Experimental Evaluation of a Membrane Micro Channel Reactor for Liquid Phase Direct Synthesis of Hydrogen Peroxide in Continuous Flow Using Nafion® Membranes for Safe Utilization of Undiluted Reactants



Abstract
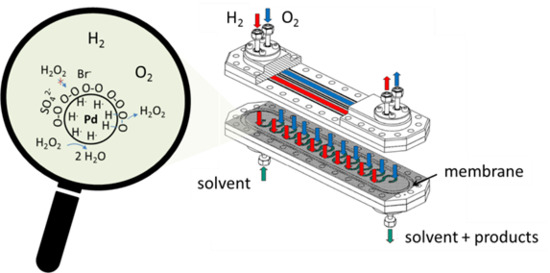
:
1. Introduction
2. Results and Discussion
2.1. Influence of Residence Time and Catalyst
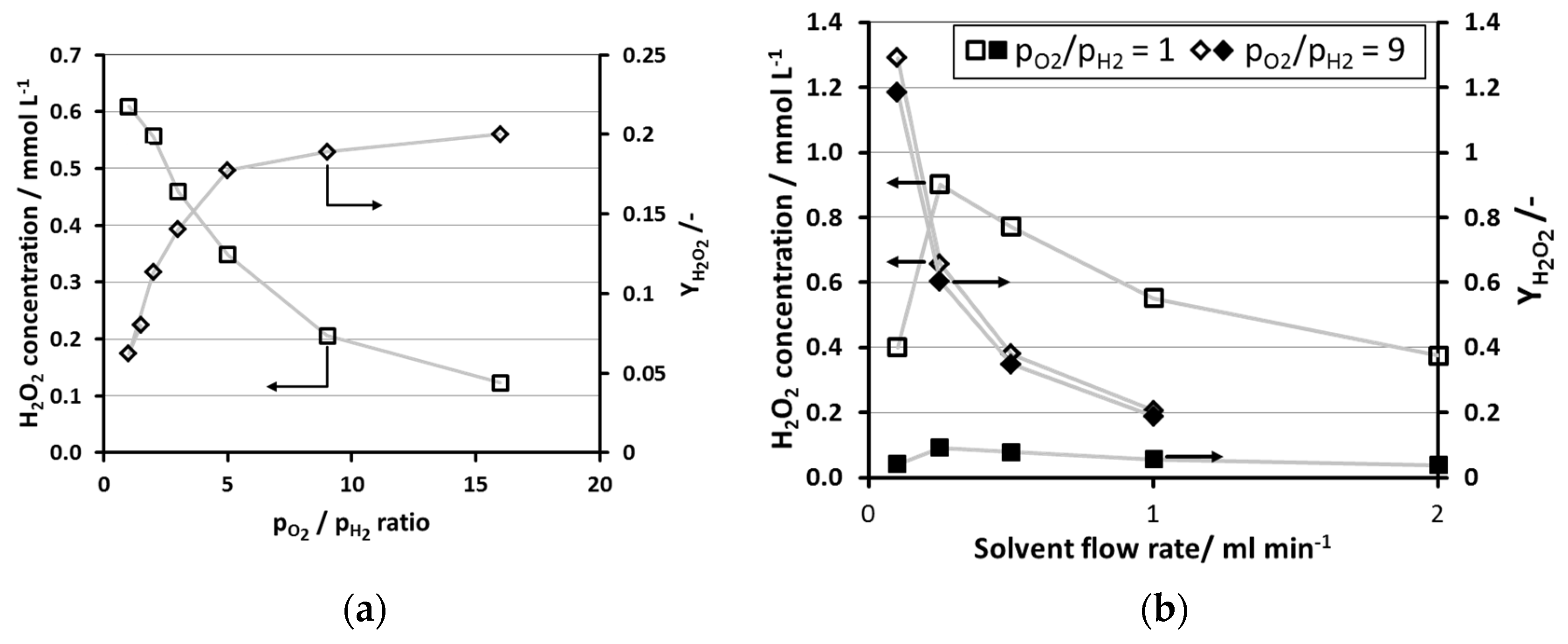
2.2. Influence of O2/H2-Ratio
2.3. Influence of System Pressure
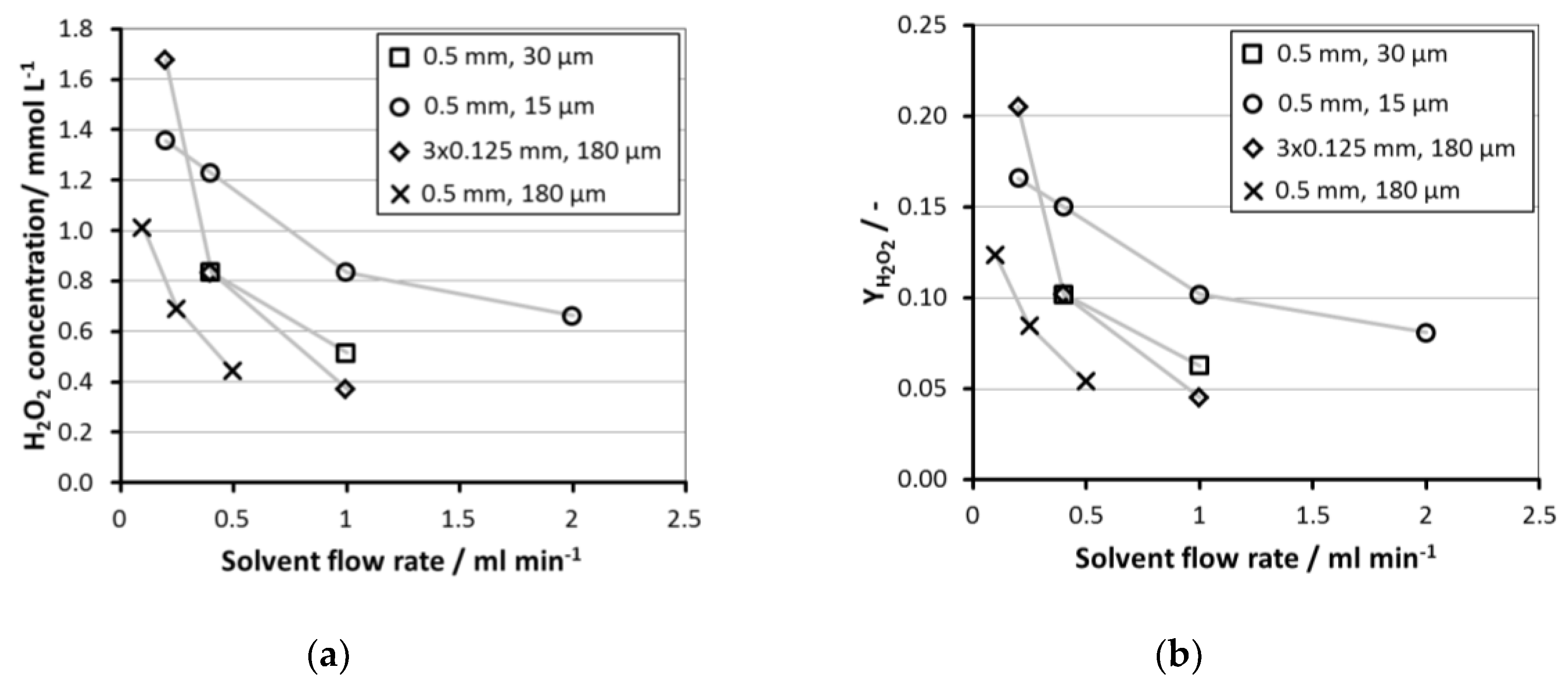
2.4. Influence of Micro Channel Depth and Membrane Thickness
2.5. Influence of Additives
2.6. System Productivity
3. Materials and Methods
3.1. The Membrane Micro Channel Reactor
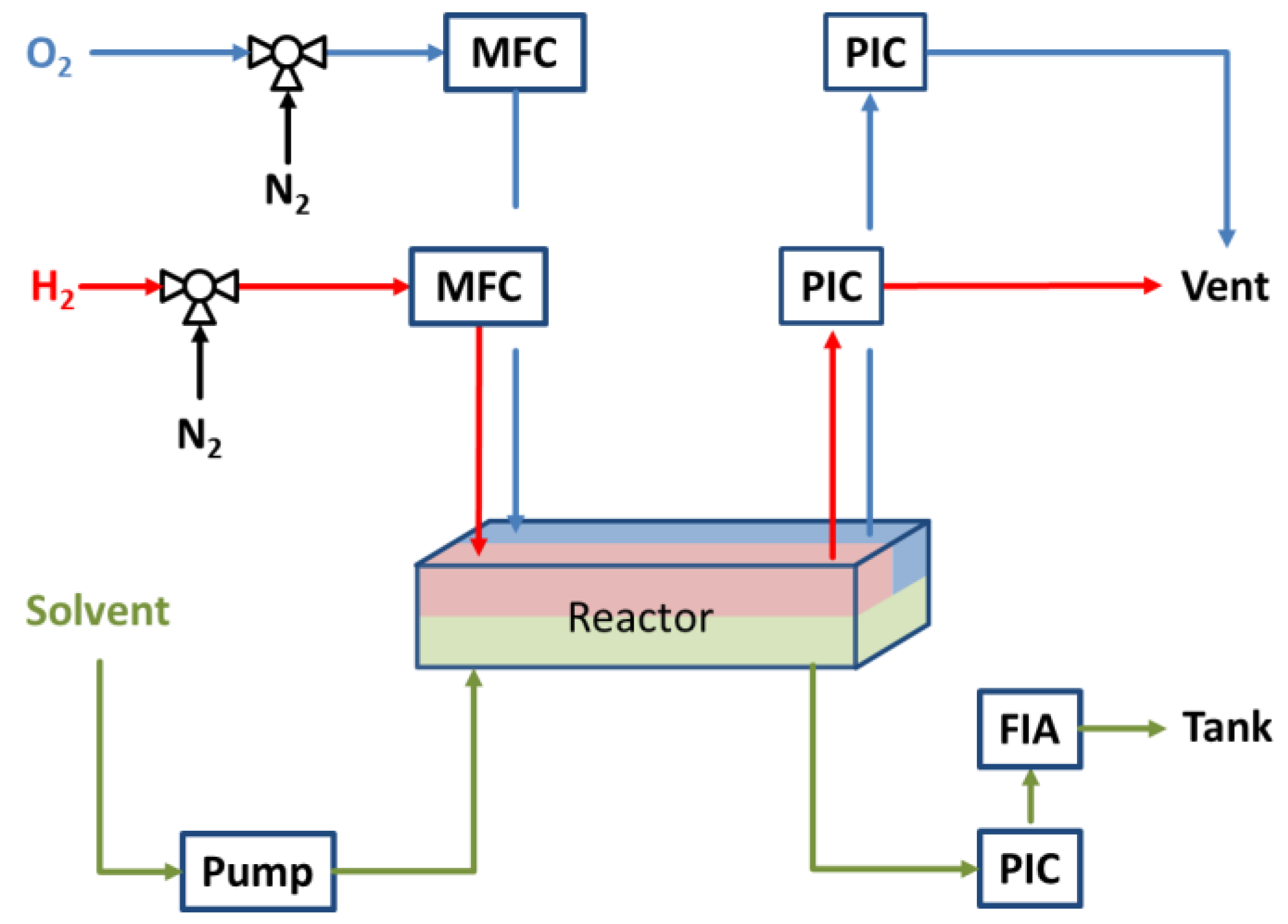
3.2. Experimental Setup for Direct Synthesis of Hydrogen Peroxide
3.3. Experimental Procedure
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dittmeyer, R.; Boeltken, T.; Piermartini, P.; Selinsek, M.; Loewert, M.; Dallmann, F.; Kreuder, H.; Cholewa, M.; Wunsch, A.; Belimov, M.; et al. Micro and micro membrane reactors for advanced applications in chemical energy conversion. Curr. Opin. Chem. Eng. 2017, 17, 108–125. [Google Scholar] [CrossRef]
