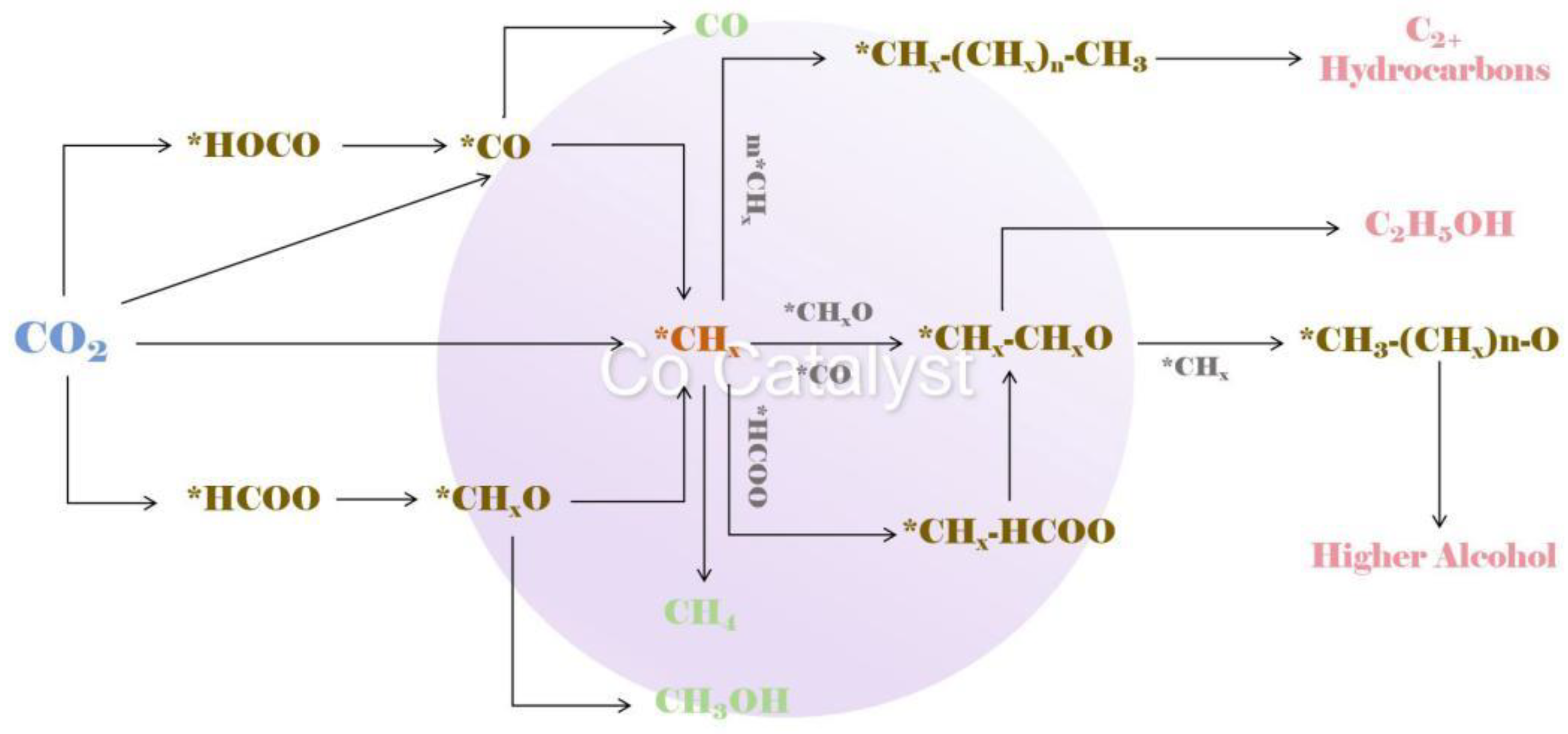
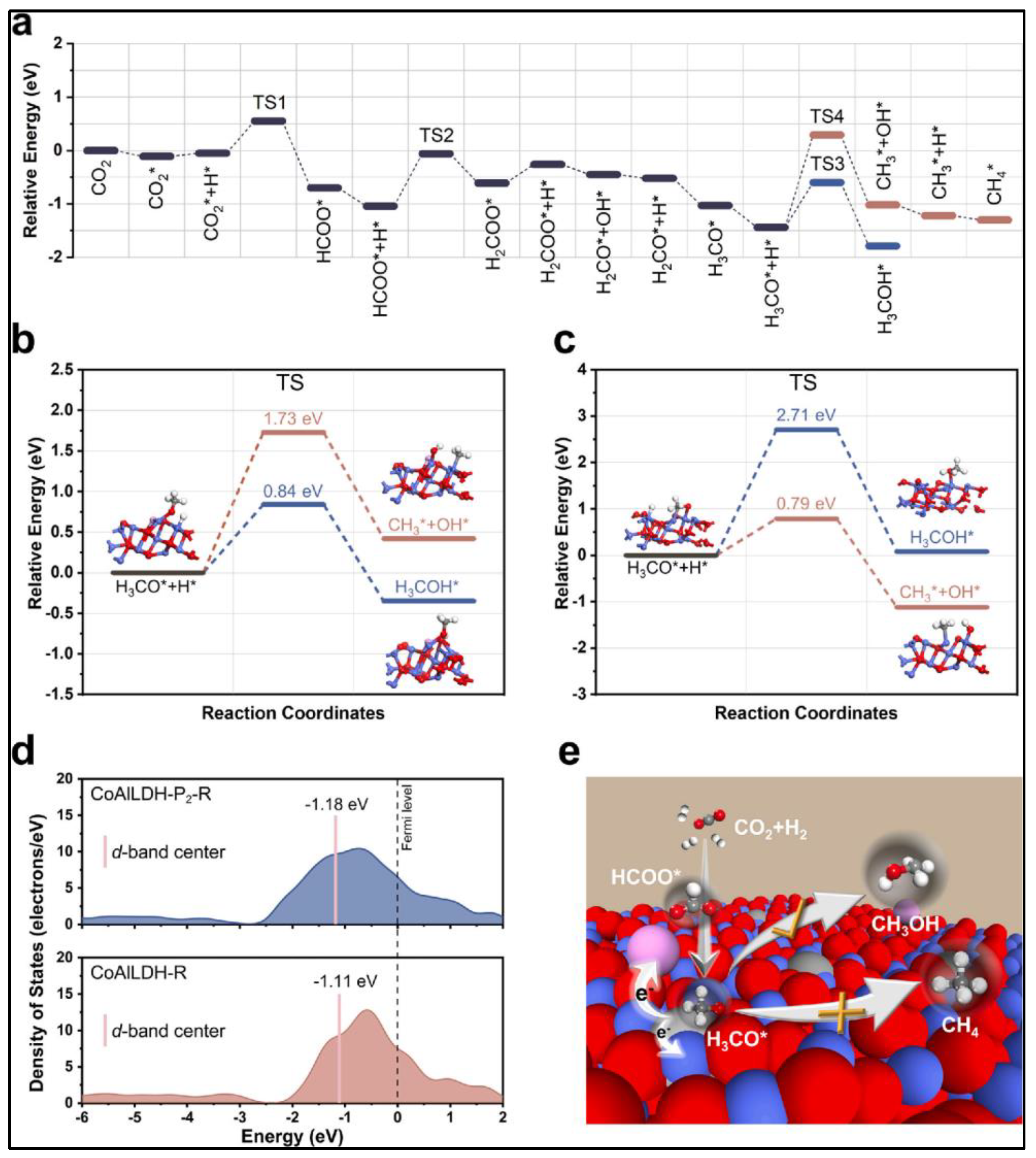
3.1. Detailed Pathways of CO2 Hydrogenation and Catalyst Behaviour
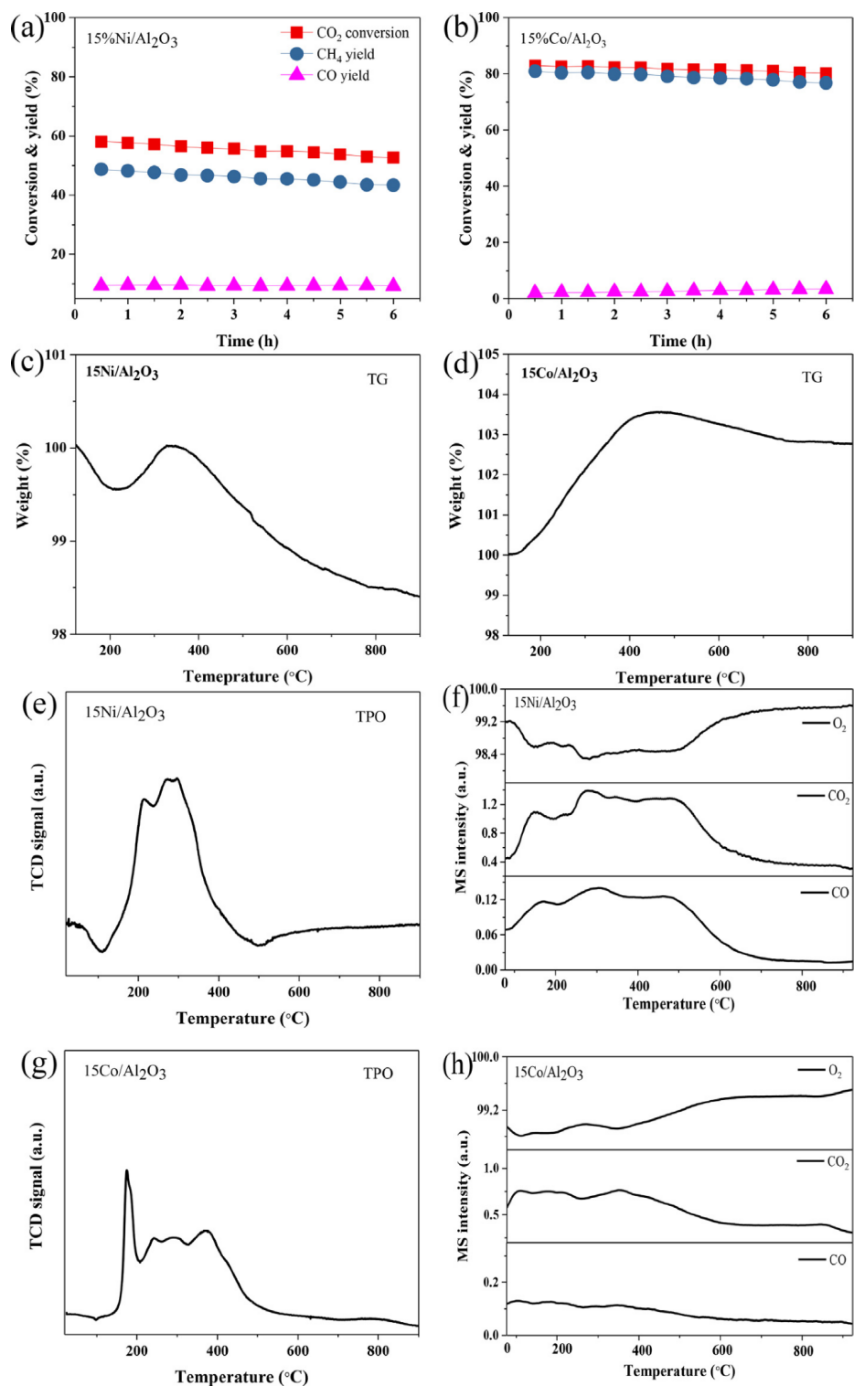
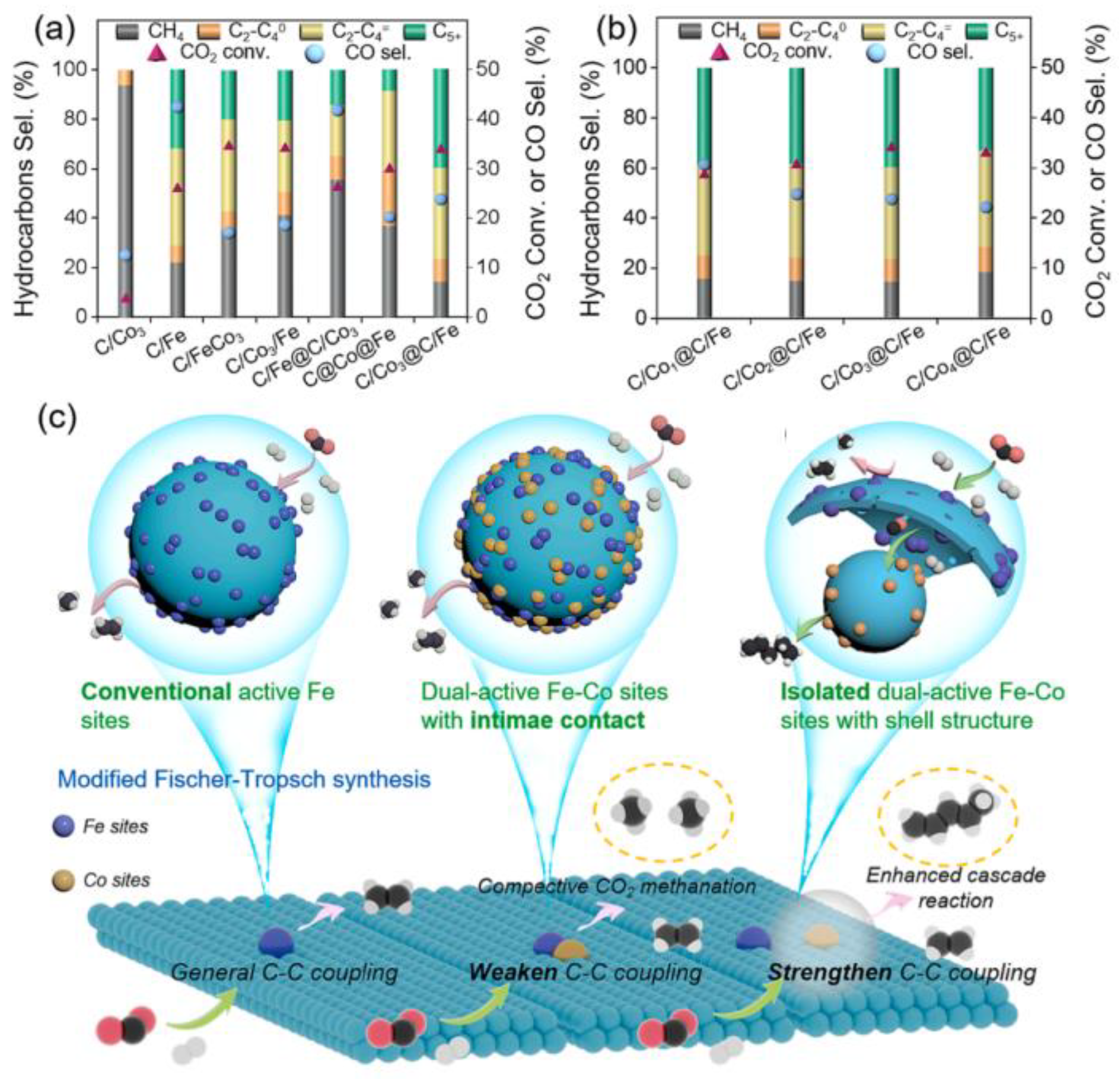
3.2. Synergistic Effects and Active Sites in CO2 Hydrogenation
4. Optimisation of Co-Based Catalysts
4.1. Detailed Pathways of CO2 Hydrogenation and Catalyst Behaviour
(ΔH°298K = −165 kJ/mol ΔG°298K = −113.6 kJ/mol)
4.2. CO2 Hydrogenation to Hydrocarbon Chemicals and Fuels
4.3. Innovations in CO2-to-Methanol Catalysis
5. Limitations and Future Perspectives
5.1. Limitations
5.2. Future Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Liu, C.; Kang, J.; Huang, Z.Q.; Song, Y.H.; Xiao, Y.S.; Song, J.; He, J.X.; Chang, C.R.; Ge, H.Q.; Wang, Y.; et al. Gallium nitride catalyzed the direct hydrogenation of carbon dioxide to dimethyl ether as primary product. Nat. Commun. 2021, 12, 2305. [Google Scholar] [CrossRef] [PubMed]
- Le Quéré, C.; Andres, R.J.; Boden, T.; Conway, T.; Houghton, R.A.; House, J.I.; Marland, G.; Peters, G.P.; van der Werf, G.R.; Ahlström, A.; et al. The global carbon budget 1959–2011. Earth Syst. Sci. Data 2013, 5, 165–185. [Google Scholar] [CrossRef]
- Lovelock, C.E.; Duarte, C.M. Dimensions of Blue Carbon and emerging perspectives. Biol. Lett. 2019, 15, 20180781. [Google Scholar] [CrossRef] [PubMed]
- Mac Dowell, N.; Fennell, P.S.; Shah, N.; Maitland, G.C. The role of CO2 capture and utilization in mitigating climate change. Nat. Clim. Chang. 2017, 7, 243–249. [Google Scholar] [CrossRef]
- Chen, S.; Liu, J.; Zhang, Q.; Teng, F.; McLellan, B.C. A critical review on deployment planning and risk analysis of carbon capture, utilization, and storage (CCUS) toward carbon neutrality. Renew. Sustain. Energy Rev. 2022, 167, 112537. [Google Scholar] [CrossRef]
- Truong, T.H.; Lin, B.-W.; Lo, C.-H.; Tung, C.-P.; Chao, C.-W. Possible pathways for low carbon transitions: Investigating the efforts of oil companies in CCUS technologies. Energy Strategy Rev. 2024, 54, 101421. [Google Scholar] [CrossRef]
- Gu, C.; Li, K.; Gao, S.; Li, J.; Mao, Y. CO2 abatement feasibility for blast furnace CCUS retrofits in BF-BOF steel plants in China. Energy 2024, 294, 130756. [Google Scholar] [CrossRef]
- Huang, Z.; Deng, S.; Zhang, Q.; Zhao, R.; Li, S.; Veselovskaya, J.; Kozlov, D.; Wang, J. Closing plastic loop with CCUS: Life cycle assessment of a novel strategy for plastic sustainable transition and negative emissions. Resour. Conserv. Recycl. 2024, 208, 107738. [Google Scholar] [CrossRef]
- Sun, B.; Fan, B.; Wu, C.; Xie, J. Exploring incentive mechanisms for the CCUS project in China’s coal-fired power plants: An option-game approach. Energy 2024, 288, 129694. [Google Scholar] [CrossRef]
- Lu, S.; Yu, Z.; Zhang, Y.; Xu, T. Review of non-isothermal processes in CCUS from a geomechanical perspective. Earth Sci. Rev. 2024, 255, 104848. [Google Scholar] [CrossRef]
- Azar, C.; Lindgren, K.; Obersteiner, M.; Riahi, K.; van Vuuren, D.P.; den Elzen, K.M.G.; Möllersten, K.; Larson, E.D. The feasibility of low CO2 concentration targets and the role of bio-energy with carbon capture and storage (BECCS). Clim. Chang. 2010, 100, 195–202. [Google Scholar] [CrossRef]
- Heuberger, C.F.; Staffell, I.; Shah, N.; Mac Dowell, N. Quantifying the value of CCS for the future electricity system. Energy Environ. Sci. 2016, 9, 2497–2510. [Google Scholar] [CrossRef]
- Nath, F.; Mahmood, M.N.; Yousuf, N. Recent advances in CCUS: A critical review on technologies, regulatory aspects and economics. Geoenergy Sci. Eng. 2024, 238, 212726. [Google Scholar] [CrossRef]
- De, S.; Dokania, A.; Ramirez, A.; Gascon, J. Advances in the design of heterogeneous catalysts and thermocatalytic processes for CO2 utilization. ACS Catal. 2020, 10, 14147–14185. [Google Scholar] [CrossRef]
- Bai, S.-T.; De Smet, G.; Liao, Y.; Sun, R.; Zhou, C.; Beller, M.; Maes, B.U.W.; Sels, B.F. Homogeneous and heterogeneous catalysts for hydrogenation of CO2 to methanol under mild conditions. Chem. Soc. Rev. 2021, 50, 4259–4298. [Google Scholar] [CrossRef]
- Liu, W.; Sahoo, B.; Junge, K.; Beller, M. Cobalt complexes as an emerging class of catalysts for homogeneous hydrogenations. Acc. Chem. Res. 2018, 51, 1858–1869. [Google Scholar] [CrossRef]
