
Comparative Assessment of First-Row 3d Transition Metals (Ti-Zn) Supported on CeO2 Nanorods for CO2 Hydrogenation

 ,
, 


Abstract

1. Introduction
2. Results
2.1. Textural and Structural Characterization (BET (Brunauer–Emmett–Teller), XRD)
2.2. Morphological Characterization (SEM/TEM)-Elemental Analysis (SEM/EDS)
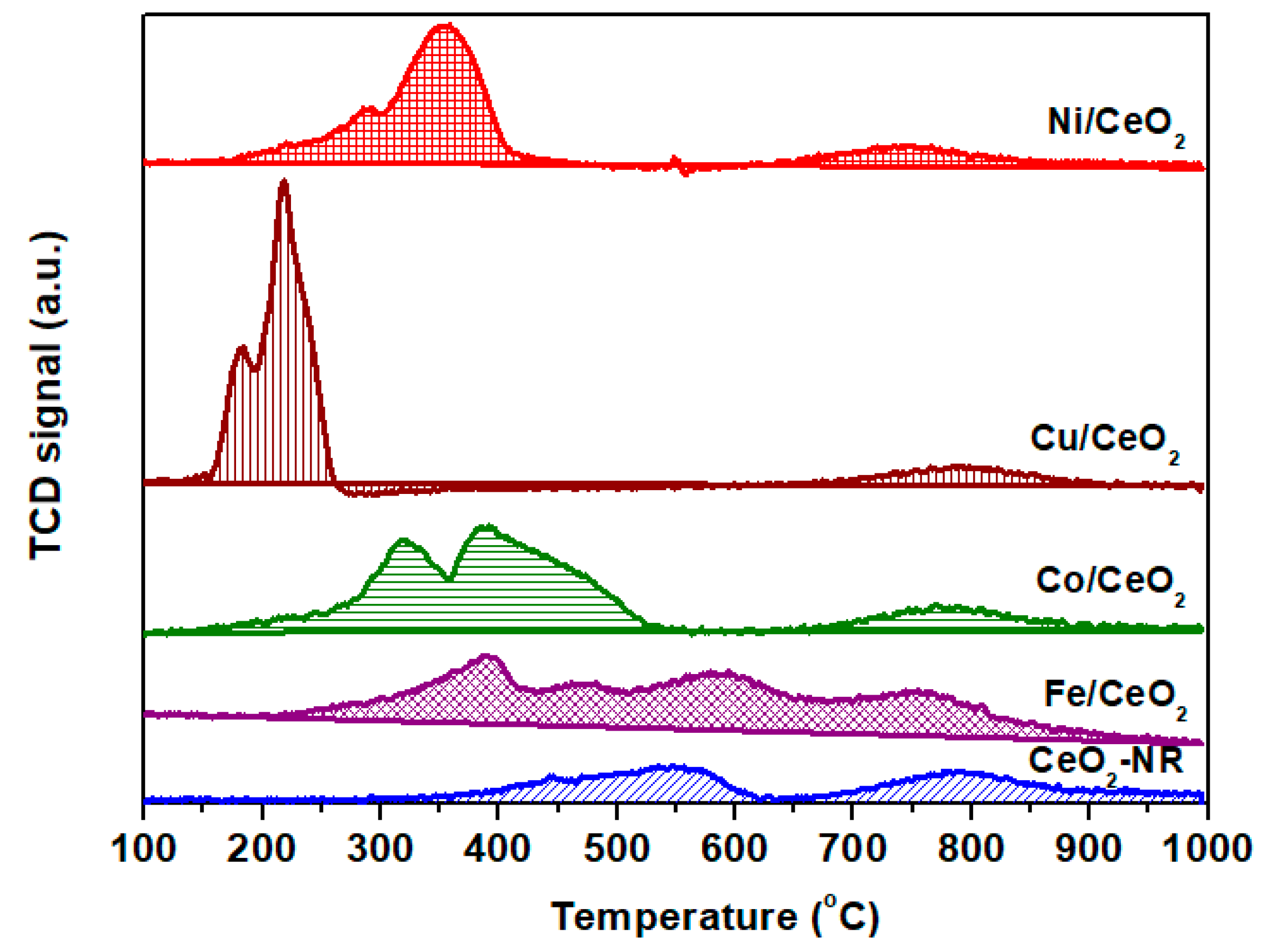