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: Oxford, UK, 2000; Volume 30. [Google Scholar]
- Edwards, J.K.; Freakley, S.J.; Lewis, R.J.; Pritchard, J.C.; Hutchings, G.J. Advances in the direct synthesis of hydrogen peroxide from hydrogen and oxygen. Catal. Today 2015, 248, 3–9. [Google Scholar] [CrossRef]
- Henkel, H.; Weber, W. Manufacture of Hydrogen Peroxide. U.S. Patent 1108752, 25 August 1914. [Google Scholar]
- Gosser, L.W. Catalytic Process for Making H2O2 from Hydrogen and Oxygen. U.S. Patent 4681751, 21 July 1987. [Google Scholar]
- Centi, G.; Perathoner, S.; Abate, S. Direct synthesis of hydrogen peroxide: Recent advances. Mod. Heterog. Oxid. Catal. Des. React. Charact. 2009, 253–287. [Google Scholar] [CrossRef]
- Haas, T.; Stochniol, G.; Rollmann, J. Direct Synthesis of Hydrogen Peroxide and Integration Thereof into Oxidation Processes. U.S. Patent 6764671, 20 July 2004. [Google Scholar]
- Haas, T.; Stochniol, G.; Rollmann, J. Direct Synthesis of Hydrogen Peroxide and Integration Thereof into Oxidation Processes. U.S. Patent 7005528, 28 February 2006. [Google Scholar]