- Sen, R.; Goeppert, A.; Surya Prakash, G.K. Homogeneous hydrogenation of CO2 and CO to methanol: The renaissance of low-temperature catalysis in the context of the methanol economy. Angew. Chem. Int. Ed Engl. 2022, 61, e202207278. [Google Scholar] [CrossRef] [PubMed]
- Onishi, N.; Himeda, Y. Homogeneous catalysts for CO2 hydrogenation to methanol and methanol dehydrogenation to hydrogen generation. Coord. Chem. Rev. 2022, 472, 214767. [Google Scholar] [CrossRef]
- Furimsky, E. CO2 hydrogenation to methanol and methane over carbon-supported catalysts. Ind. Eng. Chem. Res. 2020, 59, 15393–15423. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, L.; Wang, Z. Advances in the preparation of light alkene from carbon dioxide by hydrogenation. Fuel 2022, 324, 124503. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, L.; Zhou, Q.; Li, Y.; Liu, L.; Nie, W.; Xu, R.; Zhang, J.; Cheng, Z.; Wang, H. Carbon dioxide enabled hydrogen storage by methanol: Highly selective and efficient catalysis with well-defined heterogeneous catalysts. Coord. Chem. Rev. 2024, 508, 215775. [Google Scholar] [CrossRef]
- Liu, S.; He, Y.; Fu, W.; Chen, J.; Ren, J.; Liao, L.; Sun, R.; Tang, Z.; Mebrahtu, C.; Zeng, F. Hetero-site cobalt catalysts for higher alcohols synthesis by CO2 hydrogenation: A review. J. CO2 Util. 2023, 67, 102322. [Google Scholar] [CrossRef]
- Tu, J.; Wu, H.; Qian, Q.; Han, S.; Chu, M.; Jia, S.; Feng, R.; Zhai, J.; He, M.; Han, B. Low temperature methanation of CO2 over an amorphous cobalt-based catalyst. Chem. Sci. 2021, 12, 3937–3943. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shen, C.; Sun, K.; Jia, X.; Ye, J.; Liu, C.-J. Advances in studies of the structural effects of supported Ni catalysts for CO2 hydrogenation: From nanoparticle to single atom catalyst. J. Mater. Chem. A Mater. Energy Sustain. 2022, 10, 5792–5812. [Google Scholar] [CrossRef]
- Liang, C.; Tian, H.; Gao, G.; Zhang, S.; Liu, Q.; Dong, D.; Hu, X. Methanation of CO2 over alumina supported nickel or cobalt catalysts: Effects of the coordination between metal and support on formation of the reaction intermediates. Int. J. Hydrogen Energy 2020, 45, 531–543. [Google Scholar] [CrossRef]
- Bogdan, V.I.; Pokusaeva, Y.A.; Koklin, A.E.; Savilov, S.V.; Chernyak, S.A.; Lunin, V.V.; Kustov, L.M. Carbon dioxide reduction with hydrogen on carbon-nanotube-supported catalysts under supercritical conditions. Energy Technol. 2019, 7, 1900174. [Google Scholar] [CrossRef]
- Díez-Ramírez, J.; Sánchez, P.; Kyriakou, V.; Zafeiratos, S.; Marnellos, G.E.; Konsolakis, M.; Dorado, F. Effect of support nature on the cobalt-catalyzed CO2 hydrogenation. J. CO2 Util. 2017, 21, 562–571. [Google Scholar] [CrossRef]
- Owen, R.E.; O’Byrne, J.P.; Mattia, D.; Plucinski, P.; Pascu, S.I.; Jones, M.D. Cobalt catalysts for the conversion of CO2 to light hydrocarbons at atmospheric pressure. Chem. Commun. 2013, 49, 11683–11685. [Google Scholar] [CrossRef]
- He, Z.; Cui, M.; Qian, Q.; Zhang, J.; Liu, H.; Han, B. Synthesis of liquid fuel via direct hydrogenation of CO2. Proc. Natl. Acad. Sci. USA 2019, 116, 12654–12659. [Google Scholar] [CrossRef]
- Hwang, S.-M.; Han, S.J.; Park, H.-G.; Lee, H.; An, K.; Jun, K.-W.; Kim, S.K. Atomically alloyed Fe–co catalyst derived from a N-coordinated co single-atom structure for CO2 hydrogenation. ACS Catal. 2021, 11, 2267–2278. [Google Scholar] [CrossRef]
- Zhang, H.; Mao, D.; Zhang, J.; Wu, D. Tuning the CO2 hydrogenation path by moderately phosphating the Co-Al catalyst toward methanol synthesis. Appl. Catal. B 2024, 340, 123257. [Google Scholar] [CrossRef]
- Irshad, M.; Chun, H.-J.; Khan, M.K.; Jo, H.; Kim, S.K.; Kim, J. Synthesis of n-butanol-rich C3+ alcohols by direct CO2 hydrogenation over a stable Cu–Co tandem catalyst. Appl. Catal. B 2024, 340, 123201. [Google Scholar] [CrossRef]
- Zhang, S.; An, K.; Xin, J.; Jiang, Y.; Niu, M.; Song, P.; Wu, C.; Wang, H.; Liu, Y. Carbon modified active pairs of Co-Co2C for CO2 hydrogenation to alcohols. Chem. Eng. J. 2024, 486, 150334. [Google Scholar] [CrossRef]
- Jia, C.; Gao, J.; Dai, Y.; Zhang, J.; Yang, Y. The thermodynamics analysis and experimental validation for complicated systems in CO2 hydrogenation process. J. Energy Chem. 2016, 25, 1027–1037. [Google Scholar] [CrossRef]
