Abstract
The efficient utilization of heavy oil is of great significance to alleviating the global energy crisis. How to efficiently convert heavy oil into high-value-added light fuel oil has become a hot issue in the field of petrochemicals. As the residual part of crude oil processing, heavy oil has a complex composition and contains polycyclic aromatic hydrocarbons, long-chain alkanes, and heteroatom compounds, which makes it difficult to process directly. Zeolite, as an important type of solid acid catalyst, has a unique pore structure, adjustable acidity, and good thermal stability. It can promote the efficient cracking and conversion of heavy oil molecules, reduce coke formation, and improve the yield and quality of light oil products. This paper systematically reviews the development status of heavy oil cracking technology, focusing on the structural characteristics, acidity regulation of zeolite catalysts, and their applications in heavy oil cracking and hydrocracking. The mechanism of the cracking reaction of polycyclic aromatic hydrocarbons and long-chain alkanes is analyzed in detail, and the catalytic characteristics and modification methods of zeolite in the reaction process are explained. In addition, this paper summarizes the main challenges faced by zeolite catalysts in practical applications, including uneven acidity distribution, limited pore diffusion, and easy catalyst deactivation, and proposes targeted development strategies. Finally, this paper looks forward to the future development direction of zeolite catalysts in the field of heavy oil cracking and upgrading reactions, emphasizes the importance of structural optimization and multi-scale characterization, and provides theoretical support and practical reference for the design and industrial application of efficient zeolite catalysts.
1. Introduction
With the development of the global economy and population growth, human demand for energy continues to increase. According to a research report by the International Energy Agency (IEA) [1], the current global energy structure is still dominated by oil, accounting for 30% (Figure 1a). In addition, global oil consumption has been growing in recent years, especially in the Asia-Pacific region. This rapid increase in demand for oil may cause a shortage of oil resources (Figure 1b). At present, the global heavy oil production accounts for a large proportion (Figure 1c). Due to the rapid growth in global demand for oil energy, light crude oil is relatively easy to process, so the proportion of light crude oil in the world’s oil reserves is rapidly declining. If the abundant heavy oil components are utilized, the energy shortage problem will be greatly alleviated. It can be seen that with the increasing reduction in global light crude oil resources and the continuous growth of energy demand, unconventional oil resources such as heavy oil, extra-heavy oil and oil sands are gradually becoming an important part of the future energy supply. Therefore, how to make full use of heavy oil is very important.
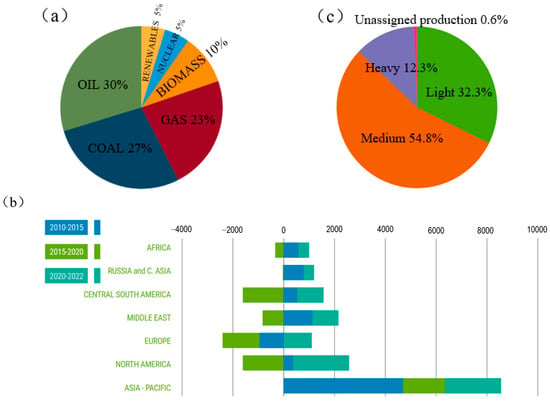
Figure 1.
Global energy production and consumption [1]: (a) global energy structure in 2022; (b) world oil consumption growth from 2010 to 2022 (million barrels per day); (c) global crude oil production in 2022.
Heavy oil is abundant on Earth, accounting for more than 50% of global oil resources [2,3], and has huge development potential. However, it is not easy to utilize this petroleum fraction with high viscosity, high boiling point, and high impurities. Its complex chemical composition and processing characteristics pose great challenges to its efficient utilization. Heavy oil has a complex composition, containing a large amount of polycyclic aromatic hydrocarbons, long-chain alkanes, colloids, and asphaltene, as well as metal impurities such as sulfur, nitrogen, nickel, and vanadium [4,5]. These substances will cause a series of problems during the processing, resulting in low conversion efficiency of heavy oil, further increasing processing costs and technical difficulties.
The viscosity of heavy oil is several times that of conventional crude oil, and its poor fluidity makes it difficult to transport. It must be heated or diluted to achieve effective transportation and processing, which increases energy consumption and economic costs. In addition, heavy oil contains a high proportion of impurities such as sulfur and nitrogen. These impurities not only reduce product quality during processing, but also easily react with catalysts, causing catalyst poisoning, seriously affecting catalytic efficiency, and causing equipment corrosion problems [6]. At the same time, polycyclic aromatic hydrocarbons and asphaltene in heavy oil are easily decomposed to form coke under high-temperature cracking conditions. A large amount of coke is deposited on the reactor surface and in the catalyst pores, which not only hinders the diffusion of reactants, but also causes reactor coking, reduced thermal efficiency, and ultimately leads to rapid catalyst deactivation [7,8]. These problems greatly limit the efficient conversion and industrial application of heavy oil. Therefore, the development of efficient and economical heavy oil upgrading technology, especially through catalytic cracking and hydrocracking and other efficient technologies, to achieve efficient utilization of heavy oil resources, has become a core issue that needs to be urgently solved in the current energy field.
Heavy oil cracking technology is an important way to convert heavy oil with high molecular weight and complex components into light oil products (such as gasoline and diesel) and high value-added chemicals, with significant economic and environmental benefits. Compared with traditional thermal cracking technology, catalytic cracking and hydrocracking technology have become the mainstream methods of heavy oil processing at present [9]. The introduction of solid acid catalysts through catalytic cracking technology can promote the cracking and recombination of large molecular hydrocarbons in heavy oil at relatively low temperatures, effectively reduce energy consumption, improve the economy of the cracking process, and significantly improve the selectivity of the target product. Catalytic cracking technology is particularly suitable for the efficient degradation of large molecular hydrocarbons in heavy oil and has important industrial application value. Secondly, hydrocracking technology relies on the synergistic effect of hydrogen and catalysts to achieve efficient breaking of C-C bonds and convert polycyclic aromatic hydrocarbons into saturated hydrocarbons through the hydrogenation ring-opening reaction, effectively reducing the content of impurities such as sulfur and nitrogen, and producing higher quality and cleaner light oil products [10,11]. In addition, the by-products such as low-carbon olefins and aromatics produced in the cracking process are important basic chemical raw materials with broad market demand, providing a new economic growth point for the petrochemical industry chain [12]. From the perspective of energy security, heavy oil cracking technology can significantly increase the utilization rate of unconventional oil resources and alleviate the supply pressure caused by the depletion of light crude oil resources worldwide. At the same time, the use of hydrocracking technology can effectively reduce the emission of pollutants such as sulfur oxides and nitrogen oxides, which meets the strict requirements of the world for clean energy and environmental protection [13]. Therefore, heavy oil cracking technology is not only an important way to solve the problem of energy supply, but also a key technology to promote green and sustainable development.
Zeolite catalysts are the core of heavy oil catalytic cracking technology. With their unique microporous structure, adjustable acidity, and good thermal stability, they play an irreplaceable role in the field of heavy oil processing [14]. Zeolites have regular micropores and supercage structures. These pore structures provide ideal reaction space for large molecular hydrocarbons in heavy oil, promoting the diffusion and efficient cracking of large molecular hydrocarbons. For example, the supercage structure of Y-type zeolite can accommodate heavy oil molecules with larger diameters, significantly improving the cracking efficiency of large molecular hydrocarbons. In addition, ZSM-5 zeolite has good shape-selective catalytic performance due to its unique ten-membered ring pore structure, which can promote the efficient generation of light hydrocarbons and aromatics [15]. In addition, the acidity of zeolite can be precisely controlled by adjusting the silicon–aluminum ratio, metal ion exchange, dealumination treatment, and other methods to optimize the acidity distribution and acid strength of the catalyst. Appropriate acidity helps to improve the selectivity of the cracking reaction and reduce the occurrence of side reactions, such as coke formation. At the same time, the acidity control of zeolite can achieve personalized optimization of catalyst performance for different types of heavy oil components [16,17]. Zeolite catalysts have good thermal stability and can maintain structural stability under high-temperature cracking conditions. They are not prone to lattice collapse and are suitable for long-term industrial operation. By introducing rare earth metals (such as La and Ce), the resistance of zeolite catalysts to metal poisoning is further enhanced, effectively resisting the poisoning effect of metal impurities such as nickel and vanadium in heavy oil, thereby extending the service life of the catalyst and reducing production costs [18].
Based on the importance of heavy oil resource utilization and the advantages of zeolite catalysts in heavy oil cracking, this paper reviews the application of zeolite catalysts in heavy oil cracking and upgrading reactions, aiming to summarize the current research status, analyze the existing challenges, and look forward to future research directions. The research framework of this paper is as follows: first, the current status and challenges of heavy oil resource utilization are introduced, and the importance of heavy oil cracking technology and the application advantages of zeolite catalysts are explained; secondly, the development status of heavy oil cracking technology is reviewed, and the characteristics and limitations of thermal cracking, catalytic cracking, and hydrocracking technologies are analyzed; thirdly, the structural characteristics and catalytic properties of Y-type zeolite, ZSM-5 zeolite, Beta zeolite, Ferrierite zeolite, and Mordenite zeolite are discussed in detail, as well as their applications in heavy oil cracking and hydrocracking; fourthly, the reaction mechanism of polycyclic aromatic hydrocarbons and long-chain alkanes cracking is analyzed, and the catalytic characteristics and reaction paths of zeolites are discussed; fifthly, the main challenges faced by zeolite catalysts in practical applications are summarized, and improvement strategies are proposed; finally, the whole paper is summarized, and the future development direction of zeolite catalysts in the field of heavy oil cracking is prospected. Through a systematic review and analysis, this paper aims to provide theoretical support and practical reference for the design and application of zeolite catalysts in heavy oil cracking.
2. Research Progress of Heavy Oil Cracking Reaction
2.1. Composition and Properties of Heavy Oil
The American Petroleum Institute gravity index (API gravity) is an important indicator for measuring crude oil types. Heavy oil is defined as crude oil with an API gravity between 10.0 and 22.3, and crude oil with an API gravity below 10.0 is defined as extra-heavy crude oil [4]. Generally speaking, the higher the API gravity value, the higher the value of the crude oil. The classification of crude oil is based on its flow characteristics, which can reflect the technical characteristics of crude oil for subsequent production, transportation, and refining.
Heavy oil is a high molecular weight component remaining in the oil processing process. Its composition and properties are complex, mainly including long-chain alkanes, polycyclic aromatic hydrocarbons, gums, and asphaltene [16,19]. The physical and chemical properties of these components directly affect the processing and upgrading efficiency of heavy oil. Long-chain alkanes are relatively simple components in heavy oil, but due to their large molecular weight and strong thermal stability, direct cracking is difficult. Polycyclic aromatic hydrocarbons are structures composed of multiple benzene rings fused together, with a highly stable conjugated system, and are not prone to cracking reactions. In addition, gums and asphaltene contain a large amount of metal impurities (such as nickel and vanadium) and heteroatoms (such as sulfur and nitrogen), which can easily lead to catalyst deactivation during the cracking process and affect the reaction efficiency [18,20].
Heavy oil usually contains more asphaltenes and resins than conventional oil [21], which directly affects the recovery, transportation, and refining processes. Table 1 shows the common composition differences between conventional oil, heavy oil, and residue [4,21]. The asphaltene content shown represents typical ranges from various crude oil sources; in some cases, heavy oils—especially from unconventional deposits—may contain more asphaltenes than residue fractions derived from lighter crudes. Asphaltene is one of the components of heavy oil and is also the most complex heavy organic compound. The hydrocarbons of asphaltene contain different components, such as polycyclic aromatic hydrocarbons, heteroaromatic compounds, bifunctional or multifunctional molecules (such as ketones, amides, phenols, carboxylic acids), and metal (such as Ni and V) complex compounds [22]. Asphaltene tends to clump and precipitate, causing equipment fouling [23]. The number of carbon atoms in the molecular chain of heavy crude oil often exceeds 15, and it has long-chain hydrocarbons, while short-chain hydrocarbons are considered light crude oil. In heavy petroleum, polycyclic aromatic hydrocarbons and long-chain alkanes account for a large proportion [24,25]. Polycyclic aromatic hydrocarbons refer to polycyclic aromatic hydrocarbons in which two carbon atoms are shared by two benzene rings.

Table 1.
Composition of conventional petroleum, heavy oil, and residual oil [4,21].
In addition, heavy oils from different sources have significant differences in composition. For example, the asphaltene content of Canadian oil sands heavy oil is as high as 30–50%, with a large amount of metal impurities, while the aromatic hydrocarbon content of Middle Eastern crude oil residues is relatively high, and the content of resins and asphaltene is low [6,16]. Therefore, a deep understanding of the composition and properties of heavy oil is an important basis for selecting appropriate processing technology and catalysts.
2.2. The Main Upgrading Methods of Heavy Oil
Heavy oil upgrading refers to the process of converting heavy oil into light oil products and high value-added chemicals by physical or chemical methods. At present, the main ways to upgrade heavy oil include thermal cracking technology, catalytic cracking technology, and hydrocracking technology. These technologies have their own characteristics and different applicability and limitations in practical applications. The main ways to upgrade heavy oil are summarized in Table 2. Thermal cracking technology generates small molecular hydrocarbons by high-temperature cracking of heavy oil molecules. However, thermal cracking technology easily leads to large amounts of coke production and difficult-to-control product distribution. Catalytic cracking technology performs cracking reactions under the action of catalysts, with mild reaction conditions, less coke production, and higher selectivity for target products. Hydrocracking technology not only achieves heavy oil cracking through the synergistic effect of hydrogen and catalysts, but also can improve the hydrogen-to-carbon ratio of the product and reduce the content of impurities such as sulfur and nitrogen. In comparison, catalytic cracking technology and hydrocracking technology have good industrial application prospects and are the focus of current research.
Table 2.
Main upgrading methods of heavy oil.
2.2.1. Thermal Cracking Technology
Thermal cracking technology is the earliest process used in heavy oil upgrading and has been used by refineries since the first half of the 20th century. The thermal cracking process has attracted much attention because it does not require expensive catalysts. It cracks in a container to produce small molecular products by pressurizing and heating, which often have lower boiling points [26,27].
Thermal cracking technology generates lighter hydrocarbon products by breaking C-C bonds in heavy oil under high-temperature conditions. The reaction temperature is generally between 500 and 700 °C, and the molecular chain is mainly broken by the action of heat [18,21]. This technology is simple and has low equipment investment, but it has many limitations. First, the reaction selectivity is poor during thermal cracking, the product distribution is wide, and the yield of light oil products is low; second, polycyclic aromatic hydrocarbons and asphaltene in heavy oil are prone to generate a large amount of coke at high temperatures, resulting in coking of the reactor and affecting normal production operations [28].
Although the thermal cracking process has high operational flexibility, its high energy consumption and low selectivity limit the promotion of industrial applications. Therefore, catalytic cracking technology combined with catalysts has gradually become the mainstream method for heavy oil upgrading.
2.2.2. Catalytic Cracking Technology
Catalytic cracking technology (FCC) is one of the most widely used heavy oil upgrading technologies. By introducing solid acid catalysts, efficient cracking and conversion of heavy oil can be achieved at relatively low temperatures. Commonly used catalysts include Y-type zeolite and ZSM-5 zeolite. They promote the breakage and recombination of C-C bonds of macromolecular hydrocarbons in heavy oil to produce light oil products by providing evenly distributed acid sites and suitable pore structures [29,30]. Y-type zeolite is the core component of FCC catalysts. It has a large supercage structure and high acidity, which is suitable for the cracking reaction of macromolecular hydrocarbons in heavy oil. ZSM-5 zeolite has a unique ten-membered ring pore structure that can improve the selectivity of light olefins and aromatics. It is often used in combination with Y-type zeolite to improve the product distribution in the catalytic cracking process [31,32]. In addition, the acid distribution and stability of zeolite catalysts can be effectively optimized through metal modification, dealumination treatment, and other means, further improving the yield and quality of light oil products.
Compared with thermal cracking technology, catalytic cracking technology can avoid coking caused by excessive temperature, and the quality and stability of the products are higher. The raw materials of catalytic cracking technology can be straight run or vacuum gas oil, or heavy raw materials, such as heavy oil or extra-heavy oil, atmospheric or vacuum residue oil, etc., or a mixture of the above substances. When the raw material is the above mixture, it is necessary to ensure that too much coke will not be produced on the catalyst under the reaction conditions. Before catalytic cracking technology is carried out, deasphalting, demetallization, and hydrotreatment or hydrocracking processes are often required to avoid excessive coking or catalyst deactivation [33,34,35]. It is worth noting that hydrotreatment of catalytic cracking technology raw materials can reduce the emission of harmful sulfur oxides and can effectively improve the quality and yield of naphtha in the product. Therefore, hydrotreatment before catalytic cracking technology can obtain considerable benefits.
Compared with non-catalytic processes such as steam cracking and thermal cracking, catalytic cracking (FCC) technology has significant advantages in product selectivity control. Through the FCC process, crude oil can be efficiently converted into light olefins, which is widely regarded as one of the most cost-effective and reliable methods for producing petrochemical feedstocks such as ethylene, propylene, and butenes. Figure 2a shows the process of directly converting crude oil into light olefins such as ethylene, propylene, 1-butene, and 2-butene through catalytic cracking [30]. In the FCC reactor, crude oil will rapidly undergo catalytic cracking after contacting with the high-temperature zeolite-based catalyst to produce a variety of different products. Figure 2b shows a typical hydrocarbon catalytic cracking and light olefin production pathway. In this process, long-chain hydrocarbons are cracked to produce straight-chain and branched alkanes and olefins. The Y-type zeolite catalyst in Figure 2b is characterized by a higher Si/Al ratio, which indicates that it has more and more stable active sites, which can significantly improve the efficiency of hydrocarbon cracking [36]. USY zeolite is the most commonly used active catalyst component in the FCC process, followed by ZSM-5 zeolite, which is also widely used in FCC catalytic cracking. USY zeolite plays a key role in cracking VGO (vacuum gas oil) and HGO (heavy gas oil) into gasoline-range hydrocarbons, while ZSM-5 zeolite relies on its unique pore size to efficiently convert naphtha fractions into light olefins through shape selectivity. The schematic diagram of Figure 2c shows the composition structure of a typical FCC catalyst and the cracking reaction process triggered by its interaction with crude oil [37].
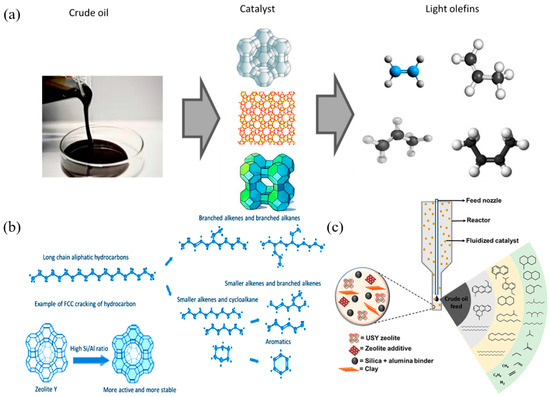
Figure 2.
(a) Illustration of the catalytic cracking of crude oil into petrochemical feedstock [30]; (b) typical catalytic cracking of hydrocarbons once in contact with the hot catalyst in the FCC reactor [36]; (c) schematic diagram of the FCC catalyst’s composition and cracking activities [37]. Reproduced with permission from Abdulkadir Tanimu et al., Energy & Fuels; published by American Chemical Society, 2022 [37].
2.2.3. Hydrocracking Technology
Hydrocracking technology is a process that uses the synergistic effect of hydrogen and catalysts to achieve the hydrogenation ring opening and C-C bond breaking of polycyclic aromatic hydrocarbons in heavy oil, thereby producing light oil products. The reaction temperature of this technology is generally between 300 and 450 °C, and commonly used catalysts include metal/zeolite composite catalysts, such as NiMo/Y zeolite, Pt/ZSM-5 zeolite, etc. [10,38,39,40]. Compared with thermal cracking, hydrocracking requires the combined action of hydrogen and a suitable catalyst and can be carried out at a lower temperature, so the energy consumption required for the reaction is lower [41].
The main advantages of hydrocracking technology are as follows: on the one hand, the introduction of hydrogen can effectively reduce the content of impurities such as sulfur and nitrogen, and improve the quality of oil products; on the other hand, polycyclic aromatic hydrocarbons are converted into saturated hydrocarbons through a hydrogenation ring-opening reaction, reducing the formation of coke [11,42]. However, the hydrocracking process has problems such as high hydrogen consumption and large equipment investment. How to reduce the hydrogen consumption in the reaction process and improve the activity and stability of the catalyst is the focus of current research on hydrocracking technology.
Figure 3 shows the classic hydrocracking mechanism of normal alkanes (n-alkanes) on a bifunctional catalyst. The catalyst has the following two main functions: 1. The metal active site is responsible for the dehydrogenation of the reactant (n-alkanes) to generate olefins and the hydrogenation of the intermediate product (olefins). The reaction reaches a balance between dehydrogenation and hydrogenation, and the generated olefins can desorb and diffuse to the acidic site. 2. The Brønsted acid sites (BAS) serves as an active site to protonate the olefin to generate a secondary carbocation. The secondary carbon cations generate cracking products through β-splitting or structural rearrangement reactions. Among the cracking products, the generation of light alkanes and branched alkanes is related to the type of carbon cation and the cracking path. This mechanism shows in detail the process of normal alkanes undergoing dehydrogenation, cracking, rearrangement, and hydrogenation steps on a bifunctional catalyst to generate light alkanes or branched alkanes. Through this catalytic effect, efficient generation of cracking products can be achieved [43].
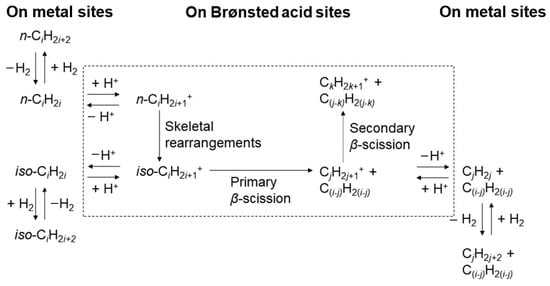
Figure 3.
Classical mechanism of hydrocracking of an n-alkane on a bifunctional catalyst including metal sites for dehydrogenation/hydrogenation and Brønsted acid sites (BAS). i stands for the carbon number of the reactant, and j and k stand the carbon number of the cracked or hydrocracked products [43]. Reproduced with permission from Jens Weitkamp et al., ChemCatChem; published by John Wiley and Sons, 2012 [43].
Figure 4 shows two schematic diagrams of processes involving alkyl aromatics. The conventional hydrocracking process (Figure 4a) describes the hydrogenation of aromatics, followed by the cracking of alkyl groups and aromatic rings. This process is achieved through a hydrogenation–dehydrogenation cycle and catalytic cracking to produce smaller hydrocarbon molecules. The process requires a large amount of hydrogen and is energy intensive, and typically produces a wide range of products, including alkanes and single-ring aromatics. The dealkylation followed by partial hydrogenation process (Figure 4b) focuses on a selective dealkylation process that removes alkyl groups from polycyclic aromatic hydrocarbons (PAHs) through BAS catalysis. Subsequently, partial hydrogenation is performed to stabilize the aromatic compounds after dealkylation. Compared with conventional hydrocracking, this method efficiently produces long-chain alkanes and single-ring aromatics (such as benzene, naphthalene, and their derivatives) with less hydrogen consumption. The two pathways compare the differences in energy consumption, selectivity, and product types between conventional hydrocracking and dealkylation technologies, highlighting the potential of dealkylation processes in the production of high-value-added fuels and petrochemical feedstocks [10].
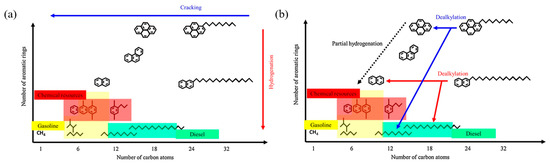
Figure 4.
Schematic drawing of (a) conventional hydrocracking and (b) dealkylation followed by partial hydrogenation of polyaromatics of alkylaromatic hydrocarbons [10]. Reproduced with permission from Naonobu Katada et al., Applied Catalysis A: General; published by Elsevier, 2017 [10].
2.3. Catalysts in the Heavy Oil Cracking Process
Several typical processes for heavy oil upgrading are introduced above. This review focuses on heavy oil cracking, which mainly includes thermal cracking, catalytic cracking, and hydrocracking. Thermal cracking generally does not require the participation of catalysts, so this subsection only discusses catalysts related to catalytic cracking and hydrocracking. When talking about catalysts, catalysts can often be divided into homogeneous catalysts or heterogeneous catalysts based on whether the reactants and catalysts are in the same phase. In addition, they can also be divided into mineral catalysts, water-soluble catalysts, oil-soluble catalysts, dispersed catalysts, or solid acid catalysts based on the solubility of the catalyst. The catalysts used in the heavy oil cracking process are summarized in Table 3.
Table 3.
Catalysts used in heavy oil cracking process.
In the heavy oil cracking process, various catalysts have been widely used in different processes due to their characteristics and applicability. Mineral catalysts use natural minerals such as clay minerals and quartz. They have attracted much attention in early applications due to their large specific surface area and rich acid sites [44]. However, the activity of this type of catalyst is limited by the reservoir temperature, and its performance is slightly insufficient compared with modern synthetic catalysts [45]. As one of the earliest cracking catalysts used [46], water-soluble catalysts rely on the catalytic action of transition metal ions (such as Ru, Fe, Mo, etc.) and have the characteristics of low cost and a significant desulfurization effect [47]. However, since water-soluble catalysts are difficult to fully contact with heavy oil, their industrial application effect is limited, especially in the in situ combustion (ISC) process involving complex reaction steps. Oil-soluble catalysts can effectively solve this problem. Oil-soluble catalysts have attracted widespread attention due to their good compatibility with heavy oil, such as nickel sulfonates [48], molybdenum oleates [49,50], and aromatic metal sulfonic acid complexes [51,52]. These catalysts can significantly reduce the viscosity of heavy oil and improve cracking efficiency, but their preparation process is complicated, and the cost is high. In addition, dispersed catalysts are composed of solid particles (heterogeneous phase) and water-soluble or oil-soluble catalysts (homogeneous phase). Their high contact area can effectively inhibit the diffusion of free radicals during the reaction, but they require complex dispersion and pretreatment processes and are difficult to regenerate [53,54].
Solid acid catalysts are the most widely used in heavy oil cracking, and their types include zeolites, superacids, and heteropolyacids [55]. These catalysts have the advantages of strong acidity, high catalytic activity, and good selectivity. They can effectively catalyze the breaking of C-C bonds at lower reaction temperatures, thereby increasing the yield of light oil products. At the same time, solid acid catalysts can remove heteroatoms such as sulfur and nitrogen in heavy oil, reduce the viscosity of heavy oil, and improve product quality. However, solid acid catalysts are prone to the clogging of pores due to coke accumulation during use, and their sensitivity to metal impurities makes them easily deactivated. Compared with other heavy oil cracking processes, hydrocracking catalysts have unique properties—that is, the catalysts used in hydrocracking must be bifunctional catalysts. The so-called bifunctionality means that the catalyst has the function of cracking heavy oil, and also needs to have the functions of hydrogenation and dehydrogenation. Bifunctional catalysts are composed of metals (such as Pd, Ni-Mo) and acidic carriers (such as ZSM-5, Al2O3). The metal component is responsible for hydrogenation and dehydrogenation, and the acidic carrier provides cracking active sites and shape-selective catalytic ability. It has both cracking and hydrogenation functions, which can significantly improve the quality of light oil products and has strong stability. However, its precious metal components are susceptible to sulfur poisoning, and the raw material purity requirements are high, so pretreatment is required to ensure performance [56,57,58].
As the core representative of solid acid catalysts, zeolite catalysts have become the core technology in the field of heavy oil catalytic cracking due to their high efficiency, selectivity, and stability, especially showing significant advantages in the production of light oil products and heavy oil upgrading. Their microporous structure not only gives them excellent shape-selective catalytic ability, but also can effectively control product distribution and improve the selectivity of light oil products. It can also reduce energy consumption and optimize the catalytic effect by adjusting the pore size and acid sites. Commonly used zeolites such as ZSM-5 and Y-type zeolite can be used for catalytic cracking and can achieve the synergistic effect of hydrogenation and cracking in a bifunctional catalyst system. In addition, zeolite catalysts are an ideal choice for economical catalysts because of their abundant resources and relatively low preparation costs. Zeolite occupies an important position in the future development and improvement of catalysts, and also provides important support for promoting green and efficient heavy oil cracking technology [59,60].
Figure 5 shows the modification of a Y-type zeolite catalyst by continuous steam, alkaline, and acid washing treatments to improve its catalytic performance in heavy oil cracking. The modification process aims to optimize the physicochemical properties of the catalyst by gradually adjusting the silicon–aluminum ratio, acid site distribution, and pore structure. The bar graph illustrates the selectivity effects of the catalysts treated with different alkaline reagents (such as NaOH, Na2CO3, and CaCO3) on various products (such as gasoline, kerosene, light gas oil, etc.), among which the CaCO3-modified catalyst shows better gasoline and kerosene yields. The modified Y-type zeolite catalyst shows higher light oil output efficiency and lower coke generation in heavy oil cracking, providing efficient technical support for heavy oil upgrading and petrochemical product optimization [61].
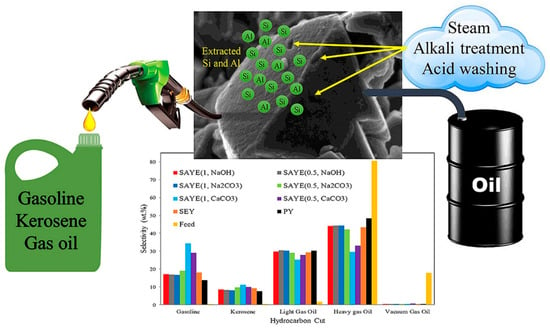
Figure 5.
Improving the physicochemical properties of Y zeolite for catalytic cracking of heavy oil via sequential steam–alkali–acid treatments [61]. Reproduced with permission from Erfan Aghaei et al., Microporous and Mesoporous Materials; published by Elsevier, 2020 [61].