2.3. Reducibility Studies (H2-TPR)
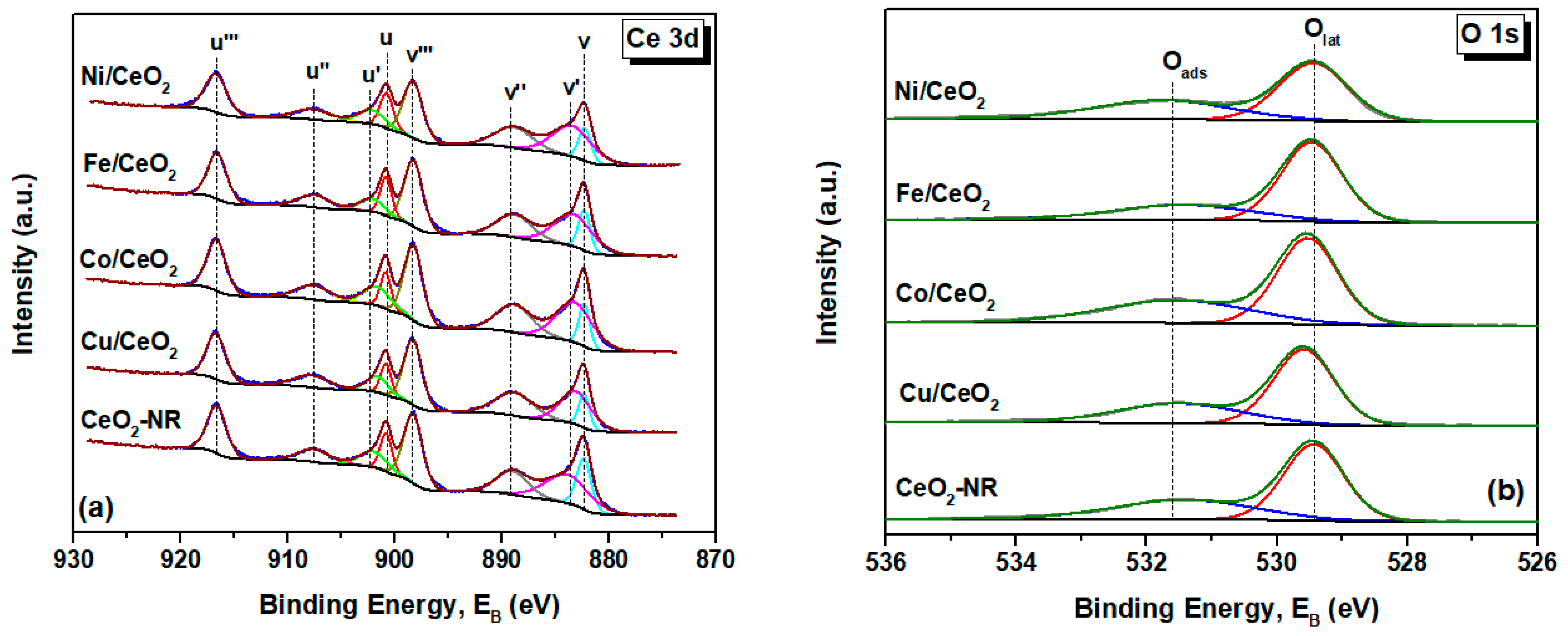
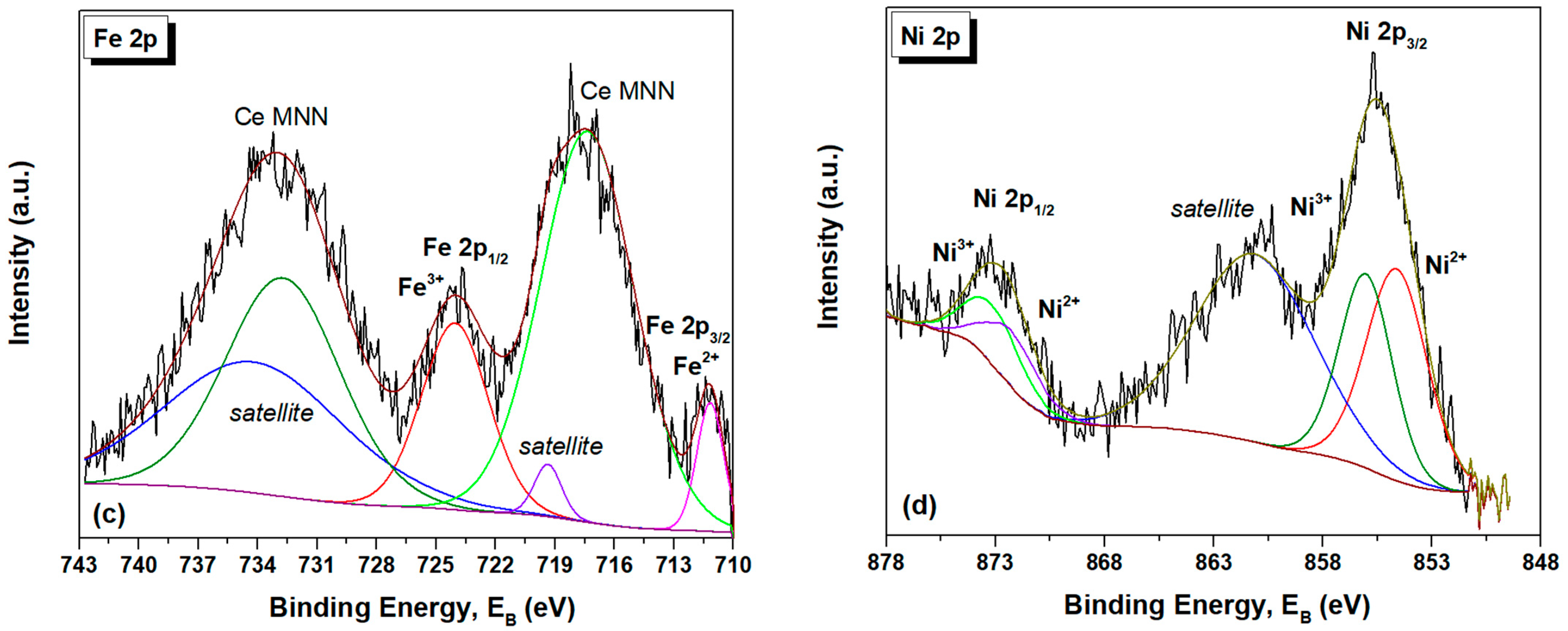
2.4. Surface Analysis (XPS)
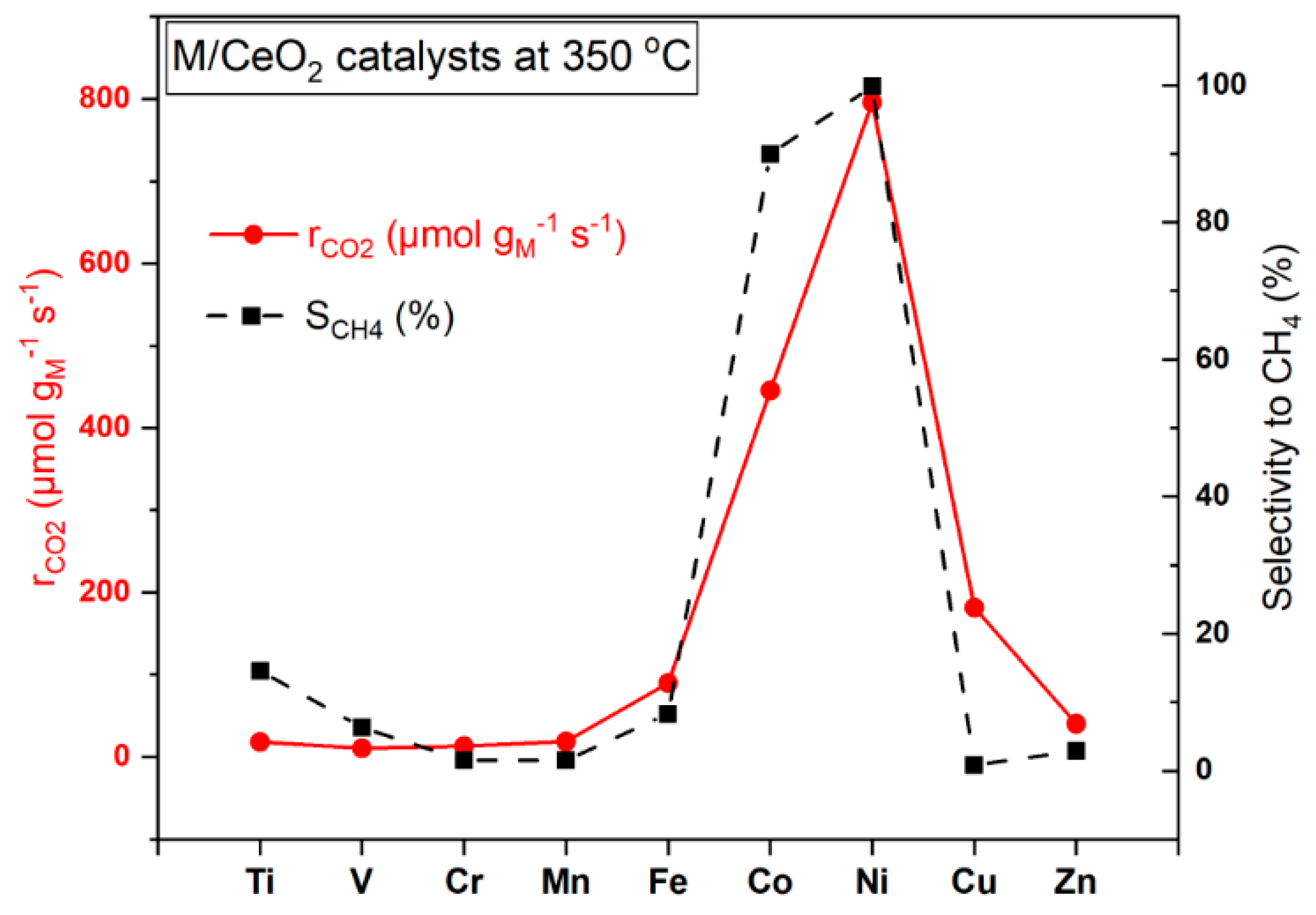
2.5. Catalytic Performance
- (i)
- The CO2 conversion of TM/CeO2 is strongly dependent on the metal entity, following the general trend: Ni > Co > Cu > Fe > Zn > Cr ≈ Ti ≈ V ≈ Mn.