- Kattel, S.; Liu, P.; Chen, J.G. Tuning selectivity of CO2 hydrogenation reactions at the metal/oxide interface. J. Am. Chem. Soc. 2017, 139, 9739–9754. [Google Scholar] [CrossRef]
- Pham, C.Q.; Bahari, M.B.; Kumar, P.S.; Ahmed, S.F.; Xiao, L.; Kumar, S.; Qazaq, A.S.; Siang, T.J.; Tran, H.-T.; Islam, A. Carbon dioxide methanation on heterogeneous catalysts: A review. Environ. Chem. Lett. 2022, 20, 3613–3630. [Google Scholar] [CrossRef]
- Gao, P.; Zhang, L.; Li, S.; Zhou, Z.; Sun, Y. Novel heterogeneous catalysts for CO2 hydrogenation to liquid fuels. ACS Cent. Sci. 2020, 6, 1657–1670. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Wang, S.; Liu, B.; Liu, J.; Wang, L.; Xiao, Y.; Xu, Y.; Liu, X. Insights into the influence of CeO2 crystal facet on CO2 hydrogenation to methanol over pd/CeO2 catalysts. ACS Catal. 2020, 10, 11493–11509. [Google Scholar] [CrossRef]
- Li, H.; Zhao, J.; Luo, L.; Du, J.; Zeng, J. Symmetry-breaking sites for activating linear carbon dioxide molecules. Acc. Chem. Res. 2021, 54, 1454–1464. [Google Scholar] [CrossRef]
- Ashok, J.; Pati, S.; Hongmanorom, P.; Tianxi, Z.; Junmei, C.; Kawi, S. A review of recent catalyst advances in CO2 methanation processes. Catal. Today 2020, 356, 471–489. [Google Scholar] [CrossRef]
- Su, X.; Xu, J.; Liang, B.; Duan, H.; Hou, B.; Huang, Y. Catalytic carbon dioxide hydrogenation to methane: A review of recent studies. J. Energy Chem. 2016, 25, 553–565. [Google Scholar] [CrossRef]
- Studt, F.; Behrens, M.; Kunkes, E.L.; Thomas, N.; Zander, S.; Tarasov, A.; Schumann, J.; Frei, E.; Varley, J.B.; Abild-Pedersen, F.; et al. The mechanism of CO and CO2 hydrogenation to methanol over Cu-based catalysts. ChemCatChem 2015, 7, 1105–1111. [Google Scholar] [CrossRef]
- Kunkes, E.L.; Studt, F.; Abild-Pedersen, F.; Schlögl, R.; Behrens, M. Hydrogenation of CO2 to methanol and CO on Cu/ZnO/Al2O3: Is there a common intermediate or not? J. Catal. 2015, 328, 43–48. [Google Scholar] [CrossRef]
- Wang, L.; Wang, L.; Zhang, J.; Liu, X.; Wang, H.; Zhang, W.; Yang, Q.; Ma, J.; Dong, X.; Yoo, S.J.; et al. Selective hydrogenation of CO2 to ethanol over cobalt catalysts. Angew. Chem. Int. Ed Engl. 2018, 57, 6104–6108. [Google Scholar] [CrossRef] [PubMed]
- Khodakov, A.Y.; Chu, W.; Fongarland, P. Advances in the development of novel cobalt Fischer-Tropsch catalysts for synthesis of long-chain hydrocarbons and clean fuels. Chem. Rev. 2007, 107, 1692–1744. [Google Scholar] [CrossRef]
- Zeng, F.; Mebrahtu, C.; Xi, X.; Liao, L.; Ren, J.; Xie, J.; Heeres, H.J.; Palkovits, R. Catalysts design for higher alcohols synthesis by CO2 hydrogenation: Trends and future perspectives. Appl. Catal. B 2021, 291, 120073. [Google Scholar] [CrossRef]
- Ren, M.; Zhang, Y.; Wang, X.; Qiu, H. Catalytic hydrogenation of CO2 to methanol: A review. Catalysts 2022, 12, 403. [Google Scholar] [CrossRef]
- Xie, C.; Chen, C.; Yu, Y.; Su, J.; Li, Y.; Somorjai, G.A.; Yang, P. Tandem catalysis for CO2 hydrogenation to C2-C4 hydrocarbons. Nano Lett. 2017, 17, 3798–3802. [Google Scholar] [CrossRef]
- Roy, S.; Cherevotan, A.; Peter, S.C. Thermochemical CO2 hydrogenation to single carbon products: Scientific and technological challenges. ACS Energy Lett. 2018, 3, 1938–1966. [Google Scholar] [CrossRef]
- Vogt, C.; Monai, M.; Kramer, G.J.; Weckhuysen, B.M. The renaissance of the Sabatier reaction and its applications on Earth and in space. Nat. Catal. 2019, 2, 188–197. [Google Scholar] [CrossRef]
- Lu, Z.; Cheng, Y.; Li, S.; Yang, Z.; Wu, R. CO2 thermoreduction to methanol on the MoS2 supported single Co atom catalyst: A DFT study. Appl. Surf. Sci. 2020, 528, 147047. [Google Scholar] [CrossRef]
- Gnanamani, M.K.; Jacobs, G.; Hamdeh, H.H.; Shafer, W.D.; Liu, F.; Hopps, S.D.; Thomas, G.A.; Davis, B.H. Hydrogenation of carbon dioxide over Co–Fe bimetallic catalysts. ACS Catal. 2016, 6, 913–927. [Google Scholar] [CrossRef]
- Liang, J.; Liu, J.; Guo, L.; Wang, W.; Wang, C.; Gao, W.; Guo, X.; He, Y.; Yang, G.; Yasuda, S.; et al. CO2 hydrogenation over Fe-Co bimetallic catalysts with tunable selectivity through a graphene fencing approach. Nat. Commun. 2024, 15, 512. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Guo, L.; Ai, P.; Wu, H.; Lu, Z.; Huang, J.; Tong, J.; Zheng, L.; Sun, S. Isolated dual-active Fe-Co sites efficiently promote CO2 hydrogenation upgrading. Fuel 2024, 364, 131054. [Google Scholar] [CrossRef]