- Parasher, S.; Rueter, M.; Zhou, B. Nanocatalyst Anchored onto Acid Functionalized Solid Support and Methods of Making and Using Same. U.S. Patent 7045481, 16 May 2006. [Google Scholar]
- Rueter, M.; Zhou, B.; Parasher, S. Process for Direct Catalytic Hydrogen Peroxide Production. U.S. Patent 7144565, 5 December 2006. [Google Scholar]
- BusinessWire Web Page. Available online: https://www.businesswire.com/news/home/20050309005685/en/DegussaHeadwaters-Pilot-Plant-Validates-New-Technology (accessed on 26 October 2018).
- Goor, G.; Glenneberg, J.; Jacobi, S. Hydrogen peroxide. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2000. [Google Scholar]
- United States Environmental Protection Agency Web Page. Available online: https://www.epa.gov/greenchemistry/presidential-green-chemistry-challenge-2007-greener-reaction-conditions-award (accessed on 26 October 2018).
- García-Serna, J.; Moreno, T.; Biasi, P.; Cocero, M.J.; Mikkola, J.-P.; Salmi, T.O. Engineering in direct synthesis of hydrogen peroxide: Targets, reactors and guidelines for operational conditions. Green Chem. 2014, 16, 2320–2343. [Google Scholar] [CrossRef]