- (ii)
- The Ni/CeO2 catalyst exhibits by far the best performance overall, in terms of maximum conversion and light-off temperature, offering ~90% conversion at 300 °C, which is close to equilibrium conversion (94%) under the employed experimental conditions. Equally importantly, Cu/CeO2 was the most active rWGS material, attaining a CO2-to-CO conversion of 47% at 450 °C, approaching equilibrium conversion (48.5%).
- (iii)
- The early 3d transition metals (Ti, V, Cr, Mn) are almost inactive (i.e., XCO2 < 10% at T < 400 °C), exhibiting an even lower conversion performance as compared to bare ceria nanorods.
- (iv)
- All TM/CeO2 systems, except for Ni and Co, are mainly selective to CO. In particular, the Ni-based catalyst is completely selective to CH4 in the whole temperature range investigated, whereas the Co-based sample exhibits intermediate selectivity values depending on temperature. All other TM/CeO2 catalysts are completely selective to CO.
- (v)
- In terms of CO2 conversion and CO/CH4 selectivity, no catalyst deactivation occurred during short-term (24 h) stability experiments over the optimum catalysts (Fe, Co, Ni, Cu) supported on ceria nanorods (not shown for brevity).
3. Discussion
4. Materials and Methods
4.1. Materials Synthesis
4.2. Materials Characterization
4.3. Catalytic Evaluation Studies
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Atsbha, T.A.; Yoon, T.; Seongho, P.; Lee, C.-J. A Review on the Catalytic Conversion of CO2 using H2 for Synthesis of CO, Methanol, and Hydrocarbons. J. CO2 Util. 2021, 44, 101413. [Google Scholar] [CrossRef]
- Hansen, J.; Sato, M.; Ruedy, R.; Lo, K.; Lea, D.W.; Medina-Elizade, M. Global temperature change. Proc. Natl. Acad. Sci. USA 2006, 103, 14288–14293. [Google Scholar] [CrossRef] [PubMed]
- Meylan, F.D.; Moreau, V.; Erkman, S. CO2 Utilization in the Perspective of Industrial Ecology, an Overview. J. CO2 Util. 2015, 12, 101–108. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Y.; Laaksonen, A.; Krook-Riekkola, A.; Yang, Z.; Lu, X.; Ji, X. Carbon Recycling—An Immense Resource and Key to a Smart Climate Engineering: A Survey of Technologies, Cost and Impurity Impact. Renew. Sustain. Energy Rev. 2020, 131, 110010. [Google Scholar] [CrossRef]
- Saeidi, S.; Amin, N.A.S.; Rahimpour, M.R. Hydrogenation of CO2 to Value-Added Products—A Review and Potential Future Developments. J. CO2 Util. 2014, 5, 66–81. [Google Scholar] [CrossRef]
- Bahmanpour, A.M.; Signorile, M.; Kröcher, O. Recent Progress in Syngas Production via Catalytic CO2 Hydrogenation Reaction. Appl. Catal. B Environ. 2021, 295, 120319. [Google Scholar] [CrossRef]
- Garba, M.D.; Usman, M.; Khan, S.; Shehzad, F.; Galadima, A.; Ehsan, M.F.; Ghanem, A.S.; Humayun, M. CO2 towards Fuels: A Review of Catalytic Conversion of Carbon Dioxide to Hydrocarbons. J. Environ. Chem. Eng. 2021, 9, 104756. [Google Scholar] [CrossRef]
- Torres-Sempere, G.; Pastor-Perez, L.; Odriozola, J.A.; Yu, J.; Duran-Olivencia, F.J.; Bobadilla, L.F.; Reina, T.R. Recent Advances on Gas-Phase CO2 Conversion: Catalysis Design and Chemical Processes to Close the Carbon Cycle. Curr. Opin. Green Sustain. Chem. 2022, 36, 100647. [Google Scholar] [CrossRef]
- Rafiee, A.; Rajab Khalilpour, K.; Milani, D.; Panahi, M. Trends in CO2 Conversion and Utilization: A Review from Process Systems Perspective. J. Environ. Chem. Eng. 2018, 6, 5771–5794. [Google Scholar] [CrossRef]
- Romeo, L.M.; Bailera, M. Design Configurations to Achieve an Effective CO2 use and Mitigation through Power to Gas. J. CO2 Util. 2020, 39, 101174. [Google Scholar] [CrossRef]
- Quintino, F.M.; Nascimento, N.; Fernandes, E.C. Aspects of Hydrogen and Biomethane Introduction in Natural Gas Infrastructure and Equipment. Hydrogen 2021, 2, 301–318. [Google Scholar] [CrossRef]
- Konsolakis, M.; Lykaki, M.; Stefa, S.; Carabineiro, S.A.C.; Varvoutis, G.; Papista, E.; Marnellos, G.E. CO2 Hydrogenation over Nanoceria-Supported Transition Metal Catalysts: Role of Ceria Morphology (Nanorods versus Nanocubes) and Active Phase Nature (Co versus Cu). Nanomaterials 2019, 9, 1739. [Google Scholar] [CrossRef] [PubMed]
- Tawalbeh, M.; Javed, R.M.N.; Al-Othman, A.; Almomani, F. The Novel Contribution of Non-Noble Metal Catalysts for Intensified Carbon Dioxide Hydrogenation: Recent Challenges and Opportunities. Energy Convers. Manag. 2023, 279, 116755. [Google Scholar] [CrossRef]
- Tawalbeh, M.; Javed, R.M.N.; Al-Othman, A.; Almomani, F.; Ajith, S. Unlocking the Potential of CO2 Hydrogenation into Valuable Products Using Noble Metal Catalysts: A Comprehensive Review. Environ. Technol. Innov. 2023, 31, 103217. [Google Scholar] [CrossRef]