- Raghav, H.; Pendem, C.; Tripathi, S.; Kumar, S.; Sarkar, B. Enhanced light olefin production from CO2 over potassium promoted Fe–Co bimetallic ZrO2 supported catalysts. Fuel 2024, 368, 131645. [Google Scholar] [CrossRef]
- Wang, D.; Xie, Z.; Porosoff, M.D.; Chen, J.G. Recent advances in carbon dioxide hydrogenation to produce olefins and aromatics. Chem 2021, 7, 2277–2311. [Google Scholar] [CrossRef]
- Wang, L.; He, S.; Wang, L.; Lei, Y.; Meng, X.; Xiao, F.-S. Cobalt–nickel catalysts for selective hydrogenation of carbon dioxide into ethanol. ACS Catal. 2019, 9, 11335–11340. [Google Scholar] [CrossRef]
- Xu, D.; Wang, Y.; Ding, M.; Hong, X.; Liu, G.; Tsang, S.C.E. Advances in higher alcohol synthesis from CO2 hydrogenation. Chem 2021, 7, 849–881. [Google Scholar] [CrossRef]
- Yao, W.; Olajide, G.; Boudreaux, C.M.; Wysocki, M.M.; Ahmed, M.K.; Qu, F.; Szilvási, T.; Papish, E.T. Cobalt(I) pincer complexes as catalysts for CO2 hydrogenation to formate. Organometallics 2024, 43, 1447–1458. [Google Scholar] [CrossRef]
- Wu, S.-B.; Zhang, T.; Chung, L.W.; Wu, Y.-D. A missing piece of the mechanism in metal-catalyzed hydrogenation: Co (−I)/Co (0)/Co (+I) catalytic cycle for Co (−I)-catalyzed hydrogenation. Org. Lett. 2019, 21, 360–364. [Google Scholar] [CrossRef]
- Yan, X.; Ge, H.; Yang, X. Hydrogenation of CO2 to methanol catalyzed by cp*co complexes: Mechanistic insights and ligand design. Inorg. Chem. 2019, 58, 5494–5502. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, X.; Shao, Z.; Wang, H.; Sun, Y. Direct CO2 hydrogenation to ethanol over supported Co2C catalysts: Studies on support effects and mechanism. J. Catal. 2020, 382, 86–96. [Google Scholar] [CrossRef]
- Have, I.C.T.; Kromwijk, J.J.G.; Monai, M.; Ferri, D.; Sterk, E.B.; Meirer, F.; Weckhuysen, B.M. Uncovering the reaction mechanism behind CoO as active phase for CO2 hydrogenation. Nat. Commun. 2022, 13, 324. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, J.; Chen, X.; Gu, S.; Wei, Y.; Wang, L.; Wan, H.; Guan, G. Development of multifunctional catalysts for the direct hydrogenation of carbon dioxide to higher alcohols. Molecules 2024, 29, 2666. [Google Scholar] [CrossRef] [PubMed]
- Gnanamani, M.K.; Jacobs, G.; Keogh, R.A.; Shafer, W.D.; Sparks, D.E.; Hopps, S.D.; Thomas, G.A.; Davis, B.H. Fischer-Tropsch synthesis: Effect of pretreatment conditions of cobalt on activity and selectivity for hydrogenation of carbon dioxide. Appl. Catal. A Gen. 2015, 499, 39–46. [Google Scholar] [CrossRef]
- Fu, W.; Liu, S.; He, Y.; Chen, J.; Ren, J.; Chen, H.; Sun, R.; Tang, Z.; Mebrahtu, C.; Zeng, F. CO2 hydrogenation over CoAl based catalysts: Effects of cobalt-metal oxide interaction. Appl. Catal. A Gen. 2024, 678, 119720. [Google Scholar] [CrossRef]
- Ding, X.; Fu, J.; Lyu, Y.; Ma, L.; Xu, Y.; Liu, X. Active sites of the cobalt catalysts controlled by surface silanols of silicalite-1 in CO2 hydrogenation to ethanol. Chem. Eng. J. 2024, 494, 152923. [Google Scholar] [CrossRef]
- Wang, L.; Guan, E.; Wang, Y.; Wang, L.; Gong, Z.; Cui, Y.; Meng, X.; Gates, B.C.; Xiao, F.-S. Silica accelerates the selective hydrogenation of CO2 to methanol on cobalt catalysts. Nat. Commun. 2020, 11, 1033. [Google Scholar] [CrossRef]
- Cui, G.; Lou, Y.; Zhou, M.; Li, Y.; Jiang, G.; Xu, C. Review of mechanism investigations and catalyst developments for CO2 hydrogenation to alcohols. Catalysts 2024, 14, 232. [Google Scholar] [CrossRef]
- Li, W.; Nie, X.; Yang, H.; Wang, X.; Polo-Garzon, F.; Wu, Z.; Zhu, J.; Wang, J.; Liu, Y.; Shi, C.; et al. Crystallographic dependence of CO2 hydrogenation pathways over HCP-Co and FCC-Co catalysts. Appl. Catal. B 2022, 315, 121529. [Google Scholar] [CrossRef]
- Fan, T.; Liu, H.; Shao, S.; Gong, Y.; Li, G.; Tang, Z. Cobalt catalysts enable selective hydrogenation of CO2 toward diverse products: Recent progress and Perspective. J. Phys. Chem. Lett. 2021, 12, 10486–10496. [Google Scholar] [CrossRef]
- Yu, Y.; Mottaghi-Tabar, S.; Iqbal, M.W.; Yu, A.; Simakov, D.S.A. CO2 methanation over alumina-supported cobalt oxide and carbide synthesized by reverse microemulsion method. Catal. Today 2020, 379, 250–261. [Google Scholar] [CrossRef]