However, catalysts still face many challenges in the heavy oil cracking process: first, polycyclic aromatic hydrocarbons and asphaltene in heavy oil easily form coke on the catalyst surface, resulting in pore blockages and catalyst deactivation; second, metal impurities such as nickel and vanadium contained in heavy oil will react irreversibly with the catalyst, causing catalyst poisoning [62,63]. In response to these problems, researchers have improved the anti-coking and anti-metal poisoning properties of catalysts by introducing mesoporous structures, rare earth metal modification, and dealuminization treatment, thereby extending the service life of catalysts and improving catalytic efficiency. Figure 6 shows the typical mechanisms of heterogeneous catalyst deactivation, which mainly include the following categories: 1. Fouling: reactants or intermediates are deposited on the catalyst surface or in the pores, blocking active sites and affecting catalytic efficiency. 2. Poisoning: certain chemicals (such as sulfur or nitrogen compounds) are strongly adsorbed on the active sites of the catalyst, hindering the catalytic reaction. 3. Vapor Formation/Leaching: the active components in the catalyst react with gas or liquid to form volatile compounds or dissolve into the liquid phase, resulting in the loss of active substances. 4. Vapor–Solid Interaction: the active components in the catalyst react with vapor to form an inactive phase, resulting in reduced activity. 5. Thermal Degradation/Sintering: high temperature causes the catalyst surface or pores to shrink, reducing the active surface area. 6. Attrition/Crushing: mechanical wear or external force causes the catalyst particles to break and lose the internal active surface area. These deactivation mechanisms are important challenges in catalyst research and industrial applications. Studying their causes and mitigation measures is of great significance for improving the life and stability of catalysts. And in industrial practice, effectively slowing down these deactivation processes is also a major challenge—especially for high-acidity zeolite catalysts operating under non-hydrogenation conditions. Therefore, proper feedstock pretreatment (such as deasphalting or demetallization) is often necessary to maintain catalyst stability and prolong service life [62].
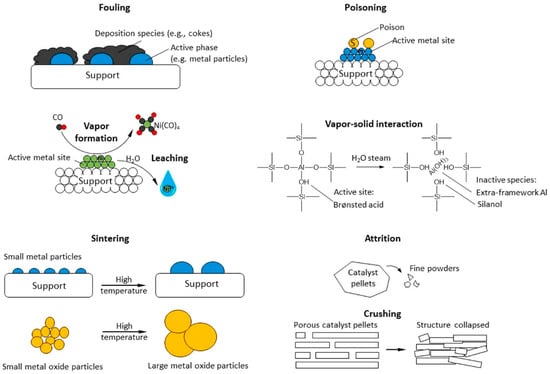
Figure 6.
Examples of heterogeneous catalyst deactivation mechanisms [62]. Reproduced with permission from Fan Lin et al., ACS Catalysis; published by American Chemical Society, 2022 [62].
3. Structure and Application of Zeolite Catalysts
Catalysts are the core of the heavy oil cracking process, and their performance directly determines the activity, selectivity, and stability of the reaction. Zeolite catalysts have significant advantages in heavy oil cracking due to their unique microporous structure and adjustable acidity [37,61]. The main functions of zeolite catalysts in heavy oil cracking include the following: providing evenly distributed acid sites, promoting the breakage and recombination of C-C bonds, and improving the selectivity of target products; improving the diffusion properties of heavy oil macromolecular hydrocarbons and reducing the occurrence of side reactions through pore structure optimization. In addition, the high thermal stability and resistance to metal poisoning of zeolite catalysts enable them to maintain high catalytic activity under high-temperature cracking conditions [15,61,64].
Zeolites are widely used as catalysts for cracking components [65,66,67,68]. The activity and selectivity for different fractions (natural gas, naphtha, kerosene, and diesel) depend on their acidity (including micropores and mesopores), which is directly related to the framework Si/Al ratio and the extra-framework Al content [66]. Most industrial applications of zeolites rely on BAS [69]. Therefore, many studies are devoted to determining the number, nature, and strength of acid sites and further studying the effect of acidity on catalytic activity [70,71,72,73]. Catalytic (hydro)cracking is an important type of acid-catalyzed reaction. The study of the effect of acid strength of zeolite catalysts on catalytic activity can guide actual industrial production and improve cracking efficiency, and has attracted widespread attention in the petrochemical field [74,75].
3.1. Structural Characteristics and Classification of Zeolites
Zeolite is a type of aluminosilicate material with a regular microporous structure. Its skeleton structure is formed by a silicon–oxygen tetrahedron (SiO4) and aluminum–oxygen tetrahedron (AlO4) connected by oxygen bridge bonds. The structural characteristics of zeolite determine its unique catalytic performance, mainly including pore structure, acidity, and thermal stability [76,77]. According to the different pore dimensions, zeolite can be divided into one-dimensional, two-dimensional, and three-dimensional pore structures. These pores provide ideal space for the diffusion of reactant molecules and catalytic reactions. Schematic diagrams of one-dimensional, two-dimensional, and three-dimensional pore structures are shown in Figure 7 [78].
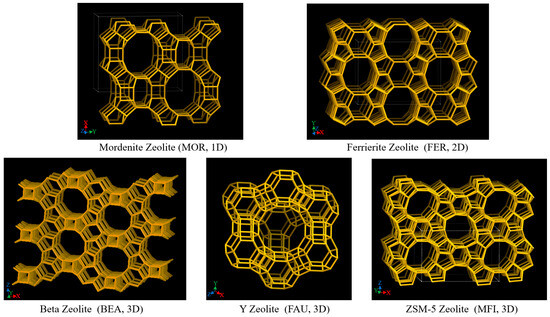
Figure 7.
Schematic diagram of one-dimensional, two-dimensional, and three-dimensional pore structures [78].
3.1.1. One-Dimensional, Two-Dimensional, and Three-Dimensional Pore Structures
Mordenite zeolite (MOR) is a typical representative of one-dimensional pore structure zeolites. Its pores are arranged linearly, and it has strong acidity and high stability, but its diffusion performance is limited [79,80]. This structure is suitable for cracking and isomerization reactions of long-chain alkanes, but it is easily affected by diffusion limitations during heavy oil cracking, which affects the reaction rate. In addition, due to the linear arrangement of the one-dimensional pores, after the reactant molecules react on the catalyst surface, the product discharge rate is slow, which makes it easy to induce side reactions, such as the formation of coke, thereby accelerating the deactivation of the catalyst [81].
Zeolites with two-dimensional pore structures, such as Ferrierite zeolite (FER), have a pore system composed of ten-membered straight pores and eight-membered cross-pores. This structure facilitates the transport of reactants and products, reduces diffusion resistance, and improves catalytic efficiency. The two-dimensional structure of FER zeolite provides a larger reaction space, improves the diffusion performance of large molecular hydrocarbons in the catalyst pores, and reduces the effect of diffusion limitation on the reaction rate. In the cracking of heavy oil and the hydrocracking of polycyclic aromatic hydrocarbons, FER zeolite exhibits high catalytic activity and good anti-deactivation performance. In addition, the pore structure and acidity distribution of FER zeolite can be further optimized through post-treatment and metal modification, thereby further improving its catalytic performance and stability [82,83].
Three-dimensional pore structures include Beta zeolite (BEA), Y-type zeolite (FAU), and ZSM-5 zeolite (MFI). These three types of zeolites show significant advantages in catalytic cracking reactions. The three-dimensional pore structure provides a larger diffusion space for the zeolite, reduces the diffusion resistance of reactants and products in the pores, and thus improves the catalytic efficiency [84]. Y-type zeolite has a supercage structure (12-membered ring), which provides a larger pore size and reaction space, and is suitable for cracking and recombination reactions of large molecular hydrocarbons, especially exhibiting excellent performance in the fluid catalytic cracking (FCC) process. ZSM-5 zeolite has excellent shape-selective catalytic performance due to its unique ten-membered ring pore structure and is suitable for the cracking and aromatization reactions of light hydrocarbons. Its high silicon–aluminum ratio gives it good hydrothermal stability, making it suitable for catalytic reactions under high-temperature conditions. In addition, Beta zeolite has a three-dimensional cross-12-membered ring macropore system with a large pore diameter (about 6.6 Å × 6.4 Å), which is conducive to the diffusion and transmission of large molecular hydrocarbons, thereby reducing diffusion limitations and improving catalytic efficiency. Beta zeolite is particularly suitable for heavy oil cracking and hydrocracking of polycyclic aromatic hydrocarbons. In addition, through post-treatment and metal modification, the acid distribution and pore structure of Beta zeolite can be further optimized, thereby enhancing its anti-deactivation performance and catalytic selectivity [85,86].
3.1.2. Common Zeolite Types and Their Applications in Heavy Oil Cracking
Zeolite catalysts are widely used in heavy oil catalytic cracking reactions due to their unique pore structure, adjustable acidity, and good thermal stability. Different types of zeolites have different structural characteristics and catalytic properties, showing significant advantages and challenges in the heavy oil cracking and upgrading process. Common zeolites mainly include Y-type zeolite, ZSM-5 zeolite, Beta zeolite, Ferrierite zeolite, and Mordenite zeolite. The comparison of the characteristics and applications of these five zeolites is shown in Table 4.
Table 4.
Comparison of properties and applications of five zeolites.
Y-Type Zeolite and Its Applications
As shown in Figure 7, Y-type zeolite is an important large-pore zeolite belonging to the FAU structure type, in which sodalite cages and hexagonal prism cages are arranged and connected according to the diamond structure, and multiple sodalite cages form a unique three-dimensional cavity structure, namely a supercage. The diameter of the supercage cavity is about 1.2 nm, and the supercages are connected to each other through a twelve-membered ring pore structure with a diameter of about 0.74 nm. Thanks to the large cavity and three-dimensional pores of Y-type zeolite, a variety of molecules can enter the supercage and react. Therefore, Y-type zeolite is an excellent solid–acid catalyst and is widely used in the field of petroleum refining [87]. Y-type zeolite can usually be synthesized by hydrothermal synthesis, hydrothermal conversion, and ion exchange. In addition, high-silicon–aluminum-ratio Y-type zeolite, heteroatom Y-type zeolite, and mesoporous Y-type zeolite have attracted much attention due to their higher stability, better stability and catalytic performance, and better transport performance, respectively [88].
The framework silicon–aluminum ratio of Y-type zeolite directly determines its acidic characteristics. Y-type zeolite with a low silicon–aluminum ratio contains a large number of BAS, which can effectively promote the breakage of C-C bonds in heavy oil, thereby improving the activity of the cracking reaction. However, strong acidity can also easily lead to coke formation, affecting the service life of the catalyst [89]. The acid distribution and strength of Y-type zeolite can be effectively regulated by dealumination treatment and rare earth metal ion exchange modification. For example, dealumination treatment can increase the silicon–aluminum ratio, reduce strong acid sites, and inhibit the formation of coke; rare earth modification can enhance the thermal stability of the acid center, thereby improving the stability and activity of the catalyst under high-temperature conditions [89,90].
Y-type zeolite is widely used in the fluidized catalytic cracking (FCC) process of heavy oil. Its supercage structure enables it to process large molecular hydrocarbons in heavy oil and produce high value-added products such as light oil and low-carbon olefins. In the FCC reaction, Y-type zeolite exhibits high activity, high selectivity, and good stability [87]. However, Y-type zeolite also faces some challenges in practical applications, such as catalyst deactivation, coke formation, and metal contamination [89]. Catalyst deactivation is mainly caused by coke deposition and heavy metal (such as Ni, V) contamination. The formation of coke is closely related to the acid strength of Y-type zeolite. Excessive acidity will lead to carbon cation polymerization and form coke that is difficult to remove. Metal contamination reacts with acid sites, resulting in poisoning of acid centers and decreased catalyst activity. Therefore, these problems can be effectively solved, and the service life of the catalyst can be extended by acidity regulation, rare earth metal modification, and the introduction of anti-metal contamination additives [90,91].
ZSM-5 Zeolite and Its Applications
As shown in Figure 7, ZSM-5 zeolite belongs to the MFI structure type and has a unique ten-membered ring pore structure with a pore size of about 0.55 nm, presenting a three-dimensional pore system. This structure gives ZSM-5 zeolite excellent shape-selective catalytic performance, enabling it to effectively improve the selectivity of light olefins and aromatics in heavy oil cracking reactions. Since its pore size is close to the diameter of small molecular hydrocarbons, ZSM-5 zeolite has unique advantages in light hydrocarbon cracking and aromatization reactions, and is particularly suitable for highly selective catalysis of target products [92]. In addition, the high silicon–aluminum ratio of ZSM-5 zeolite gives it good hydrothermal stability, which can maintain structural integrity under high-temperature conditions and is suitable for high-temperature catalytic cracking reactions [37]. In addition, the acidic characteristics of ZSM-5 zeolite are mainly determined by the silicon–aluminum ratio in the framework. High silicon–aluminum ratio ZSM-5 has weaker acidity, which is suitable for selective catalytic reactions and reduces the occurrence of side reactions. The microporous structure of ZSM-5 zeolite is suitable for the generation of small molecular products but has certain limitations on the diffusion of large molecular hydrocarbons. To solve this problem, researchers have introduced mesoporous structures through post-treatment methods such as alkali treatment to improve the diffusion properties of reactants and products, thereby increasing the overall activity of the catalyst [93].
Due to its excellent shape-selective catalytic performance, ZSM-5 zeolite exhibits high activity and selectivity in the cracking reaction of long-chain alkanes. In the cracking process of long-chain alkanes, ZSM-5 zeolite can promote the uniform breaking of C-C bonds to produce high value-added products such as low-carbon olefins and aromatics. However, the strong acid sites of ZSM-5 zeolite may lead to side reactions and promote the formation of coke, thereby affecting the stability and service life of the catalyst. In order to improve this problem, the formation of coke can be effectively reduced by regulating the acid strength, introducing metal additives (such as Pt, Ni), and optimizing the pore structure, further improving the conversion rate and selectivity of long-chain alkane cracking, and extending the service life of the catalyst [94,95].
Beta Zeolite and Its Applications
As shown in Figure 7, Beta zeolite belongs to the *BEA structure type and has a three-dimensional cross-porous macroporous structure with a pore size of about 0.66 nm. This structure not only facilitates the diffusion and transport of macromolecular hydrocarbons, but also gives Beta zeolite good catalytic performance, making it particularly suitable for the cracking and hydrogenation of polycyclic aromatic hydrocarbons and macromolecular hydrocarbons. Beta zeolite exhibits high catalytic efficiency in heavy oil hydrocracking, which is beneficial for improving the yield of light oil products [96]. In addition, Beta zeolite has high acidity and strong thermal stability, which enables it to exhibit excellent catalytic performance under high-temperature conditions.
Polycyclic aromatic hydrocarbons are important components of heavy oil and have a highly stable structure that is difficult to crack under normal conditions. Beta zeolite, with its three-dimensional large pore structure and strong acidity, can effectively promote the cracking reaction of polycyclic aromatic hydrocarbons to produce light oil products and low-carbon olefins [96]. By regulating the acid distribution and pore structure of Beta zeolite through metal loading and post-treatment modification, its catalytic performance, catalytic selectivity, and anti-deactivation ability can be further improved, while effectively reducing coke formation and extending the service life of the catalyst [97].
Ferrierite Zeolite and Its Applications
As shown in Figure 7, Ferrierite zeolite belongs to the FER structure type and is a zeolite with a two-dimensional pore structure. It has a unique ten-membered ring straight pore (pore diameter of about 0.54 nm) and an eight-membered ring cross pore (pore diameter of about 0.42 nm), forming a dual-pore system. The unique pore structure of Ferrierite zeolite gives it excellent shape-selective catalytic performance, which can effectively and selectively catalyze small molecular reactants and perform well in isomerization reactions and light hydrocarbon conversions, but it has certain obstacles to the diffusion of large molecular hydrocarbons and polycyclic aromatic hydrocarbons [98,99].
Ferrierite zeolite is widely used in reactions such as light hydrocarbon isomerization and long-chain alkane conversion due to its unique pore structure and strong acidity. In light hydrocarbon isomerization, Ferrierite zeolite exhibits excellent shape-selective catalytic performance and can effectively promote the isomerization reaction of straight-chain alkanes to generate branched alkanes to increase the octane number. For example, Ferrierite zeolite exhibits high activity and selectivity for the isomerization of n-butene, while inhibiting the formation of by-products [100]. In the conversion of long-chain alkanes, Ferrierite zeolite exhibits good diffusion performance and thermal stability in the cracking reaction of long-chain alkanes and is suitable for the production of high-value-added products such as light olefins and aromatics. In addition, by introducing precious metal additives such as Pt and Pd, Ferrierite zeolite can be endowed with hydrogenation function, further improving the efficiency of the cracking reaction and the yield of the target product [101].
Although Ferrierite zeolite performs well in isomerization and alkane conversion, it still faces some challenges, such as pore diffusion limitation, catalyst deactivation, and metal contamination. Pore diffusion can be improved by alkali treatment or template-assisted synthesis of mesoporous structures to improve the diffusion efficiency of reactants and products. The catalyst’s anti-deactivation ability can be enhanced by adding metal additives and anti-coking technology, and the catalyst’s stability and durability can be improved. Green synthesis technology can be achieved by promoting environmentally friendly solvents and renewable templates, and sustainable synthesis and application of Ferrierite zeolite can be achieved [102].
Mordenite Zeolite and Its Applications
As shown in Figure 7, Mordenite zeolite belongs to the MOR structure type, has a one-dimensional linear pore structure, a pore size of about 0.65 nm, and has strong acidity. Due to its unique pore structure, Mordenite zeolite has high activity in heavy oil cracking and hydrocarbon isomerization reactions [103]. However, due to its relatively linear pore structure and poor diffusion performance, it is easy for reactants and products to be retained in the pores, affecting the catalytic efficiency.
Mordenite zeolite exhibits excellent catalytic performance in the isomerization and cracking reactions of long-chain alkanes. Its strong acid sites can effectively promote the breaking of C-C bonds to generate high-octane isohydrocarbons and low-carbon olefins. However, its strong acidity can also easily cause coke formation, resulting in catalyst deactivation [81]. However, by introducing mesoporous structures, rare earth metal ions, and other post-treatment methods, the diffusion properties, pore structure, acid distribution, and anti-coke formation ability of Mordenite zeolite can be improved, thereby improving its catalytic performance and stability in heavy oil cracking reactions [104,105].
In general, Y-type zeolite, ZSM-5 zeolite, Beta zeolite, Ferrierite zeolite, and Mordenite zeolite have their own characteristics and have different application advantages in different catalytic cracking reactions. Y-type zeolite and Beta zeolite are suitable for processing large molecular hydrocarbons in heavy oil and are widely used in cracking and hydrogenation reactions. ZSM-5 zeolite has outstanding performance in the generation of low-carbon olefins and aromatics due to its excellent shape-selective catalytic performance. Ferrierite zeolite and Mordenite zeolite are more suitable for the selective catalytic reaction of small molecules and long-chain alkanes. Different zeolites have differentiated application values in heavy oil cracking due to their unique pore structure and acidic characteristics. By rationally selecting and modifying these zeolites, the catalyst performance can be optimized, and the target product can be efficiently produced for different heavy oil components and reaction conditions.
3.2. Acidic Characteristics of Zeolites
The acidity of zeolites comes from Brønsted acid sites (BAS) and Lewis acid sites (LAS). BAS are formed by the combination of framework aluminum atoms and protons and can provide protons to participate in catalytic cracking reactions. LAS are mainly derived from extra-framework aluminum or metal cations and can accept electron pairs. The acidity strength, acid site distribution, and pore structure of zeolites directly affect their catalytic performance. The two acid sites, BAS and LAS, generally work synergistically to jointly determine the catalytic activity, selectivity, and stability of zeolites. The strength, quantity, distribution, and type of zeolite acidity are closely related to its silicon–aluminum ratio, framework structure, and subsequent modification methods. The regulation of zeolite acidity is an important way to optimize catalytic performance and adapt to different reaction conditions. For the complex reaction process of heavy oil cracking, the rational regulation of acid characteristics can not only help improve the selectivity of the target product, but also inhibit the formation of side reactions such as coke [106].
3.2.1. Brønsted Acid Sites (BAS) and Lewis Acid Sites (LAS)
There are two main sources of zeolite acidity. One is the BAS formed by the combination of framework aluminum atoms and hydroxyl groups, which can donate protons to the outside. The other is the LAS formed by cations outside the framework or unsaturated aluminum atoms on the framework, which can accept external electron pairs. BAS and LAS can be converted into each other under certain conditions.
BAS are negatively charged sites generated by the aluminum–oxygen tetrahedrons in the zeolite framework, which are formed by combining with protons (H+). These acid sites are mainly distributed in the micropores of zeolites and are one of the most important active centers in catalytic cracking reactions. BAS provide protons to protonate hydrocarbon molecules in heavy oil to form highly active carbon cations. This intermediate is unstable and will rapidly undergo cracking or recombination reactions to produce target products such as light oil products and low-carbon olefins [107]. At the same time, the lower the silicon–aluminum ratio of the zeolite, the higher the aluminum content in the framework, and the greater the number of BAS. However, too many acid sites will lead to overreaction, increase the probability of side reactions, and ultimately generate a large amount of coke, which will block the pores of the zeolite and reduce the activity and life of the catalyst [108]. Katsuki et al. [109] used NH3 infrared mass spectrometry and programmed temperature desorption (TPD) technology to study the acid strength of in situ-prepared HNaY zeolite [Si/Al = 2.55, NaNH4Y (96% NH4+/4% Na+)] and found that NH3 molecules were adsorbed at the -O2H position, while ammonium ions (NH4+) were formed at the -O1H, -O3H, and -O4H positions, and that the formation of NH4+ was more favorable than the adsorption of NH3 molecules from an energetic point of view. Dedecek et al. [110] found that Al/Si substitution reduced the 29Si chemical shift of the nearest neighbor Si atom and increased the 27Al chemical shift of the next nearest neighbor Al atom (NNN-Al), while the effect of more distant Al/Si substitution on acid strength was not obvious. Huang et al. [111] found, based on the cluster model at the BLYP/DNP level, that the order of acid strength of the acidic sites determined by the alkaline probe molecules (NH3 and pyridine) is different from the order of the intrinsic acid strength (OH deprotonation energy), and that the alkaline probe molecules generally tend to adsorb on the acidic sites in the supercage.
LAS are mainly formed by non-framework aluminum or exchange cations, such as metal ions, defect sites generated by dealumination treatment, etc. The characteristic of LAS is that they can accept electron pairs, especially playing a key role in hydrocracking and isomerization reactions. In particular, in zeolites modified by metal ion exchange, LAS can synergize with metal active centers to promote the occurrence of hydrogenation reactions. For example, when metals such as Ni, Mo, and Pt are introduced into zeolites, LAS can achieve hydrogenation ring-opening reactions of polycyclic aromatic hydrocarbons in the presence of these metals, significantly reducing the content of unsaturated components in the product and improving the quality and yield of light oil products [112].
In addition, BAS and LAS can be converted into each other under certain specific zeolite modification conditions. For example, through metal ion exchange or high-temperature calcination, some BAS will be converted into LAS. This acidity regulation can optimize the activity and selectivity of the catalyst to meet the needs of different reaction conditions [113].
People have long recognized that the acidity of zeolites is affected by their structure—that is, the distribution of Al. The aluminum content of zeolites can be controlled by post-synthesis treatment (such as dealumination) or by changing the synthesis conditions. The following uses HY zeolite as an example to introduce the formation of acid sites and intrinsic catalytic performance of Y-type zeolites.

The OH group of HY zeolite is the acid center and can be expressed by the following equilibrium formula:

When the temperature rises, the equilibrium of the above formula moves to the right, resulting in a decrease in the number of OH groups, so the intensity of its infrared band decreases. When the temperature is above 770 K, LAS centers begin to appear, which are associated with tri-coordinated aluminum atoms and are formed by further dehydration of H-Y.
The extra-framework aluminum ions will strengthen the acid sites and form LAS center, as shown in Figure 8:
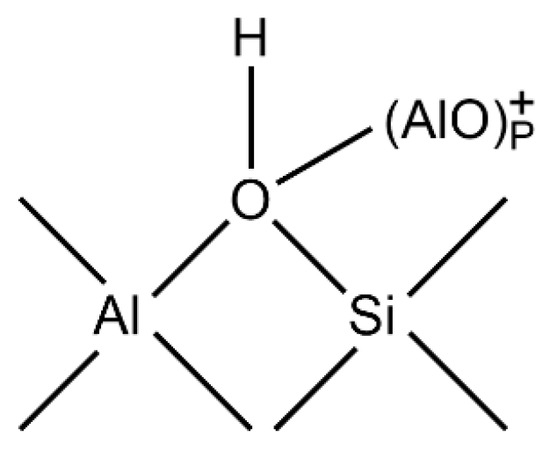
Figure 8.
Schematic diagram of the structure of extra-framework aluminum ions.
Multivalent cations may also produce OH acid sites, such as [Ca(OH2)]2+ → [Ca(OH)]+ + H+.
Reduction in transition metal ions can also form acid centers, such as Cu2+ + H2 → Cu0 + 2H+ and Ag+ + 1/2H2 → Ag0 + H+.
3.2.2. Effect of Acidity on Catalytic Performance
Hydrocracking catalysts are typically bifunctional, combining metal hydrogenation sites with zeolite acidic sites to enable the breaking and transformation of heavy hydrocarbon molecules. The Brønsted acid sites (BAS) in zeolites play a vital role in facilitating C-C bond cleavage and determining product distribution [7,114,115]. Arribas et al. [116] demonstrated that the BAS acidity of Pt/USY zeolite significantly influenced both the yield and selectivity in the ring-opening hydrogenation of 1-methylnaphthalene. Dealuminated forms such as HTY and HTY-Al were superior to NaY and HY zeolites in producing light aromatics due to their optimized acid strength and wider pore size distribution [117]. Additionally, Lemberton et al. [118] found that the cleavage of the saturated central ring in dihydrophenanthrene may occur through an acidic mechanism. These studies highlighted the significant effect of zeolite acidity on hydrocracking catalytic activity, which was confirmed by similar studies by other researchers [10,119,120,121].
A key parameter affecting catalytic activity in alkane cracking is the framework aluminum content, which directly determines the number and strength of acid sites. Studies have shown that increasing framework Al in Y-type zeolite or ZSM-5 correlates with increased catalytic activity for cracking hexane and isobutane [122,123,124,125,126]. Moreover, acid strength influences not only activity but also the selectivity of products like light olefins [127,128]. For example, Bai et al. [129] established the relationship between the reaction pathway of naphtha catalytic pyrolysis and catalyst acid strength, showing that reducing BAS acidity suppresses secondary reactions of light olefins—such as hydrogen transfer, oligomerization, cyclization, and aromatization—and significantly enhances light olefin yield. Similarly, Zhai et al. [130] investigated n-hexane catalytic pyrolysis over HZSM-5 and found that both the Si/Al ratio and catalyst-to-oil ratio positively correlated with propylene selectivity, while lower acid density favored unimolecular cracking. In line with these findings, Xue et al. [131] employed 1-hexene cracking as a probe reaction to evaluate the acid site distribution in Sn-modified ZSM-5, demonstrating that tailored Al siting and acid strength significantly influence cracking pathways and the selectivity of hydrocarbon products.
However, overly strong acidity can accelerate side reactions including coke formation and excessive cracking, particularly in the processing of heavy oils rich in polycyclic aromatics and long-chain alkanes. This leads to rapid catalyst deactivation due to pore blockage. Therefore, regulating the strength, amount, and distribution of acidity is crucial to maintain catalyst efficiency, selectivity, and longevity [89].
To address this, several strategies have been proposed: Increasing Si/Al ratio: reduces acid strength, suppresses coke formation, and enhances selectivity—commonly applied in Y-type zeolites via dealumination [89]. Acid or alkali post-treatment: acid leaching removes framework aluminum, moderating strong acid sites and improving metal tolerance; alkali treatment creates mesoporosity, reducing diffusion barriers and enhancing activity [93]. Metal ion exchange and heteroatom introduction: rare earth metals like La or Ce, and noble metals such as Pt and Pd, can stabilize acid centers and improve resistance to deactivation. Acid site distribution optimization: achieved through nanocrystallization or mesoporous engineering to reduce side reactions and improve product selectivity [91,132]. In summary, the tailored regulation of acidity—through structural modification, post-treatment, and metal loading—is fundamental to optimizing the catalytic performance of zeolites for complex reactions such as heavy oil cracking and hydrocracking.
Figure 9 shows the improvement of catalytic performance after rare earth cerium (Ce) modification of HZSM-5 zeolite by the hydrothermal method. Two modification methods are shown in the figure, namely 1. Ce3+ modification: Ce3+ ions interact with silanol groups (Si-OH) on the surface of zeolite to generate new BAS and repair defective sites in zeolite. This modification method significantly enhances the acid strength and acid density of the catalyst. 2. CeO2 modification: CeO2 clusters are introduced into the pore structure of zeolite to form stable CeOx clusters. These clusters further enhance the acid stability of zeolite and the stability of the framework structure. Both modification methods can significantly improve the selectivity of HZSM-5 zeolite for ethylene (C2H4) and propylene (C3H6) in the catalytic cracking reaction of n-heptane, as well as the reaction stability. At the same time, the modified catalyst has a lower coke deposition rate, which prolongs the service life of the catalyst. These modified catalysts significantly increased the yield of light olefins in the cracking reaction by optimizing the strong acid/weak acid and BAS/LAS (B/L) ratios and improved the catalyst stability and resistance to deactivation [106].
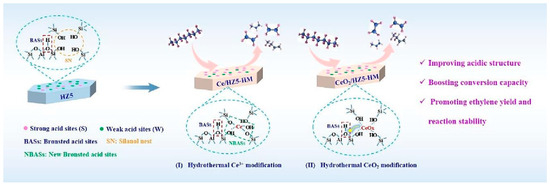
Figure 9.