- Ebrahimi, P.; Kumar, A.; Khraisheh, M. A Review of CeO2 Supported Catalysts for CO2 Reduction to CO through the Reverse Water Gas Shift Reaction. Catalysts 2022, 12, 1101. [Google Scholar] [CrossRef]
- Chen, X.; Chen, Y.; Song, C.; Ji, P.; Wang, N.; Wang, W.; Cui, L. Recent Advances in Supported Metal Catalysts and Oxide Catalysts for the Reverse Water-Gas Shift Reaction. Front. Chem. 2020, 8, 709. [Google Scholar] [CrossRef]
- Lykaki, M.; Mandela, E.; Varvoutis, G.; Lampropoulos, A.; Marnellos, G.E.; Konsolakis, M. State-of-the-Art Thermocatalytic Systems for CH4 and CO Production via CO2 Hydrogenation: Critical Comparison, Mechanistic Considerations and Structure-Performance Insights. Discov. Chem. Eng. 2024, 4, 11. [Google Scholar] [CrossRef]
- Choi, Y.; Sim, G.D.; Jung, U.; Park, Y.; Youn, M.H.; Chun, D.H.; Rhim, G.B.; Kim, K.Y.; Koo, K.Y. Copper Catalysts for CO2 Hydrogenation to CO through Reverse Water–Gas Shift Reaction for e-Fuel Production: Fundamentals, Recent Advances, and Prospects. Chem. Eng. J. 2024, 492, 152283. [Google Scholar] [CrossRef]
- Gao, X.; Wang, Z.; Huang, Q.; Jiang, M.; Askari, S.; Dewangan, N.; Kawi, S. State-of-Art Modifications of Heterogeneous Catalysts for CO2 Methanation—Active Sites, Surface Basicity and Oxygen Defects. Catal. Today 2022, 402, 88–103. [Google Scholar] [CrossRef]
- Lee, W.J.; Li, C.; Prajitno, H.; Yoo, J.; Patel, J.; Yang, Y.; Lim, S. Recent Trend in Thermal Catalytic Low Temperature CO2 Methanation: A Critical Review. Catal. Today 2021, 368, 2–19. [Google Scholar] [CrossRef]
- Ashok, J.; Pati, S.; Hongmanorom, P.; Tianxi, Z.; Junmei, C.; Kawi, S. A Review of Recent Catalyst Advances in CO2 Methanation Processes. Catal. Today 2020, 356, 471–489. [Google Scholar] [CrossRef]
- Memon, M.A.; Jiang, Y.; Hassan, M.A.; Ajmal, M.; Wang, H.; Liu, Y. Heterogeneous Catalysts for Carbon Dioxide Methanation: A View on Catalytic Performance. Catalysts 2023, 13, 1514. [Google Scholar] [CrossRef]
- Bacariza, M.C.; Spataru, D.; Karam, L.; Lopes, J.M.; Henriques, C. Promising Catalytic Systems for CO2 Hydrogenation into CH4: A Review of Recent Studies. Processes 2020, 8, 1646. [Google Scholar] [CrossRef]
- Hussain, I.; Tanimu, G.; Ahmed, S.; Aniz, C.U.; Alasiri, H.; Alhooshani, K. A Review of the Indispensable Role of Oxygen Vacancies for Enhanced CO2 Methanation Activity over CeO2-Based Catalysts: Uncovering, Influencing, and Tuning Strategies. Int. J. Hydrog. Energy 2023, 48, 24663–24696. [Google Scholar] [CrossRef]
- Konsolakis, M.; Lykaki, M. Facet-Dependent Reactivity of Ceria Nanoparticles Exemplified by CeO2-Based Transition Metal Catalysts: A Critical Review. Catalysts 2021, 11, 452. [Google Scholar] [CrossRef]
- Yang, C.; Lu, Y.; Zhang, L.; Kong, Z.; Yang, T.; Tao, L.; Zou, Y.; Wang, S. Defect Engineering on CeO2-Based Catalysts for Heterogeneous Catalytic Applications. Small Struct. 2021, 2, 2100058. [Google Scholar] [CrossRef]
- Manan, W.N.; Wan Isahak, W.N.R.; Yaakob, Z. CeO2-Based Heterogeneous Catalysts in Dry Reforming Methane and Steam Reforming Methane: A Short Review. Catalysts 2022, 12, 452. [Google Scholar] [CrossRef]
- Dong, C.; Zong, X.; Jiang, W.; Niu, L.; Liu, Z.; Qu, D.; Wang, X.; Sun, Z. Recent Advances of Ceria-Based Materials in the Oxidation of Carbon Monoxide. Small Struct. 2021, 2, 2000081. [Google Scholar] [CrossRef]
- Fan, L.; Zhang, J.; Ma, K.; Zhang, Y.; Hu, Y.-M.; Kong, L.; Jia, A.; Zhang, Z.; Huang, W.; Lu, J.-Q. Ceria Morphology-Dependent Pd-CeO2 Interaction and Catalysis in CO2 Hydrogenation into Formate. J. Catal. 2021, 397, 116–127. [Google Scholar] [CrossRef]
- Jiang, F.; Wang, S.; Liu, B.; Liu, J.; Wang, L.; Xiao, Y.; Xu, Y.; Liu, X. Insights into the Influence of CeO2 Crystal Facet on CO2 Hydrogenation to Methanol over Pd/CeO2 Catalysts. ACS Catal. 2020, 10, 11493–11509. [Google Scholar] [CrossRef]
- Martin, N.M.; Velin, P.; Skoglundh, M.; Bauer, M.; Carlsson, P.-A. Catalytic Hydrogenation of CO2 to Methane over Supported Pd, Rh and Ni Catalysts. Catal. Sci. Technol. 2017, 7, 1086–1094. [Google Scholar] [CrossRef]
- Khobragade, R.; Roškarič, M.; Žerjav, G.; Košiček, M.; Zavašnik, J.; Van de Velde, N.; Jerman, I.; Tušar, N.N.; Pintar, A. Exploring the Effect of Morphology and Surface Properties of Nanoshaped Pd/CeO2 Catalysts on CO2 Hydrogenation to Methanol. Appl. Catal. A Gen. 2021, 627, 118394. [Google Scholar] [CrossRef]
- Wang, C.; Lu, Y.; Zhang, Y.; Fu, H.; Sun, S.; Li, F.; Duan, Z.; Liu, Z.; Wu, C.; Wang, Y.; et al. Ru-Based Catalysts for Efficient CO2 Methanation: Synergistic Catalysis between Oxygen Vacancies and Basic Sites. Nano Res. 2023, 16, 12153–12164. [Google Scholar] [CrossRef]
- Chen, M.; Liu, L.; Chen, X.; Qin, X.; Li, K.; Zhang, J.; Bao, X.; Ma, L.; Zhang, C. Effects of Ru Particle Size over TiO2 on the Catalytic Performance of CO2 Hydrogenation. Appl. Surf. Sci. 2024, 654, 159640. [Google Scholar] [CrossRef]