- Jacobs, G.; Das, T.K.; Zhang, Y.; Li, J.; Racoillet, G.; Davis, B.H. Fischer–Tropsch synthesis: Support, loading, and promoter effects on the reducibility of cobalt catalysts. Appl. Catal. A Gen. 2002, 233, 263–281. [Google Scholar] [CrossRef]
- Ra, E.C.; Kim, K.Y.; Kim, E.H.; Lee, H.; An, K.; Lee, J.S. Recycling carbon dioxide through catalytic hydrogenation: Recent key developments and perspectives. ACS Catal. 2020, 10, 11318–11345. [Google Scholar] [CrossRef]
- Deng, K.; Lin, L.; Rui, N.; Vovchok, D.; Zhang, F.; Zhang, S.; Senanayake, S.D.; Kim, T.; Rodriguez, J.A. Studies of CO2 hydrogenation over cobalt/ceria catalysts with in situ characterization: The effect of cobalt loading and metal–support interactions on the catalytic activity. Catal. Sci. Technol. 2020, 10, 6468–6482. [Google Scholar] [CrossRef]
- Liu, S.; Yang, C.; Zha, S.; Sharapa, D.; Studt, F.; Zhao, Z.-J.; Gong, J. Moderate surface segregation promotes selective ethanol production in CO2 hydrogenation reaction over CoCu catalysts. Angew. Chem. Int. Ed Engl. 2022, 61, e202109027. [Google Scholar] [CrossRef]
- Fan, W.K.; Tahir, M. Recent trends in developments of active metals and heterogenous materials for catalytic CO2 hydrogenation to renewable methane: A review. J. Environ. Chem. Eng. 2021, 9, 105460. [Google Scholar] [CrossRef]
- Schaaf, T.; Grünig, J.; Schuster, M.R.; Rothenfluh, T.; Orth, A. Methanation of CO2—Storage of renewable energy in a gas distribution system. Energy Sustain. Soc. 2014, 4, 2. [Google Scholar] [CrossRef]
- Fayisa, B.A.; Yang, Y.; Zhen, Z.; Wang, M.-Y.; Lv, J.; Wang, Y.; Ma, X. Engineered chemical utilization of CO2 to methanol via direct and indirect hydrogenation pathways: A review. Ind. Eng. Chem. Res. 2022, 61, 10319–10335. [Google Scholar] [CrossRef]
- Wei, J.; Ge, Q.; Yao, R.; Wen, Z.; Fang, C.; Guo, L.; Xu, H.; Sun, J. Directly converting CO2 into a gasoline fuel. Nat. Commun. 2017, 8, 15174. [Google Scholar] [CrossRef]
- Gao, P.; Li, S.; Bu, X.; Dang, S.; Liu, Z.; Wang, H.; Zhong, L.; Qiu, M.; Yang, C.; Cai, J.; et al. Direct conversion of CO2 into liquid fuels with high selectivity over a bifunctional catalyst. Nat. Chem. 2017, 9, 1019–1024. [Google Scholar] [CrossRef]
- Frontera, P.; Macario, A.; Ferraro, M.; Antonucci, P. Supported catalysts for CO2 methanation: A review. Catalysts 2017, 7, 59. [Google Scholar] [CrossRef]
- Zhou, W.; Cheng, K.; Kang, J.; Zhou, C.; Subramanian, V.; Zhang, Q.; Wang, Y. New horizon in C1 chemistry: Breaking the selectivity limitation in transformation of syngas and hydrogenation of CO2 into hydrocarbon chemicals and fuels. Chem. Soc. Rev. 2019, 48, 3193–3228. [Google Scholar] [CrossRef]
- Sahebdelfar, S.; Takht Ravanchi, M. Carbon dioxide utilization for methane production: A thermodynamic analysis. J. Pet. Sci. Eng. 2015, 134, 14–22. [Google Scholar] [CrossRef]
- Li, W.; Liu, Y.; Mu, M.; Ding, F.; Liu, Z.; Guo, X.; Song, C. Organic acid-assisted preparation of highly dispersed Co/ZrO2 catalysts with superior activity for CO2 methanation. Appl. Catal. B 2019, 254, 531–540. [Google Scholar] [CrossRef]
- Parastaev, A.; Muravev, V.; Huertas Osta, E.; van Hoof, A.J.F.; Kimpel, T.F.; Kosinov, N.; Hensen, E.J.M. Boosting CO2 hydrogenation via size-dependent metal–support interactions in cobalt/ceria-based catalysts. Nat. Catal. 2020, 3, 526–533. [Google Scholar] [CrossRef]
- Zhen, W.; Li, B.; Lu, G.; Ma, J. Enhancing catalytic activity and stability for CO2 methanation on Ni@MOF-5 via control of active species dispersion. Chem. Commun. 2015, 51, 1728–1731. [Google Scholar] [CrossRef] [PubMed]
- Jampaiah, D.; Damma, D.; Chalkidis, A.; Venkataswamy, P.; Bhargava, S.K.; Reddy, B.M. MOF-derived ceria-zirconia supported Co3O4 catalysts with enhanced activity in CO2 methanation. Catal. Today 2020, 356, 519–526. [Google Scholar] [CrossRef]
- Li, Z.; Chen, J.; Xie, Y.; Wen, J.; Weng, H.; Wang, M.; Zhang, J.; Cao, J.; Tian, G.; Zhang, Q.; et al. Zonal activation of molecular carbon dioxide and hydrogen over dual sites Ni-Co-MgO catalyst for CO2 methanation: Synergistic catalysis of Ni and Co species. J. Energy Chem. 2024, 91, 213–225. [Google Scholar] [CrossRef]
- Pastor-Pérez, L.; Patel, V.; Le Saché, E.; Reina, T.R. CO2 methanation in the presence of methane: Catalysts design and effect of methane concentration in the reaction mixture. J. Energy Inst. 2020, 93, 415–424. [Google Scholar] [CrossRef]