Hydrothermally Ce-modified HZSM-5 zeolite enhancing its strong acidity and BAS/LAS ratio: stably boosting ethylene/propylene ratio for cracking n-heptane [106]. Reproduced with permission from Qi Liu et al., Fuel; published by Elsevier, 2024 [106].
Figure 10a depicts the correlation between the catalytic performance of modified Y zeolite catalysts in the Meerwein–Ponndorf–Verley (MPV) reaction and the LAS concentration. The red line in the figure represents the LAS concentration (measured by pyridine-FTIR), while the blue line represents the initial rate of the reaction. As the concentration of extra-framework aluminum (EFAl) in the catalyst increases, the LAS concentration increases significantly, and at the same time, the reaction rate also increases synchronously, indicating the key role of LAS in the reaction. Isopropyl alcohol (iPrOH) and ketone compounds (such as 4-tert-butylcyclohexanone) are catalyzed by LAS to produce alcohols (such as 4-tert-butylcyclohexanol) through MPV reaction. This improvement in catalytic performance is mainly attributed to the extra-framework aluminum species introduced into the catalyst after ion exchange, which significantly enhance the LAS acidity of the catalyst. The figure further indicates that the generation of these extra-framework aluminum species is achieved through simple aluminum ion exchange, which does not destroy the zeolite framework and significantly improves the LAS catalytic activity, providing potential high-efficiency catalysts for important industrial reactions such as fluid catalytic cracking (FCC) and biomass conversion. Figure 10b shows the mechanism of LAS acidity induced by aluminum species in different coordination states before and after ion exchange. Before ion exchange, the framework-associated aluminum (Al(VIa)) of Y zeolite mainly exists in octahedral coordination, and these aluminum species provide the basis for the formation of LAS. After ion exchange, the LAS acidity is further increased by introducing additional extra-framework aluminum (Al(VIb)). These extra-framework aluminums mainly exist in the form of neutral aluminum oxide or aluminum hydroxide clusters, indicating that their octahedral coordination structure has strong catalytic activity. Aluminum ion exchange not only increases the number of LAS, but also retains the original BAS. This feature optimizes the acid distribution of the catalyst, thereby significantly enhancing the catalytic performance. In addition, the figure shows that after appropriate ion exchange, the zeolite structure remains intact, which lays the foundation for its application in catalytic reactions with high activity and stability. These modified LAS show high reaction rates and efficiencies in catalytic reactions including the Meerwein–Ponndorf–Verley (MPV) reduction reaction. In this way, aluminum can be introduced into Y zeolite through a simple post-treatment process, thereby developing more efficient LAS catalysts [113].
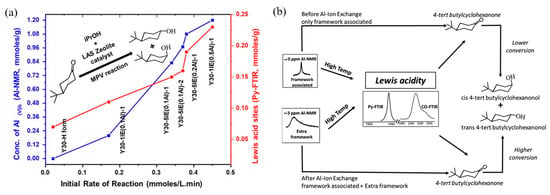
Figure 10.
(a) Correlating the concentration of extra-framework Al(VIb) species to catalytic rate of reaction and total content of LAS obtained from pyridine-probed FTIR quantitative analysis; (b) summary of results illustrating aluminum species in different coordinations responsible for LAS acidity before and after aluminum ion-exchange [113]. Reproduced with permission from Syeda R. Batool et al., Journal of Catalysis; published by Elsevier, 2022 [113].
3.2.3. Catalytic Performance of Low-Acidity Aluminosilicates
While zeolites with strong acidity are widely used for heavy oil cracking due to their high activity, they also suffer from rapid deactivation caused by coke formation and sensitivity to metal impurities. In this context, low-acidity aluminosilicates have emerged as promising alternatives, particularly for processing feeds with high concentrations of heteroatoms and heavy metals. These materials—such as amorphous silica-alumina (ASA), mesoporous Al-MCM-41, and other low-crystallinity aluminosilicates—exhibit moderate acidity that allows sufficient catalytic activity for cracking reactions while greatly enhancing resistance to coke deposition and deactivation.
Figure 11 illustrates the relationship between the acidic properties and pore structures of various zeolitic and mesoporous aluminosilicate catalysts, and their influence on the conversion and product distribution during Fischer–Tropsch (F-T) wax cracking. The catalysts are classified into three groups. Group 1 includes freshly calcined zeolites such as H-Y, H-ZSM-5, and H-Beta, characterized by high acidity and crystallinity. These catalysts demonstrate high F-T wax conversion rates and significant gasoline yields, along with a notable proportion of light olefins. Group 2 consists of severely steamed zeolites (e.g., metal-modified steam-treated types), which undergo partial dealumination, leading to reduced BAS acidity. Despite this, they maintain considerable cracking activity, with a product distribution that favors light olefins and light cycle oil (LCO). Group 3 encompasses low-crystallinity or amorphous mesoporous materials such as low-crystallinity ZSM-5, amorphous Beta-derived structures, ordered mesoporous materials (such as Al-MCM-41), and commercial amorphous silica–alumina (ASA). Although these materials exhibit weaker acidity, their large pore structures enhance diffusion of bulky hydrocarbon molecules and reduce deactivation. They tend to favor LCO formation and suppress excessive secondary cracking, highlighting their advantages in scenarios where product selectivity and catalyst stability are prioritized over maximum conversion. Overall, Figure 11 underscores that both acid strength and pore architecture are critical factors influencing catalytic efficiency and product selectivity in F-T wax upgrading processes [89].
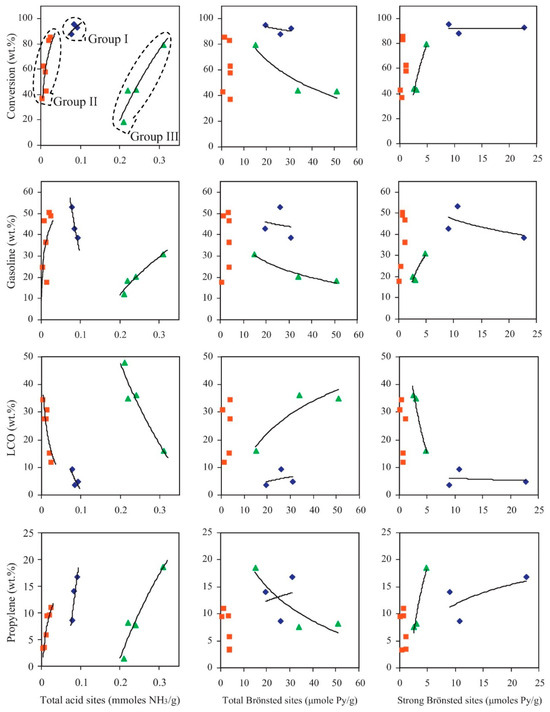
Figure 11.
Correlation between F–T wax conversion and product yields with the acidic properties of the various zeolitic and mesoporous aluminosilicate catalytic materials (group I: fresh-calcined zeolites H-Y, H-ZSM-5, and H-Beta; group II: severely steamed zeolites (including the metal modified steamed zeolites); group III: 3% crystalline ZSM-5-based, amorphous Beta-based, ordered mesoporous Al-MCM-41, and commercial ASA catalyst) [89]. Reproduced with permission from V.G. Komvokis et al., Catalysis Today; published by Elsevier, 2012 [89].
Notably, Yang et al. [133] found that the acidity distribution of aluminosilicate catalysts is largely governed by the Si/Al ratio, and catalysts with increased porosity and reduced acidity exhibited lower coke deposition during lignin pyrolysis. Another notable development involves embedding alumina into HZSM-5 to create mesoporous matrices with moderate acidity, enhancing thermal stability and improving conversion of bulky bio-oils [134]. In addition, Hou et al. [135] reported that acid-modified kaolin, a natural aluminosilicate, exhibited promising performance in the upgrading of coal pyrolysis volatiles, offering a cost-effective and thermally stable alternative to traditional zeolites, which are often limited by high cost and coke deposition.
Overall, low-acidity aluminosilicates represent a valuable class of materials in the evolving landscape of catalytic systems for heavy oil upgrading, offering a trade-off between activity and operational stability. Their role is particularly pronounced in integrated petrochemical refining systems aiming for longer cycle life and cleaner fuel production.
3.3. Synthesis and Modification of Zeolites
The synthesis and modification of zeolites are key means to improve their catalytic performance, mainly including hydrothermal synthesis methods, post-treatment and modification methods, as well as metal ion exchange and heteroatom introduction. By regulating the structure, acidity, and stability of zeolites, their application performance in reactions such as catalytic cracking and hydrocracking can be significantly improved to meet the needs under different reaction conditions. The synthesis and modification methods of zeolites are summarized in Table 5, revealing the optimization effects and applicable scenarios of different methods. Dealumination and metal ion exchange are essential for improving the thermal stability and heavy oil conversion performance of catalysts, while alkali treatment and heteroatom introduction can significantly improve the diffusion performance of macromolecules and product selectivity. The combined application of these modification strategies can achieve precise regulation of catalyst performance according to industrial needs, thereby meeting production requirements under different reaction conditions.
Table 5.
Summary of zeolite synthesis and modification methods.
3.3.1. Hydrothermal Synthesis Method
The hydrothermal synthesis method is a classic method for preparing zeolites and is widely used to synthesize different types of zeolites, such as Y-type, ZSM-5, Beta, etc. This method uses a silicon source (such as silica sol, sodium silicate), an aluminum source (such as sodium aluminate, aluminum sulfate), a template (such as tetrapropylammonium hydroxide), and an alkali source (such as sodium hydroxide, potassium hydroxide) to undergo a crystallization reaction under high-temperature and high-pressure conditions to form a zeolite material with a regular pore structure [136,137].
During the hydrothermal synthesis process, multiple factors have an important influence on the structure and properties of zeolites. First, the reaction temperature is an important parameter for controlling the crystallization rate and crystal size of zeolites. Too high a temperature may lead to excessive crystal growth and increased pore structure defects; too low a temperature may delay the crystallization process and affect the purity and crystallinity of the product [138]. Secondly, the pH value has a significant effect on the formation of the silicon–aluminum ratio of the zeolite framework. Under alkaline conditions, it is conducive to the introduction of framework aluminum to form zeolites with high aluminum content, but too high an aluminum content may lead to uneven acidity distribution. In addition, the silicon–aluminum ratio is an important factor in regulating the acidity of zeolites. Zeolites with a high silicon–aluminum ratio have lower acid strength and are suitable for catalytic reactions with lower acidity requirements [139].
The selection and dosage of templates are crucial to the pore structure of zeolites. Different templates will lead to the formation of different pore structures and crystal morphologies. For example, tetrapropylammonium hydroxide (TPAOH) is often used to synthesize ZSM-5 zeolite, which can induce the formation of a ten-membered ring pore structure. However, excessive use of templates not only increases costs but also affects environmental friendliness. Therefore, in recent years, green synthesis technology has gradually attracted attention, and sustainable synthesis is achieved by introducing recyclable templates and regulating reaction conditions [140].
Figure 12a shows the effect of adjusting the Si/Al ratio and crystallization time on the crystal morphology during the synthesis of ZSM-5 zeolite, revealing how these factors determine the final structure and performance of the zeolite under hydrothermal synthesis conditions. The figure shows the role of silicon source, aluminum source, and template in the hydrothermal synthesis process to produce ZSM-5 zeolites with different Si/Al ratios. Under low Si/Al ratio conditions, ZSM-5 exhibits a rougher surface and dispersed particles, while under high Si/Al ratio conditions, a more dense and regular particle morphology is formed. This difference is due to the effect of Si/Al ratio on crystal morphology and acid site distribution, which ultimately affects the performance of the catalyst. This study explored the effect of Si/Al ratio and crystallization time on the morphology and acidic properties of ZSM-5 zeolite, revealing that at a low Si/Al ratio, the increase in aluminum content significantly slows down the crystallization rate and leads to significant changes in particle morphology. The results of the study provide theoretical guidance for optimizing the synthesis process of ZSM-5 catalysts to achieve efficient methanol conversion reactions and selective production of target products [139]. Figure 12b shows the differences in physicochemical properties, surface nitrate species and acidity, and catalytic hydrothermal stability of Cu/SSZ-13 zeolite catalysts with different Si/Al ratios from three perspectives. 1. Physicochemical properties: The XPS (X-ray photoelectron spectroscopy) data in the upper left corner of the figure show that the coordination environment and dispersion state of Cu2+ species vary with the Si/Al ratio. This is reflected in the ratio of isolated framework Cu2+ ions to aggregated CuO-like surface species, which significantly affects catalytic activity and stability. 2. Surface nitrate species and acidity: the bar chart in the upper right corner of the figure shows the changes in the absorption intensity of nitrate species on the surface of fresh (Fresh) and hydrothermally aged (Aged) catalysts, indicating that the higher the Si/Al ratio, the more significant the reduction in the number of acidic and nitrate species after hydrothermal aging, which will affect the intermediate generation and reaction rate in the NH3-SCR reaction. 3. Catalytic hydrothermal stability performance: The curve below the figure shows the relationship between NOx conversion rate and temperature, indicating that the catalyst with a Si/Al ratio of 6.5 has the best NOx conversion rate (over 90%) and better hydrothermal stability in the range of 200–500 °C, while the conversion efficiency and stability of the catalyst with a high Si/Al ratio are significantly reduced. Although this study primarily evaluated the Cu/SSZ-13 catalysts for NH3-SCR, the influence of Si/Al ratio on acidity, Cu dispersion, and hydrothermal stability is also highly relevant to hydrocracking of petroleum hydrocarbons. The enhanced acidity and structural stability at lower Si/Al ratios benefit both NOx reduction and hydrocarbon conversion, offering cross-applicable insights for designing zeolite catalysts in heavy oil upgrading [138].
Figure 12.
(a) Origin of morphology change and effect of crystallization time and Si/Al ratio during synthesis of zeolite ZSM-5 [139]. Reproduced with permission from Jan J. Weigand et al., ChemCatChem; published by John Wiley and Sons, 2022 [139]; (b) Cu/SSZ-13 zeolites prepared by in situ hydrothermal synthesis method as NH3-SCR catalysts: influence of the Si/Al ratio on the activity and hydrothermal properties [138]. Reproduced with permission from Han Jiang et al., Fuel; published by Elsevier, 2019 [138].
3.3.2. Post-Treatment and Modification Methods
Post-treatment and modification of zeolite are important ways to optimize its acidity, pore structure, and catalytic performance, mainly including dealumination, acid treatment, and alkali treatment. These methods can improve the activity and stability of the catalyst by finely regulating the acidity and pore characteristics according to the structural characteristics and reaction requirements of zeolite.
Dealumination is mainly carried out by removing aluminum from the zeolite framework through acid treatment or steam treatment, thereby increasing the silicon–aluminum ratio and reducing the acid strength, reducing the presence of overly strong acid sites, and effectively inhibiting the formation of coke and extending the service life of the catalyst. In addition, dealumination can also improve the thermal stability of zeolite, making it show higher stability and selectivity in high-temperature cracking reactions [141]. Common methods of dealumination include hydrothermal dealumination, chemical dealumination, and composite dealumination.
Hydrothermal dealumination was first proposed by McDaniel et al. in 1970 [142]. By treating ammonium-exchanged NaY zeolite with high-temperature steam, part of the framework aluminum will be hydrolyzed and removed. Then, high-temperature calcination will cause the silicon inside the Y-type zeolite to migrate and rearrange, thereby forming ultrastable Y-type zeolite (USY) with a high silicon content. Yurtaeva et al. [143] dealuminated Y-type zeolite under high-temperature steam. As the silicon–aluminum ratio increased, the concentration of acid sites decreased, but the acidity also increased. The effect of silicon–aluminum ratio on catalytic performance was studied by HTVGO cracking. The results showed that as the silicon–aluminum ratio increased, the yield of C2-C4 olefins increased from 25.3 wt% to 27.1 wt%, which was due to the reduced contribution of hydrogen transfer reaction due to the reduced acidity. Although the hydrothermal dealumination method can form silicon-rich Y-type zeolite, it will lead to an increase in non-framework aluminum, which will have an adverse effect on catalytic performance [144].
Chemical dealumination utilizes the interaction between acid, salt, or a chelating agent and zeolite to dealuminate the framework. López-Fonseca et al. [145] controlled the aluminum content of commercial HY zeolite by changing the ratio of (NH4)2SiF6 to zeolite. In the experiment, 16%, 32%, 50%, and 64% of the aluminum in the zeolite was removed, respectively. The results showed that when the degree of dealumination was less than 50%, the zeolite structure was almost unaffected; but when the degree of dealumination reached 64%, a large amount of aluminum was removed from the zeolite framework, resulting in obvious structural collapse of the zeolite. The Y-type zeolite modified by dealumination showed higher catalytic activity than the zeolite before dealumination. The increase in activity was related to the strong acidity generated by dealumination.
Since the hydrothermal dealumination method will lead to an increase in the non-framework aluminum content, which will have a certain impact on the catalytic performance, the chemical dealumination method can remove non-framework aluminum, but the structure of the zeolite after dealumination may be unstable. Therefore, the composite dealumination method combining hydrothermal dealumination and chemical dealumination is more advantageous. It can not only produce Y-type zeolite with a high silicon–aluminum ratio, but also improve the stability of the zeolite and remove non-framework aluminum. Wang et al. [146] used hexaalkyltrimethylammonium bromide (CTAB) as a surfactant and used a composite dealumination method to post-treat Y-type zeolite to improve its silicon–aluminum ratio. The experimental results show that a small amount of surfactant can significantly improve the pore structure, silicon–aluminum ratio, and catalytic performance in hydrocracking of Y-type zeolite. The prepared high silicon–aluminum ratio Y-type zeolite was used to hydrocrack vacuum gas oil. The results showed that compared with the commercial catalyst, the hydrocracking performance and intermediate distillate oil yield of the prepared Y-type zeolite were increased by 2.42% and 4.20%, respectively.
Acid treatment removes framework aluminum and non-framework aluminum in zeolite through the action of dilute acid, optimizes the acid distribution and reduces the acid strength, thereby reducing the probability of side reactions. Acid treatment can not only improve the structural defects of zeolite, but also improve the selectivity and stability of the catalyst, making it more adaptable in heavy oil cracking reactions [147,148].
Unlike acid treatment, alkali treatment uses alkaline solution (such as NaOH) to modify zeolite, mainly by introducing a mesoporous structure to improve diffusion performance and alleviate the diffusion limitation of reactants and products in the pores during the reaction. Zeolite treated with alkali has a higher specific surface area and pore volume, which is conducive to the transmission and conversion of large molecular hydrocarbons, thereby significantly improving the activity and selectivity of the catalyst. In the heavy oil cracking reaction, zeolite treated with alkali exhibits higher catalytic efficiency, further meeting the needs of industrial production [149,150].
In Figure 13a, we can see the characterization of HZSM-5 (MFI) zeolite and its role in the cracking reaction of 1-butene. The figure shows two different modification methods: treating the catalyst with H3PO4 (phosphoric acid) and KOH (potassium hydroxide). The figure shows that the use of phosphoric acid can extend the life of the catalyst, while potassium hydroxide improves the selectivity for propylene. Specifically, H3PO4 modification helps to reduce the number of active sites on the catalyst, thereby inhibiting the occurrence of side reactions and delaying the deposition of coke, thereby improving the stability of the catalyst. On the other hand, KOH modification can improve the selectivity of propylene by increasing the basicity of the catalyst surface and reducing the occurrence of side reactions such as aromatization. With this modification, the catalyst can more efficiently and selectively produce propylene rather than other olefins or aromatic products when cracking 1-butene. Through these different modification methods, this study not only improved the performance of the catalyst, but also conducted an in-depth discussion on the catalytic mechanism of MFI zeolite [151]. Figure 13b shows the effects of different treatments on the crystal structure, catalytic performance, and crystallinity of Beta zeolite. The right side of the picture shows the comparison of the crystallinity and pore structure of Beta zeolite after alkaline treatment with NaOH and NaOH&TBAOH (tetrabutylammonium hydroxide). The parent Beta zeolite has 100% crystallinity and shows high catalytic activity. The crystal structure of Beta zeolite treated with NaOH was destroyed, and the crystallinity dropped to 66%, but at the same time, more heterogeneous mesoporous structures were formed. The crystallinity of Beta zeolite treated with NaOH&TBAOH remained at 100%, and a uniform mesoporous structure was formed, combined with good microporous properties. The bar graph on the left shows that NaOH and NaOH&TBAOH treatments significantly improved the catalytic performance of zeolite in the cracking reactions of triisopropylbenzene (TIPB), n-decane, and vacuum gas oil (VGO); especially, the NaOH&TBAOH-treated sample performed best in terms of propylene and middle distillate selectivity. This study explored the effect of optimizing the alkaline treatment method on Beta zeolite. In particular, the hierarchical pore structure formed by the combined alkaline treatment of NaOH and TBAOH can improve the overall activity and selectivity of the catalyst without destroying the acidic sites of the zeolite. This result is of great significance for industrial gas–oil cracking reactions, helping to increase the yield of propylene and light oil while reducing the formation of by-product coke [150]. The left side of Figure 13c shows the structural changes of the IM-5 zeolite and AT-2h samples obtained after alkaline treatment (desilication). In the IM-5 sample, silicon (Si) and aluminum (Al) atoms are arranged in a certain proportion in the framework, while the sample after alkaline treatment shows the removal of silicon and changes in the pore structure. In this process, the removal of Al and Si leads to changes in the surface morphology and the generation of more mesoporous structures. The right side shows the performance comparison of IM-5 and AT-2h samples in catalytic cracking reactions. The results show that the AT-2h sample exhibits higher propylene and isobutane yields. Compared with IM-5, the AT-2h sample has better molecular diffusion performance during catalytic cracking due to its larger mesopore volume, thereby improving the cracking efficiency. This enhanced molecular diffusion ability effectively improves the yield of light olefins. These results show that the high-mesoporous IM-5 sample generated by alkali treatment can show better product selectivity in catalytic cracking reactions, especially in terms of increasing propylene yield, showing significant advantages compared with traditional IM-5 [149].
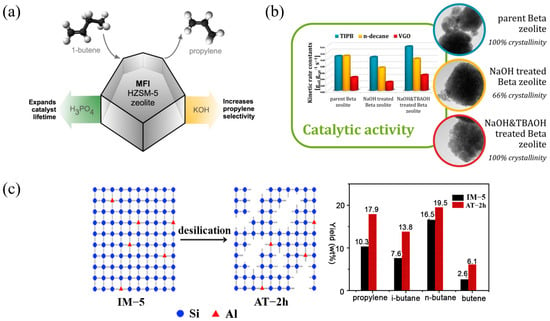
Figure 13.
(a) Controlling coke deactivation and cracking selectivity of MFI zeolite by H3PO4 or KOH modification [151]. Reproduced with permission from Eva Epelde et al., Applied Catalysis A: General; published by Elsevier, 2015 [151]; (b) catalytic cracking performance of alkaline-treated Beta zeolite in the terms of acid sites properties and their accessibility [150]. Reproduced with permission from K. Tarach et al., Journal of Catalysis; published by Elsevier, 2014 [150]; (c) highly mesoporous IM-5 zeolite prepared by alkaline treatment and its catalytic cracking performance [149]. Reproduced with permission from Qianqian Yu et al., Microporous and Mesoporous Materials; published by Elsevier, 2019 [149].
3.3.3. Metal Ion Exchange and Heteroatom Introduction
Metal ion exchange and heteroatom introduction are two of the important methods for zeolite modification. By introducing metal ions or heteroatoms, the acidity and catalytic properties of zeolite can be effectively regulated, and new active centers can be given to zeolite, thereby further improving its catalytic effect.
Metal ion exchange is the process of improving the catalytic performance of zeolites by introducing rare earth metal (such as La, Ce) or noble metal (such as Pt, Pd) cations to replace the original cations in zeolites. For example, the combination of rare earth metals and Y-type zeolites can effectively enhance the stability of acid sites, significantly improve the thermal stability and resistance to metal poisoning of zeolites, and enable them to exhibit higher activity and light oil yield in catalytic cracking reactions [152]. In addition, the introduction of metal ions can also give zeolites new active functions. For example, the introduction of noble metals such as Pt and Pd can play a role in hydrogenation and dearomatization in hydrocracking reactions, further improving the quality and yield of the target product.
Figure 14a shows the role of rare earth elements (RE3+) in partially exchanged REY zeolite, especially in the process of generating new acid sites (BAS). The figure depicts the comparison of the structures of two zeolites, USY (unmodified) and REY (after rare earth element exchange). In USY zeolite, there are strong acid sites (strong BAS), which are mainly composed of bridging Si–O(H)–Al groups and usually have strong acidity. In REY zeolite, the introduction of rare earth elements leads to the formation of new acid sites, which are located outside the framework and are usually mild (moderate acidic strength). These new acid sites are mainly derived from the RE–O(H)–Al bridging structure, which is different from the Al–O(H)–Si groups within the framework. The figure clearly shows that rare earth elements enhance the catalytic performance of zeolites by changing the distribution of acid sites, especially in terms of selectivity and stability in catalytic reactions. The figure shows how the acidity of the zeolite can be adjusted by partial exchange of rare earth elements to generate both strong and mild acid sites, thereby optimizing its catalytic performance. Figure 14b shows the role of rare earth elements (RE3+) in ultrastable REY zeolite, especially their effect on the catalytic performance of Ni-Mo/REY+Al2O3 catalyst in the vacuum gas oil hydrocracking reaction. The inset on the left shows how the addition of RE3+ affects the acid sites (BAS) of the zeolite. In this structure, the strong acid sites (i.e., the bridging Si–O(H)–Al groups) play an important role in the cracking reaction of the catalyst. By exchanging rare earth ions, especially elements such as La, Ce, and Nd, the number and strength of these acid sites can be adjusted, thereby changing the cracking selectivity of the catalyst. The graph on the right shows the selectivity of REY zeolite catalysts with different RE contents in the vacuum gas oil hydrocracking reaction. As the RE content increases, the selectivity of the catalyst for middle distillates (such as diesel and jet fuel) increases significantly, while the selectivity for light hydrocarbons (such as gas and naphtha) decreases. This is because the addition of RE reduces the concentration of strong acid sites, reduces side reactions, and thus improves the selectivity of the target product. This study shows that by optimizing the loading amount of rare earth elements, the acidic properties of REY zeolite can be effectively regulated, thereby improving the cracking performance of the catalyst, especially in the process of producing high-quality middle distillates [153].
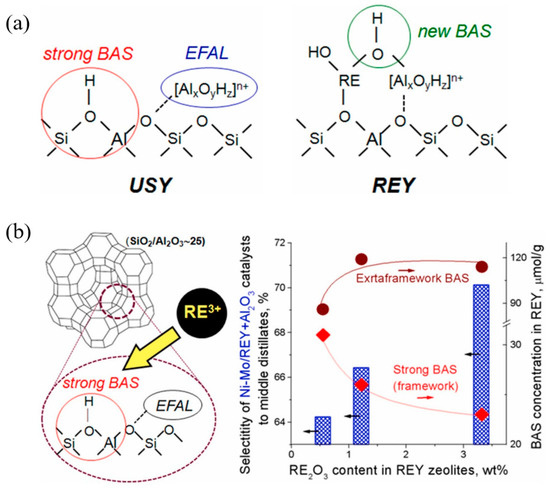
Figure 14.
(a) Formation of new BAS with moderate acidic strength for partially exchanged REY zeolite; (b) effect of rare earths on acidity of high-silica ultrastable REY zeolites and catalytic performance of NiMo/REY+Al2O3 catalysts in vacuum gas oil hydrocracking [153]. Reproduced with permission from I.G. Danilova et al., Microporous and Mesoporous Materials; published by Elsevier, 2022 [153].
On the other hand, the acidity strength and distribution of zeolites can be effectively regulated by introducing heteroatoms such as phosphorus and boron, reducing the occurrence of side reactions, and improving the selectivity of catalytic reactions. For example, phosphorus doping can reduce the strength of BAS, optimize the selectivity of catalysts, and reduce the formation of coke, while boron doping can improve the framework structure of zeolites and enhance their thermal stability and resistance to deactivation [154].
Figure 15 shows the process of preparing three-dimensional ordered porous carbon materials (3D CMs) by using metal-ion-exchanged Y-type zeolite (NaY) as a hard template and combining ethylene gas as a carbon source. The main steps include the following: 1. Metal ion exchange of NaY: the sodium ions in the NaY template are replaced by metal ions (such as Co2+, Mn2+, Cu2+ and Ni2+) to form M-Y (M is a metal ion). 2. Carbonization and graphitization: the metal ions catalyze ethylene to form a carbon framework through the chemical vapor deposition (CVD) process of ethylene and water vapor at 600 °C, and then graphitize it at 850 °C to form an ordered graphite structure. 3. Template removal: the zeolite template is removed by alkali (NaOH) treatment, followed by the use of acid (HCl) to remove the residual metal species and amorphous silicates, and finally form 3D CMs. The SEM image on the right shows the microstructure of the 3D carbon material. The granular morphology shown above retains the morphology of the template, while the high-resolution image below shows the porous characteristics and fine structure of the material. The 3D carbon materials have a high specific surface area and porous structure and can achieve in situ introduction of transition metal substances by adjusting the synthesis conditions. They have shown excellent performance in catalysis such as the oxygen reduction reaction (ORR), with comparable electrochemical stability and activity to commercial Pt/C (20 wt%) catalysts, and have great potential in energy applications such as fuel cells [154].
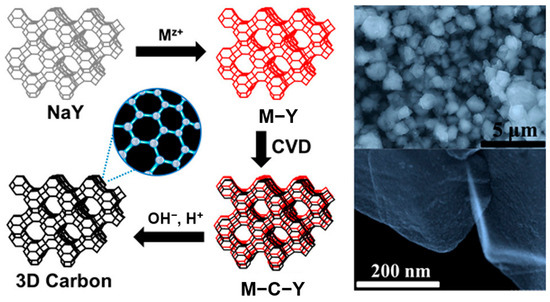
Figure 15.
Structural engineering of 3D carbon materials from transition metal ion-exchanged Y zeolite templates [154]. Reproduced with permission from Gun-hee Moon et al., Chemistry of Materials; published by American Chemical Society, 2018 [154].