- Akter, N.; Zhang, S.; Lee, J.; Kim, D.H.; Boscoboinik, J.A.; Kim, T. Selective Catalytic Reduction of NO by Ammonia and NO Oxidation Over CoOx/CeO2 Catalysts. Mol. Catal. 2020, 482, 110664. [Google Scholar] [CrossRef]
- Bian, Z.; Chan, Y.M.; Yu, Y.; Kawi, S. Morphology Dependence of Catalytic Properties of Ni/CeO2 for CO2 Methanation: A Kinetic and Mechanism Study. Catal. Today 2020, 347, 31–38. [Google Scholar] [CrossRef]
- Konsolakis, M.; Lykaki, M. Recent Advances on the Rational Design of Non-Precious Metal Oxide Catalysts Exemplified by CuOx/CeO2 Binary System: Implications of Size, Shape and Electronic Effects on Intrinsic Reactivity and Metal-Support Interactions. Catalysts 2020, 10, 160. [Google Scholar] [CrossRef]
- Xiao, M.; Zhang, X.; Yang, Y.; Cui, X.; Chen, T.; Wang, Y. M (M = Mn, Co, Cu)-CeO2 Catalysts to Enhance Their CO Catalytic Oxidation at a Low Temperature: Synergistic Effects of the Interaction between Ce3+-Mx+-Ce4+ and the Oxygen Vacancy Defects. Fuel 2022, 323, 124379. [Google Scholar] [CrossRef]
- Cargnello, M.; Fornasiero, P.; Gorte, R.J. Opportunities for Tailoring Catalytic Properties through Metal-Support Interactions. Catal. Lett. 2012, 142, 1043–1048. [Google Scholar] [CrossRef]
- Liu, C.; Cundari, T.R.; Wilson, A.K. CO2 Reduction on Transition Metal (Fe, Co, Ni, and Cu) Surfaces: In Comparison with Homogeneous Catalysis. J. Phys. Chem. C 2012, 116, 5681–5688. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, X.; Zheng, Z. A Review of Transition Metal Oxygen-Evolving Catalysts Decorated by Cerium-Based Materials: Current Status and Future Prospects. CCS Chem. 2022, 4, 31–53. [Google Scholar] [CrossRef]
- Varvoutis, G.; Lykaki, M.; Marnellos, G.E.; Konsolakis, M. Recent Advances on Fine-Tuning Engineering Strategies of CeO2-Based Nanostructured Catalysts Exemplified by CO2 Hydrogenation Processes. Catalysts 2023, 13, 275. [Google Scholar] [CrossRef]
- Varvoutis, G.; Lykaki, M.; Stefa, S.; Binas, V.; Marnellos, G.E.; Konsolakis, M. Deciphering the Role of Ni Particle Size and Nickel-Ceria Interfacial Perimeter in the Low-Temperature CO2 Methanation Reaction over Remarkably Active Ni/CeO2 Nanorods. Appl. Catal. B Environ. 2021, 297, 120401. [Google Scholar] [CrossRef]
- Varvoutis, G.; Lykaki, M.; Stefa, S.; Papista, E.; Carabineiro, S.A.C.; Marnellos, G.E.; Konsolakis, M. Remarkable Efficiency of Ni Supported on Hydrothermally Synthesized CeO2 Nanorods for Low-Temperature CO2 Hydrogenation to Methane. Catal. Commun. 2020, 142, 106036. [Google Scholar] [CrossRef]
- Podrojková, N.; Sans, V.; Oriňak, A.; Oriňaková, R. Recent Developments in the Modelling of Heterogeneous Catalysts for CO2 Conversion to Chemicals. ChemCatChem 2020, 12, 1802–1825. [Google Scholar] [CrossRef]
- Jangam, A.; Das, S.; Dewangan, N.; Hongmanorom, P.; Hui, W.M.; Kawi, S. Conversion of CO2 to C1 Chemicals: Catalyst Design, Kinetics and Mechanism Aspects of the Reactions. Catal. Today 2020, 358, 3–29. [Google Scholar] [CrossRef]
- Saeidi, S.; Najari, S.; Fazlollahi, F.; Nikoo, M.K.; Sefidkon, F.; Klemeš, J.J.; Baxter, L.L. Mechanisms and Kinetics of CO2 Hydrogenation to Value-Added Products: A Detailed Review on Current Status and Future Trends. Renew. Sustain. Energy Rev. 2017, 80, 1292–1311. [Google Scholar] [CrossRef]
- Su, X.; Yang, X.; Zhao, B.; Huang, Y. Designing of Highly Selective and High-Temperature Endurable RWGS Heterogeneous Catalysts: Recent Advances and the Future Directions. J. Energy Chem. 2017, 26, 854–867. [Google Scholar] [CrossRef]
- Zhang, W.; Sun, J.; Wang, H.; Cui, X. Recent Advances in Hydrogenation of CO2 to CO with Heterogeneous Catalysts Through the RWGS Reaction. Chem. Asian J. 2024, 19, e202300971. [Google Scholar] [CrossRef]
- Triviño, M.L.T.; Arriola, N.C., Jr.; Seok Kang, Y.; Gil Seo, J. Transforming CO2 to Valuable Feedstocks: Emerging Catalytic and Technological Advances for the Reverse Water Gas Shift Reaction. Chem. Eng. J. 2024, 487, 150369. [Google Scholar] [CrossRef]
- Xie, Y.; Wen, J.; Li, Z.; Chen, J.; Zhang, Q.; Ning, P.; Chen, Y.; Hao, J. Progress in Reaction Mechanisms and Catalyst Development of Ceria-Based Catalysts for Low-Temperature CO2 Methanation. Green Chem. 2023, 25, 130–152. [Google Scholar] [CrossRef]
- Alam, M.I.; Cheula, R.; Moroni, G.; Nardi, L.; Maestri, M. Mechanistic and Multiscale Aspects of Thermo-Catalytic CO2 conversion to C1 products. Catal. Sci. Technol. 2021, 11, 6601–6629. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Cai, P.; Wang, Z.; Lv, X.; Kawi, S. Surface Acidity/Basicity and Oxygen Defects of Metal Oxide: Impacts on Catalytic Performances of CO2 Reforming and Hydrogenation Reactions. Top. Catal. 2023, 66, 299–325. [Google Scholar] [CrossRef]