- Zhou, G.; Wu, T.; Zhang, H.; Xie, H.; Feng, Y. Carbon dioxide methanation on ordered mesoporous Co/KIT-6 catalyst. Chem. Eng. Commun. 2014, 201, 233–240. [Google Scholar] [CrossRef]
- Razzaq, R.; Li, C.; Usman, M.; Suzuki, K.; Zhang, S. A highly active and stable Co4N/γ-Al2O3 catalyst for CO and CO2 methanation to produce synthetic natural gas (SNG). Chem. Eng. J. 2015, 262, 1090–1098. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, K.; Wang, Y.; Liu, Z.; Gao, X.; Zhang, J.; Ma, Q.; Fan, S.; Zhao, T.-S.; Yao, M. Recent advances in thermocatalytic hydrogenation of carbon dioxide to light olefins and liquid fuels via modified Fischer-Tropsch pathway. J. CO2 Util. 2023, 67, 102321. [Google Scholar] [CrossRef]
- Ghosh, I.K.; Iqbal, Z.; van Heerden, T.; van Steen, E.; Bordoloi, A. Insights into the unusual role of chlorine in product selectivity for direct hydrogenation of CO/CO2 to short-chain olefins. Chem. Eng. J. 2021, 413, 127424. [Google Scholar] [CrossRef]
- Cheng, K.; Kang, J.; King, D.L.; Subramanian, V.; Zhou, C.; Zhang, Q.; Wang, Y. Advances in catalysis for syngas conversion to hydrocarbons. In Advances in Catalysis; Elsevier: Amsterdam, The Netherlands, 2017; pp. 125–208. [Google Scholar] [CrossRef]
- Guo, L.; Sun, J.; Ji, X.; Wei, J.; Wen, Z.; Yao, R.; Xu, H.; Ge, Q. Directly converting carbon dioxide to linear α-olefins on bio-promoted catalysts. Commun. Chem. 2018, 1, 11. [Google Scholar] [CrossRef]
- Li, W.; Zhang, A.; Jiang, X.; Janik, M.J.; Qiu, J.; Liu, Z.; Guo, X.; Song, C. The anti-sintering catalysts: Fe–Co–Zr polymetallic fibers for CO2 hydrogenation to C2= –C4= –rich hydrocarbons. J. CO2 Util. 2018, 23, 219–225. [Google Scholar] [CrossRef]
- Russell, W.W.; Miller, G.H. Catalytic hydrogenation of carbon dioxide to higher hydrocarbons1. J. Am. Chem. Soc. 1950, 72, 2446–2454. [Google Scholar] [CrossRef]
- Li, W.; Zhang, G.; Jiang, X.; Liu, Y.; Zhu, J.; Ding, F.; Liu, Z.; Guo, X.; Song, C. CO2 hydrogenation on unpromoted and M-promoted co/TiO2 catalysts (M = Zr, K, cs): Effects of crystal phase of supports and metal–support interaction on tuning product distribution. ACS Catal. 2019, 9, 2739–2751. [Google Scholar] [CrossRef]
- Shi, Z.; Yang, H.; Gao, P.; Chen, X.; Liu, H.; Zhong, L.; Wang, H.; Wei, W.; Sun, Y. Effect of alkali metals on the performance of CoCu/TiO2 catalysts for CO2 hydrogenation to long-chain hydrocarbons. Chin. J. Catalysis 2018, 39, 1294–1302. [Google Scholar] [CrossRef]
- Zhang, Y.-X.; Guo, X.-Y.; Liu, B.; Zhang, J.-L.; Gao, X.-H.; Ma, Q.-X.; Fan, S.-B.; Zhao, T.-S. Cellulose modified iron catalysts for enhanced light olefins and linear C5+ α-olefins from CO hydrogenation. Fuel 2021, 294, 120504. [Google Scholar] [CrossRef]
- Numpilai, T.; Chanlek, N.; Poo-Arporn, Y.; Wannapaiboon, S.; Cheng, C.K.; Siri-Nguan, N.; Sornchamni, T.; Kongkachuichay, P.; Chareonpanich, M.; Rupprechter, G.; et al. Pore size effects on physicochemical properties of Fe-Co/K-Al2O3 catalysts and their catalytic activity in CO2 hydrogenation to light olefins. Appl. Surf. Sci. 2019, 483, 581–592. [Google Scholar] [CrossRef]
- Kim, K.Y.; Lee, H.; Noh, W.Y.; Shin, J.; Han, S.J.; Kim, S.K.; An, K.; Lee, J.S. Cobalt ferrite nanoparticles to form a catalytic co–Fe alloy carbide phase for selective CO2 hydrogenation to light olefins. ACS Catal. 2020, 10, 8660–8671. [Google Scholar] [CrossRef]
- Xu, Q.; Xu, X.; Fan, G.; Yang, L.; Li, F. Unveiling the roles of Fe-Co interactions over ternary spinel-type ZnCoxFe2-xO4 catalysts for highly efficient CO2 hydrogenation to produce light olefins. J. Catal. 2021, 400, 355–366. [Google Scholar] [CrossRef]
- Owen, R.E.; Plucinski, P.; Mattia, D.; Torrente-Murciano, L.; Ting, V.P.; Jones, M.D. Effect of support of Co-Na-Mo catalysts on the direct conversion of CO2 to hydrocarbons. J. CO2 Util. 2016, 16, 97–103. [Google Scholar] [CrossRef]
- Scarfiello, C.; Durupt, A.; Tison, Y.; Minh, D.P.; Soulantica, K.; Serp, P. Effects of Pd and Co intimacy in Pd-modified Co/TiO2 catalysts for direct CO2 hydrogenation to fuels: The closer not the better. Catal. Sci. Technol. 2024, 14, 2896–2907. [Google Scholar] [CrossRef]
- Riedel, T.; Schulz, H.; Schaub, G.; Jun, K.-W.; Hwang, J.-S.; Lee, K.-W. Fischer–tropsch on iron with H2/CO and H2/CO2as synthesis gases: The episodes of formation of the Fischer–tropsch regime and construction of the catalyst. Top. Catal. 2003, 26, 41–54. [Google Scholar] [CrossRef]