In summary, the synthesis and modification of zeolites are the key to improving their catalytic performance. Through hydrothermal synthesis, post-treatment and modification, metal ion exchange, and heteroatom introduction, the precise control of zeolite structure and acidity can be achieved, and its application performance in heavy oil cracking and hydrocracking reactions can be improved, so as to better meet the needs of industrial production. The hydrothermal synthesis method uses high-temperature and high-pressure conditions to crystallize and generate a zeolite framework with a regular pore structure and controllable acid sites. It can not only accurately control the silicon–aluminum ratio and crystal size, but also meet the requirements of various industrial reactions for catalyst structure and performance. It is the core technology for preparing high-efficiency catalytic materials (such as Y-type zeolite for FCC cracking and ZSM-5 zeolite for light olefin production) and provides an important guarantee for improving industrial catalytic efficiency. Dealumination treatment (especially composite dealumination) effectively reduces excessively strong acid sites by increasing the silicon–aluminum ratio, thereby reducing coke formation and significantly improving the thermal stability of the catalyst. This is particularly important for treating heavy-oil-containing heavy metals, and can significantly extend the life of the catalyst, making it suitable for high-temperature heavy oil cracking. Acid treatment dissolves non-framework aluminum and excess acid sites in zeolite with dilute acid, effectively optimizing the acid distribution, improving the selectivity and stability of the catalyst, and reducing the probability of side reactions, so that it can reduce the generation of by-products and increase the yield of target products in heavy oil cracking reactions, but excessive treatment should be avoided to protect the integrity of the framework structure. Alkali treatment solves the diffusion limitation problem of large molecular hydrocarbons in micropores by introducing mesoporous structures, especially in the cracking of polycyclic aromatic hydrocarbons (such as residual oil), which can greatly increase the yield of light oil products and low-carbon olefins, and is an important means of upgrading heavy oil and residual oil. Metal ion exchange enhances the ability to resist metal contamination by introducing rare earth metals (such as La and Ce), while the introduction of precious metals (such as Pt and Pd) gives the catalyst hydrogenation function, significantly improving the efficient conversion ability of vacuum wax oil, making it occupy an important position in the production of light oil products. The introduction of heteroatoms (such as phosphorus and boron) improves the selectivity of the catalyst by regulating the strength of the acidic sites, reducing side reactions and coke formation, and performing particularly well in the isomerization of light hydrocarbons and the conversion of long-chain alkanes. It can effectively produce high-octane gasoline and light components, significantly improving economic benefits.
4. Study on the Reaction Mechanism of Heavy Oil Cracking
Heavy oil cracking reaction is a highly complex catalytic process involving multiple reaction pathways and the generation of intermediates. Due to the complex composition, large molecular weight, and strong chemical stability of heavy oil, the study of its reaction mechanism has important guiding significance for catalyst design and optimization. Hydrocracking of polycyclic aromatic hydrocarbons and catalytic cracking of long-chain alkanes are the two main pathways of heavy oil cracking, and their reaction processes are affected by many factors such as catalyst acidity distribution, metal active center characteristics, and reaction conditions. A deep understanding of the mechanisms of these two reactions will help achieve more efficient heavy oil conversion and generate high-value-added light oil products and low-carbon hydrocarbons.
4.1. Hydrocracking Reaction Mechanism of Condensed Aromatics
Polycyclic aromatic hydrocarbons are the components with the highest content and the most stable structure in heavy oil. Their polycyclic aromatic hydrocarbon structure gives them extremely high thermal stability and cracking resistance. The hydrocracking of polycyclic aromatic hydrocarbons usually includes steps such as hydrogenation, dealkylation, and ring opening. In the hydrocracking reaction, the conversion of polycyclic aromatic hydrocarbons requires the synergistic effect of metal–acid bifunctional catalysts. Polycyclic aromatic hydrocarbons undergo a hydrogenation saturation reaction through the metal center, and then the C-C bond is broken and rearranged at the acidic site of the zeolite, ultimately generating light oil products and gaseous hydrocarbons. The acidity and the metal activity of the catalyst have an important influence on the reaction pathway. By regulating the acidity of the zeolite, the hydrocracking pathway of polycyclic aromatic hydrocarbons can be optimized and the selectivity of the target product can be improved [155].
4.1.1. Hydrogen Activation and Metal–Acid Synergy
The activation of hydrogen is a key step in the hydrocracking reaction of polycyclic aromatic hydrocarbons. Metal active centers (such as Pt, Ni, Mo, etc.) can adsorb hydrogen molecules and promote the dissociation of hydrogen molecules to generate active hydrogen atoms. Under the action of the metal center, hydrogen atoms combine with the aromatic rings in the polycyclic aromatic hydrocarbon molecules, so that the aromatic rings are gradually hydrogenated and reduced to cycloalkanes. This process not only reduces the chemical stability of polycyclic aromatic hydrocarbons, but also provides the reaction prerequisite for the subsequent C-C bond cleavage [156].
Metal–acid synergy is crucial in the hydrocracking of polycyclic aromatic hydrocarbons. The metal center provides hydrogenation activity, while the zeolite acid sites are responsible for activating the carbon chain and promoting the cracking reaction. The BAS play a key role in the formation of carbocation intermediates, which are further cracked to form low-molecular-weight hydrocarbons through β-scission reactions or rearrangement reactions at the acid sites [155]. The efficient synergistic effect of metal and acid can greatly improve the reaction rate and product selectivity while reducing coke formation.
In addition, the activity and selectivity of hydrocracking catalysts are affected by the metal and acid distribution. The size and dispersion of metal particles and their interaction with the zeolite acid sites have a significant impact on the catalytic performance. Therefore, by optimizing the metal loading and acid distribution, efficient synergistic effects of the catalysts can be achieved [157].
Figure 16 provides a comprehensive visual summary of the study on selective hydrocracking of 1-methylnaphthalene (1-MN) to benzene, toluene, and xylene (BTX) over NiW/Beta bifunctional catalysts, with a focus on how variations in metal–acid balance (both in terms of ratio and proximity) influence catalytic performance. Panel (a) outlines the conceptual framework, emphasizing the synergistic role of metal and acid functionalities in bifunctional catalysts. It shows how the metal–acid ratio and proximity are critical factors in optimizing the conversion of 1-MN into valuable BTX products via hydrocracking. Here, metal sites (for hydrogenation/dehydrogenation) and acid sites (for isomerization/cracking) cooperate on a zeolite-based support to facilitate the reaction. Panel (b) illustrates four catalyst configurations with different metal–acid proximities: (1) NiW/A-B(60), where metal and acid sites are separated by quartz wool (centimeter-scale distance); (2) NiW/A+B(60), where metal and acid particles are physically mixed (millimeter-scale); (3) NiW/AB(60), where the two components are ground and mixed (micrometer-scale); and (4) NiW/Beta(60), where metal and acid functions are integrated into the same support (nanometer-scale). Panel (c) presents a series of graphs (A1–D) showing how product selectivity evolves with increasing 1-MN conversion over three NiW/AB(x) catalysts with different metal–acid ratios: AB(25), AB(60), and AB(150). As conversion increases, BTX selectivity is enhanced particularly in NiW/AB(60), which achieves a balance between excessive cracking and insufficient transformation. Meanwhile, the gas product selectivity (Panel D) indicates that a lower nM/nA value leads to higher production of light alkanes such as butane and propane due to enhanced cracking activity. Panel (d) compares product selectivity across catalysts with varying nM/nA values at equivalent conversion levels. It reveals that an optimal nM/nA of ~0.40 yields the highest BTX selectivity, while higher or lower values lead to a shift toward either incomplete transformation (excessive intermediates) or over-cracking (gas and low-molecular-weight products). The red dashed line shows 1-MN conversion, supporting the finding that both activity and selectivity are maximized at this intermediate metal–acid balance. In summary, Figure 16 supports the conclusion that achieving an optimal metal–acid ratio (nM/nA ≈ 0.40) and ensuring nanoscale proximity between metal and acid sites significantly enhances BTX selectivity and overall catalytic performance during 1-MN hydrocracking [155].
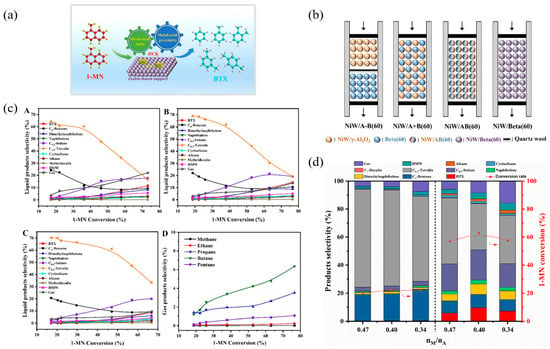
Figure 16.
(a) Selective hydrocracking of 1-methylnaphthalene to benzene/toluene/xylene (BTX) over NiW/Beta bifunctional catalyst: effects of metal–acid balance. (b) Schematic diagram of the catalysts with different metal–acid proximities. (c) 1-MN conversion versus product selectivity over (A). NiW/AB(25), (B). NiW/AB(60), and (C). NiW/AB(150) catalysts. And (D). 1-MN conversion versus gas product selectivity over NiW/AB(60) catalyst. (d) Product selectivity over NiW/AB(x) catalysts at similar conversion of 1-MN [155]. Reproduced with permission from Zunlong Hu et al., Fuel; published by Elsevier, 2024 [155].
4.1.2. Hydrogenation Ring Opening and C-C Bond Cleavage Pathways
The hydrocracking reaction process of polycyclic aromatic hydrocarbons mainly includes two stages: hydrogenation ring opening and C-C bond cleavage. In the hydrogenation ring opening stage, the aromatic rings of polycyclic aromatic hydrocarbons are gradually hydrogenated and saturated under the action of metal active centers to form cycloalkane intermediates. The reaction activity of this process is affected by the selectivity of the metal catalyst and the hydrogen partial pressure. A higher hydrogen partial pressure helps to accelerate the hydrogenation reduction in the aromatic ring [43].
The cycloalkane intermediates after hydrogenation and saturation are protonated at the acidic sites of the zeolite to form carbon cation intermediates. The carbon cation preferentially cleaves the weak C-C bonds in the molecule through β-fragmentation or rearrangement reactions to produce light oil products and low-carbon hydrocarbon products [158]. It is worth noting that the distribution and strength of the acidic sites have an important influence on the cracking path. Strong acidity is conducive to the formation of carbon cations, but too strong acidity will also lead to the rapid formation of coke, thereby causing catalyst deactivation.
Figure 17 presents the classification of β-scission reactions of alkylcarbenium ions based on the minimum carbon number (denoted as i) required in the parent n-alkane molecule for each scission type to occur. In general, a higher i value reflects greater steric feasibility and energetic favorability, leading to more stable carbocations and thus faster, more selective β-scission reactions. According to the different cleavage pathways, β-cleavage reactions are divided into four types: type A, type B, type C, and type D. β-cleavage in type A has the fastest reaction rate among all cleavage types. The reaction starts with a tertiary carbenium ion and generates another tertiary carbenium ion. For type A cleavage to occur, the carbenium ion needs at least eight carbon atoms and three branches, so it is the most common cleavage pathway, especially in catalytic cleavage processes, where it dominates. β-cleavage in type B is further subdivided into type B1 and type B2. In type B1 cleavage, the secondary carbenium ion is cleaved into a tertiary carbenium ion; while in type B2, the tertiary carbenium ion is cleaved into a secondary carbenium ion. This type of cleavage reaction requires at least seven carbon atoms and two branches, and the cleavage rate is slower than that of type A, but it is still a common cleavage pathway. β-cleavage in type C starts with a secondary carbenium ion and eventually generates another secondary carbenium ion. This type of cleavage usually requires at least six carbon atoms and a branched chain, has a slow reaction rate, and usually occurs at a higher conversion rate. The reaction conditions for this type of cleavage are relatively mild, so the reaction rate is low. β-cleavage in type D is the least common type, which is characterized by cleavage starting from the secondary carbon cation to generate the primary carbon cation. However, due to the high energy of the primary carbon cation, this cleavage reaction usually does not occur under ideal catalytic cleavage conditions. Therefore, type D cleavage reactions are rare. In general, the types of β-cleavage reactions are classified in detail from the perspective of reaction rate and reaction conditions, among which type A cleavage reactions are the fastest and most common, while types B, C, and D cleavage reactions are suitable for slower cleavage pathways and higher conversion rates, respectively. This classification aids in identifying dominant β-scission pathways and their selectivity trends across alkanes of different chain lengths [43].
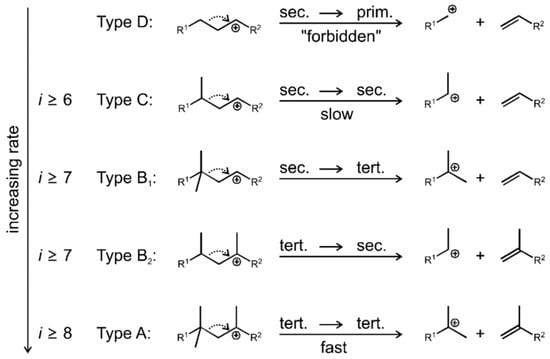
Figure 17.
Classification of β-scission reactions of alkylcarbenium ions according to the minimum carbon number of the reactant molecule (i) and branching structure required [43]. Reproduced with permission from Jens Weitkamp et al., ChemCatChem; published by John Wiley and Sons, 2012 [43].
Figure 18 systematically illustrates the hydrocracking of long-chain n-alkanes on bifunctional catalysts, focusing on the reaction pathways, product distributions, and β-scission mechanisms. In Figure 18a, the proposed pathway shows how long-chain n-alkanes (n-CiH2i+2) are converted on bifunctional catalysts like Pt/zeolite. The reaction involves dehydrogenation to alkenes, protonation to form carbocations, followed by skeletal rearrangement and β-scission. Under ideal conditions, this leads mainly to Type A β-scission, which requires tribranched carbocations and produces two branched fragments. Figure 18b compares product distributions from n-nonane on two catalysts. The large-pore Pt/Ca-Y shows a symmetrical product pattern, typical of Type A β-scission. In contrast, the medium-pore Pt/H-ZSM-5 shows more C3 and C6 products, indicating that Type B1, B2, and Type C β-scissions dominate due to spatial limitations that prevent the formation of bulky intermediates. Figure 18c shows how cracking efficiency depends on alkane chain length. On Pt/H-ZSM-5, cracking becomes significant from n-heptane, supporting that Type B and C scissions (which need less branching) are active. On Pt/Ca-Y, higher carbon numbers (like n-octane or more) are needed, in line with Type A β-scission. Figure 18d illustrates Type B1 and B2 β-scissions, where cleavage occurs between secondary and tertiary carbocations. These dominate in medium-pore catalysts. Figure 18e shows Type C β-scission, involving cleavage between two secondary centers. In summary, this hydrocracking reaction uses n-alkanes (e.g., n-nonane or longer) as reactants and produces shorter alkanes. Under hydrogen and moderate temperature, the dominant β-scission pathway depends on catalyst pore size: Type A β-scission for large-pore catalysts (Pt/Ca-Y), Type B1, B2, and Type C β-scissions for medium-pore catalysts (Pt/H-ZSM-5) due to shape selectivity.
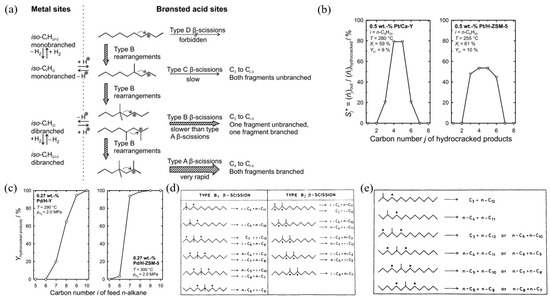
Figure 18.
(a) Proposed pathway of ideal hydrocracking of a long-chain n-alkane n-CiH2i+2 on a bifunctional catalyst. (b) Carbon number distributions of the hydrocracked products from n-nonane on bifunctional catalysts based on a large-pore (Pt/Ca-Y) and a medium-pore (Pt/H-ZSM-5) zeolite. (c) Yields of hydrocracked products from n-alkanes with five to ten carbon atoms on a large-pore (left) and a medium-pore (right) zeolite catalyst. For a given catalyst, the reaction conditions are identical for all six n-alkane reactants [43]. Reproduced with permission from Jens Weitkamp et al., ChemCatChem; published by John Wiley and Sons, 2012 [43]. (d) Hypothetical type B1 and B2 β-scissions of pentadecyl cations (the positive charge is located at either of the carbon atoms marked by an asterisk). (e) Type C β-scission of pentadecyl cations (the positive charge is located at either of the carbon atoms marked by an asterisk) [159]. Reproduced with permission from Jens Weitkamp et al., Applied Catalysis; published by Elsevier, 1983 [159].
By regulating the metal loading and acidity distribution of the catalyst, the cracking pathway of polycyclic aromatic hydrocarbons can be optimized, the occurrence of side reactions can be reduced, and the selectivity and yield of the target product can be improved. In recent years, the development of bifunctional catalysts, such as Pt-Y zeolite and NiMo/ZSM-5 zeolite, has provided more efficient catalytic materials for the hydrocracking of polycyclic aromatic hydrocarbons [160].
4.1.3. Examples of Typical Hydrocracking Reaction Mechanisms of Condensed Aromatics
Polycyclic aromatic hydrocarbons are the main components of heavy oil and must be cracked into monocyclic or bicyclic aromatic compounds before they can be used [161,162]. The production of liquid fuels with a high H/C ratio using polycyclic aromatic hydrocarbons as raw materials is an important part of the hydrocracking reaction. The following will explore the reaction mechanism of hydrocracking using polycyclic aromatic hydrocarbons as the research object. According to the U.S. EPA [163], polycyclic aromatic hydrocarbons (PAHs) are defined as compounds containing two or more fused aromatic rings. Research on the hydrocracking mechanism of polycyclic aromatic hydrocarbons can help improve the yield of the target product and guide production practice.
The simplest polycyclic aromatic hydrocarbons are naphthalene, anthracene, phenanthrene, etc., among which naphthalene is a bicyclic aromatic hydrocarbon, and anthracene and phenanthrene are tricyclic aromatic hydrocarbons. Figure 19a shows the reaction mechanism of the naphthalene hydrocracking system [164]. It can be seen from the figure that in order to produce low-carbon aromatics such as benzene, toluene, and xylene (BTX) from naphthalene hydrocracking, naphthalene must be hydrogenated to tetralin. Then, tetralin is converted into benzene, toluene, and xylene by hydrocracking. However, in this process, there may be side reactions in which tetralin is further hydrogenated to decalin and finally cracked into diesel, liquefied petroleum gas (LPG), and naphtha. Corma et al. [165] studied the hydrocracking of tetralin, as shown in Figure 19b. The results showed that the hydrocracking process of tetralin involves multiple steps, such as dealkylation, transalkylation, isomerization, cracking, and hydrogen transfer steps. The hydrocracking reaction of tetralin can be initiated by unimolecular and bimolecular mechanisms. The formation of phenylbutyltetralin (PBT) in the reaction can be explained by the bimolecular mechanism. At high reaction temperatures, the unimolecular mechanism is dominant, including the attack on the BAS on aromatic hydrocarbons and cycloalkanes. In addition, the ring opening of cycloalkanes to form alkenyl and alkylbenzenes can be explained by the formation of carbocations, β-fragmentation of carbocations, and proton cleavage reactions.
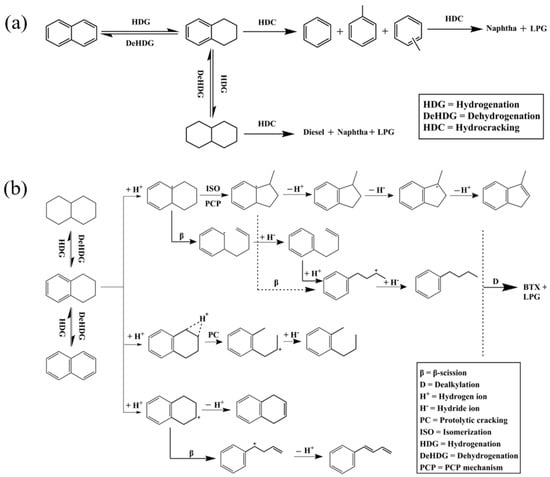
Figure 19.
Reaction mechanism of (a) naphthalene hydrocracking [164], reproduced with permission from Yeseul Choi et al., Applied Catalysis A: General; published by Elsevier, 2015 [164], and (b) tetralin hydrocracking [165], reproduced with permission from A Corma et al., Journal of Catalysis; published by Elsevier, 2001 [165].
The above typical reactions of naphthalene and tetralin are carried out in steps. Similarly, the hydrocracking process of anthracene and its homologues is currently generally considered to be carried out in steps, as shown in Figure 20. The hydrocracking of anthracene is believed to have three reaction pathways [166,167]: the hydrocracking of the intermediate ring to form biphenyl, the hydrocracking of the side ring to form naphthalene, and the hydrocracking of the intermediate ring to form alkylbenzenes. Among these three pathways, the hydrocracking of the intermediate ring consumes the least hydrogen. If the catalytic process of anthracene can be controlled so that it proceeds along the path of the hydrocracking of the intermediate ring as much as possible, then the energy consumption can be greatly reduced—that is, the amount of hydrogen used can be reduced.
Figure 20.
Reaction mechanism of anthracene hydrocracking, reproduced with permission from Honglei Fan et al., ChemCatChem; published by John Wiley and Sons, 2011 [166], reproduced with permission from J.L. Pinilla et al., Fuel; published by Elsevier, 2013 [167].
The hydrocracking process of phenanthrene and its homologues is currently generally considered to be carried out in steps. As shown in Figure 21, there are currently three views on the hydrocracking reaction mechanism of phenanthrene: reaction network 1 proposed by Beltramone et al. [168], reaction network 2 proposed by Korre et al. [169], and reaction network 3 proposed by Qian et al. [170]. Studies have shown that the hydrogenation reaction of phenanthrene is mutually converted among the five substances shown on the left side of Figure 21, and the reaction is a reversible reaction. Cornejo et al. [171] studied three hydrocracking reaction pathways of phenanthrene: (1) hydrocracking of the intermediate ring; (2) complete hydroisomerization, ring opening, and cracking of phenanthrene; and (3) hydrocracking of the side ring. The results show that phenanthrene preferentially enters the side ring for hydrocracking; that is, one side ring of phenanthrene is hydrogenated and cracked to form dicyclic aromatics, or both end rings are hydrogenated and cracked to form monocyclic aromatics (path on the right side of Figure 21).
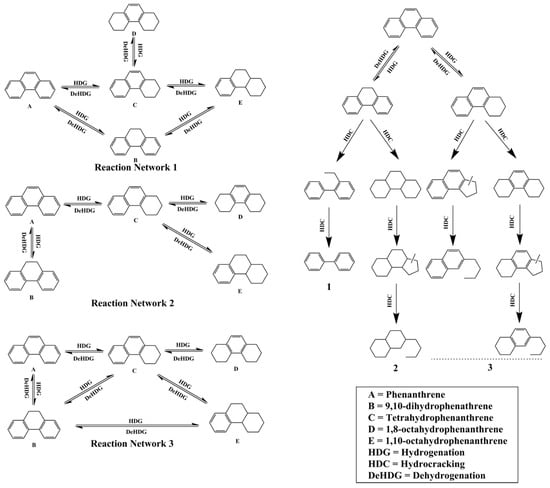
Figure 21.
Reaction mechanism of phenanthrene hydrocracking [170]. Reproduced with permission from Weihua Qian et al., Applied Catalysis A: General; published by Elsevier, 1999 [170].
Polycyclic aromatic hydrocarbons (PAHs) with four and more than four condensed aromatic rings, such as pyrene and coronene, present significant challenges for catalytic conversion due to their high thermal stability and bulky molecular structures. Figure 22 shows the possible hydrocracking reaction pathway of pyrene under the action of a Y-type zeolite catalyst, which clearly reflects the process of converting polycyclic aromatic hydrocarbons into BTEXN (benzene, toluene, ethylbenzene, xylene, and naphthalene)-type light aromatic hydrocarbons. This pathway first undergoes hydrogenation saturation reaction of aromatic rings, followed by ring cleavage reaction under the action of strong acid sites of the zeolite catalyst to generate aromatic intermediates with smaller structures, and finally generates light aromatic hydrocarbons through rupture and rearrangement. The intermediates include some hydrogenation products (such as tetrahydropyrene), isomeric aromatics (such as phenanthrene), and dicyclic products (such as naphthalene), which indicate that the cracking process of large molecular PAHs has strong structural selectivity and multi-step reaction characteristics. This study verifies that PAHs with larger molecular size can also achieve effective catalytic cracking under the synergistic effect of suitable acidity and pore structure, providing a reference model for in-depth understanding of the cracking pathway of PAHs with more condensed aromatic rings (such as coronene), and emphasizes the key role of regulating zeolite pore structure and acidity in the activation and selective conversion of large molecular PAHs.
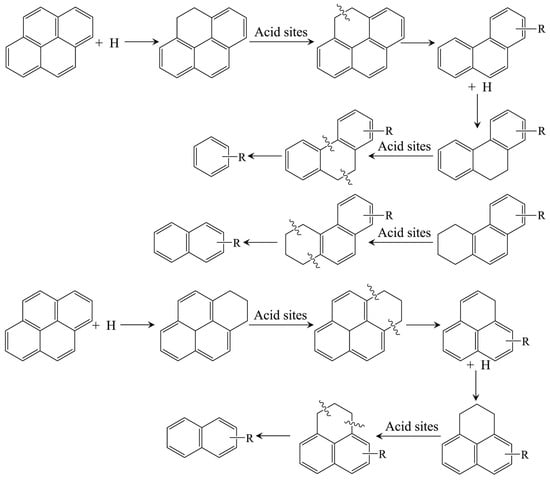
Figure 22.
Possible path of BTEXN formation from pyrene catalytic pyrolysis over Y-type zeolites [172]. Reproduced with permission from Peng Lv et al., Journal of the Energy Institute; published by Elsevier, 2020 [172].
Dibenzothiophene (DBT), a representative sulfur-containing polycyclic aromatic hydrocarbon in coal tar, undergoes complex transformation during catalytic pyrolysis over Y-type zeolites. The reaction mechanism proposed in Figure 23 involves two main pathways. In the first route, a benzene ring of DBT is initially hydrogenated and subsequently cleaved at the acid sites of the zeolite, forming benzothiophene derivatives. These intermediates undergo further hydrogenation and ring-opening reactions, ultimately yielding light aromatic compounds such as benzene, toluene, and phenylmercaptan. In the second pathway, the cleavage of the sulfur–carbon bond in DBT produces biphenyl and fluorene-type intermediates. These species are further cracked and stabilized by hydrogen donors within the zeolite pores to form BTEXN components. This mechanism closely resembles hydrodesulfurization (HDS) behavior and underscores the dual function of Y-type zeolites—providing both acidity and hydrogen transfer capability—for the efficient conversion of sulfur-rich aromatic heterocycles into valuable light aromatics. The hierarchical pore structure of the modified zeolites further facilitates the diffusion of bulky DBT molecules, improving accessibility to active sites and enhancing overall catalytic performance.
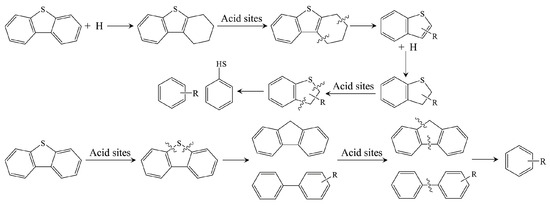
Figure 23.
Possible path of BTEXN formation from dibenzothiophene catalytic pyrolysis over Y-type zeolites [172]. Reproduced with permission from Peng Lv et al., Journal of the Energy Institute; published by Elsevier, 2020 [172].
4.2. Catalytic Cracking Reaction Mechanism of Long-Chain Alkanes
The catalytic cracking of long-chain alkanes is an important reaction pathway in the catalytic cracking of heavy oil. It mainly breaks the C-C bond through the acidic sites of zeolite to produce light oil products and low-carbon olefins. According to the reaction conditions and catalyst type, the catalytic cracking pathway of long-chain alkanes can be divided into two mechanisms: unimolecular cracking and bimolecular cracking. The catalytic cracking of long-chain alkanes mainly proceeds through the carbon cation mechanism, including steps such as protonation, intermediate generation, and β-fragmentation. Studies have shown that the central C-C bond is easier to break than the terminal C-C bond, and the acid strength of the catalyst has a significant effect on the reaction activity [173].
4.2.1. Unimolecular and Bimolecular Cracking Mechanisms
In the unimolecular cracking mechanism, long-chain alkane molecules are first protonated at the acidic sites of the zeolite to form carbon cation intermediates. The carbon cations selectively break the weak C-C bonds in the molecules through β-scission reactions to produce low-carbon olefins and light oil products [174]. The unimolecular cracking mechanism usually occurs under high-temperature conditions and is suitable for catalysts with weak acidity, such as ZSM-5 zeolite with a high silicon–aluminum ratio. This reaction pathway has high selectivity, but the reaction rate is relatively low.
In contrast, the bimolecular cracking mechanism involves the interaction between two alkane molecules. In this mechanism, one alkane molecule is protonated at the acidic site to form a carbocation intermediate; the carbocation then undergoes a hydrogen transfer reaction with another alkane molecule to form a new carbocation, which results in molecular cracking to produce light olefins and gaseous hydrocarbons [175]. The bimolecular cracking mechanism is suitable for catalysts with stronger acidity, such as Y-type zeolite with a low silicon–aluminum ratio, and has a faster reaction rate, but is prone to produce coke byproducts.
4.2.2. Effect of Zeolite Acidity on Reaction Pathway
The acidity of zeolite is a key factor in determining the cracking path of long-chain alkanes. The strength of the acidity and the distribution of acidic sites directly affect the generation of carbon cations and the progress of the cracking reaction. Stronger acidity is conducive to the progress of bimolecular cracking reactions with high reaction rates, but it is also easy to cause side reactions and rapid generation of coke, which can cause catalyst deactivation [151,176]. Weaker acidity is more suitable for unimolecular cracking reactions, which is conducive to improving the selectivity of the reaction and reducing the formation of coke.