- Etim, U.J.; Zhang, C.; Zhong, Z. Impacts of the Catalyst Structures on CO2 Activation on Catalyst Surfaces. Nanomaterials 2021, 11, 3265. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.; Deo, G. A Potential Descriptor for the CO2 Hydrogenation to CH4 over Al2O3 Supported Ni and Ni-Based Alloy Catalysts. Appl. Catal. B Environ. 2017, 218, 525–537. [Google Scholar] [CrossRef]
- Feibelman, P.J.; Hamann, D.R. Electronic Structure of a “Poisoned” Transition-Metal Surface. Phys. Rev. Lett. 1984, 52, 61–64. [Google Scholar] [CrossRef]
- Escaño, M.C.; Nguyen, T.Q.; Nakanishi, H.; Kasai, H. Another way of looking at bonding on bimetallic surfaces: The role of spin polarization of surface metal d states. J. Phys. Condens. Matter 2009, 21, 492201. [Google Scholar] [CrossRef]
- Sun, J.; Zhao, H.; Fang, X.; Zhai, S.; Zhai, D.; Sun, L.; Deng, W. Theoretical Studies on the Catalytic Hydrogenation of Carbon Dioxide by 3d Transition Metals Single-Atom Catalyst Supported on Covalent Triazine Frameworks. Mol. Catal. 2021, 508, 111581. [Google Scholar] [CrossRef]
- Sebastian, R.; Swapna, M.S.; Sankararaman, S. Thermal Lens Study of Absolute Porosity in Ceria: A Sankar–Loeb Model Approach. SN Appl. Sci. 2020, 2, 1145. [Google Scholar] [CrossRef]
- Deori, K.; Gupta, D.; Saha, B.; Awasthi, S.K.; Deka, S. Introducing Nanocrystalline CeO2 as Heterogeneous Environmental Friendly Catalyst for the Aerobic Oxidation of Para-Xylene to Terephthalic Acid in Water. J. Mater. Chem. A 2013, 1, 7091–7099. [Google Scholar] [CrossRef]
- Peyrovi, P.; Gillot, S.; Dacquin, J.-P.; Granger, P.; Dujardin, C. The Activity of CeVO4-Based Catalysts for Ammonia-SCR: Impact of Surface Cerium Enrichment. Catal. Lett. 2021, 151, 1003–1012. [Google Scholar] [CrossRef]
- Mosleh, M.; Mahinpour, A. Sonochemical Synthesis and Characterization of Cerium Vanadate Nanoparticles and Investigation of Its Photocatalyst Application. J. Mater. Sci. Mater. Electron. 2016, 27, 8930–8934. [Google Scholar] [CrossRef]
- Rao, G.R. Influence of Metal Particles on the Reduction Properties of Ceria-Based Materials Studied by TPR. Bull. Mater. Sci. 1999, 22, 89–94. [Google Scholar] [CrossRef]
- Kammert, J.; Moon, J.; Wu, Z. A Review of the Interactions between Ceria and H2 and the Applications to Selective Hydrogenation of Alkynes. Chin. J. Catal. 2020, 41, 901–914. [Google Scholar] [CrossRef]
- Sudarsanam, P.; Hillary, B.; Amin, M.H.; Rockstroh, N.; Bentrup, U.; Brückner, A.; Bhargava, S.K. Heterostructured Copper-Ceria and Iron-Ceria Nanorods: Role of Morphology, Redox, and Acid Properties in Catalytic Diesel Soot Combustion. Langmuir 2018, 34, 2663–2673. [Google Scholar] [CrossRef] [PubMed]
- Bayram, B.; Soykal, I.I.; Von Deak, D.; Miller, J.T.; Ozkan, U.S. Ethanol Steam Reforming over Co-Based Catalysts: Investigation of Cobalt Coordination Environment under Reaction Conditions. J. Catal. 2011, 284, 77–89. [Google Scholar] [CrossRef]
- Guo, X.; Zhou, R. A New Insight into the Morphology Effect of Ceria on CuO/CeO2 Catalysts for CO Selective Oxidation in Hydrogen-Rich Gas. Catal. Sci. Technol. 2016, 6, 3862–3871. [Google Scholar] [CrossRef]
- Liu, H.-X.; Li, S.-Q.; Wang, W.-W.; Yu, W.-Z.; Zhang, W.-J.; Ma, C.; Jia, C.-J. Partially Sintered Copper–ceria as Excellent Catalyst for the High-Temperature Reverse Water Gas Shift Reaction. Nat. Commun. 2022, 13, 867. [Google Scholar] [CrossRef]
- Oo, W.; Park, J.H.; Sonia, Z.A.; Win, M.Z.; Cho, D.; Yi, K.B. Modification of Copper-Ceria Catalyst via Reverse Microemulsion Method and Study of the Effects of Surfactant on WGS Catalyst Activity. Catalysts 2023, 13, 951. [Google Scholar] [CrossRef]
- Zhao, P.; Qin, F.; Huang, Z.; Sun, C.; Shen, W.; Xu, H. Morphology-Dependent Oxygen Vacancies and Synergistic Effects of Ni/CeO2 Catalysts for N2O Decomposition. Catal. Sci. Technol. 2018, 8, 276–288. [Google Scholar] [CrossRef]
- Serafin, J.; Llorca, J. Nanoshaped Cerium Oxide with Nickel as a Non-Noble Metal Catalyst for CO2 Thermochemical Reactions. Molecules 2023, 28, 2926. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, P.; Koberstein, J.; Khalid, S.; Chan, S.-W. Cerium Oxidation State in Ceria Nanoparticles Studied with X-Ray Photoelectron Spectroscopy and Absorption near Edge Spectroscopy. Surf. Sci. 2004, 563, 74–82. [Google Scholar] [CrossRef]
- Sohn, H.; Celik, G.; Gunduz, S.; Dogu, D.; Zhang, S.; Shan, J.; Tao, F.F.; Ozkan, U.S. Oxygen Mobility in Pre-Reduced Nano- and Macro-Ceria with Co Loading: An AP-XPS, In-Situ DRIFTS and TPR Study. Catal. Lett. 2017, 147, 2863–2876. [Google Scholar] [CrossRef]