- Jo, H.; Khan, M.K.; Irshad, M.; Arshad, M.W.; Kim, S.K.; Kim, J. Unraveling the role of cobalt in the direct conversion of CO2 to high-yield liquid fuels and lube base oil. Appl. Catal. B 2022, 305, 121041. [Google Scholar] [CrossRef]
- Shi, Z.; Yang, H.; Gao, P.; Li, X.; Zhong, L.; Wang, H.; Liu, H.; Wei, W.; Sun, Y. Direct conversion of CO2 to long-chain hydrocarbon fuels over K–promoted CoCu/TiO2 catalysts. Catal. Today 2018, 311, 65–73. [Google Scholar] [CrossRef]
- Olah, G.A.; Goeppert, A.; Prakash, G.K.S. Beyond Oil and Gas: The Methanol Economy; Wiley: Hoboken, NJ, USA, 2009; ISBN 978352732422. [Google Scholar] [CrossRef]
- Dalena, F.; Senatore, A.; Basile, M.; Knani, S.; Basile, A.; Iulianelli, A. Advances in methanol production and utilization, with particular emphasis toward hydrogen generation via membrane reactor technology. Membranes 2018, 8, 98. [Google Scholar] [CrossRef]
- Christensen, E.; Yanowitz, J.; Ratcliff, M.; McCormick, R.L. Renewable oxygenate blending effects on gasoline properties. Energy Fuels 2011, 25, 4723–4733. [Google Scholar] [CrossRef]
- Khan, M.U.; Wang, L.; Liu, Z.; Gao, Z.; Wang, S.; Li, H.; Zhang, W.; Wang, M.; Wang, Z.; Ma, C.; et al. Pt3Co octapods as superior catalysts of CO2 hydrogenation. Angew. Chem. Int. Ed Engl. 2016, 55, 9548–9552. [Google Scholar] [CrossRef] [PubMed]
- Konsolakis, M.; Lykaki, M. Recent advances on the rational design of non-precious metal oxide catalysts exemplified by CuOx/CeO2 binary system: Implications of size, shape and electronic effects on intrinsic reactivity and metal-support interactions. Catalysts 2020, 10, 160. [Google Scholar] [CrossRef]
- Li, C.-S.; Melaet, G.; Ralston, W.T.; An, K.; Brooks, C.; Ye, Y.; Liu, Y.-S.; Zhu, J.; Guo, J.; Alayoglu, S.; et al. High-performance hybrid oxide catalyst of manganese and cobalt for low-pressure methanol synthesis. Nat. Commun. 2015, 6, 6538. [Google Scholar] [CrossRef]
- Pustovarenko, A.; Dikhtiarenko, A.; Bavykina, A.; Gevers, L.; Ramírez, A.; Russkikh, A.; Telalovic, S.; Aguilar, A.; Hazemann, J.-L.; Ould-Chikh, S.; et al. Metal–organic framework-derived synthesis of cobalt indium catalysts for the hydrogenation of CO2 to methanol. ACS Catal. 2020, 10, 5064–5076. [Google Scholar] [CrossRef]
- Lian, Y.; Fang, T.; Zhang, Y.; Liu, B.; Li, J. Hydrogenation of CO2 to alcohol species over Co@Co3O4/C-N catalysts. J. Catal. 2019, 379, 46–51. [Google Scholar] [CrossRef]
- Zheng, J.-N.; Kang, A.; Wang, J.-M.; Jing, L.; Yuan, L. Effect of support nature on the cobalt-catalyzed CO2 hydrogenation Co/La-Ga-O composite oxide catalyst. J. Fuel Chem. Technol. 2019, 47, 697–708. [Google Scholar] [CrossRef]
- Ouyang, B.; Xiong, S.; Zhang, Y.; Liu, B.; Li, J. The study of morphology effect of Pt/Co3O4 catalysts for higher alcohol synthesis from CO2 hydrogenation. Appl. Catal. A Gen. 2017, 543, 189–195. [Google Scholar] [CrossRef]
- Liu, B.; Ouyang, B.; Zhang, Y.; Lv, K.; Li, Q.; Ding, Y.; Li, J. Effects of mesoporous structure and Pt promoter on the activity of Co-based catalysts in low-temperature CO2 hydrogenation for higher alcohol synthesis. J. Catal. 2018, 366, 91–97. [Google Scholar] [CrossRef]
- Andrews, J.W. Hydrogen production and carbon sequestration by steam methane reforming and fracking with carbon dioxide. Int. J. Hydrogen Energy 2020, 45, 9279–9284. [Google Scholar] [CrossRef]
- Sarmah, M.K.; Singh, T.P.; Kalita, P.; Dewan, A. Sustainable hydrogen generation and storage—A review. RSC Adv. 2023, 13, 25253–25275. [Google Scholar] [CrossRef]
- Li, H.; Qiu, C.; Ren, S.; Dong, Q.; Zhang, S.; Zhou, F.; Liang, X.; Wang, J.; Li, S.; Yu, M. Na+-gated water-conducting nanochannels for boosting CO2 conversion to liquid fuels. Science 2020, 367, 667–671. [Google Scholar] [CrossRef]
- Saeidi, S.; Amin, N.A.S.; Rahimpour, M.R. Hydrogenation of CO2 to value-added products—A review and potential future developments. J. CO2 Util. 2014, 5, 66–81. [Google Scholar] [CrossRef]
- Samimi, F.; Karimipourfard, D.; Rahimpour, M.R. Green methanol synthesis process from carbon dioxide via reverse water gas shift reaction in a membrane reactor. Chem. Eng. Res. Des. 2018, 140, 44–67. [Google Scholar] [CrossRef]








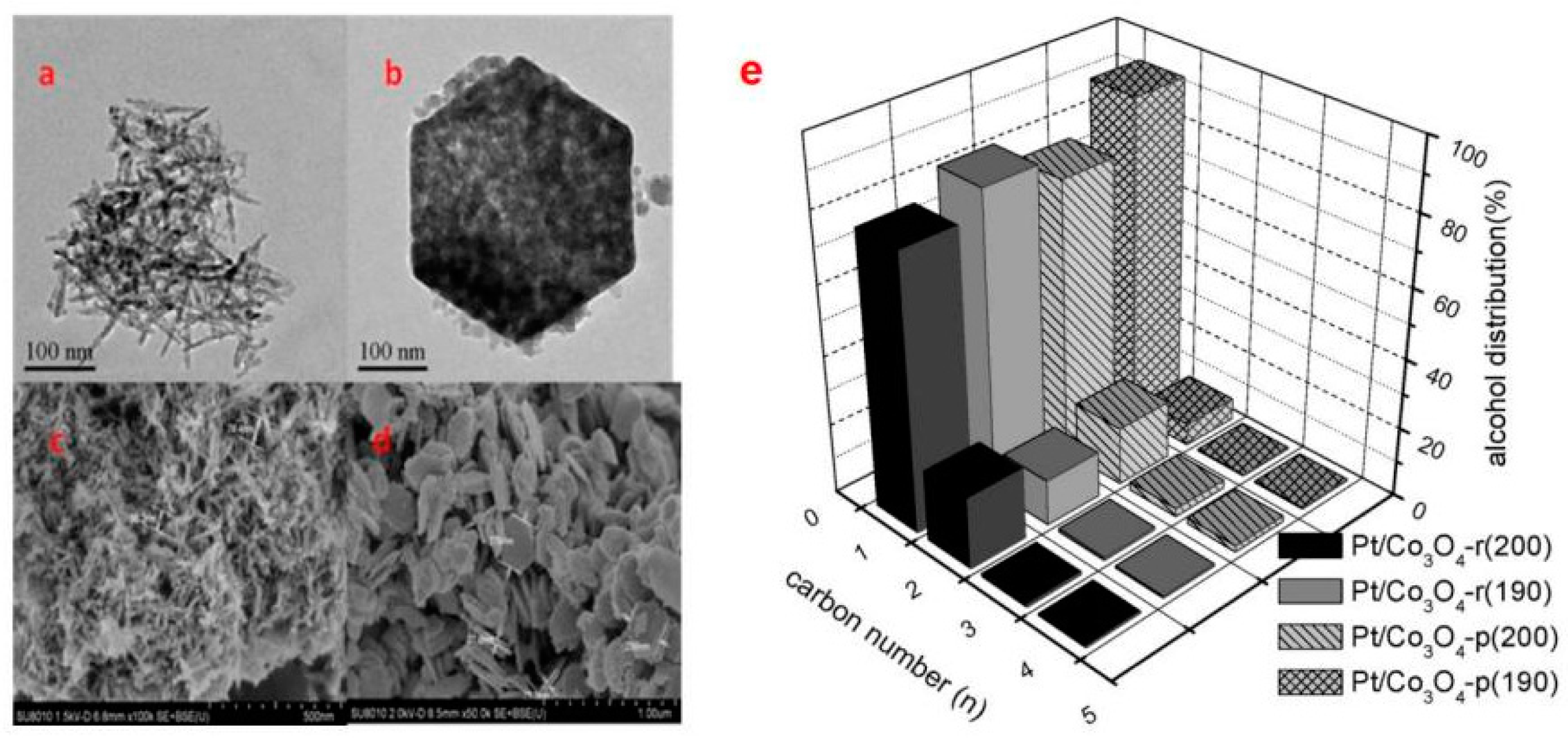

