
Tailoring the Oxygen Evolution Activity and Stability Using Defect Chemistry

 , and
, and Abstract
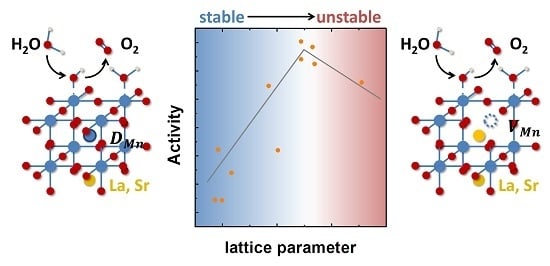
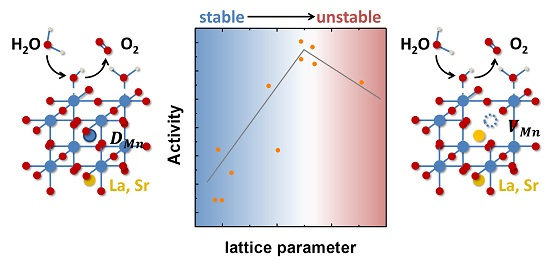{kind=link}
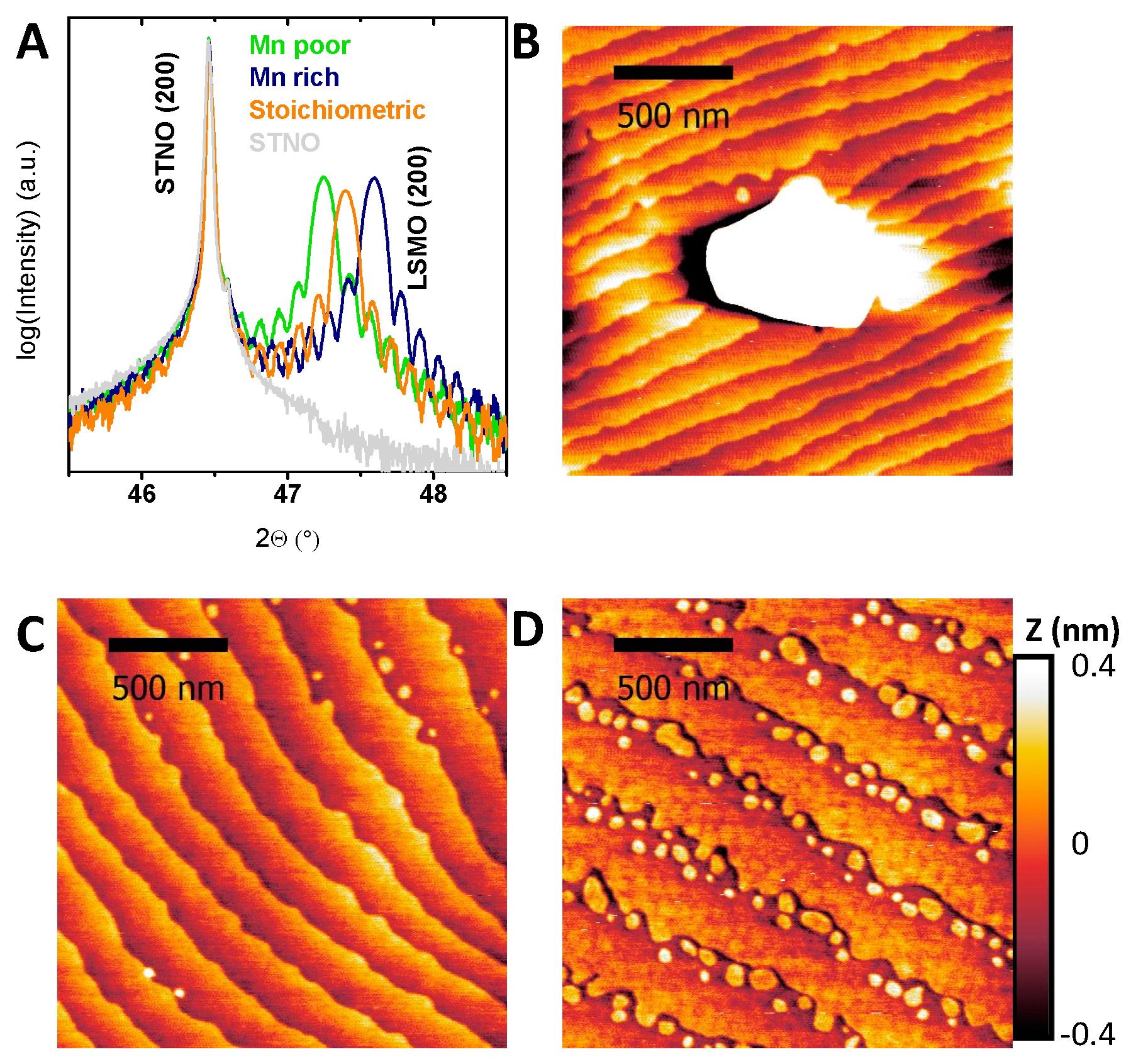{kind=link}