- Xu, J.; Harmer, J.; Li, G.; Chapman, T.; Collier, P.; Longworth, S.; Tsang, S.C. Size Dependent Oxygen Buffering Capacity of Ceria Nanocrystals. Chem. Commun. 2010, 46, 1887–1889. [Google Scholar] [CrossRef]
- Alkhoori, A.A.; Elmutasim, O.; Dabbawala, A.A.; Vasiliades, M.A.; Petallidou, K.C.; Emwas, A.-H.; Anjum, D.H.; Singh, N.; Baker, M.A.; Charisiou, N.D.; et al. Mechanistic Features of the CeO2-Modified Ni/Al2O3 Catalysts for the CO2 Methanation Reaction: Experimental and Ab Initio Studies. ACS Appl. Energy Mater. 2023, 6, 8550–8571. [Google Scholar] [CrossRef]
- Szamosvölgyi, Á.; Rajkumar, T.; Sápi, A.; Szenti, I.; Ábel, M.; Gómez-Pérez, J.F.; Baán, K.; Fogarassy, Z.; Dodony, E.; Pécz, B.; et al. Interfacial Ni Active Sites Strike Solid Solutional Counterpart in CO2 Hydrogenation. Environ. Technol. Innov. 2022, 27, 102747. [Google Scholar] [CrossRef]
- Kuan, W.-F.; Chung, C.-H.; Lin, M.M.; Tu, F.-Y.; Chen, Y.-H.; Yu, W.-Y. Activation of Carbon Dioxide with Surface Oxygen Vacancy of Ceria Catalyst: An Insight from in-Situ X-Ray Absorption near Edge Structure Analysis. Mater. Today Sustain. 2023, 23, 100425. [Google Scholar] [CrossRef]
- Martin, N.M.; Hemmingsson, F.; Schaefer, A.; Ek, M.; Merte, L.R.; Hejral, U.; Gustafson, J.; Skoglundh, M.; Dippel, A.-C.; Gutowski, O.; et al. Structure-Function Relationship for CO2 Methanation over Ceria Supported Rh and Ni Catalysts under Atmospheric Pressure Conditions. Catal. Sci. Technol. 2019, 9, 1644–1653. [Google Scholar] [CrossRef]
- Cao, F.; Xiao, Y.; Zhang, Z.; Li, J.; Xia, Z.; Hu, X.; Ma, Y.; Qu, Y. Influence of Oxygen Vacancies of CeO2 on Reverse Water Gas Shift Reaction. J. Catal. 2022, 414, 25–32. [Google Scholar] [CrossRef]
- Jenkinson, K.; Spadaro, M.C.; Golovanova, V.; Andreu, T.; Morante, J.R.; Arbiol, J.; Bals, S. Direct Operando Visualization of Metal Support Interactions Induced by Hydrogen Spillover During CO2 Hydrogenation. Adv. Mater. 2023, 35, 2306447. [Google Scholar] [CrossRef]
- Ye, H.; Na, W.; Gao, W.; Wang, H. Carbon-Modified CuO/ZnO Catalyst with High Oxygen Vacancy for CO2 Hydrogenation to Methanol. Energy Technol. 2020, 8, 2000194. [Google Scholar] [CrossRef]
- Jiang, D.; Wang, W.; Zhang, L.; Zheng, Y.; Wang, Z. Insights into the Surface-Defect Dependence of Photoreactivity over CeO2 Nanocrystals with Well-Defined Crystal Facets. ACS Catal. 2015, 5, 4851–4858. [Google Scholar] [CrossRef]
- Hu, J.; Wei, F.; Hu, X.; Xu, J.; Deng, W. Synthesis of CuO-Loaded Ceria Hollow Spheres for Catalytic CO Oxidation. ChemistrySelect 2022, 7, e202103476. [Google Scholar] [CrossRef]
- Konsolakis, M.; Sgourakis, M.; Carabineiro, S.A.C. Surface and Redox Properties of Cobalt-Ceria Binary Oxides: On the Effect of Co Content and Pretreatment Conditions. Appl. Surf. Sci. 2015, 341, 48–54. [Google Scholar] [CrossRef]
- Zhan, Y.; Liu, Y.; Peng, X.; Zhao, W.; Zhang, Y.; Wang, X.; Au, C.; Jiang, L. Molecular-Level Understanding of Reaction Path Optimization as a Function of Shape Concerning the Metal-Support Interaction Effect of Co/CeO2 on Water-Gas Shift Catalysis. Catal. Sci. Technol. 2019, 9, 4928–4937. [Google Scholar] [CrossRef]
- Lykaki, M.; Stefa, S.; Carabineiro, S.A.C.; Pandis, P.K.; Stathopoulos, V.N.; Konsolakis, M. Facet-Dependent Reactivity of Fe2O3/CeO2 Nanocomposites: Effect of Ceria Morphology on CO Oxidation. Catalysts 2019, 9, 371. [Google Scholar] [CrossRef]
- Yogendra, K.; Sitaramulu, P.; Nazeer, S.; Kumar, P.M.; Reddy, B.M.; Rao, T.V. Influence of Iron Doping in Mesoporous Ceria on the Physicochemical Properties and Catalytic Activity in Styrene Oxidation. Appl. Surf. Sci. 2024, 676, 160971. [Google Scholar] [CrossRef]
- Wang, H.; Jin, B.; Wang, H.; Ma, N.; Liu, W.; Weng, D.; Wu, X.; Liu, S. Study of Ag Promoted Fe2O3@CeO2 as Superior Soot Oxidation Catalysts: The Role of Fe2O3 Crystal Plane and Tandem Oxygen Delivery. Appl. Catal. B Environ. 2018, 237, 251–262. [Google Scholar] [CrossRef]
- Liu, F.; Wang, Z.; Wang, D.; Chen, D.; Chen, F.; Li, X. Morphology and Crystal-Plane Effects of Fe/W-CeO2 for Selective Catalytic Reduction of NO with NH3. Catalysts 2019, 9, 288. [Google Scholar] [CrossRef]
- Liu, W.; Wang, W.; Tang, K.; Guo, J.; Ren, Y.; Wang, S.; Feng, L.; Yang, Y. The Promoting Influence of Nickel Species in the Controllable Synthesis and Catalytic Properties of Nickel-Ceria Catalysts. Catal. Sci. Technol. 2016, 6, 2427–2434. [Google Scholar] [CrossRef]
- Tang, K.; Liu, W.; Li, J.; Guo, J.; Zhang, J.; Wang, S.; Niu, S.; Yang, Y. The Effect of Exposed Facets of Ceria to the Nickel Species in Nickel-Ceria Catalysts and Their Performance in a NO + CO Reaction. ACS Appl. Mater. Interfaces 2015, 7, 26839–26849. [Google Scholar] [CrossRef] [PubMed]