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | T (°C) | P (MPa) | GHSV b | WHSV c | SV b | CO2 Conv. (%) | CH4 Sel. (%) | H2/CO2 | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Co-Zr0.1-B-O a | 180 | 8.0 | - | - | - | - | 97.8 | 1.0 | [23] |
| 2 | 15 wt% Co/Al2O3 | 400 | 1 atm | 16,000 | - | - | 82 | 80 | 4.0 | [26] |
| 3 | 25 wt% Ni/Al2O3 | 400 | 1 atm | 16,000 | - | - | 75 | 69 | 4.0 | [25] |
| 4 | Co(10)/CNT | 250 | 8.0 | - | - | - | 24 | 100 | 1.0 | [27] |
| 5 | Ni(10)/CNT | 250 | 8.0 | - | - | - | 4 | 100 | 1.0 | [27] |
| 6 | Ni-Co-MgO | 290 | - | 20,000 | - | - | 80.5 | 99.6 | 4.0 | [90] |
| 7 | Ni-Co/CeO2-ZrO2 | 350 | - | - | 12,000 | - | 61 | 97 | 4.0 | [91] |
| 8 | 2%Co-ZrO2 | 400 | 3.0 | - | - | 7200 | 85 | 99 | 4.0 | [86] |
| 9 | Co/KIT-6 | 280 | - | 22,000 | - | - | 48.9 | 100 | 4.6 | [92] |
| 10 | Co/meso-SiO2 | 300 | - | 22,000 | - | - | 28 | 68.1 | 4.6 | [89] |
| 11 | 20 wt%Co/CZ-MOF | 320 | 1.5 | - | - | 1500 | 81.2 | 100 | 3.0 | [73] |
| 12 | Co2C/γ-Al2O3 | 300 | 0.3 | 60,000 | - | - | 89 | 99 | 4.0 | [93] |
| 13 | 20Co4N/γ-Al2O3 | 300 | 1.5 | 5000 | - | - | 98 | 98 | 4.0 | [93] |
| 14 | Co/CeO2 | 300 | 0.1 | - | - | - | 97.0 | ≈100 | 9.0 | [28] |
| 15 | 10CoCZ700 | 225 | 0.1 | - | - | - | 50 | ≈46 | 4.0 | [87] |
| Entry | Catalyst | T (°C) | P (MPa) | CO2 Conv. (%) | CO Sel. (%) | Product Selectivity (%) | H2/ CO2 | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CH4 | C=2–4 | C2–4 | C5+ | ||||||||
| 1 | C/Co3@C/Fe | 320 | 2.0 | 33.4 | 23.8 | 14.4 | 36.8 | 9.1 | 39.7 | 3.0 | [55] |
| 2 | Na-CoCu/TiO2 | 250 | 5.0 | 18.4 | 30.2 | 26.1 | 31.8 | 42.1 | 3.0 | [101] | |
| 3 | K-CoCu/TiO2 | 250 | 5.0 | 13.0 | 35.1 | 34.1 | 30.8 | 35.1 | 3.0 | [110] | |
| 4 | Na-Co-Mo/SiO2-TiO2 | 200 | 0.1 | 13.5 | 66.7 | 23.1 | 37.3 | 15.4 | 24.2 | 3.0 | [106] |
| 5 | Na-CoFe2O4/CNT | 340 | 1.0 | 34.4 | 18.6 | 14.8 | 5.5 | 38.8 | 40.9 | 3.0 | [104] |
| 6 | ZnCo0.5Fe1.5O4 | 320 | 2.5 | 49.6 | 5.8 | 18.9 | 6.2 | 36.1 | 38.7 | 3.0 | [105] |
| 7 | FeK/Co-NC | 300 | 2.5 | 51.7 | 2.8 | 22.2 | 34.2 | 43.6 | 3.0 | [31] | |
| 8 | Co6/MnOx a | 200 | 8.0 | 15.3 | 0.4 | 46.6 | 53.4 | 1.0 | [30] | ||
| 9 | 10Co/TiO2* + 1Pd/TiO2* | 220 | 2.0 | 19.4 | 0.8 | 66.4 | 12.7 | 20.1 | 2.0 | [107] | |
| 10 | CeO2-Pt@mSiO2-Co | 250 | 0.62 | 24 | 74 | 60 | 40 | 0 | 3.0 | [49] | |
| 11 | Mn-Co@CoOx/Co2C | 270 | 4.0 | 64.7 | 0.2 | 44.2 | 2.0 | 17.8 | 36.0 | 3.0 | [109] |
| Entry | Catalyst | T (°C) | P (MPa) | CO2 Conv. (%) | Product Selectivity (%) | H2/CO2 | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|
| CO | CH4 | CH3OH | C2+OH | |||||||
| 1 | Pt3Co a | 150 | 3.2 | - | Produced methanol = 17.3mmol(5h) | 3.0 | [114] | |||
| 2 | MnOx/m-Co3O4 | 250 | 0.4 | - | SMethanol = 30%, SEthylene = 10%, SDimeyhyi = 5% | 3.2 | [116] | |||
| 3 | Co@Si0.95 | 320 | 2.0 | 8.6 | 8 | 21.5 | 70.5 | - | 3.0 | [69] |
| 4 | In2O3@Co3O4 | 250 | 5.0 | - | 11 | 2.0 | 87 | - | 4.0 | [117] |
| 5 | Co@Co3O4/C-N a | 220 | 2.0 | 18.6 | 0 | 79.5 | 18.3 | 1.2 | 3.0 | [118] |
| 6 | CoAlOx-600 a | 140 | 4.0 | - | - | - | - | >92.1 | 3.0 | [45] |
| 7 | Pt/Co3O4-r | 200 | 2.0 | 27.8 | 0 | 60.0 | 14.8 | 3.0 | [120] | |
| 8 | Pt/Co3O4-p | 200 | 2.0 | 22.4 | 0 | 77.8 | 19.2 | 3.0 | [120] | |
| 9 | Pt/Co3O4-m | 200 | 2.0 | 10.7 | 28.3 | 14.9 | 47.2 | 3.0 | [120] | |
| 10 | LaCo0.7Ga0.3O3 | 240 | 3.0 | 9.8 | 0 | 23.1 | 74.7 | 3.0 | [119] | |
| 11 | Na-CuCo-9 | 330 | 4.0 | 22.1 | 20.5 | 18.0 | 4.1 | SC3+OH = 27.4% | 1.0 | [121] |
| 12 | 600-CDM (CoGa1.0Al1.0O4/SiO2) | 270 | 3.0 | 3.7 | - | - | 29.5 | 16.9 C-mol% | - | [34] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, X.; Wang, X.; Xu, H. Advancements in Cobalt-Based Catalysts for Enhanced CO2 Hydrogenation: Mechanisms, Applications, and Future Directions: A Short Review. Catalysts 2024, 14, 560. https://doi.org/10.3390/catal14090560
He X, Wang X, Xu H. Advancements in Cobalt-Based Catalysts for Enhanced CO2 Hydrogenation: Mechanisms, Applications, and Future Directions: A Short Review. Catalysts. 2024; 14(9):560. https://doi.org/10.3390/catal14090560
Chicago/Turabian StyleHe, Xixue, Xinyu Wang, and Hao Xu. 2024. "Advancements in Cobalt-Based Catalysts for Enhanced CO2 Hydrogenation: Mechanisms, Applications, and Future Directions: A Short Review" Catalysts 14, no. 9: 560. https://doi.org/10.3390/catal14090560