By adjusting the silicon–aluminum ratio of zeolite and introducing rare earth metal additives (such as La, Ce) and precious metals (such as Pt, Pd), the acidity distribution of zeolite can be effectively optimized, and the stability and anti-deactivation performance of the catalyst can be improved. For example, rare earth-modified Y-type zeolite exhibits higher cracking selectivity for long-chain alkanes, effectively improving the yield of light oil products and low-carbon olefins [153].
Figure 24 shows the control of different acid strengths on the reaction pathways in the catalytic cracking of 1-pentene. The figure illustrates the different effects of strong and weak acid sites on the cracking pathways of 1-pentene. Under the catalytic action of strong acid sites, cracking pathways I′ and II′-1 are more active, where pathway I′ is the cracking of C5= to C2= and C3=, while pathway II′-1 is the cracking of C6= to C2= and C4=. Conversely, weak acid sites tend to promote pathway II′ (2C5= generates C10+, which is then cracked to C4= and C6=) and pathway II′-2 (C6= is cracked to 2C3=). The two main reaction pathways (I′ and II′) shown in the figure show that strong acid sites promote more β-cleavage reactions, while weak acid sites favor the cracking of more stable secondary and tertiary carbocation intermediates, thus affecting the selectivity and product distribution of the reaction. By adjusting the acid strength on the ZSM-5 catalyst, the selectivity for propylene and the propylene/ethylene ratio can be significantly improved, thereby optimizing the efficiency of the catalytic cracking process [174].
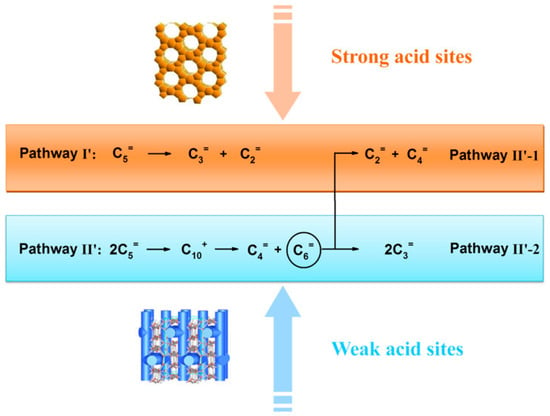
Figure 24.
Acid strength controlled reaction pathways for the catalytic cracking of 1-pentene to propene over ZSM-5 [174]. Reproduced with permission from Long F. Lin et al., ACS Catalysis; published by American Chemical Society, 2015 [174].
4.2.3. Examples of Typical Catalytic Cracking Reaction Mechanisms of Long-Chain Alkanes
Long-chain alkanes are also the main components of heavy oil and must be cracked into short-chain small molecules before they can be used. The production of liquid fuels with a high H/C ratio using long-chain alkanes as raw materials is also an important part of catalytic cracking reactions. Although it is easy for catalytic cracking of alkanes to occur, there are huge challenges in studying the cracking reaction mechanism [177]. The number of carbon atoms has a significant effect on the reactivity of alkanes. Generally speaking, the reactivity of normal alkanes increases with the number of carbon atoms [178,179]. In addition, branched alkanes are more reactive than straight-chain alkanes when the number of carbon atoms is the same [180,181]. The cracking products of alkanes under mild conditions can generally be divided into two types: one is hydrocarbons produced by chain isomerization, and the other is lighter hydrocarbons produced by carbon–carbon bond cleavage [177]. The carbon cation mechanism can be used to explain the catalytic cracking of alkanes under the action of strong acids [180,182]. The following uses long-chain alkanes as research objects to explore the reaction mechanism of catalytic cracking.
As shown in Figure 25, the reaction mechanism of the catalytic cracking of n-pentane on the BAS of zeolite mainly has two pathways [183], namely, the formation of product ethane–propylene (PROD) through transition state 2 (TS2) and the formation of product ethylene–propane (PROD′) through transition state 2′ (TS2′). The experimental results show that both pathways show major contributions, proving that ethane, ethylene, propane, and propylene are the main products. The catalytic cracking of n-pentane is based on a unimolecular reaction mechanism—that is, it includes two steps: the cleavage of n-pentane protons to form pentene intermediates and the dissociation of pentene intermediates to form ethane–propylene and ethylene–propane. As shown in Figure 25, n-pentane is initially adsorbed on the BAS of the zeolite (ADS), and then the proton on the zeolite surface is transferred to the C2-C3 bond of the pentane molecule (TS1), and then the pentene intermediate (INT) is generated. Thereafter, the pentene intermediate decomposes into ethane–propylene or ethylene–propane. For the ethane–propylene pathway, ethane is formed by accepting a proton from a BAS, and propylene is formed by returning a proton to the zeolite framework (TS2). In the ethylene–propane pathway, ethylene is formed by transferring a proton back to the zeolite framework, and propane is formed by accepting a proton from the zeolite surface (TS2′). Finally, ethane–propylene and ethylene–propane products (PROD and PROD′) are formed on the zeolite surface.
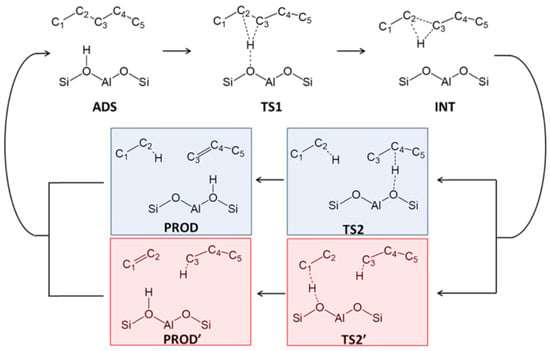
Figure 25.
Reaction mechanism of catalytic cracking of n-pentane into ethane–propylene (PROD) and ethylene–propane products (PROD′) [183]. Reproduced with permission from Anawat Thivasasith et al., Physical chemistry chemical physics; published by Royal Society of Chemistry, 2019 [183].
Figure 26 shows the monomolecular protonation cracking mechanism of n-hexane. In this reaction process, n-hexane molecules react with the BAS of zeolite, and protons (H+) react with the C-C or C-H bonds of n-hexane to generate transition intermediates, which are eventually cracked into small molecular products such as ethane and methane. In the figure, proton attacks on different carbon–carbon bonds (for example, C3-C4 or C2-C3) will lead to different cracking products, indicating that the cracking pathways and products have a certain diversity. This mechanism reveals the key steps of protonation cracking and the effect of proton attacks at different positions on product selectivity. For the protonation cracking of n-hexane, the energy difference between the proton and different C-C bonds determines the distribution of cracking products. In some cases, the reaction may generate a transition state carbenium ion, which is then decomposed into products such as light alkanes or hydrogen [184].
Figure 26.
Monomolecular protonation cracking mechanism of n-hexane [184]. Reproduced with permission from Dongdong Chen et al., Applied Catalysis A: General; published by Elsevier, 2023 [184].
Figure 27 shows the main reaction pathways and product distribution of n-heptane thermal cracking under the conditions of 973 K, 0.5 MPa, and 2 mL/min. The thick arrows indicate the reaction pathways that contribute significantly to the formation of the main products. During the cracking process, n-heptane mainly undergoes carbon–carbon bond cleavage reactions, as well as key steps such as hydrogen abstraction and free radical recombination, and eventually produces small molecular products such as ethylene (C2H4), propylene (C3H6), and methane (CH4). In the reaction pathway, n-heptane (n-C7H16) first undergoes hydrogen abstraction reactions to generate alkyl radicals at different positions, such as C7H15-2, C7H15-3, etc. These radicals then undergo β-cleavage reactions to generate smaller olefins and alkanes. For example, C5H11-1 is cracked to produce ethylene (C2H4) and propylene (C3H6). Similarly, short-chain alkyl radicals, such as nC3H7 and pC4H9, undergo further β-scission to form ethylene and other major products. This pathway indicates that ethylene is the main product in the cracking process of n-heptane, and its production pathway mainly comes from the cleavage of carbon–carbon bonds and free radical reactions. In addition, since temperature and pressure have an important influence on the rate of free radical reactions, the reaction pathway and product distribution may vary under different conditions. This also explains why changes in pressure and temperature can significantly affect the yield and selectivity of alkanes and alkenes [185].
Figure 27.
The main reaction pathway of n-heptane pyrolysis under 973 K, 0.5 MPa, and 2 mL/min (thick arrows denote the primary contribution) [185]. Reproduced with permission from Yong Wu et al., Combustion and Flame; published by Elsevier, 2018 [185].
As shown in Figure 28, there are two mechanisms for the catalytic cracking of n-octane, namely, the unimolecular catalytic cracking and the bimolecular catalytic cracking of n-octane [186]. The unimolecular catalytic cracking mechanism of n-octane generally undergoes two steps. The first step is the protonation of n-octane, and the second step is the decomposition of the protonated intermediate to produce n-hexane and ethylene. The bimolecular catalytic cracking mechanism of n-octane includes the hydride transfer between the adsorbed carbon cations and alkanes, and the subsequent isomerization and β-fragmentation. The bimolecular cracking of n-octane is believed to undergo four steps: hydride transfer, isomerization, intermolecular hydrogen transfer, and β-fragmentation. The products of the bimolecular cracking of n-octane are isobutylene and n-butane.
Figure 28.
Unimolecular and bimolecular cracking mechanisms of n-octane on zeolites [186]. Reproduced with permission from Jia Fu et al., The Journal of Physical Chemistry C; published by American Chemical Society, 2018 [186].
The hydrocracking of pristane (2,6,10,14-tetramethylpentadecane) reveals unique mechanistic characteristics that distinguish it from conventional linear or lightly branched alkanes. As shown in Figure 29, specific β-scission pathways dominate the conversion of pristane into C9 and C10 fragments, especially under low-conversion conditions over bifunctional catalysts such as Pt/SiO2–Al2O3. These pathways involve the initial formation of secondary alkylcarbenium ions at branching sites, followed by methyl shifts and selective cleavage of carbon–carbon bonds adjacent to internal methyl groups. Two distinct reaction routes are illustrated: pathway A leads to 2,6-dimethylheptane (2,6-DMC7) and 2-methylnonene (2-MC9) via a single β-scission; pathway B, enabled by additional methyl shifts, generates 2-methyloctane (2-MC8) and 2,7-dimethyloctane (2,7-DMC8). Notably, the preference for cracking near inner branching points rather than terminal methyl groups reflects the influence of carbenium ion stability and steric accessibility. This mechanistic insight highlights the need to revise traditional cracking models—typically based on linear alkanes—when dealing with highly branched, long-chain hydrocarbons derived from renewable or complex feedstocks. The findings emphasize that branching configuration and molecular symmetry play pivotal roles in determining both the scission position and the distribution of cracking products.
Figure 29.
Examples of reaction pathways leading to the cracking of pristane into C9 and C10 fragments. (A). illustrates a β-scission mechanism following methyl shifts resulting in 2,6-dimethylheptane (2,6-DMC₇) and 2-methylnonane (2-MC₉); (B). depicts an alternative cracking pathway involving multiple methyl shifts and cyclization of a tribranched carbocation, leading to 2-methyloctane (2-MC₈) and 2,7-dimethyloctene (2,7-DMC₈). Positive charges are omitted when two possibilities are present [187]. Reproduced with permission from G. Burnens et al., Journal of Catalysis; published by Elsevier, 2011 [187].
The hydroisomerization and hydrocracking of n-hexadecane (n-C16) are strongly influenced by the spatial intimacy between metal and acid sites in bifunctional catalysts. As illustrated in Figure 30, the Pt/48-A (atomic intimacy), Pt/A-48 (nanoscale intimacy), and Pt/A+48 (microscale intimacy) catalysts offer distinct mechanistic pathways. In Pt/48-A, where Pt is deposited directly within the microporous ZSM-48 channels, intermediates are formed via pore-mouth or key-lock mechanisms and rearranged through protonated cyclopropane (PCP) pathways. Due to diffusion limitations in the zeolite channels, n-C16 often undergoes type C β-scission, yielding linear hydrocracking products. In Pt/A-48, the metal and acid sites are in nanoscale proximity on separate supports, facilitating balanced transport and intermediate conversion. This favors type B β-scission via di-branched carbenium ions, producing both high mono-branched isomer selectivity and moderate hydrocracking. In contrast, Pt/A+48, with physically mixed components and microscale intimacy, allows for extensive acid-site engagement, leading to over-cracking via type A β-scission and reduced isomer yields. The figure captures how acid-step count (nas), spatial confinement, and branching patterns collectively shape product selectivity and reaction efficiency. Importantly, the nanoscale metal–acid intimacy in Pt/A-48 offers the optimal balance—maximizing isomer formation while minimizing undesired cracking—representing a promising strategy for the shape-selective conversion of long-chain alkanes into high-value products.
Figure 30.
The main reaction network and mechanism for hydroisomerization/hydrocracking of n-C16 over Pt/48-A, Pt/A-48, and Pt/A+48 catalysts; the solid line arrows represent the main reaction and the dotted line arrows represent the possible reactions [188]. Reproduced with permission from Wenli Zhao et al., Fuel; published by Elsevier, 2023 [188].
4.3. Catalytic Cracking Reaction Mechanism of Resins and Asphaltenes
Resins and asphaltenes represent the most complex and refractory components in heavy oil. These macromolecular species are enriched with heteroatoms (such as N, S, and O), transition metals (Ni, V), and multi-ring aromatic structures, contributing significantly to catalyst fouling, coke formation, and deactivation. Their cracking behavior differs substantially from that of long-chain alkanes or polycyclic aromatic hydrocarbons due to their structural complexity and strong intermolecular associations [21,189].
The catalytic cracking of asphaltenes involves multiple reaction steps, typically initiated by thermal activation or hydrogenation of the fused aromatic rings, followed by ring-opening, dealkylation, and fragmentation into lighter aromatics such as benzene, toluene, and naphthalene, and intermediates like phenanthrene and fluorene. The large molecular size and rigidity of asphaltenes pose diffusion limitations, which are mitigated by catalysts with hierarchical pore structures or mesoporosity, such as modified Y-type or Beta zeolites. The high acidity of conventional zeolites promotes cracking but also accelerates coke formation, necessitating the use of moderately acidic or dual-functional catalysts for improved stability [190,191,192,193,194,195].
Resins, being structurally less condensed than asphaltenes but still polar and heavy, exhibit somewhat higher reactivity. Their catalytic cracking typically yields light cycle oil (LCO) and middle distillates, and under optimal pore and acidity configurations, can also contribute to light aromatics. Nevertheless, resins serve as precursors to coke, particularly when they accumulate near active acid sites without sufficient diffusion or hydrogenation. Catalysts with mesoporosity or moderate acidity, such as amorphous silica–alumina (ASA) or Al-MCM-41, have demonstrated improved tolerance to resin-derived fouling and prolonged catalytic activity [190,191,192,193,194,195].
Figure 31 illustrates the catalytic hydrothermal upgrading mechanism of heavy oil using nano-Fe2O3 within a porous medium, simulating reservoir conditions. Part (a) presents a schematic of the in situ reaction pathway, where water undergoes dissociation facilitated by thermally activated Fe3+ species. This leads to hydrogen generation via the water–gas shift reaction (WGSR), producing active H2 and OH− species. These intermediates participate in hydrogen transfer reactions that cleave heteroatom-containing bonds (e.g., C-S, C-O, C-N), thus removing nitrogen, sulfur, and oxygen functionalities from complex polycyclic aromatic structures found in crude oil. The reaction occurs within a sand-packed porous system embedded with Fe2O3 nanoparticles, which not only catalyze bond cleavage but are also stabilized by quartz surfaces, maintaining their dispersion and catalytic activity. The end result is the conversion of high-viscosity crude oil into a low-viscosity upgraded product rich in saturated and light aromatic hydrocarbons. Part (b) complements this mechanistic overview through FTIR characterization of resins and asphaltenes at various treatment temperatures. The spectra reveal progressive changes in functional group vibrations as the temperature increases, including the weakening of peaks corresponding to polyaromatic C=C and aliphatic C-H bonds, alongside the appearance of new bands indicative of oxidative and desulfurization transformations. These findings support the conclusion that thermal and catalytic processes disrupt the macromolecular frameworks of heavy fractions, leading to dealkylation, ring opening, and the generation of smaller, more desirable hydrocarbons. Therefore, Figure 31 confirms the efficacy of nano-Fe2O3-catalyzed hydrothermal processes in achieving in situ heavy oil upgrading, highlighting the dual role of iron oxide nanoparticles in facilitating electron transfer reactions and promoting heterolytic bond cleavage under reservoir-like conditions [190].
Figure 31.
(a) In situ hydrothermal upgrading and mechanism of heavy oil with nano-Fe2O3 in the porous media. (b) The FTIR characterization of resin and asphaltene [190]. Reproduced with permission from Jingjing Li et al., Journal of Analytical and Applied Pyrolysis; published by Elsevier, 2024 [190].
Figure 32 offers a comprehensive view of the hydrothermal cracking pathways of various model compounds representing functional groups found in heavy oil, particularly resins and asphaltenes. Panel (a) shows the GC-MS results of six model compounds: thiophene, fluorenone, diphenyl ether, dodecylbenzene, phenanthrene, and quinoline, before and after catalytic hydrothermal treatment. These represent typical heteroatom-containing and aromatic structures in heavy oil. After treatment with nano-Fe2O3, the original compounds undergo significant transformation, indicating bond cleavage and structural simplification. For instance, thiophene shows desulfurization, and dodecylbenzene shows side-chain cracking. Panel (b) proposes reaction mechanisms corresponding to these observations: dibenzothiophene undergoes C-S bond cleavage to form biphenyl and H2S; fluorenone is hydrogenated and deoxygenated to fluorene and simpler aromatic structures; diphenyl ether undergoes hydrodeoxygenation, eventually yielding benzene derivatives; dodecylbenzene experiences C-C bond scission, producing alkylbenzenes of varying lengths; phenanthrene undergoes ring hydrogenation; quinoline remains mostly intact due to the high bond energy of the C=N bond. These findings reinforce that nano-Fe2O3 effectively promotes the cleavage of heteroatom bonds (C-S, C-O, C=O, C-N) and side chains in heavy fractions under hydrothermal conditions. The electron transfer between Fe3+/Fe2+ and the catalytic role of H+ generated from water dissociation are central to these mechanisms, enabling depolymerization and hydrogenation of complex aromatics in heavy oil [190].
Figure 32.
(a) The GC-MS of model molecular compounds: (A). thiophene, (B). fluorenone, (C). diphenyl ether, (D). dodecylbenzene, (E). phenanthrene, and (F). quinoline. (b) The reaction mechanism of dibenzothiophene, fluorenone, diphenyl ether, dodecyl benzene, phenanthrene, and quinoline [190]. Reproduced with permission from Jingjing Li et al., Journal of Analytical and Applied Pyrolysis; published by Elsevier, 2024 [190].
Figure 33 illustrates the catalytic aquathermolysis mechanism of heavy oil using water-soluble aluminum-based catalysts, Al(CH3COO)3 and Al2(SO4)3, under CO2 and N2 atmospheres. Panel (a) schematically demonstrates how these catalysts hydrolyze in water at elevated temperatures, forming AlxOy(AlOH) species that promote catalytic cracking via acid sites. This facilitates C-C and C–heteroatom bond cleavage, dehydrogenation, and isomerization reactions. Panel (b) presents FTIR spectra of heavy oil and asphaltene fractions, revealing that after treatment, the intensity of aliphatic (CH2, CH3) bands increases while aromatic C=C and sulfoxide-associated bands decrease—indicating hydrogenation, desulfurization, and aromatics reduction. Panel (c) shows compositional shifts: saturates increase, while aromatics, resins, and asphaltenes significantly decrease—especially under CO2 atmosphere and in the presence of Al2(SO4)3. This highlights the enhanced efficiency of CO2 in promoting structural breakdown and upgrading, with the sulfate catalyst demonstrating superior performance in reducing heavy fractions [191].
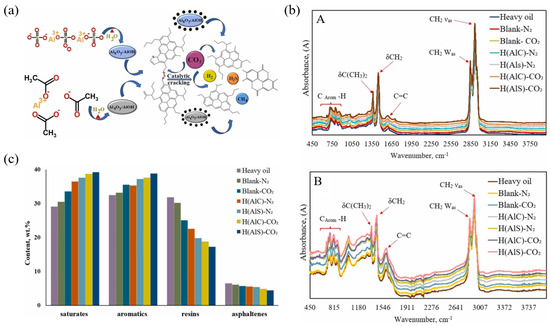
Figure 33.
(a) This schematically presents the mechanism of catalytic aquathermolysis of heavy oil. (b) IR spectra of (A). heavy oil and (B). asphaltene fractions before and after aquathermolysis. (c) Group composition of heavy oil samples before and after catalytic aquathermolysis [191]. Reproduced with permission from Yasser I.I. Abdelsalam et al., Applied Catalysis A: General; published by Elsevier, 2025 [191].
The successful transformation of resins and asphaltenes requires a balance between acid site strength, pore accessibility, and thermal stability. Bifunctional catalysts that incorporate metallic hydrogenation sites (e.g., Ni, Mo) with acid supports can further enhance performance by pre-treating the polyaromatics through partial hydrogenation, thus increasing cracking susceptibility and reducing coke formation. Therefore, understanding the molecular behavior of these heavy fractions under catalytic conditions is key to designing selective and robust catalysts for residue upgrading.
The main components of heavy oil, such as polycyclic aromatic hydrocarbons, long-chain alkanes, resins, and asphaltene, have different cracking reaction pathways and challenges. Through in-depth research on their hydrocracking and catalytic cracking mechanisms, combined with the optimization design of catalyst metal active centers, acid distribution, and pore structure, the heavy oil upgrading efficiency and light oil output rate can be significantly improved, thus providing theoretical guidance and technical support for the efficient and clean utilization of heavy oil resources.
5. Challenges and Optimization Strategies of Zeolite Catalysts
Zeolite catalysts show excellent catalytic performance in heavy oil cracking and hydrocracking reactions, but they still face many challenges in industrial applications, including catalyst deactivation, uneven acidity regulation, limited diffusion, insufficient metal loading synergy, and lack of combination of theoretical calculations and experimental data. The existence of these problems limits the performance stability and application life of zeolite catalysts and affects the heavy oil conversion efficiency and product quality. Therefore, in-depth research and improvement on the deactivation mechanism, acidity regulation, structural optimization, metal modification, and combination of theoretical calculations and experimental data of zeolite catalysts have important practical significance and application value.
5.1. Catalyst Deactivation Mechanism and Anti-Deactivation Modification
Zeolite catalyst deactivation is one of the most critical issues in heavy oil cracking reactions, which is mainly manifested in three aspects: coke deposition, metal contamination, and loss of acid sites. These deactivation mechanisms interact with each other during heavy oil processing, gradually reducing the activity of the catalyst, thereby affecting the selectivity and conversion efficiency of the reaction.
Coke deposition is one of the main causes of catalyst deactivation, especially on strongly acidic zeolites [196]. Since zeolites have a large number of BAS, hydrocarbon molecules easily form carbon cation intermediates on the acid sites. These intermediates further polymerize at high temperatures to form coke that is insoluble in the reaction system. Coke deposition blocks the pores of zeolites and reduces the effective utilization of active sites, leading to catalyst deactivation. In addition, excessive acidity accelerates the occurrence of side reactions and increases the rate of coke formation. Therefore, acidity regulation and structural modification, such as dealumination and introduction of rare earth metals, can effectively inhibit the formation of coke and delay catalyst deactivation.
Metal contamination is another significant catalyst deactivation mechanism during heavy oil cracking. Heavy oil often contains high concentrations of heavy metals such as nickel (Ni) and vanadium (V). These metals are deposited on the surface or in the pores of zeolite under high-temperature reaction conditions, causing acid site poisoning and catalyst pore blockage [197]. In response to the metal contamination problem, researchers have proposed a variety of strategies to resist metal poisoning, such as introducing metal scavengers (such as phosphides and rare earth metal oxides) into the catalyst. These additives can react with heavy metal ions, thereby reducing the impact of heavy metals on acid sites. In addition, by optimizing the surface structure of zeolite and reducing the adsorption rate of metal ions, the catalyst’s ability to resist metal contamination can also be effectively improved.
The loss of acid sites is mainly related to the detachment of aluminum from the zeolite framework under high-temperature reaction conditions. During long-term use of zeolite, framework aluminum atoms are easily detached to form non-framework aluminum, which not only reduces the number of acid sites but also affects the structural stability of the zeolite [198]. To this end, researchers have introduced heteroatoms (such as phosphorus and boron) or rare earth metals that stabilize the framework structure to enhance the thermal stability of zeolites, thereby improving the catalyst’s resistance to deactivation.
Figure 34a shows a schematic diagram of the coke deposition behavior on the ZSM-5 catalyst with different defect contents and pore structures. The figure shows the changes in the generation and deposition position of coke precursors under three different mesoporosity (low, medium, and high) and defect sites. The formation of coke is significantly affected by the pore structure and internal defect sites. Specifically, when the mesoporosity is low and the defect sites are high (such as internal silanol groups), the coke deposition inside the catalyst is more significant, which accelerates the deactivation of the catalyst. In the case of high mesoporosity and low defect sites, the coke is deposited more on the outer surface, which helps to slow down the deactivation of the catalyst. It shows that the ZSM-5 catalyst with low mesoporosity and high defect sites can easily generate coke precursors inside, and through the promotion of internal defect sites, heavier coke is formed. This internal coke has a greater impact on the catalyst activity. In contrast, for catalysts with higher mesoporosity and fewer defect sites, although the total amount of coke is similar, the coke is deposited more on the external surface, which slows down the accumulation of internal coke and inhibits the deactivation of the catalyst to a certain extent. Through the optimization of this structural design, studies have shown that the active life of ZSM-5 catalysts in aromatization reactions can be significantly extended by controlling the pore structure and the density of defect sites [196]. Figure 34b shows the coke yield of aluminum nitride FCC catalysts at different residual BAS concentrations. The data show that with the increase in residual BAS concentration (mmol/g), the coke yield gradually increases, and different metal contents (3.0, 3.5, 4.0) have different effects on coke yield. The experiment in this figure shows that the change in the acid sites of aluminum nitride catalysts under metal contamination directly affects the amount of coke generated. When the residual BAS concentration is high, the acid sites of the FCC catalyst increase, resulting in more coke formation during the catalytic reaction. In particular, with the increase in BAS concentration, more acid sites on the catalyst surface participate in the reaction, thereby increasing the amount of coke generated [197].

Figure 34.
(a) Schematic illustration of coke deposition behaviors on zeolite catalysts with different defect contents and porous structures. Dark blue squares indicate areas with coke deposition; light blue squares represent coke precursors; blue dots mark defect sites. The combination of mesoporosity and low defect sites (far right) leads to the least internal coke formation [196]. Reproduced with permission from Kyungho Lee et al., Journal of Catalysis; published by Elsevier, 2017 [196]; (b) coke yield against residual BAS concentration for vanadium-poisoned catalysts [197]. Reproduced with permission from U.J. Etim et al., Microporous and Mesoporous Materials; published by Elsevier, 2019 [197].
5.2. Acidity Control and Structure Optimization
The acidity of zeolite directly determines its activity and selectivity in catalytic cracking reactions. Reasonable acidity regulation can effectively balance the reaction rate and product selectivity and reduce side reactions and coke formation. The acidity of zeolite mainly comes from BAS and LAS, which have different action mechanisms in catalytic cracking reactions [108].
By adjusting the silicon–aluminum ratio, the acidity strength and quantity of zeolites can be optimized. Zeolites with a high silicon–aluminum ratio have weaker acidity, which helps to inhibit excessive polymerization of carbon cations and reduce coke formation, making them suitable for catalytic cracking reactions with higher selectivity. Zeolites with a low silicon–aluminum ratio have stronger acidity, which can accelerate the cracking reaction rate, but are also prone to side reactions and coke formation [199].
Structural optimization is mainly achieved through post-treatment methods, including dealumination, acid treatment, and alkali treatment. Dealumination can improve the acid distribution and strength and reduce the coke formation rate by removing overly strong acid sites; acid treatment can remove non-framework aluminum, increase the silicon–aluminum ratio of zeolite, enhance thermal stability and resistance to metal contamination; alkali treatment partially dissolves the framework, introduces a mesoporous structure, alleviates diffusion limitations, and improves the overall activity of the catalyst [200,201].
In recent years, the development of dual-acid catalysts has provided a new path for acid regulation. By introducing metal active components to form a metal–acid synergistic catalytic system, it is possible to achieve hydrogenation activity and regulate the cracking reaction path, thereby improving the overall performance of the catalyst.
Figure 35 shows a schematic diagram of the formation of BAS on the surface of γ-Al2O3 after partial fluorination treatment, and the effect of fluorination on the acid sites on the catalyst surface is detailed in the figure. In the figure, the unmodified γ-Al2O3 surface mainly contains LAS, while after partial fluorination treatment, BAS are generated on the surface, which is beneficial for hydrocarbon conversion reactions. Through the incorporation of fluorine, some hydroxyl groups are replaced by fluorine to form BAS. These acid sites contribute to the cracking of hydrocarbon molecules during the catalytic process, thereby improving the reaction efficiency. Partially fluorinated γ-Al2O3 improves the catalytic performance by replacing some of the hydroxyl groups of the hexacoordinated aluminum atoms to form fluorides. In particular, this surface fluorination treatment significantly enhances the BAS acidity while reducing the LAS acidity, thereby suppressing the formation of excessive coke and improving the oil conversion rate, especially in the cracking of heavy oil (VGO). The fluorinated catalysts (such as USY/Kaolin/BF-H2) showed a brighter color than the untreated γ-Al2O3, and the color of the catalyst produced after the reaction was also lighter, indicating that it had a better catalytic effect and lower coke production [108].