- Musab Ahmed, S.; Ren, J.; Ullah, I.; Lou, H.; Xu, N.; Abbasi, Z.; Wang, Z. Ni-Based Catalysts for CO2 Methanation: Exploring the Support Role in Structure-Activity Relationships. ChemSusChem 2024, 17, e202400310. [Google Scholar] [CrossRef] [PubMed]
- Boaro, M.; Colussi, S.; Trovarelli, A. Ceria-Based Materials in Hydrogenation and Reforming Reactions for CO2 Valorization. Front. Chem. 2019, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, D.; Zhao, H.; Wang, C.; Xu, Y.; Li, L.; Li, Z.; Wang, H.; Li, K. Boosting CO2 Methanation Activity by Tuning Ni Crystal Plane and Oxygen Vacancy in Ni/CeO2 Catalyst. Chem. Eng. J. 2024, 494, 153004. [Google Scholar] [CrossRef]
- Dietz, L.; Piccinin, S.; Maestri, M. Mechanistic Insights into CO2 Activation via Reverse Water—Gas Shift on Metal Surfaces. J. Phys. Chem. C 2015, 119, 4959–4966. [Google Scholar] [CrossRef]
- Wang, S.-G.; Liao, X.-Y.; Cao, D.-B.; Huo, C.-F.; Li, Y.-W.; Wang, J.; Jiao, H. Factors Controlling the Interaction of CO2 with Transition Metal Surfaces. J. Phys. Chem. C 2007, 111, 16934–16940. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Waghmare, U.V.; Lee, S.-C. An improved d-band model of the catalytic activity of magnetic transition metal surfaces. Sci. Rep. 2016, 6, 35916. [Google Scholar] [CrossRef]
- Lee, S.M.; Lee, Y.H.; Moon, D.H.; Ahn, J.Y.; Nguyen, D.D.; Chang, S.W.; Kim, S.S. Reaction Mechanism and Catalytic Impact of Ni/CeO2-X Catalyst for Low-Temperature CO2 Methanation. Ind. Eng. Chem. Res. 2019, 58, 8656–8662. [Google Scholar] [CrossRef]
- Huang, W.; Gao, Y. Morphology-Dependent Surface Chemistry and Catalysis of CeO2 Nanocrystals. Catal. Sci. Technol. 2014, 4, 3772–3784. [Google Scholar] [CrossRef]
- Li, L.; Jiang, L.; Li, D.; Yuan, J.; Bao, G.; Li, K. Enhanced Low-Temperature Activity of CO2 Methanation over Ni/CeO2 Catalyst: Influence of Preparation Methods. Appl. Catal. O Open 2024, 192, 206956. [Google Scholar] [CrossRef]
- Lykaki, M.; Pachatouridou, E.; Carabineiro, S.A.C.; Iliopoulou, E.; Andriopoulou, C.; Kallithrakas-Kontos, N.; Boghosian, S.; Konsolakis, M. Ceria nanoparticles shape effects on the structural defects and surface chemistry: Implications in CO oxidation by Cu/CeO2 catalysts. Appl. Catal. B Environ. 2018, 230, 18–28. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Nominal Metal Loading | EDS Analysis | BET Analysis | XRD Analysis | TEM Analysis | ||
|---|---|---|---|---|---|---|---|
| Atomic Ratio M/Ce | Metal Content (wt%) | SBET (m2/g) | Average Crystallite Size (nm) | MxOy Particle Size (nm) | |||
| CeO2 | MxOy | ||||||
| CeO2-NR | - | - | - | 79 | 15 | - | - |
| Ti/CeO2 | 6.5 | 0.24 | 6.3 | - | 11 | 20 | 16 |
| V/CeO2 | 6.9 | 0.28 | 7.6 | - | 14 | 45 | 28 |
| Cr/CeO2 | 7.0 | 0.26 | 7.2 | - | 11 | - 1 | 10 |
| Mn/CeO2 | 7.4 | 0.22 | 6.5 | - | 11 | - 1 | 15 |
| Fe/CeO2 | 7.5 | 0.21 | 6.3 | 69 | 10 | 7 | 11 |
| Co/CeO2 | 7.9 | 0.26 | 8.1 | 72 | 14 | 16 | 15 |
| Ni/CeO2 | 7.9 | 0.25 | 7.8 | 72 | 14 | 23 | 10 |
| Cu/CeO2 | 8.5 | 0.25 | 8.6 | 75 | 12 | 43 | 16 |
| Zn/CeO2 | 8.7 | 0.24 | 8.3 | 76 | 12 | 44 | 41 |
| Sample | H2 Consumption (mmol H2 g−1) 1 | Theoretical H2 Consumption (mmol H2 g−1) 2 | Peak Temperature (°C) | |||
|---|---|---|---|---|---|---|
| CeO2-NR | 0.6 | - | - | 545 | - | 788 |
| Fe/CeO2 | 1.6 | 1.9 | 390 | 465 | 588 | 759 |
| Cu/CeO2 | 1.8 | 1.3 | 181 | - | 217 | 793 |
| Ni/CeO2 | 1.8 | 1.3 | 220 | 288 | 353 | 747 |
| Co/CeO2 | 2.4 | 1.7 | 318 | - | 388 | 789 |
| Sample | Oads/Olat | Ce3+ (%) |
|---|---|---|
| CeO2-NR | 0.59 | 27.2 |
| Fe/CeO2 | 0.43 | 26.9 |
| Cu/CeO2 | 0.54 | 25.5 |
| Co/CeO2 | 0.63 | 29.9 |
| Ni/CeO2 | 0.65 | 30.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lykaki, M.; Stefa, S.; Varvoutis, G.; Binas, V.D.; Marnellos, G.E.; Konsolakis, M. Comparative Assessment of First-Row 3d Transition Metals (Ti-Zn) Supported on CeO2 Nanorods for CO2 Hydrogenation. Catalysts 2024, 14, 611. https://doi.org/10.3390/catal14090611
Lykaki M, Stefa S, Varvoutis G, Binas VD, Marnellos GE, Konsolakis M. Comparative Assessment of First-Row 3d Transition Metals (Ti-Zn) Supported on CeO2 Nanorods for CO2 Hydrogenation. Catalysts. 2024; 14(9):611. https://doi.org/10.3390/catal14090611
Chicago/Turabian StyleLykaki, Maria, Sofia Stefa, Georgios Varvoutis, Vassilios D. Binas, George E. Marnellos, and Michalis Konsolakis. 2024. "Comparative Assessment of First-Row 3d Transition Metals (Ti-Zn) Supported on CeO2 Nanorods for CO2 Hydrogenation" Catalysts 14, no. 9: 611. https://doi.org/10.3390/catal14090611
APA StyleLykaki, M., Stefa, S., Varvoutis, G., Binas, V. D., Marnellos, G. E., & Konsolakis, M. (2024). Comparative Assessment of First-Row 3d Transition Metals (Ti-Zn) Supported on CeO2 Nanorods for CO2 Hydrogenation. Catalysts, 14(9), 611. https://doi.org/10.3390/catal14090611