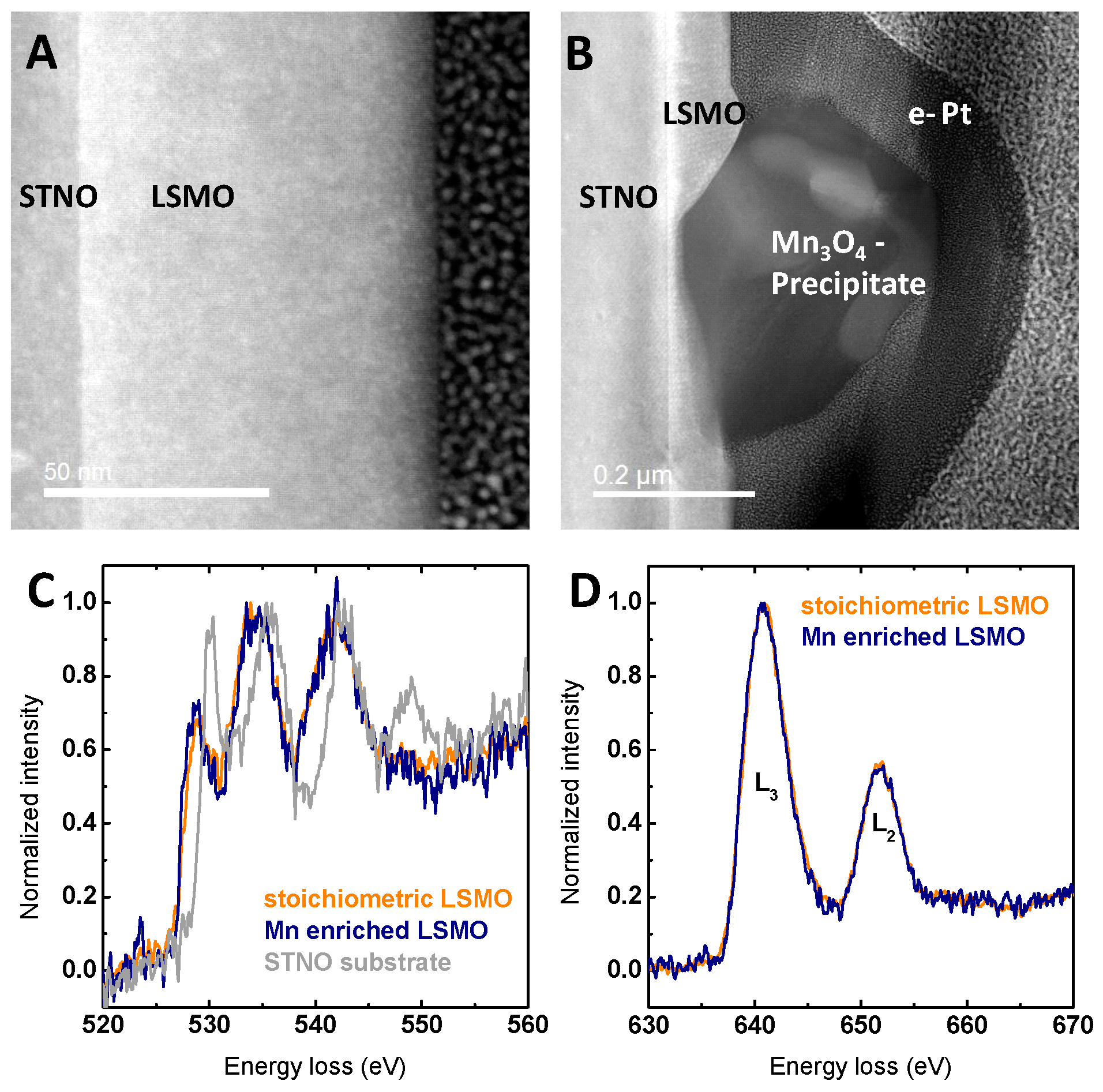{kind=link}
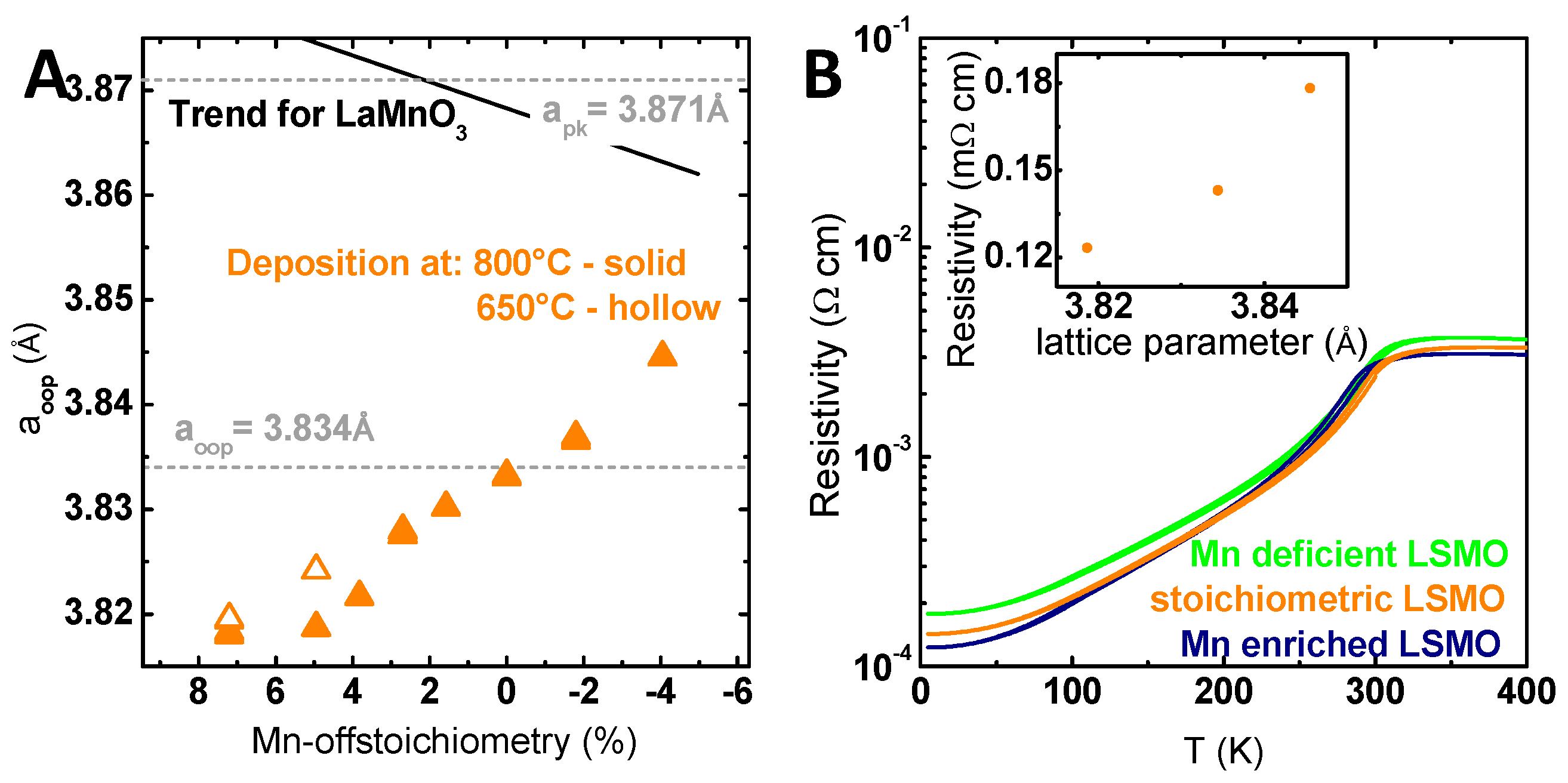{kind=link}
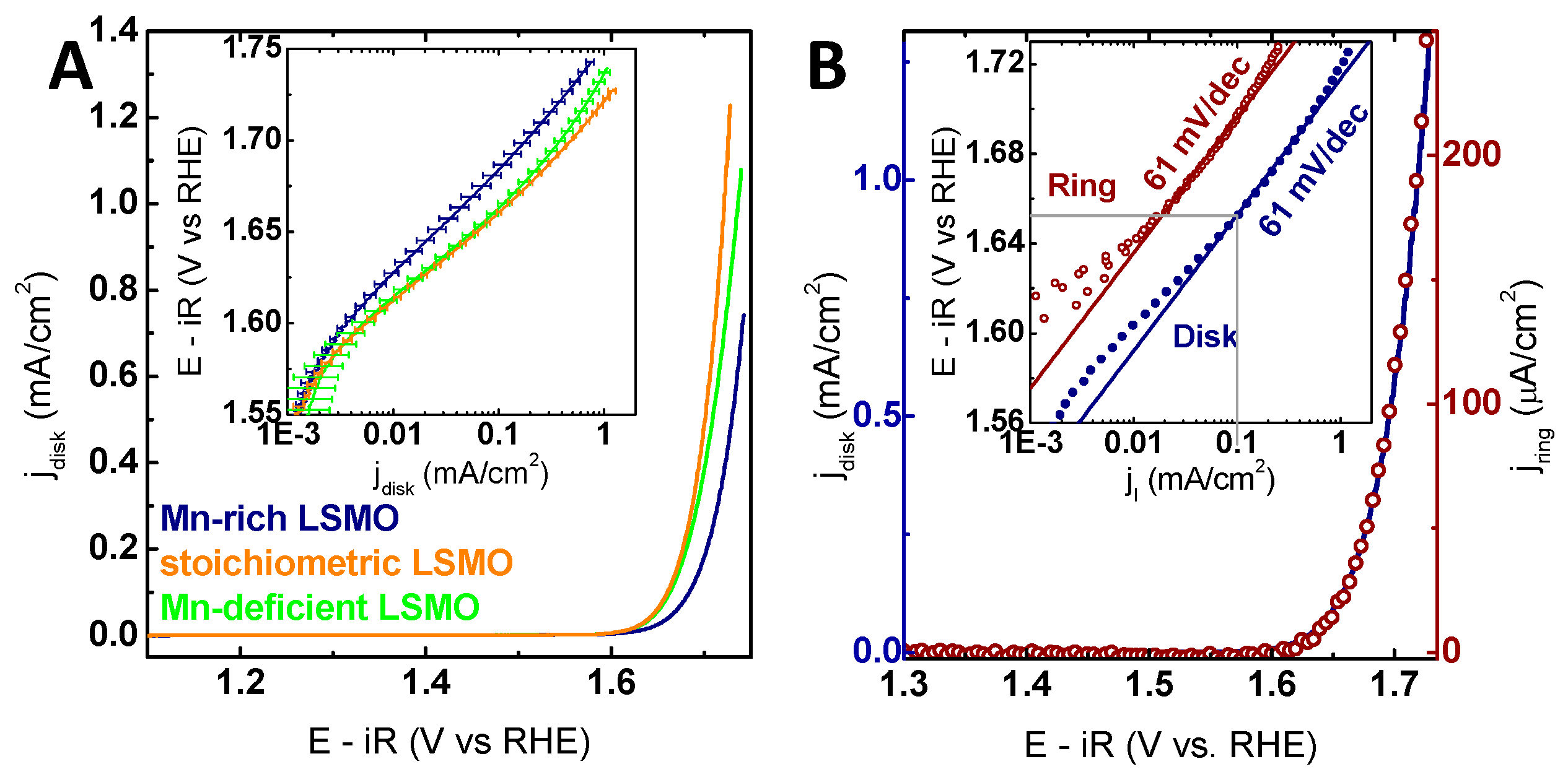{kind=link}
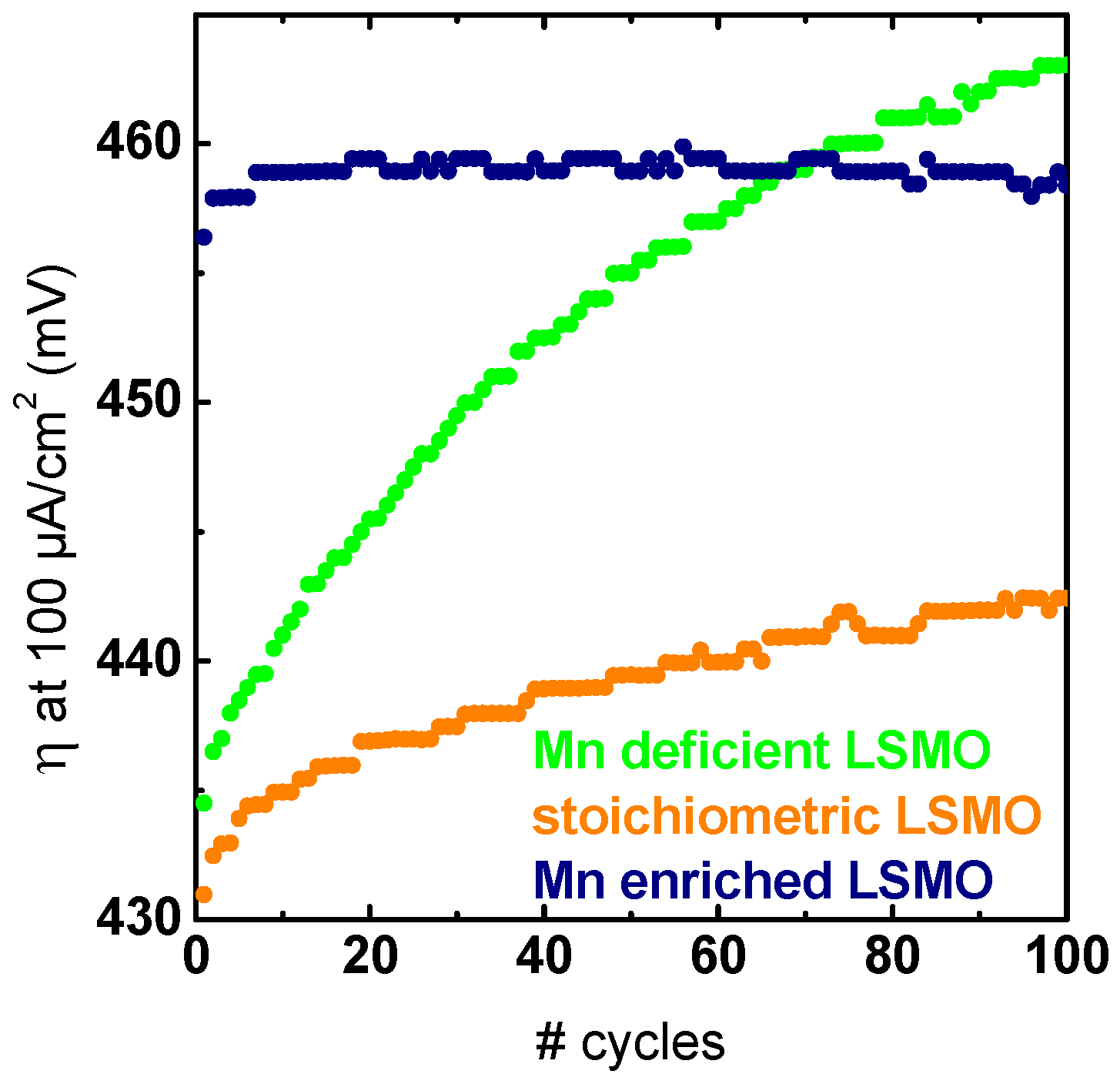{kind=link}
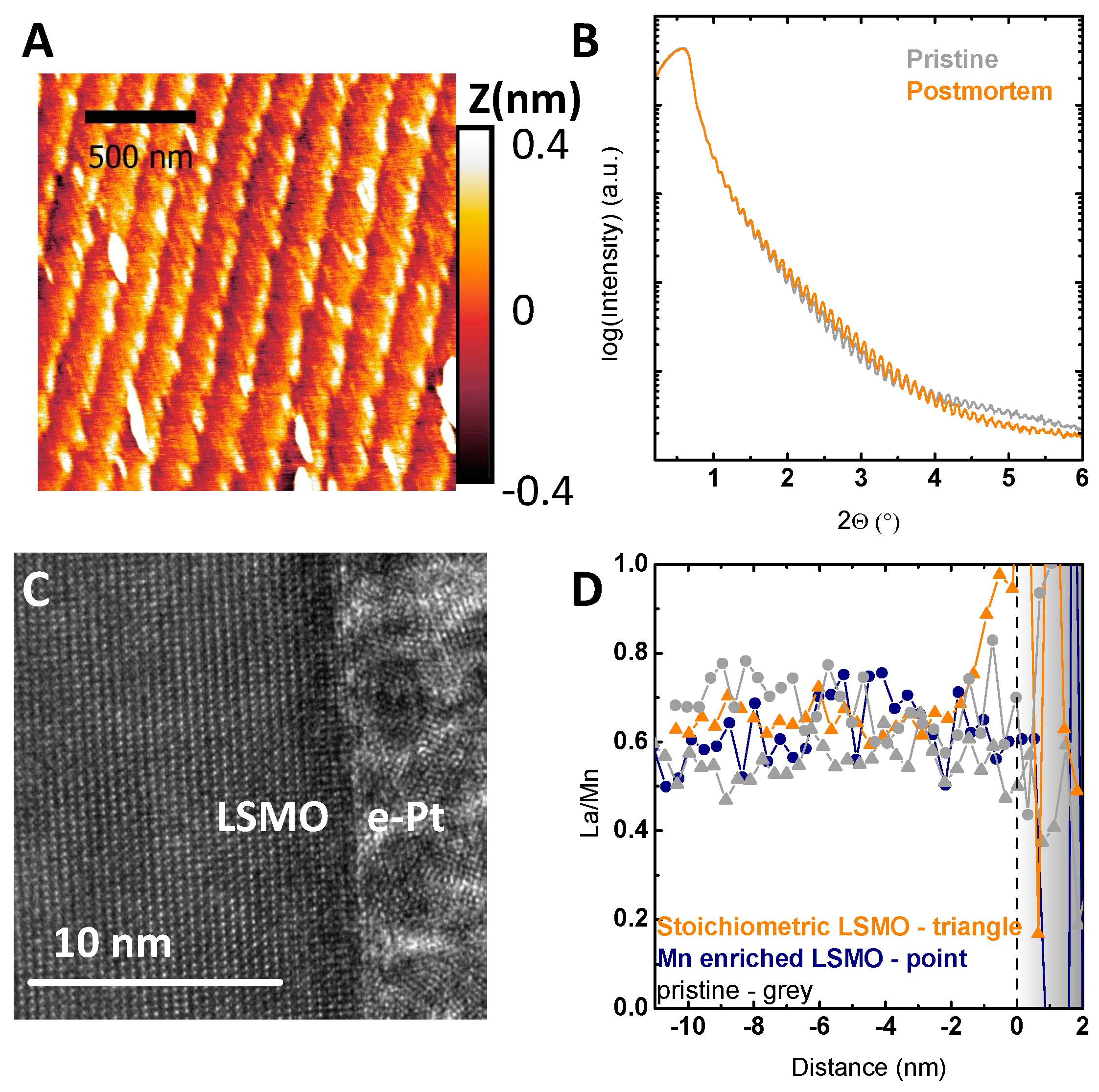{kind=link}
{kind=link}
Share and Cite
Scholz, J.; Risch, M.; Wartner, G.; Luderer, C.; Roddatis, V.; Jooss, C. Tailoring the Oxygen Evolution Activity and Stability Using Defect Chemistry. Catalysts 2017, 7, 139. https://doi.org/10.3390/catal7050139
Scholz J, Risch M, Wartner G, Luderer C, Roddatis V, Jooss C. Tailoring the Oxygen Evolution Activity and Stability Using Defect Chemistry. Catalysts. 2017; 7(5):139. https://doi.org/10.3390/catal7050139
Chicago/Turabian StyleScholz, Julius, Marcel Risch, Garlef Wartner, Christoph Luderer, Vladimir Roddatis, and Christian Jooss. 2017. "Tailoring the Oxygen Evolution Activity and Stability Using Defect Chemistry" Catalysts 7, no. 5: 139. https://doi.org/10.3390/catal7050139
APA StyleScholz, J., Risch, M., Wartner, G., Luderer, C., Roddatis, V., & Jooss, C. (2017). Tailoring the Oxygen Evolution Activity and Stability Using Defect Chemistry. Catalysts, 7(5), 139. https://doi.org/10.3390/catal7050139