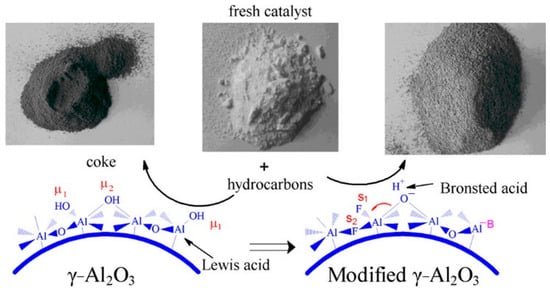
Figure 35.
Schematic diagram for the formation of a BAS on the surface of γ-Al2O3 after partial fluorination and its favorable effect in hydrocarbon conversions: the color of USY/Kaolin/BF-H2 is lighter than that of USY/Kaolin/γ-Al2O3 after reactions with VGO [108]. Reproduced with permission from Rui Feng et al., The Journal of Physical Chemistry C; published by American Chemical Society, 2014 [108].
5.3. Mesoporation Modification and Diffusion Performance Improvement
The microporous structure of zeolite easily causes diffusion restriction during the heavy oil cracking process; especially for macromolecular hydrocarbon reactants, the microporous structure hinders their access to the active sites, reducing the catalytic efficiency. To this end, researchers have proposed mesoporous modification technology, which introduces mesoporous structure into zeolite to improve diffusion performance, thereby improving the overall activity and selectivity of the catalyst [202].
Mesoporous modification mainly includes alkali treatment, the template method, and the hard template method. Alkali treatment is the most commonly used mesoporous method. It partially dissolves the zeolite framework with NaOH solution, introduces a mesoporous structure, and increases the specific surface area and pore volume [203]. The template method forms a mesoporous structure during the synthesis of zeolite by introducing an organic template. It is often used to modify Beta zeolite and ZSM-5 zeolite. The hard template method forms a regular mesoporous structure by coating a hard template (such as carbon nanotubes) with a silicon source after the template is removed.
Mesoporous modification not only improves the diffusion performance of reactants and products, but also increases the exposure rate of the active sites of the catalyst, further improving the activity of the catalyst. For example, mesoporous Beta zeolite exhibits excellent catalytic performance in the cracking reaction of polycyclic aromatic hydrocarbons, effectively increasing the yield of light oil products [204].
OZ5 is a standard ZSM-5 catalyst without modification and mainly has a microporous structure, while AZ5-3 introduces a mesoporous structure through alkali treatment, which has higher molecular diffusion capacity and improved catalytic performance, especially showing better catalytic effect when processing larger molecular reactants. Figure 36a shows the change in the Cumene adsorption amount of OZ5 and AZ5 over time at 308 K. The AZ5 series catalysts (especially AZ5-3) showed a higher adsorption rate in the early stage, indicating that Cumene molecules diffused rapidly on the surface and/or in the mesopores of AZ5-3. When the adsorption amount increased to a certain extent, the adsorption rate of AZ5-3 gradually decreased, which is closely related to the phase change and molecular rearrangement process of Cumene molecules in the mesoporous structure. In contrast, the adsorption rate of OZ5 rose rapidly in the early stage, but soon stabilized, showing the limitation of its microporous structure, which made it difficult for molecules to diffuse into deeper parts. Figure 36b is a linear data-fitting diagram of AZ5-3 and OZ5, which is used to calculate the activation energy (Ea) and exponential factor (A) of diffusion–adsorption. Using the Arrhenius equation, the results show that the diffusion–adsorption activation energy of AZ5-3 is significantly lower than that of OZ5, decreasing from 65 kJ/mol to 14 kJ/mol, indicating that the energy required for the diffusion process of molecules is significantly reduced after the introduction of the mesoporous structure, which helps to improve the activity of the catalyst. Figure 36c shows the adsorption heat curves of OZ5 and AZ5-3 at different Cumene loadings. With the increase in Cumene loading, the adsorption heat of OZ5 shows a downward trend, which is related to the saturation of active surface sites. AZ5-3 shows a higher adsorption heat, especially at medium loading, which may be due to the phase transition in the mesopores and the enhanced interaction between molecules, resulting in an increase in the adsorption heat. This shows that the mesoporous structure of AZ5-3 promotes the molecular rearrangement and phase transition of Cumene, thereby improving its adsorption capacity and thermodynamic stability [202]. Figure 36d shows the product yield diversity of different types of zeolite catalysts (MZ5 and Ga-MS1 with mesoporous structures and Z5 and Ga-S1 with microporous structures) in the catalytic increase in coal cracking volatiles. The figure shows that the mesoporous catalysts MZ5 and Ga-MS1 show higher yields of aromatics (such as BTEXN, decalin, and polycyclic aromatic hydrocarbons (PAHs)) and effective reduction in phenolic compounds compared with the traditional microporous catalysts Z5 and Ga-S1. This is because the mesoporous catalyst can reduce the resistance of micropore diffusion and provide more active external surface sites, which is conducive to the diffusion of larger reactant and product molecules [204].
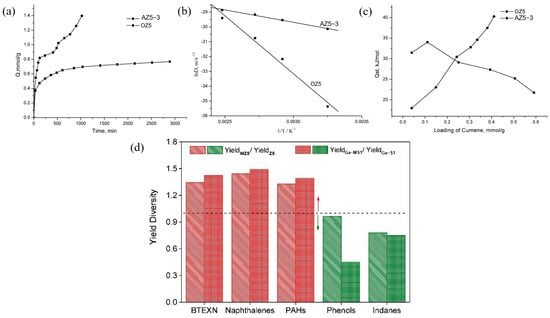
Figure 36.
(a) The adsorption amount of Cumene on OZ5 and AZ5 versus time by IGA at 308 K; (b) data linear fitting of the OZ5 and AZ5-3 to calculate Ea and A; (c) the adsorption heat curve under different loading of the OZ5 and AZ5-3 [202]. Reproduced with permission from Liang Zhao et al., Journal of Catalysis; published by Elsevier, 2008 [202]; (d) the diversity in product yields between mesoporous zeolite catalysts (MZ5 and Ga-MS1) and microporous zeolite catalysts (Z5 and Ga-S1) [204]. Reproduced with permission from Chenyao Bi et al., Fuel; published by Elsevier, 2020 [204].
5.4. Metal Loading and Synergistic Catalysis
Metal loading is one of the important means of optimizing zeolite catalysts. By introducing metal active components, metal–acid synergistic catalysis can be achieved, significantly improving the hydrogenation activity and selectivity of the catalyst. In the heavy oil hydrocracking process, the metal center (such as Pt, Ni, Mo) is responsible for the activation and hydrogenation reaction of hydrogen, while the acidic site of zeolite is responsible for the cracking of the carbon skeleton. The synergistic effect of the two is the core of an efficient hydrocracking reaction [155,205,206].
Precious metals such as Pt and Pd have excellent hydrogenation activity and are often used in the modification of heavy oil hydrocracking catalysts. Pt loaded onto Y-type zeolite or ZSM-5 zeolite can significantly increase the hydrogenation saturation rate of polycyclic aromatic hydrocarbons, reduce coke formation, and increase the yield of light oil products [207]. In addition, rare earth metals (such as La and Ce) combined with zeolites can enhance the stability of acidic centers, inhibit metal contamination, and improve the catalyst’s anti-deactivation performance.
By optimizing the matching of metal distribution, loading, and acid sites, the synergistic catalytic effect of metal and acid can be achieved. For example, the NiMo/ZSM-5 catalyst exhibits excellent catalytic performance in heavy oil hydrocracking reactions, which can effectively improve the selectivity and conversion rate of products and meet the needs of industrial production [86].
Figure 37a shows that in the catalytic hydrodeoxygenation (HDO) process of lignin-derived phenolic compounds, the metal/acid bifunctional catalyst achieves efficient C-O bond breaking and hydrogenation reaction through the synergistic effect of metal sites and acid sites. The metal site is mainly responsible for the activation of hydrogen and the hydrogenation reaction, while the BAS and LAS jointly promote the deoxygenation process, in which the BAS promotes dehydration by protonation, and the LAS stabilizes the intermediate and reduces the activation energy of the reaction. Performance optimization depends on surface modification strategies, including metal carbonization, oxygen defect regulation, nitrogen/phosphorus doping, etc. These measures regulate the electronic structure and active site distribution of the catalyst. By rationally controlling the metal particle size and the spatial distribution of the acid sites, the synergistic effect of the hydrogenation and deoxygenation pathways is enhanced, the selectivity and stability of the reaction are greatly improved, and it provides important support for the efficient utilization of biomass resources and the green chemical industry [205]. Figure 37b shows the key role and reaction pathway of the metal/zeolite bifunctional catalyst in the bio-oil upgrading process. The figure visually illustrates how metals and zeolites work together to remove oxygen atoms and reorganize carbon chain structures to produce high-quality fuels and chemicals. On the left side of the figure, bio-oil and hydrogen (H2) enter the reaction system together. Metal catalysts (such as Pt, Ni, etc.) mainly participate in hydrogenation reactions in the initial stage, effectively removing oxygen atoms in bio-oil, converting oxygen-containing compounds into saturated hydrocarbons, such as alkanes and aromatics. At the same time, this process is also accompanied by the cracking of carboxyl and other oxygen groups. Subsequently, the reactants undergo carbon chain reorganization and dehydration reactions on the zeolite catalyst. The acidic sites of zeolites (such as BAS and LAS) optimize the carbon chain structure of the target product by catalyzing reactions such as carbon chain breakage, isomerization, and cracking, meeting the requirements of fuel viscosity, distillation range, freezing point, etc. Ultimately, the bifunctional catalyst efficiently converts bio-oil into high-quality fuels and chemicals, including gasoline, diesel, and chemical products, through the synergistic effect of metal–acids. The core of this process lies in optimizing the dynamic balance between metal-catalyzed hydrogenation and carbon chain rearrangement at the zeolite acid sites to achieve high conversion and selectivity with minimal by-product formation [206].
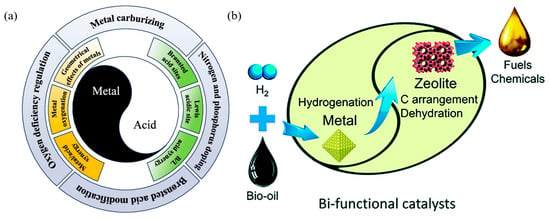
Figure 37.
(a) Hydrodeoxygenation of lignin phenol derivatives to aromatic hydrocarbons: a mini-review of metal/acid bifunctional catalysts [205]. Reproduced with permission from Wenlong Cao et al., Energy & Fuels; published by American Chemical Society, 2024 [205]; (b) bifunctional catalysts for bio-oil upgrading [206]. Reproduced with permission from Yanchun Shi et al., Catalysis Science & Technology; published by Royal Society of Chemistry, 2017 [206].
5.5. High Integration of Theoretical Calculations and Experimental Data
The existing integration of theoretical calculations and experimental verification is not high, and there is a lack of in-depth understanding of the reaction mechanism. The combination of theoretical calculations and experimental data plays a vital role in the design and optimization of zeolite catalysts. Through computational chemistry methods, researchers can gain a deep understanding of the reaction mechanism, catalytic active sites, and the interaction between reactants and catalysts, and then predict the results of the catalytic reaction and the performance of the catalyst. This combination of calculations and experiments can not only guide the synthesis and modification of catalysts, but also effectively improve the performance of catalysts [208].
The reaction mechanism of zeolite catalysts is usually complex, involving processes such as protonation, the generation and transfer of carbon cations, etc. Using computational methods such as density functional theory (DFT), researchers can simulate these reaction steps and predict the reaction pathways under different catalyst structures. For example, in the hydrocracking reaction, theoretical calculations can reveal the synergistic effect between metals and acid sites and predict the optimal metal loading and acidity regulation strategy. These computational results can provide guidance for experimental design and help optimize catalyst performance [209].
Theoretical calculations can be used to predict the effects of the acidity distribution and pore structure of zeolite catalysts with different structures on reaction performance. For example, researchers calculated the effects of different Si/Al ratios on the distribution of acid sites and verified their catalytic performance with experimental data, thus providing a theoretical basis for the structural design of the catalyst. In addition, by computationally simulating the effects of different metal components, researchers were able to optimize the metal loading position and ratio, further improving the catalyst’s hydrocracking performance and resistance to deactivation [210,211].
Although theoretical calculations provide valuable guidance for catalyst design, the final catalyst performance still needs to be verified by experimental data. Through experimental data feedback, researchers can adjust the assumptions and parameters of the computational model and optimize the catalyst design. For example, in the design of metal-supported catalysts, experimental data can reveal actual influencing factors such as metal dispersion and the effectiveness of acidic sites, thereby guiding further theoretical calculations and experimental optimization [212,213].
The combination of theoretical calculations and experimental data is not just a one-way verification process but can form effective two-way feedback between the two. Theoretical calculations can help design more reasonable catalyst structures, while experimental data can verify the accuracy of the calculations and reveal the underlying reaction mechanism. Through this two-way feedback optimization process, researchers can develop catalysts with higher performance and longer service life to meet the needs of industrial applications such as heavy oil cracking and hydrocracking [214].
In the future, with the improvement of computing power and the advancement of experimental methods, the combination of theoretical calculations and experimental data will become closer, providing more precise support for the efficient design and application of zeolite catalysts.
Figure 38 shows the performance of the H-FAU and EFAl/H-FAU zeolite models in propane cracking reactions [215]. The structures of the two zeolite models are first given in the figure (Figure 38a). The EFAl/H-FAU model contains a trinuclear [Al3O4H3]4+ complex in the sodalite cage of the zeolite, and these metal species play a key role in the hydrogenation reaction. The reaction mechanism diagram (Figure 38b) describes the rate-determining step of single-molecule propane cracking, namely the protonation of the C-C bond. The propane molecule is first protonated under the catalysis of the acidic site to form a carbon cation intermediate, which is then decomposed into methane and ethylene. Figure 38c shows the local structures of the adsorption complex (Ads), transition state (TS), and reaction intermediate (Int) of propane cracking in the H-FAU and EFAl/H-FAU models. The calculation results show that the presence of EFAl changes the structure of the transition state and intermediate. In the EFAl/H-FAU model, the protonated C-C bond is 0.14 Å shorter than that in the H-FAU model, while in the carbon cation intermediate, the C-C bond in the EFAl/H-FAU model is 0.15 Å longer than that in the H-FAU model. The reaction energy diagram in Figure 38d shows that in the EFAl/H-FAU model, the reaction energy barrier of propane cracking is reduced from 200 kJ/mol in the H-FAU model to 149 kJ/mol. The presence of EFAl significantly stabilizes the transition state and reaction intermediates in the cracking process. This result indicates that the multinuclear EFAl species can enhance the activity of the acidic sites and reduce the reaction energy barrier, thereby improving the catalytic efficiency of propane cracking.
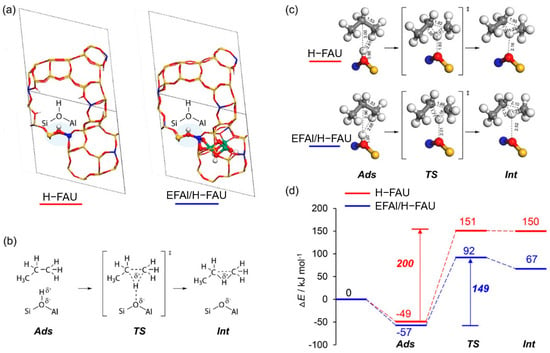
Figure 38.
(a) EFAl-free (H-FAU) and EFAl-containing (EFAl/H-FAU) faujasite models used for propane cracking reactions. (b) Reaction mechanism of the rate-determining step of monomolecular propane cracking by protonation of the C-C bond. (c) Local structures of adsorption complexes (Ads), transition states (TS), and reaction intermediates (Int) for propane cracking in H-FAU and EFAl/H-FAU models. (d) Reaction energy diagram of propane cracking in faujasite zeolites [215]. Reproduced with permission from Chong Liu et al., ACS Catalysis; published by American Chemical Society, 2015 [215].
Figure 39a shows the free energy diagram of the methane-assisted reforming of p-cresol, which shows the reaction paths on pure Mo2C and transition metal (TM)-doped Mo2C surfaces. The figure analyzes the free energy changes of each reaction step during the methane-assisted reforming process at 1073 K. The reaction steps involve the adsorption, protonation, C-O bond cleavage, and formation of the final product of p-cresol molecules. The reaction steps (a–j) describe the free energy changes of each reaction stage, including adsorption (Ads), transition state (TS), and intermediate (Int). The free energy diagram shows that the cleavage of C-O bonds and the desorption energies of C and O on all doped TM (such as Fe and Ni) and Mo2C composite surfaces are significantly lower than those on the pure Mo2C surface. For example, Fe- and Ni-doped Mo2C surfaces exhibit lower energy barriers in the desorption reactions of C and O, especially Ni-r-Mo2C, which has the lowest desorption barrier (2.47 eV), significantly reducing the difficulty of methane-reforming reactions. The figure also shows the catalytic performance of different metal surfaces in the reaction of methane with p-methylphenol, indicating that metal doping helps to reduce the reaction energy barrier and optimize the product ratio of H2 and CO. These results highlight the importance of bifunctional catalysts, especially in the activation of methane and the deoxygenation of p-cresol, where TM-doped Mo2C surfaces exhibit higher catalytic activity and better selectivity. Overall, the figure shows a way to optimize the methane-assisted p-cresol-reforming reaction by regulating the surface properties of Mo2C by doping with transition metals [209]. Figure 39b shows the key elements of catalyst design under dynamic reaction conditions and highlights the challenges in this field. The dynamics loop in the center of the diagram connects reactor design and catalyst design and shows that these two fields require multidisciplinary collaborations such as theory, kinetics, and spectroscopy to achieve optimal catalytic performance. The relevant factors listed in the figure include changes in reaction conditions (e.g., concentration, temperature, pressure, etc.) and catalyst surface states, which affect the conversion rate, selectivity, and product composition of the catalytic reaction. In energy storage and chemical conversion processes, catalytic reactions often face dynamically changing conditions, especially when using renewable energy. These changes require catalysts to have higher adaptability and tolerance, which not only depends on the design of the catalyst but is also closely related to the design of the reactor. In order to meet these challenges, catalysts need to be able to maintain stable reaction efficiency under changing energy supply and reactant composition. This requires researchers not only to optimize the stability of the catalyst, but also to consider more flexible and modular structures when designing reactors to cope with fluctuations in energy and raw material supply. To solve these problems, it is necessary to rely on the combined use of advanced technologies such as operando spectroscopy, theoretical modeling, and kinetics simulation to gain a deep understanding of the performance of catalysts under dynamic reaction conditions and promote the innovation of new catalyst and reactor designs [213].
Figure 39.
(a) Free energy (E-TS) diagram of the methane-assisted p-cresol-reforming reaction over pure and TM-doped Mo2C(001) surfaces supported over GNS (temperature = 1073 K). Elementary reaction steps in the CH4-assisted p-cresol-reforming pathway are described above the free energy diagram (a–j) [209]. “*” denotes surface-adsorbed species (e.g., CH4*, OH*, C*, etc.). Reproduced with permission from Amoolya D. Lalsare et al., ACS Catalysis; published by American Chemical Society, 2021 [209]; (b) future challenges in heterogeneous catalysis: understanding catalysts under dynamic reaction conditions [213]. Reproduced with permission from Jan-Dierk Grunwaldt et al., ChemCatChem; published by John Wiley and Sons, 2016 [213].
In summary, the optimization strategies for zeolite catalysts include catalyst deactivation modification, acidity regulation, mesoporation modification, metal synergistic catalysis, and the combination of theoretical calculations and experimental data. Through multi-level and multi-angle optimization design, the limitations of zeolite catalysts in practical applications can be effectively overcome, their catalytic activity and stability can be improved, and the development of heavy oil efficient conversion technology can be promoted.
6. Conclusions and Outlook
Zeolite catalysts have shown great application potential in the field of heavy oil cracking and hydrocracking due to their unique microporous structure, adjustable acidity, and good thermal stability. This paper systematically reviews the structural characteristics, acidity regulation, deactivation mechanism, and optimization strategy of zeolite catalysts, and deeply discusses the application and mechanism analysis of Y-type zeolite, ZSM-5 zeolite, Beta zeolite, Ferrierite zeolite, and Mordenite zeolite in heavy oil catalytic cracking. The acidity and pore structure of different types of zeolites are closely related to their catalytic performance. By regulating the structure and acidity of zeolites, the catalytic efficiency of heavy oil cracking can be significantly improved. In the future, through the combination of theory and experiment, the development of efficient zeolite catalysts will be further promoted, providing strong support for the efficient utilization of heavy oil resources. In addition, in the face of many challenges in industrial applications, further optimization and application research of zeolite catalysts remain the key directions in the future.
6.1. Research Summary
Through a systematic analysis of the application of zeolite catalysts in heavy oil cracking reactions, the following main conclusions can be drawn. The structure and acidity characteristics of zeolite are the key factors that determine its catalytic performance. Y-type zeolite, with its supercage structure, can provide sufficient reaction space for the catalytic cracking of macromolecular hydrocarbons and exhibit significant catalytic activity and stability. ZSM-5 zeolite has excellent shape-selective catalytic performance due to its unique ten-membered ring pore structure, which means it shows high selectivity in the cracking of long-chain alkanes and can significantly increase the yield of low-carbon olefins and aromatics. Beta zeolite, with its large three-dimensional pore structure and strong acidity, is particularly suitable for the cracking of condensed aromatics and hydrocarbon isomerization reactions and shows important application value in improving conversion efficiency and selectivity. Ferrierite zeolite, with its unique two-dimensional pore structure and strong acidity, exhibits excellent catalytic performance in the conversion of medium-chain alkanes and aromatics. Mordenite zeolite has a one-dimensional pore structure and strong acidity, which is suitable for the cracking of high molecular weight hydrocarbons, especially in the cracking of long-chain alkanes, which significantly improves the yield of cracking products.
The analysis of the reaction mechanism further reveals the catalytic characteristics and pathways of cracking of polycyclic aromatic hydrocarbons and long-chain alkanes. In the hydrocracking process of polycyclic aromatic hydrocarbons, metal–acid synergistic catalysis plays a core role. The metal active center is responsible for the activation of hydrogen and the hydrogenation saturation of the aromatic ring, while the acidic site promotes the cracking of carbon–carbon bonds, ultimately generating light oil products and low-molecular-weight hydrocarbons. For the catalytic cracking of long-chain alkanes, the reaction pathway mainly includes two mechanisms: unimolecular cracking and bimolecular cracking. Unimolecular cracking is mainly achieved through the β-fragmentation of carbon cation intermediates and is suitable for catalysts with weaker acidity, while bimolecular cracking involves hydrogen transfer and intermolecular reactions, and often occurs on catalysts with stronger acidity. The acid strength, distribution, and pore structure of zeolites have a significant effect on the reaction pathway and product distribution. By regulating these factors, the reaction selectivity can be optimized, and the yield of the target product can be increased.
Regarding the problem of catalyst deactivation, optimization strategy is the key to improving the industrial application life of zeolites. Coke deposition, metal contamination, and acid loss are the main causes of catalyst deactivation. Coke deposition mainly comes from the polymerization of side reactions caused by strong acid sites, while metal contamination comes from the enrichment of impurity ions such as nickel and vanadium in heavy oil on the catalyst surface, which further leads to catalyst poisoning and deactivation. These problems can be effectively solved by regulating the acid distribution of zeolites, introducing mesoporous structures and optimizing metal loading, and significantly improving the stability and service life of catalysts. For example, acid regulation can inhibit coke formation; the introduction of mesoporous structures helps to improve the diffusion properties of reactants and products and reduce diffusion limitations; metal loading modification can achieve more efficient catalytic cracking under the synergistic effect of metal–acids.
In general, zeolite catalysts play an irreplaceable role in the catalytic cracking and hydrocracking reactions of heavy oil. Their unique structure and acidic characteristics, catalytic deactivation and optimization strategies, and analysis of reaction mechanisms provide an important theoretical and practical basis for the further development of catalysts. However, current zeolite catalysts still face certain challenges in terms of structural regulation, deactivation prevention, and reaction path optimization, which require further breakthroughs and innovations in future research. In response to these issues, future research will focus on greening the synthesis process, multi-scale analysis of the reaction mechanism, structural functionalization modification, and industrial applications, thereby promoting the widespread application of zeolite catalysts in the fields of efficient upgrading of heavy oil and sustainable development.
6.2. Future Development Direction of Zeolite Catalysts
With the advancement of science and technology, the development of zeolite catalysts in the field of heavy oil cracking will mainly focus on efficient synthesis, mechanism analysis, structural innovation, and sustainable application.
Efficient synthesis and green preparation are two of the important trends in the development of zeolite catalysts. Although the traditional hydrothermal synthesis method is mature, it has problems such as high energy consumption and template pollution. Future research should focus on the development of green synthesis technology, such as the use of recyclable templates, template-free synthesis, and low-temperature synthesis, to reduce production costs and environmental impact [216]. In addition, by introducing high-throughput screening and self-assembly synthesis technology, precise regulation of zeolite structure and performance can be achieved. For example, new technologies such as low-cost and high-efficiency template design and the sol-gel method have provided new approaches for the large-scale industrial production of zeolite catalysts [217].
Multiscale research and reaction mechanism analysis are the key to understanding the performance of zeolite catalysts. By combining macroscopic experiments with microscopic characterization techniques, the relationship between the structure and catalytic performance of zeolites can be deeply explored. For example, using in situ characterization techniques (such as in situ infrared spectroscopy, in situ XRD, XAFS, etc.), the structural evolution and active site changes of the catalyst during the reaction can be monitored in real time [218,219]. In addition, theoretical calculations and molecular simulations (such as density functional theory DFT and molecular dynamics simulation) provide powerful tools for reaction mechanism research. By simulating the reaction path and energy barrier changes, the microscopic mechanism of hydrogenation cracking of polycyclic aromatic hydrocarbons and catalytic cracking of long-chain alkanes can be revealed, thereby guiding the design and optimization of catalysts [220,221].
The development of zeolite catalysts with new structures and functional modifications is an effective means to improve the efficiency of heavy oil cracking. In recent years, mesoporous zeolites, layered zeolites, and composite porous structure zeolites have gradually become research hotspots. Mesoporous zeolites can significantly improve the diffusion performance of macromolecules and enhance catalytic efficiency by introducing mesoporous structures [222]. In addition, functional modification strategies, such as the introduction of rare earth metals, precious metals, and heteroatoms (such as P, B, etc.), can further regulate the acid distribution and catalytic activity of zeolites. For example, Pt-Y zeolite and NiMo/ZSM-5 zeolite exhibit excellent synergistic catalytic performance in hydrocracking reactions, effectively improving the yield and selectivity of light oil products [223].
The industrial application and sustainable development of zeolite catalysts are one of the important research directions for the future. In the field of heavy oil catalytic cracking, the high efficiency and long life of industrial catalysts are the core to achieve sustainable production. By developing high-performance catalysts that are resistant to deactivation and metal pollution, production costs can be effectively reduced and the service life of catalysts can be extended [224]. At the same time, the regeneration and recycling technology of zeolite catalysts will also become a key research direction. Through physical regeneration, chemical regeneration, and biological regeneration, waste catalysts can be recycled and reused, reducing environmental pollution and promoting green and sustainable development. In addition, the application of zeolite catalysts in fields such as biomass cracking and carbon dioxide conversion also provides new ways and application prospects for their sustainable development [225].
Zeolite catalysts have broad application prospects in the field of heavy oil catalytic cracking and hydrocracking, but they still face challenges in structural regulation, deactivation prevention and control, and sustainable development. Future research needs to make comprehensive breakthroughs in efficient synthesis, reaction mechanism analysis, new structure development, and industrial application. Through multidisciplinary cross-cutting and technological innovation, zeolite catalysts will play a more important role in the field of efficient heavy oil upgrading and green energy conversion, providing strong technical support for the sustainable development of the energy industry.
Author Contributions
Conceptualization, L.W. and H.X.; methodology, L.W.; software, L.W.; validation, L.W., H.W. and Q.D.; formal analysis, L.W., H.W. and Q.D.; investigation, L.W.; resources, H.X.; data curation, L.W.; writing—original draft preparation, L.W.; writing—review and editing, H.X.; visualization, L.W.; supervision, H.X.; project administration, H.X.; funding acquisition, Y.L. and H.X. All authors have read and agreed to the published version of the manuscript.
Funding
This work is supported by National Key R&D Program of China (2022YFB4101201).
Data Availability Statement
Data will be made available on request.
Acknowledgments
The authors are grateful for the facilities and financial support from Synfuels China, Co., Ltd.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- World Energy Review. 2023. Available online: https://www.eni.com/ (accessed on 5 March 2024).
- Tong, X.; Zhang, G.; Wang, Z.; Wen, Z.; Tian, Z.; Wang, H.; Ma, F.; Wu, Y. Distribution and potential of global oil and gas resources. Pet. Explor. Dev. 2018, 45, 779–789. [Google Scholar] [CrossRef]
- Zou, C.; Zhai, G.; Zhang, G.; Wang, H.; Zhang, G.; Li, J.; Wang, Z.; Wen, Z.; Ma, F.; Liang, Y.; et al. Formation, distribution, potential and prediction of global conventional and unconventional hydrocarbon resources. Pet. Explor. Dev. 2015, 42, 14–28. [Google Scholar] [CrossRef]
- Santos, R.; Loh, W.; Bannwart, A.; Trevisan, O. An overview of heavy oil properties and its recovery and transportation methods. Braz. J. Chem. Eng. 2014, 31, 571–590. [Google Scholar] [CrossRef]
- Rana, M.S.; Sámano, V.; Ancheyta, J.; Diaz, J. A review of recent advances on process technologies for upgrading of heavy oils and residua. Fuel 2007, 86, 1216–1231. [Google Scholar] [CrossRef]
- Corma, A. Inorganic solid acids and their use in acid-catalyzed hydrocarbon reactions. Chem. Rev. 1995, 95, 559–614. [Google Scholar] [CrossRef]
- Ding, L.; Sitepu, H.; Al-Bogami, S.A.; Yami, D.; Tamimi, M.; Shaik, K.; Sayed, E. Effect of zeolite-Y modification on crude-oil direct hydrocracking. ACS Omega 2021, 6, 28654–28662. [Google Scholar] [CrossRef]
- Uzcátegui, G.; de Klerk, A. Causes of deactivation of an amorphous silica-alumina catalyst used for processing of thermally cracked naphtha in a bitumen partial upgrading process. Fuel 2021, 293, 120479. [Google Scholar] [CrossRef]
- Miao, P.; Zhu, X.; Guo, Y.; Miao, J.; Yu, M.; Li, C. Combined mild hydrocracking and fluid catalytic cracking process for efficient conversion of light cycle oil into high-quality gasoline. Fuel 2021, 292, 120364. [Google Scholar] [CrossRef]
- Katada, N.; Kawaguchi, Y.; Takeda, K.; Matsuoka, T.; Uozumi, N.; Kanai, K.; Fujiwara, S.; Kinugasa, K.; Nakamura, K.; Suganuma, S.; et al. Dealkylation of alkyl polycyclic aromatic hydrocarbon over silica monolayer solid acid catalyst. Appl. Catal. A 2017, 530, 93–101. [Google Scholar] [CrossRef]
- Prajapati, R.; Kohli, K.; Maity, S.K. Slurry phase hydrocracking of heavy oil and residue to produce lighter fuels: An experimental review. Fuel 2021, 288, 119686. [Google Scholar] [CrossRef]
- Amghizar, I.; Vandewalle, L.A.; Van Geem, K.M.; Marin, G.B. New trends in olefin production. Engineering 2017, 3, 171–178. [Google Scholar] [CrossRef]
- Zhao, F.; Xu, T.; Zhu, G.; Wang, K.; Xu, X.; Liu, L. A review on the role of hydrogen donors in upgrading heavy oil and bitumen. Sustain. Energy Fuels 2022, 6, 1866–1890. [Google Scholar] [CrossRef]
- Huo, Q.; Gong, Y.; Dou, T.; Zhao, Z.; Pan, H.; Deng, F. Novel micro- and mesoporous composite molecular sieve assembled by zeolite L nanocrystal and its performance for the hydrodesulfurization (HDS) of fluid catalytic cracking (FCC) gasoline. Energy Fuels 2010, 24, 3764–3771. [Google Scholar] [CrossRef]
- Pan, M.; Zheng, J.; Liu, Y.; Ning, W.; Tian, H.; Li, R. Construction and practical application of a novel zeolite catalyst for hierarchically cracking of heavy oil. J. Catal. 2019, 369, 72–85. [Google Scholar] [CrossRef]
- Leyva, C.; Rana, M.S.; Trejo, F.; Ancheyta, J. On the use of acid-base-supported catalysts for hydroprocessing of heavy petroleum. Ind. Eng. Chem. Res. 2007, 46, 7448–7466. [Google Scholar] [CrossRef]
- Silaghi, M.-C.; Chizallet, C.; Raybaud, P. Challenges on molecular aspects of dealumination and desilication of zeolites. Microporous Mesoporous Mater. 2014, 191, 82–96. [Google Scholar] [CrossRef]
- Ikyereve, R.E. Investigations into the Pre-Treatment Methods for the Removal of Nickel (II) and Vanadium (IV) from Crude Oil. Ph.D. Thesis, Loughborough University, Loughborough, UK, 2014. [Google Scholar]
- Orea, M.; Ranaudo, M.A.; Lugo, P.; López, L. Retention of alkane compounds on asphaltenes. Insights about the nature of asphaltene–alkane interactions. Energy Fuels 2016, 30, 8098–8113. [Google Scholar] [CrossRef]
- Primerano, K.; Mirwald, J.; Hofko, B. Asphaltenes and maltenes in crude oil and bitumen: A comprehensive review of properties, separation methods, and insights into structure, reactivity and aging. Fuel 2024, 368, 131616. [Google Scholar] [CrossRef]
- Speight, J.G. The Chemistry and Technology of Petroleum; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- Demirbaş, A. Asphaltene yields from five types of fuels via different methods. Energy Convers. Manag. 2002, 43, 1091–1097. [Google Scholar] [CrossRef]
- Mousavi-Dehghani, S.A.; Riazi, M.R.; Vafaie-Sefti, M.; Mansoori, G.A. An analysis of methods for determination of onsets of asphaltene phase separations. J. Pet. Sci. Eng. 2004, 42, 145–156. [Google Scholar] [CrossRef]
- Yu, Z.X.; Wu, B.; Wu, Q.; Chen, L.; Zhu, Z.R. Catalytic hydrogenation and hydrocracking of PRO residual oil over USY-zeolite catalyst. Adv. Mater. Res. 2012, 347, 3710–3715. [Google Scholar] [CrossRef]
- Han, L. Basic Research on Composition, Structure and Efficient Conversion of Heavy Oils. Ph.D. Thesis, China University of Mining and Technology, Xuzhou, China, 2008. [Google Scholar]
- Gary, J.H.; Handwerk, J.H.; Kaiser, M.J.; Geddes, D. Petroleum Refining: Technology and Economics; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Hsu, C.S.; Robinson, P.R. Practical Advances in Petroleum Processing; Springer: New York, NY, USA, 2006; Volume 1. [Google Scholar]
- Wang, F.; Wei, Q.; Li, K.; Biney, B.W.; Liu, H.; Chen, K.; Wang, Z.; Guo, A. Coke formation of heavy oil during thermal cracking: New insights into the effect of olefinic-bond-containing aromatics. Fuel 2023, 336, 127138. [Google Scholar] [CrossRef]
- Komvokis, V.; Tan, L.X.L.; Clough, M.; Pan, S.S.; Yilmaz, B. Zeolites in fluid catalytic cracking (FCC). In Zeolites in Sustainable Chemistry: Synthesis, Characterization and Catalytic Applications; Xiao, F.-S., Meng, X., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 271–297. [Google Scholar]
- Emberru, R.E.; Patel, R.; Mujtaba, I.M.; John, Y.M. A review of catalyst modification and process factors in the production of light olefins from direct crude oil catalytic cracking. Sci 2024, 6, 11. [Google Scholar] [CrossRef]
- Talebian-Kiakalaieh, A.; Tarighi, S. Synthesis of hierarchical Y and ZSM-5 zeolites using post-treatment approach to maximize catalytic cracking performance. J. Ind. Eng. Chem. 2020, 88, 167–177. [Google Scholar] [CrossRef]
- Alotibi, M.F.; Alshammari, B.A.; Alotaibi, M.H.; Alotaibi, F.M.; Alshihri, S.; Navarro, R.M.; Fierro, J.L.G. ZSM-5 zeolite based additive in FCC process: A review on modifications for improving propylene production. Catal. Surv. Asia 2020, 24, 1–10. [Google Scholar] [CrossRef]
- Speight, J.G. The Desulfurization of Heavy Oils and Residua; CRC Press: Boca Raton, FL, USA, 1999. [Google Scholar]
- Speight, J.G. New approaches to hydroprocessing. Catal. Today 2004, 98, 55–60. [Google Scholar] [CrossRef]
- Speight, J.G.; Ozum, B. Petroleum Refining Processes; CRC Press: Boca Raton, FL, USA, 2001. [Google Scholar]
- Miteva, A.; Stoyanova, V. Zeolites application in terrestrial and space industry—A review. Aerosp. Res. Bulg. 2020, 32, 209–223. [Google Scholar] [CrossRef]
- Tanimu, A.; Tanimu, G.; Alasiri, H.; Aitani, A. Catalytic cracking of crude oil: Mini review of catalyst formulations for enhanced selectivity to light olefins. Energy Fuels 2022, 36, 5152–5166. [Google Scholar] [CrossRef]
- Suganuma, S.; Katada, N. Innovation of catalytic technology for upgrading of crude oil in petroleum refinery. Fuel Process. Technol. 2020, 208, 106518. [Google Scholar] [CrossRef]
- Kotikalapudi, C.M. Selective Ring Opening of Naphthenes Present in Heavy Gas Oil Derived from Athabasca Bitumen; University of Saskatchewan: Saskatoon, SK, Canada, 2009. [Google Scholar]
- Žula, M.; Grilc, M.; Likozar, B. Hydrocracking, hydrogenation and hydro-deoxygenation of fatty acids, esters and glycerides: Mechanisms, kinetics and transport phenomena. Chem. Eng. J. 2022, 444, 136564. [Google Scholar] [CrossRef]
- Speight, J.G. Heavy and Extra-Heavy Oil Upgrading Technologies; Gulf Professional Publishing: Oxford, UK, 2013. [Google Scholar]
- Du, H.; Liu, D.; Liu, H.; Gao, P.; Lv, R.; Li, M.; Lou, B.; Yang, Y. Role of hydrogen pressure in slurry-phase hydrocracking of venezuela heavy oil. Energy Fuels 2015, 29, 2104–2110. [Google Scholar] [CrossRef]
- Weitkamp, J. Catalytic hydrocracking—Mechanisms and versatility of the process. ChemCatChem 2012, 4, 292–306. [Google Scholar] [CrossRef]
- Clark, P.D.; Hyne, J.B.; Tyrer, J.D. Chemistry of organosulphur compound types occurring in heavy oil sands: 1. High temperature hydrolysis and thermolysis of tetrahydrothiophene in relation to steam stimulation processes. Fuel 1983, 62, 959–962. [Google Scholar] [CrossRef]
- Kozlowski, M.; Punase, A.; Nasr-El-Din, H.; Hascakir, B. The catalytic effect of clay on in-situ combustion performance. In Proceedings of the SPE Latin America and Caribbean Petroleum Engineering Conference, Quito, Ecuador, 18–20 November 2015; p. D031S030R003. [Google Scholar]
- Clark, P.D.; Hyne, J.B. Studies on the chemical reactions of heavy oils under steam stimulation condition. Aostra J. Res. 1990, 6, 29–39. [Google Scholar] [CrossRef]
- Maity, S.K.; Ancheyta, J.; Marroquín, G. Catalytic aquathermolysis used for viscosity reduction of heavy crude oils: A review. Energy Fuels 2010, 24, 2809–2816. [Google Scholar] [CrossRef]
- Zhao, X.F.; Tan, X.H.; Liu, Y.J. Behaviors of oil-soluble catalyst for aquathermolysis of heavy oil. Ind. Catal. 2008, 11, 31–34. [Google Scholar]
- Wen, S.; Zhao, Y.; Liu, Y.; Hu, S. A study on catalytic aquathermolysis of heavy crude oil during steam stimulation. In Proceedings of the SPE International Symposium on Oilfield Chemistry, Houston, TX, USA, 28 February–2 March 2007; p. SPE-106180-MS. [Google Scholar]
- Yusuf, A.; Al-Hajri, R.S.; Al-Waheibi, Y.M.; Jibril, B.Y. In-situ upgrading of Omani heavy oil with catalyst and hydrogen donor. J. Anal. Appl. Pyrolysis 2016, 121, 102–112. [Google Scholar] [CrossRef]
- Chao, K.; Chen, Y.; Li, J.; Zhang, X.; Dong, B. Upgrading and visbreaking of super-heavy oil by catalytic aquathermolysis with aromatic sulfonic copper. Fuel Process. Technol. 2012, 104, 174–180. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, Y.; Wu, C.; Xia, F. Laboratory experiments and field tests of an amphiphilic metallic chelate for catalytic aquathermolysis of heavy oil. Energy Fuels 2008, 22, 1502–1508. [Google Scholar] [CrossRef]
- Liu, D.; Li, M.; Deng, W.; Que, G. Reactivity and composition of dispersed Ni catalyst for slurry-phase residue hydrocracking. Energy Fuels 2010, 24, 1958–1962. [Google Scholar] [CrossRef]
- Ortiz-Moreno, H.; Ramírez, J.; Cuevas, R.; Marroquín, G.; Ancheyta, J. Heavy oil upgrading at moderate pressure using dispersed catalysts: Effects of temperature, pressure and catalytic precursor. Fuel 2012, 100, 186–192. [Google Scholar] [CrossRef]
- Zhao, F.; Liu, Y.; Lu, N.; Xu, T.; Zhu, G.; Wang, K. A review on upgrading and viscosity reduction of heavy oil and bitumen by underground catalytic cracking. Energy Rep. 2021, 7, 4249–4272. [Google Scholar] [CrossRef]
- Fahim, M.A.; Al-Sahhaf, T.A.; Elkilani, A. Fundamentals of Petroleum Refining; Elsevier: Oxford, UK, 2009. [Google Scholar]
- Hoek, A.; Huizinga, T.; Esener, A.; Maxwell, I.; Stork, W.; Van de Meerakker, F.; Sy, O. New catalysts improves heavy feedstock hydro-cracking. Oil Gas J. 1991, 89, 16. [Google Scholar]
- El-Sawy, M.S.; Hanafi, S.A.; Ashour, F.; Aboul-Fotouh, T.M. Co-hydroprocessing and hydrocracking of alternative feed mixture (vacuum gas oil/waste lubricating oil/waste cooking oil) with the aim of producing high quality fuels. Fuel 2020, 269, 117437. [Google Scholar] [CrossRef]
- Rahmawati, S.; Wahyuni, S.; Rusdi, M.; Priyanti, N.A.; Nisa, L.C.; Muttaqin, M.Z.; Fatimah, S. Review journal: The effect of heterogeneous zeolite catalyst and modified zeolite catalysts on biogaseline yields. Int. J. Sustain. Engl. Lang. Educ. Sci. 2024, 1, 49–56. [Google Scholar] [CrossRef]
- Yim, H.; Valizadeh, S.; Cho, K.; Park, Y.-K. Production of low-aromatic oil from catalytic pyrolysis of waste plastics-derived wax. J. Anal. Appl. Pyrolysis 2025, 186, 106964. [Google Scholar] [CrossRef]
- Aghaei, E.; Karimzadeh, R.; Godini, H.R.; Gurlo, A.; Gorke, O. Improving the physicochemical properties of Y zeolite for catalytic cracking of heavy oil via sequential steam-alkali-acid treatments. Microporous Mesoporous Mater. 2020, 294, 109854. [Google Scholar] [CrossRef]
- Lin, F.; Xu, M.; Ramasamy, K.K.; Li, Z.; Klinger, J.L.; Schaidle, J.A.; Wang, H. Catalyst deactivation and its mitigation during catalytic conversions of biomass. ACS Catal. 2022, 12, 13555–13599. [Google Scholar] [CrossRef]
- Akah, A. The role of rare earths in fluid catalytic cracking (FCC) catalyst. Rare Earth A Tribut. Late Mr. Rare Earth Profr. Karl Gschneidner 2024, 164, 298–342. [Google Scholar]
- Xu, Z.; Zhu, Y.; Gong, M.; Jiao, N.; Zhang, T.; Wang, H. Review on the poisoning behavior of typical metals on cracking catalysts for chemicals production from petroleum and anti-poisoning strategies. Appl. Catal. A 2024, 685, 119897. [Google Scholar] [CrossRef]
- Rigutto, M.S.; van Veen, R.; Huve, L. Zeolites in hydrocarbon processing. Stud. Surf. Sci. Catal. 2007, 168, 855–913, XXIII–XXVI. [Google Scholar] [CrossRef]
- Martínez, C.; Corma, A. Inorganic molecular sieves: Preparation, modification and industrial application in catalytic processes. Coord. Chem. Rev. 2011, 255, 1558–1580. [Google Scholar] [CrossRef]
- Di Renzo, F.; Fajula, F. Introduction to molecular sieves: Trends of evolution of the zeolite community. Stud. Surf. Sci. Catal. 2005, 157, 1–12. [Google Scholar]
- Yilmaz, B.; Müller, U. Catalytic applications of zeolites in chemical industry. Top. Catal. 2009, 52, 888–895. [Google Scholar] [CrossRef]
- Boronat, M.; Corma, A. Factors controlling the acidity of zeolites. Catal. Lett. 2015, 145, 162–172. [Google Scholar] [CrossRef]
- Van Bokhoven, J.A.; Williams, B.A.; Ji, W.; Koningsberger, D.C.; Kung, H.H.; Miller, J.T. Observation of a compensation relation for monomolecular alkane cracking by zeolites: The dominant role of reactant sorption. J. Catal. 2004, 224, 50–59. [Google Scholar] [CrossRef]
- Xu, D. Study on the Deactivated Titanosilicate Zeolite as a Catalyst for Cracking of Pentene. Master’s Thesis, East China Normal University, Shanghai, China, 2023. [Google Scholar]
- Chu, G. Effects of Different Pore Structures and Acidity Zeolites on the Alkylation of Isobutane/Butene. Master’s Thesis, Taiyuan University of Technology, Taiyuan, China, 2022. [Google Scholar]
- Yi, F.; Chen, H.; Yang, Y.; Cao, J. Mechanisms of Brönsted and Lewis acids of zeolite on pentenes conversion by in situ DRIFTS. J. Fuel Chem. Technol. 2023, 51, 625–634. [Google Scholar] [CrossRef]
- Xu, R.; Pang, W.; Huo, Q. Molecular Sieve and Porous Material Chemistry; Science Press: Beijing, China, 2004. [Google Scholar]
- Zheng, A. Theoretical Calculation of Molecular Sieve Catalysis: From Basic to Application; Science Press: Beijing, China, 2020. [Google Scholar]
- Contini, A. Structural and Ion Exchange Study of Nanostructured Materials. Ph.D. Thesis, Keele University, Keele, UK, 2024. [Google Scholar]
- Minkiewicz, J. Solvothermal Stability Studies of Porous Inorganic and Metal-Organic Materials. Ph.D. Thesis, Cardiff University, Cardiff, UK, 2021. [Google Scholar]
- Baerlocher, C.; McCusker, L.B. Database of Zeolite Structures. Available online: http://www.iza-structure.org/databases/ (accessed on 25 February 2025).
- Shelyapina, M.G. NMR relaxation to probe zeolites: Mobility of adsorbed molecules, surface acidity, pore size distribution and connectivity. Molecules 2024, 29, 5432. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, J.; Corma, A. Applications of zeolites to C1 chemistry: Recent advances, challenges, and opportunities. Adv. Mater. 2020, 32, 2002927. [Google Scholar] [CrossRef]
- del Campo, P.; Martínez, C.; Corma, A. Activation and conversion of alkanes in the confined space of zeolite-type materials. Chem. Soc. Rev. 2021, 50, 8511–8595. [Google Scholar] [CrossRef]
- Palčić, A.; Valtchev, V. Analysis and control of acid sites in zeolites. Appl. Catal. A 2020, 606, 117795. [Google Scholar] [CrossRef]
- Wang, Y.; Tong, C.; Liu, Q.; Han, R.; Liu, C. Intergrowth zeolites, synthesis, characterization, and catalysis. Chem. Rev. 2023, 123, 11664–11721. [Google Scholar] [CrossRef]
- He, Y.; Chen, J.; Li, D.; Zhang, Q.; Liu, D.; Liu, J.; Ma, X.; Wang, T. Efficient production of aromatics by catalytic pyrolysis of fruit waste over zeolites with 3D pore topologies. Energy 2021, 223, 120046. [Google Scholar] [CrossRef]
- Hu, Z.; Wu, T.; Xie, H.; Zhang, Y.; Ge, S.; Wu, Z. Synthesis and consequence of three-dimensionally ordered macroporous Beta zeolite supported NiW catalyst for efficient hydrocracking of 1-methylnaphthalene to BTX. Chem. Eng. J. 2024, 499, 155761. [Google Scholar] [CrossRef]
- Kostyniuk, A.; Bajec, D.; Likozar, B. Hydrocracking, hydrogenation and isomerization of model biomass tar in a packed bed reactor over bimetallic NiMo zeolite catalysts: Tailoring structure/acidity. Appl. Catal. A 2021, 612, 118004. [Google Scholar] [CrossRef]
- Cui, W.; Zhu, D.; Tan, J.; Chen, N.; Fan, D.; Wang, J.; Han, J.; Wang, L.; Tian, P.; Liu, Z. Synthesis of mesoporous high-silica zeolite Y and their catalytic cracking performance. Chin. J. Catal. 2022, 43, 1945–1954. [Google Scholar] [CrossRef]
- Zang, J.; Yu, H.; Liu, G.; Hong, M.; Liu, J.; Chen, T. Research progress on modifications of zeolite Y for improved catalytic properties. Inorganics 2023, 11, 22. [Google Scholar] [CrossRef]
- Komvokis, V.G.; Karakoulia, S.; Iliopoulou, E.F.; Papapetrou, M.C.; Vasalos, I.A.; Lappas, A.A.; Triantafyllidis, K.S. Upgrading of Fischer–Tropsch synthesis bio-waxes via catalytic cracking: Effect of acidity, porosity and metal modification of zeolitic and mesoporous aluminosilicate catalysts. Catal. Today 2012, 196, 42–55. [Google Scholar] [CrossRef]
- Li, Y.; Liu, N.; Shi, L.; Wang, X.; Meng, X. Enhancing aniline condensation reaction efficiency using alkali and rare earth element modified USY zeolite catalyst: Synergistic effects and mechanistic insights. Ind. Eng. Chem. Res. 2024, 63, 17477–17491. [Google Scholar] [CrossRef]
- Zhan, W.; Guo, Y.; Gong, X.; Guo, Y.; Wang, Y.; Lu, G. Current status and perspectives of rare earth catalytic materials and catalysis. Chin. J. Catal. 2014, 35, 1238–1250. [Google Scholar] [CrossRef]
- Chen, N.; Garwood, W. Industrial application of shape-selective catalysis. Catal. Rev. Sci. Eng. 1986, 28, 185–264. [Google Scholar] [CrossRef]
- Li, J.; Gao, M.; Yan, W.; Yu, J. Regulation of the Si/Al ratios and Al distributions of zeolites and their impact on properties. Chem. Sci. 2023, 14, 1935–1959. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; He, X.; Chen, X.; Qiao, C.; Wang, H.; Diao, Z.; Liu, G. Transition metal modified hierarchical ZSM-5 nanosheet for catalytic cracking of n-pentane to light olefins. Fuel 2024, 363, 130902. [Google Scholar] [CrossRef]
- Liu, H.; Ma, J.; Yuan, H.; Shi, X. Catalytic cracking of oleic acid to generate aviation kerosene using platinum–modified ZSM–5 nanosheets. J. Chem. Technol. Biotechnol. 2024, 99, 2259–2269. [Google Scholar] [CrossRef]
- Wang, F.; Ge, H.; Guo, M.; Zhang, P.; Guo, S.; Tang, M. Hydrocracking of tetrahydronaphthalene for preparation of benzene and alkylbenzene catalyzed by Mo/Beta sulfide catalyst. Ind. Eng. Chem. Res. 2024, 63, 21708–21718. [Google Scholar] [CrossRef]
- Dong, Q.; Li, R.; Jiao, H. Zeolite catalysts for selective hydrocracking of polycyclic aromatic hydrocarbons—Structures and mechanisms. ChemCatChem 2024, 16, e202400117. [Google Scholar] [CrossRef]
- Kadja, G.T.M.; Ilmi, M.M.; Azhari, N.J.; Khalil, M.; Fajar, A.T.N.; Subagjo; Makertihartha, I.G.B.N.; Gunawan, M.L.; Rasrendra, C.B.; Wenten, I.G. Recent advances on the nanoporous catalysts for the generation of renewable fuels. J. Mater. Res. Technol. 2022, 17, 3277–3336. [Google Scholar] [CrossRef]
- Costa, C.S.; Dao Thi, H.; Van Geem, K.M.; Rosário Ribeiro, M.; Silva, J.M. Assessment of acidity and the zeolite porous structure on hydrocracking of HDPE. Sustain. Energy Fuels 2022, 6, 3611–3625. [Google Scholar] [CrossRef]
- Hebisch, K.L.; Chmielniak, P.A.; Cutler, V.A.; Watson, R.B.; Gołąbek, K.; Sievers, C. Promoting role of residual organic structure directing agent in skeletal butene isomerization over ferrierite. J. Catal. 2025, 441, 115847. [Google Scholar] [CrossRef]
- Mäki-Arvela, P.; Kaka khel, T.A.; Azkaar, M.; Engblom, S.; Murzin, D.Y. Catalytic hydroisomerization of long-chain hydrocarbons for the production of fuels. Catalysts 2018, 8, 534. [Google Scholar] [CrossRef]
- Liu, X.; Huang, S.; Liu, Y.; Liu, H.; Zhang, X.; Lv, J.; Qi, J.; Wang, Y.; Wang, S.; Ma, X. Enhanced catalytic performance of ferrierite zeolites via finned morphology for carbonylation of dimethyl ether. Chem. Eng. Sci. 2024, 297, 120247. [Google Scholar] [CrossRef]
- Hussain, H.M.; Mohammed, A.A. Experimental study of Iraqi light naphtha isomerization over Ni-Pt/H-mordenite. Iraqi J. Chem. Pet. Eng. 2019, 20, 61–66. [Google Scholar] [CrossRef]
- Blay, V.; Louis, B.; Miravalles, R.; Yokoi, T.; Peccatiello, K.A.; Clough, M.; Yilmaz, B. Engineering zeolites for catalytic cracking to light olefins. ACS Catal. 2017, 7, 6542–6566. [Google Scholar] [CrossRef]
- Maghrebi, R.; Buffi, M.; Bondioli, P.; Chiaramonti, D. Isomerization of long-chain fatty acids and long-chain hydrocarbons: A review. Renew. Sustain. Energy Rev. 2021, 149, 111264. [Google Scholar] [CrossRef]
- Liu, Q.; Yang, Z.; Chen, Z.; Xiao, P.; Ge, Y.; Piao, Y.; Gong, Y. Hydrothermally Ce modified HZSM-5 zeolite enhancing its strong acidity and Brønsted/Lewis acid ratio: Stably boosting ethylene/propylene ratio for cracking n-heptane. Fuel 2024, 368, 131632. [Google Scholar] [CrossRef]
- Qureshi, Z.S.; Siddiqui, M.A.; Tanimu, A.; Aitani, A.; Akah, A.C.; Xu, Q.; AlHerz, M. Steam catalytic cracking of crude oil over novel hierarchical zeolite–containing mesoporous silica–alumina core-shell catalysts. J. Anal. Appl. Pyrolysis 2022, 166, 105621. [Google Scholar] [CrossRef]
- Feng, R.; Liu, S.; Bai, P.; Qiao, K.; Wang, Y.; Al-Megren, H.A.; Rood, M.J.; Yan, Z. Preparation and characterization of γ-Al2O3 with rich Brønsted acid sites and its application in the fluid catalytic cracking process. J. Phys. Chem. C 2014, 118, 6226–6234. [Google Scholar] [CrossRef]
- Suzuki, K.; Katada, N.; Niwa, M. Detection and quantitative measurements of four kinds of OH in HY zeolite. J. Phys. Chem. C 2007, 111, 894–900. [Google Scholar] [CrossRef]
- Dedecek, J.; Sklenak, S.; Li, C.; Gao, F.; Brus, J.; Zhu, Q.; Tatsumi, T. Effect of Al/Si substitutions and silanol nests on the local geometry of Si and Al framework sites in silicone-rich zeolites: A combined high resolution 27Al and 29Si NMR and density functional theory/molecular mechanics study. J. Phys. Chem. C 2009, 113, 14454–14466. [Google Scholar] [CrossRef]
- Huang, Z.; Li, Q.; Mo, Z.; Song, L. Study on the Bronsted acidity of Y zeolite by density fhnctional theory. Petrochem. Technol. Appl. 2014, 32, 398–403. [Google Scholar]
- Alrawi, U. Hydroconversion of n-Hexane over Platinum Supported Zeolite Catalysts Prepared by Super Critical Technique. Ph.D. Thesis, Nahrain University, Baghdad, Iraq, 2008. [Google Scholar]
- Batool, S.R.; Sushkevich, V.L.; van Bokhoven, J.A. Correlating Lewis acid activity to extra-framework aluminum species in zeolite Y introduced by Ion-exchange. J. Catal. 2022, 408, 24–35. [Google Scholar] [CrossRef]
- Ji, B.; Wu, T.; Zhou, K.; Yan, Y.; Li, S. Progresses in research for hydrocracking mechanism and catalysts of polycyclic aromatic hydrocarbons. Petrochem. Technol. 2016, 45, 1263–1271. [Google Scholar]
- Guo, Q.; Deng, Y.; Duan, A.; Zhao, Z.; Niu, H. Research advance on hydrocracking technology and the relative catalysts. Ind. Catal. 2011, 19, 21–27. [Google Scholar]
- Arribas, M.A.; Martinez, A. The influence of zeolite acidity for the coupled hydrogenation and ring opening of 1-methylnaphthalene on Pt/USY catalysts. Appl. Catal. A 2002, 230, 203–217. [Google Scholar] [CrossRef]
- Liu, Y.; Yan, L.; Bai, Y.; Li, F. Catalytic upgrading of volatile from coal pyrolysis over faujasite zeolites. J. Anal. Appl. Pyrolysis 2018, 132, 184–189. [Google Scholar] [CrossRef]
- Lemberton, J.L.; Guisnet, M. Phenanthrene hydroconversion as a potential test reaction for the hydrogenating and cracking properties of coal hydroliquefaction catalysts. Appl. Catal. 1984, 13, 181–192. [Google Scholar] [CrossRef]
- Nakano, F.; Goma, T.; Suganuma, S.; Tsuji, E.; Katada, N. Selective dealkylation of alkyl polycyclic aromatic hydrocarbons towards innovative upgrading process of practical heavy oil. Catal. Sci. Technol. 2021, 11, 239–249. [Google Scholar] [CrossRef]
- Hiramatsu, Y.; Aita, Y.; Umeki, T. Effect of acid properties of catalysts on fluid catalytic cracking of residual oil. J. Jpn. Pet. Inst. 2012, 55, 319–325. [Google Scholar] [CrossRef][Green Version]
- Graça, I.; Fernandes, A.; Lopes, J.M.; Ribeiro, M.F.; Laforge, S.; Magnoux, P.; Ramôa Ribeiro, F. Effect of phenol adsorption on HY zeolite for n-heptane cracking: Comparison with methylcyclohexane. Appl. Catal. A 2010, 385, 178–189. [Google Scholar] [CrossRef]
- Fritz, P.O.; Lunsford, J.H. The effect of sodium poisoning on dealuminated Y-type zeolites. J. Catal. 1989, 118, 85–98. [Google Scholar] [CrossRef]
- Lónyi, F.; Lunsford, J.H. The development of strong acidity in hexafluorosilicate-modified Y-type zeolites. J. Catal. 1992, 136, 566–577. [Google Scholar] [CrossRef]
- Beyerlein, R.; McVicker, G.; Yacullo, L.; Ziemiak, J. The influence of framework and nonframework aluminum on the acidity of high-silica, proton-exchanged FAU-framework zeolites. J. Phys. Chem. 1988, 92, 1967–1970. [Google Scholar] [CrossRef]
- Olson, D.; Haag, W.; Lago, R. Chemical and physical properties of the ZSM-5 substitutional series. J. Catal. 1980, 61, 390–396. [Google Scholar] [CrossRef]
- Haag, W.; Lago, R.; Weisz, P. The active site of acidic aluminosilicate catalysts. Nature 1984, 309, 589–591. [Google Scholar] [CrossRef]
- Xue, N.; Vjunov, A.; Schallmoser, S.; Fulton, J.L.; Sanchez-Sanchez, M.; Hu, J.Z.; Mei, D.; Lercher, J.A. Hydrolysis of zeolite framework aluminum and its impact on acid catalyzed alkane reactions. J. Catal. 2018, 365, 359–366. [Google Scholar] [CrossRef]
- Ji, Y.; Yang, H.; Yan, W. Effect of alkali metal cations modification on the acid/basic properties and catalytic activity of ZSM-5 in cracking of supercritical n-dodecane. Fuel 2019, 243, 155–161. [Google Scholar] [CrossRef]
- Bai, Y.; Liu, D.; Zhao, L.; Gao, J.; Xu, C.; Pang, H.; Gao, X. Tuning the concentrations of acid sites on ZSM-5 zeolite for improving light olefin production in catalytic pyrolysis of paraffin. Ind. Eng. Chem. Res. 2022, 61, 15842–15855. [Google Scholar]
- Peng, Z.; Zheng, J.; Zhang, J.-y.; Huan, W.; Qin, Y.-c.; Liu, H.-h.; Song, L.-j. Insight into reaction path and mechanism of catalytic cracking of n-hexane in HZSM-5 zeolites. J. Fuel Chem. Technol. 2021, 49, 1522–1530. [Google Scholar]
- Xue, Y.; Li, J.; Wang, P.; Cui, X.; Zheng, H.; Niu, Y.; Dong, M.; Qin, Z.; Wang, J.; Fan, W. Regulating Al distribution of ZSM-5 by Sn incorporation for improving catalytic properties in methanol to olefins. Appl. Catal. B 2021, 280, 119391. [Google Scholar] [CrossRef]
- Liu, P.; Yao, Y.; Zhang, X.; Wang, J. Rare earth metals ion-exchanged β-zeolites as supports of platinum catalysts for hydroisomerization of n-Heptane. Chin. J. Chem. Eng. 2011, 19, 278–284. [Google Scholar] [CrossRef]
- Yang, H.; Han, T.; Yang, W.; Sandström, L.; Jönsson, P.G. Influence of the porosity and acidic properties of aluminosilicate catalysts on coke formation during the catalytic pyrolysis of lignin. J. Anal. Appl. Pyrolysis 2022, 165, 105536. [Google Scholar] [CrossRef]
- Crespo, I.; Hertzog, J.; Carré, V.; Aubriet, F.; Valle, B. Alumina-embedded HZSM-5 with enhanced behavior for the catalytic cracking of biomass pyrolysis bio-oil: Insights into the role of mesoporous matrix in the deactivation by coke. J. Anal. Appl. Pyrolysis 2023, 172, 106009. [Google Scholar] [CrossRef]
- Hou, Y.; Bai, Z.; Lu, H.; Feng, Z.; Zhang, T.; Jia, Y.; Guo, Z.; Kong, L.; Bai, J.; Li, W. In-situ catalytic upgrading of Hami coal pyrolysis volatiles over acid-modified kaolin. Fuel 2023, 331, 125660. [Google Scholar] [CrossRef]
- Wang, G.; Liu, Y.; Zheng, J.; Pan, M.; Zhang, H.; Li, B.; Yuan, S.; Yi, Y.; Tian, H.; Li, R. Zeolite–zeolite composite fabricated by polycrystalline Y zeolite crystals parasitizing ZSM-5 zeolite. J. Mater. Res. 2015, 30, 2434–2446. [Google Scholar] [CrossRef]
- Kamil, M.S.M.; Cheralathan, K.K. Facile synthesis of hydrothermally stable mesoporous ZSM-5 zeolite from Al- SBA-16 via steam assisted crystallization. J. Porous Mater. 2020, 27, 587–601. [Google Scholar] [CrossRef]
- Jiang, H.; Guan, B.; Lin, H.; Huang, Z. Cu/SSZ-13 zeolites prepared by in situ hydrothermal synthesis method as NH3-SCR catalysts: Influence of the Si/Al ratio on the activity and hydrothermal properties. Fuel 2019, 255, 115587. [Google Scholar] [CrossRef]
- Jonscher, C.; Seifert, M.; Kretzschmar, N.; Marschall, M.S.; Le Anh, M.; Doert, T.; Busse, O.; Weigand, J.J. Origin of morphology change and effect of crystallization time and Si/Al ratio during synthesis of zeolite ZSM-5. ChemCatChem 2022, 14, e202101248. [Google Scholar] [CrossRef]
- Li, X.; Han, S.; Xu, J.; Jiang, N. Green synthesis of nano-H-ZSM-5 zeolite single-crystal aggregates via an in situ reconstruction of the topology of natural clay. Microporous Mesoporous Mater. 2023, 350, 112441. [Google Scholar] [CrossRef]
- Lee, H.C.; Woo, H.C.; Ryoo, R.; Lee, K.H.; Lee, J.S. Skeletal isomerization of n-butenes to isobutene over acid-treated natural clinoptilolite zeolites. Appl. Catal. A 2000, 196, 135–142. [Google Scholar] [CrossRef]
- Maher, P.K.; Mcdaniel, C.V. Preparation of Thermal Stable Synthetic Zeolites. US3518051A, 30 June 1970. [Google Scholar]
- Yurtaeva, A.S.; Sorokina, T.P.; Plekhova, K.S.; Potapenko, O.V.; Gulyaeva, T.I.; Talsi, V.P.; Doronin, V.P. Effect of modification conditions on the physicochemical characteristics of Y zeolite as a component of a petrochemical cracking catalyst. Pet. Chem. 2021, 61, 325–331. [Google Scholar] [CrossRef]
- Beyerlein, R.A.; McVicker, G.B. Defect structure and acid catalysis of high silica, FAU-framework zeolites: Effects of aluminum removal and of basic metal oxide addition. Stud. Surf. Sci. Catal. 2001, 134, 3–40. [Google Scholar]
- López-Fonseca, R.; de Rivas, B.; Gutiérrez-Ortiz, J.I.; Aranzabal, A.; González-Velasco, J.R. Enhanced activity of zeolites by chemical dealumination for chlorinated VOC abatement. Appl. Catal. B 2003, 41, 31–42. [Google Scholar] [CrossRef]
- Wang, W.; Liu, B.; Gao, S.; Li, M. A modified molecular sieve Y for hydrocracking. J. Fuel Chem. Technol. 2009, 37, 454–458. [Google Scholar]
- Wang, Q.L.; Giannetto, G.; Guisnet, M. Dealumination of zeolites III. Effect of extra-framework aluminum species on the activity, selectivity, and stability of Y zeolites in n-heptane cracking. J. Catal. 1991, 130, 471–482. [Google Scholar] [CrossRef]
- Qin, W.; Zhou, Y.; Rimer, J.D. Deleterious effects of non-framework Al species on the catalytic performance of ZSM-5 crystals synthesized at low temperature. React. Chem. Eng. 2019, 4, 1957–1968. [Google Scholar] [CrossRef]
- Yu, Q.; Sun, H.; Sun, H.; Li, L.; Zhu, X.; Ren, S.; Guo, Q.; Shen, B. Highly mesoporous IM-5 zeolite prepared by alkaline treatment and its catalytic cracking performance. Microporous Mesoporous Mater. 2019, 273, 297–306. [Google Scholar] [CrossRef]
- Tarach, K.; Góra-Marek, K.; Tekla, J.; Brylewska, K.; Datka, J.; Mlekodaj, K.; Makowski, W.; Igualada López, M.C.; Martínez Triguero, J.; Rey, F. Catalytic cracking performance of alkaline-treated zeolite Beta in the terms of acid sites properties and their accessibility. J. Catal. 2014, 312, 46–57. [Google Scholar] [CrossRef]
- Epelde, E.; Santos, J.I.; Florian, P.; Aguayo, A.T.; Gayubo, A.G.; Bilbao, J.; Castaño, P. Controlling coke deactivation and cracking selectivity of MFI zeolite by H3PO4 or KOH modification. Appl. Catal. A 2015, 505, 105–115. [Google Scholar] [CrossRef]
- Guo, L.; Cui, Y.; Li, H.; Fang, Y.; Prasert, R.; Wu, J.; Yang, G.; Yoneyama, Y.; Tsubaki, N. Selective formation of linear-alpha olefins (LAOs) by CO2 hydrogenation over bimetallic Fe/Co-Y catalyst. Catal. Commun. 2019, 130, 105759. [Google Scholar] [CrossRef]
- Danilova, I.G.; Dik, P.P.; Sorokina, T.P.; Gabrienko, A.A.; Kazakov, M.O.; Paukshtis, E.A.; Doronin, V.P.; Klimov, O.V.; Noskov, A.S. Effect of rare earths on acidity of high-silica ultrastable REY zeolites and catalytic performance of NiMo/REY+Al2O3 catalysts in vacuum gas oil hydrocracking. Microporous Mesoporous Mater. 2022, 329, 111547. [Google Scholar] [CrossRef]
- Moon, G.-h.; Bähr, A.; Tüysüz, H. Structural engineering of 3D carbon materials from transition metal ion-exchanged Y zeolite templates. Chem. Mater. 2018, 30, 3779–3788. [Google Scholar] [CrossRef]
- Hu, Z.; Hu, P.; Wang, X.; Wu, T.; Ge, S.; Xie, H.; Liu, B.; Chen, S.; Wu, Z. Selective hydrocracking of 1-methylnaphthalene to benzene/toluene/xylenes (BTX) over NiW/Beta bifunctional catalyst: Effects of metal–acid balance. Fuel 2024, 363, 130947. [Google Scholar] [CrossRef]
- Cao, Z.; Zhang, X.; Guo, R.; Ding, S.; Mei, J.; Wang, X.; Zheng, P.; Fan, J.; Xu, C.; Duan, A. Oriented hydrocracking of naphthalene into high-value light aromatics over difunctional catalysts: Effect of hydrogen spillover and utilization of hydroreaction characteristics for different active metals. ACS Catal. 2020, 10, 12342–12353. [Google Scholar] [CrossRef]
- Wu, T.; Chen, S.-L.; Yuan, G.-m.; Pan, X.; Du, J.; Zhang, Y.; Zhang, N. High metal–acid balance and selective hydrogenation activity catalysts for hydrocracking of 1-Methylnaphthalene to benzene, toluene, and xylene. Ind. Eng. Chem. Res. 2020, 59, 5546–5556. [Google Scholar] [CrossRef]
- Buchanan, J.S.; Santiesteban, J.G.; Haag, W.O. Mechanistic considerations in acid-catalyzed cracking of olefins. J. Catal. 1996, 158, 279–287. [Google Scholar] [CrossRef]
- Weitkamp, J.; Jacobs, P.A.; Martens, J.A. Isomerization and hydrocracking of C9 through C16 n-alkanes on Pt/HZSM-5 zeolite. Appl. Catal. 1983, 8, 123–141. [Google Scholar] [CrossRef]
- Chiarello, L.M.; Porto, T.G.; Wienhage, G.H.; Botton, V.; Wiggers, V.R. Pyrolysis of triglycerides for fuels and chemical production. In Handbook of Biomass Valorization for Industrial Applications; Wiley: Hoboken, NJ, USA, 2022; pp. 107–128. [Google Scholar]
- Ortega Garcia, F.; Muñoz Arroyo, J.; Flores Sánchez, P.; Mar Juárez, E.; Dominguez Esquivel, J. Hydrocracking kinetics of a heavy crude oil on a liquid catalyst. Energy Fuels 2017, 31, 6794–6799. [Google Scholar] [CrossRef]
- Zhang, N.; Ma, J.; Li, R.; Jiao, H. Hydrocracking of fused aromatic hydrocarbons catalyzed by Al-substituted HZSM-5—A case study of 9,10-dihydroanthracene. ACS Catal. 2020, 10, 9215–9226. [Google Scholar] [CrossRef]
- Epa, U. Polycyclic Aromatic Hydrocarbons (PAHs)-EPA Fact Sheet. Available online: https://archive.epa.gov/epawaste/hazard/wastemin/web/pdf/pahs.pdf (accessed on 5 April 2025).
- Choi, Y.; Lee, J.; Shin, J.; Lee, S.; Kim, D.; Lee, J.K. Selective hydroconversion of naphthalenes into light alkyl-aromatic hydrocarbons. Appl. Catal. A 2015, 492, 140–150. [Google Scholar] [CrossRef]
- Corma, A.; González-Alfaro, V.; Orchillés, A.V. Decalin and tetralin as probe molecules for cracking and hydrotreating the light cycle oil. J. Catal. 2001, 200, 34–44. [Google Scholar] [CrossRef]
- Fan, H.; Han, B.; Jiang, T.; Guo, J.; Wang, Q.; Cheng, Y.; Wu, S. Hydrocracking of anthracene to ethyl biphenyl promoted by coupling supercritical water and cracking catalysts. ChemCatChem 2011, 3, 1474–1479. [Google Scholar] [CrossRef]
- Pinilla, J.; Arcelus-Arrillaga, P.; Puron, H.; Millan, M. Reaction pathways of anthracene selective catalytic steam cracking using a NiK/Al2O3 catalyst. Fuel 2013, 109, 303–308. [Google Scholar] [CrossRef]
- Beltramone, A.R.; Resasco, D.E.; Alvarez, W.E.; Choudhary, T.V. Simultaneous hydrogenation of multiring aromatic compounds over NiMo catalyst. Ind. Eng. Chem. Res. 2008, 47, 7161–7166. [Google Scholar] [CrossRef]
- Korre, S.C.; Klein, M.T.; Quann, R.J. Polynuclear aromatic hydrocarbons hydrogenation. 1. Experimental reaction pathways and kinetics. Ind. Eng. Chem. Res. 1995, 34, 101–117. [Google Scholar] [CrossRef]
- Qian, W.; Yoda, Y.; Hirai, Y.; Ishihara, A.; Kabe, T. Hydrodesulfurization of dibenzothiophene and hydrogenation of phenanthrene on alumina-supported Pt and Pd catalysts. Appl. Catal. A 1999, 184, 81–88. [Google Scholar] [CrossRef]
- Celis-Cornejo, C.M.; Pérez-Martínez, D.J.; Orrego-Ruiz, J.A.; Baldovino-Medrano, V.G. Identification of refractory weakly basic nitrogen compounds in a deeply hydrotreated vacuum gas oil and assessment of the effect of some representative species over the performance of a Ni–MoS2/y-zeolite–alumina catalyst in phenanthrene hydrocracking. Energy Fuels 2018, 32, 8715–8726. [Google Scholar] [CrossRef]
- Lv, P.; Yan, L.; Liu, Y.; Wang, M.; Bao, W.; Li, F. Catalytic conversion of coal pyrolysis vapors to light aromatics over hierarchical Y-type zeolites. J. Energy Inst. 2020, 93, 1354–1363. [Google Scholar] [CrossRef]
- Jentoft, F.C.; Gates, B.C. Solid-acid-catalyzed alkane cracking mechanisms: Evidence from reactions of small probe molecules. Top. Catal. 1997, 4, 1–13. [Google Scholar] [CrossRef]
- Lin, L.F.; Zhao, S.F.; Zhang, D.W.; Fan, H.; Liu, Y.M.; He, M.Y. Acid strength controlled reaction pathways for the catalytic cracking of 1-Pentene to propene over ZSM-5. ACS Catal. 2015, 5, 4048–4059. [Google Scholar] [CrossRef]
- Meusinger, J.; Corma, A. Influence of zeolite composition and structure on hydrogen transfer reactions from hydrocarbons and from hydrogen. J. Catal. 1996, 159, 353–360. [Google Scholar] [CrossRef]
- Cumming, K.A.; Wojciechowski, B.W. Hydrogen transfer, coke formation, and catalyst decay and their role in the chain mechanism of catalytic cracking. Catal. Rev. 1996, 38, 101–157. [Google Scholar] [CrossRef]
- Kissin, Y.V. Chemical mechanisms of catalytic cracking over solid acidic catalysts: Alkanes and alkenes. Catal. Rev. 2001, 43, 85–146. [Google Scholar] [CrossRef]
- Flinn, R.; Larson, O.; Beuther, H. The mechanism of catalytic hydrocracking. Ind. Eng. Chem. 1960, 52, 153–156. [Google Scholar] [CrossRef]
- Abbot, J.; Wojciechowski, B. Catalytic reactions of branched paraffins on HY zeolite. J. Catal. 1988, 113, 353–366. [Google Scholar] [CrossRef]
- Greensfelder, B.; Voge, H.; Good, G. Catalytic and thermal cracking of pure hydrocarbons: Mechanisms of reaction. Ind. Eng. Chem. 1949, 41, 2573–2584. [Google Scholar] [CrossRef]
- Nace, D.M. Catalytic cracking over crystalline aluminosilicates. II. Application of microreactor technique to investigation of structural effects of hydrocarbon reactants. Ind. Eng. Chem. Prod. Res. Dev. 1969, 8, 31–38. [Google Scholar] [CrossRef]
- Thomas, C.L. Chemistry of cracking catalysts. Ind. Eng. Chem. 1949, 41, 2564–2573. [Google Scholar] [CrossRef]
- Thivasasith, A.; Maihom, T.; Pengpanich, S.; Wattanakit, C. Nanocavity effects of various zeolite frameworks on n-pentane cracking to light olefins: Combination studies of DFT calculations and experiments. Phys. Chem. Chem. Phys. 2019, 21, 22215–22223. [Google Scholar] [CrossRef]
- Chen, D.; Liu, D.; Wei, J.; Bai, Y.; Zhao, L.; Gao, J.; Xu, C. The effect of acid strength on the mechanism of catalytic pyrolysis reaction of n-hexane in ZSM5: A DFT study. Appl. Catal. A 2023, 665, 119389. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, X.; Song, Q.; Zhao, L.; Su, H.; Li, H.; Zeng, X.; Zhao, D.; Xu, J. The effect of temperature and pressure on n-heptane thermal cracking in regenerative cooling channel. Combust. Flame 2018, 194, 233–244. [Google Scholar] [CrossRef]
- Fu, J.; Feng, X.; Liu, Y.; Shan, H.; Yang, C. Mechanistic insights into the pore confinement effect on bimolecular and monomolecular cracking mechanisms of n-octane over HY and HZSM-5 zeolites: A DFT study. J. Phys. Chem. C 2018, 122, 12222–12230. [Google Scholar] [CrossRef]
- Burnens, G.; Bouchy, C.; Guillon, E.; Martens, J.A. Hydrocracking reaction pathways of 2,6,10,14-tetramethylpentadecane model molecule on bifunctional silica–alumina and ultrastable Y zeolite catalysts. J. Catal. 2011, 282, 145–154. [Google Scholar] [CrossRef]
- Zhao, W.; Liu, L.; Niu, X.; Yang, X.; Sun, J.; Wang, Q. Reaction pathways control of long-chain alkanes hydroisomerization and hydrocracking via tailoring the metal-acid sites intimacy. Fuel 2023, 349, 128703. [Google Scholar] [CrossRef]
- Ancheyta, J.; Speight, J.G. Hydroprocessing of Heavy Oils and Residua; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Li, J.; Yang, Z.; Deng, G.; Yang, F.; Wang, S.; Tang, X. In-situ hydrothermal upgrading and mechanism of heavy oil with nano-Fe2O3 in the porous media. J. Anal. Appl. Pyrolysis 2024, 183, 106757. [Google Scholar] [CrossRef]
- Abdelsalam, Y.I.I.; Galiakhmetova, L.K.; Sharifullin, A.V.; Tajik, A.; Mukhamatdinova, R.E.; Davletshin, R.R.; Vakhin, A.V. Comparative study of the catalytic effects of Al(CH3COO)₃ and Al₂(SO₄)₃ on heavy oil aquathermolysis in CO2 and N2 atmospheres. Appl. Catal. A 2025, 699, 120247. [Google Scholar] [CrossRef]
- Imanbayev, Y.; Tileuberdi, Y.; Ongarbayev, Y.; Mansurov, Z.; Batyrbayev, A.; Akkazin, Y.; Krivtsov, E.; Golovko, A.; Rudyk, S. Changing the Structure of Resin-Asphaltenes Molecules in Cracking. Eurasian Chem.-Technol. J. 2017, 19, 147–154. [Google Scholar] [CrossRef]
- Fusheng, O.; Yongqian, W.; Qiao, L. A lumped kinetic model for heavy oil catalytic cracking FDFCC process. Pet. Sci. Technol. 2016, 34, 335–342. [Google Scholar] [CrossRef]
- Voronetskaya, N.; Pevneva, G. Structural transformations of heavy oil resins and asphaltenes upon thermal cracking. Solid Fuel Chem. 2021, 55, 165–170. [Google Scholar] [CrossRef]
- Yang, J.P. Study of catalytic cracking of asphaltene in near-critical water. Adv. Mater. Res. 2014, 860, 1021–1025. [Google Scholar] [CrossRef]
- Lee, K.; Lee, S.; Jun, Y.; Choi, M. Cooperative effects of zeolite mesoporosity and defect sites on the amount and location of coke formation and its consequence in deactivation. J. Catal. 2017, 347, 222–230. [Google Scholar] [CrossRef]
- Etim, U.J.; Bai, P.; Liu, X.; Subhan, F.; Ullah, R.; Yan, Z. Vanadium and nickel deposition on FCC catalyst: Influence of residual catalyst acidity on catalytic products. Microporous Mesoporous Mater. 2019, 273, 276–285. [Google Scholar] [CrossRef]
- Feng, B.; Wei, Y.-C.; Song, W.-Y.; Xu, C.-M. A review on the structure-performance relationship of the catalysts during propane dehydrogenation reaction. Pet. Sci. 2022, 19, 819–838. [Google Scholar] [CrossRef]
- Bibby, D.M.; Howe, R.F.; McLellan, G.D. Coke formation in high-silica zeolites. Appl. Catal. A 1992, 93, 1–34. [Google Scholar] [CrossRef]
- Wang, C.; Leng, S.; Guo, H.; Cao, L.; Huang, J. Acid and alkali treatments for regulation of hydrophilicity/hydrophobicity of natural zeolite. Appl. Surf. Sci. 2019, 478, 319–326. [Google Scholar] [CrossRef]
- Ogura, M.; Shinomiya, S.-Y.; Tateno, J.; Nara, Y.; Nomura, M.; Kikuchi, E.; Matsukata, M. Alkali-treatment technique—New method for modification of structural and acid-catalytic properties of ZSM-5 zeolites. Appl. Catal. A 2001, 219, 33–43. [Google Scholar] [CrossRef]
- Zhao, L.; Shen, B.; Gao, J.; Xu, C. Investigation on the mechanism of diffusion in mesopore structured ZSM-5 and improved heavy oil conversion. J. Catal. 2008, 258, 228–234. [Google Scholar] [CrossRef]
- Tao, Y.; Kanoh, H.; Abrams, L.; Kaneko, K. Mesopore-modified zeolites: Preparation, characterization, and applications. Chem. Rev. 2006, 106, 896–910. [Google Scholar] [CrossRef]
- Bi, C.; Wang, X.; You, Q.; Liu, B.; Li, Z.; Zhang, J.; Hao, Q.; Sun, M.; Chen, H.; Ma, X. Catalytic upgrading of coal pyrolysis volatiles by Ga-substituted mesoporous ZSM-5. Fuel 2020, 267, 117217. [Google Scholar] [CrossRef]
- Cao, W.; Li, Z.; Zhang, Y.; Fu, P. Hydrodeoxygenation of lignin phenol derivatives to aromatic hydrocarbons: A mini-review of metal/acid bifunctional catalysts. Energy Fuels 2024, 38, 23246–23267. [Google Scholar] [CrossRef]
- Shi, Y.; Xing, E.; Wu, K.; Wang, J.; Yang, M.; Wu, Y. Recent progress on upgrading of bio-oil to hydrocarbons over metal/zeolite bifunctional catalysts. Catal. Sci. Technol. 2017, 7, 2385–2415. [Google Scholar] [CrossRef]
- Idris, N.A.B. Catalytic Hydroprocessing of Fatty Acid to Renewable Diesel Using Lanthanum-Modified Zeolite-Based Catalysts; The University of Newcastle: Callaghan, Australia, 2022. [Google Scholar]
- Van der Mynsbrugge, J.; Bell, A.T. Challenges for the theoretical description of the mechanism and kinetics of reactions catalyzed by zeolites. J. Catal. 2021, 404, 832–849. [Google Scholar] [CrossRef]
- Lalsare, A.D.; Khan, T.S.; Leonard, B.; Vukmanovich, R.; Tavazohi, P.; Li, L.; Hu, J. Graphene-supported Fe/Ni, β-Mo2C nanoparticles: Experimental and DFT integrated approach to catalyst development for synergistic hydrogen production through lignin-rich biomass reforming and reduced shale gas flaring. ACS Catal. 2021, 11, 364–382. [Google Scholar] [CrossRef]
- Li, S.-C.; Lin, Y.-C.; Li, Y.-P. Understanding the catalytic activity of microporous and mesoporous zeolites in cracking by experiments and simulations. Catalysts 2021, 11, 1114. [Google Scholar] [CrossRef]
- He, X.; Tian, Y.; Qiao, C.; Liu, G. Acid-driven architecture of hierarchical porous ZSM–5 with high acidic quantity and its catalytic cracking performance. Chem. Eng. J. 2023, 473, 145334. [Google Scholar] [CrossRef]
- Bond, G.C. Supported metal catalysts: Some unsolved problems. Chem. Soc. Rev. 1991, 20, 441–475. [Google Scholar] [CrossRef]
- Kalz, K.F.; Kraehnert, R.; Dvoyashkin, M.; Dittmeyer, R.; Gläser, R.; Krewer, U.; Reuter, K.; Grunwaldt, J.-D. Future challenges in heterogeneous catalysis: Understanding catalysts under dynamic reaction conditions. ChemCatChem 2017, 9, 17–29. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Rossmeisl, J.; Christensen, C.H. Towards the computational design of solid catalysts. Nat. Chem. 2009, 1, 37–46. [Google Scholar] [CrossRef]
- Liu, C.; Li, G.; Hensen, E.J.; Pidko, E.A. Nature and catalytic role of extraframework aluminum in faujasite zeolite: A theoretical perspective. ACS Catal. 2015, 5, 7024–7033. [Google Scholar] [CrossRef]
- Pan, T.; Wu, Z.; Yip, A.C.K. Advances in the green synthesis of microporous and hierarchical zeolites: A short review. Catalysts 2019, 9, 274. [Google Scholar] [CrossRef]
- Mallette, A.J.; Seo, S.; Rimer, J.D. Synthesis strategies and design principles for nanosized and hierarchical zeolites. Nat. Synth. 2022, 1, 521–534. [Google Scholar] [CrossRef]
- Liu, X.; Wang, C.; Zhou, J.; Liu, C.; Liu, Z.; Shi, J.; Wang, Y.; Teng, J.; Xie, Z. Molecular transport in zeolite catalysts: Depicting an integrated picture from macroscopic to microscopic scales. Chem. Soc. Rev. 2022, 51, 8174–8200. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, J.-M.; Rey, J.; Bignaud, C.; Bucko, T.s.; Raybaud, P.; Moscovici-Mirande, M.; Portejoie, F.; James, C.; Bouchy, C.; Chizallet, C. Multiscale modeling as a tool for the prediction of catalytic performances: The case of n-heptane hydroconversion in a large-pore zeolite. ACS Catal. 2022, 12, 1068–1081. [Google Scholar] [CrossRef]
- Arribas, D. Surface-Catalysed Dehydrogenation and Aromatisation Reactions of Hydrocarbons; International Council on Mining and Metals: London, UK, 2024. [Google Scholar]
- Li, J.; Zheng, D.; Yao, Z.; Wang, S.; Xu, R.; Deng, S.; Chen, B.; Wang, J. Formation mechanism of monocyclic aromatic hydrocarbons during pyrolysis of styrene butadiene rubber in waste passenger car tires. ACS Omega 2022, 7, 42890–42900. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Peng, C.; Wang, J.; Liu, H.; Gao, X.; Liu, H.; Xu, C. Facile synthesis of mesoporous zeolite Y with improved catalytic performance for heavy oil fluid catalytic cracking. Ind. Eng. Chem. Res. 2014, 53, 3406–3411. [Google Scholar] [CrossRef]
- Gutiérrez, A.; Arandes, J.M.; Castaño, P.; Olazar, M.; Bilbao, J. Preliminary studies on fuel production through LCO hydrocracking on noble-metal supported catalysts. Fuel 2012, 94, 504–515. [Google Scholar] [CrossRef]
- Xie, Y.; Zhang, Y.; He, L.; Jia, C.Q.; Yao, Q.; Sun, M.; Ma, X. Anti-deactivation of zeolite catalysts for residue fluid catalytic cracking. Appl. Catal. A 2023, 657, 119159. [Google Scholar] [CrossRef]
- Qazi, U.Y.; Javaid, R.; Ikhlaq, A.; Khoja, A.H.; Saleem, F. A comprehensive review on zeolite chemistry for catalytic conversion of biomass/waste into green fuels. Molecules 2022, 27, 8578. [Google Scholar] [CrossRef]







































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).