- Evonik Industries AG Web Page. Available online: https://corporate.evonik.com/en/media/press_releases/Pages/news-details.aspx?newsid=45021 (accessed on 25 October 2018).
- Ciriminna, R.; Albanese, L.; Meneguzzo, F.; Pagliaro, M. Hydrogen peroxide: A Key chemical for today’s sustainable development. ChemSusChem 2016, 9, 3374–3381. [Google Scholar] [CrossRef] [PubMed]
- Lunsford, J.H. The direct formation of H2O2 from H2 and O2 over palladium catalysts. J. Catal. 2003, 216, 455–460. [Google Scholar] [CrossRef]
- Samanta, C. Direct synthesis of hydrogen peroxide from hydrogen and oxygen: An overview of recent developments in the process. Appl. Catal. A Gen. 2008, 350, 133–149. [Google Scholar] [CrossRef]
- Dittmeyer, R.; Grunwaldt, J.-D.; Pashkova, A. A review of catalyst performance and novel reaction engineering concepts in direct synthesis of hydrogen peroxide. Catal. Today 2015, 248, 149–159. [Google Scholar] [CrossRef]
- Campos-Martin, J.M.; Blanco-Brieva, G.; Fierro, J.L. Hydrogen peroxide synthesis: An outlook beyond the anthraquinone process. Angew. Chem. Int. Ed. 2006, 45, 6962–6984. [Google Scholar] [CrossRef] [PubMed]
- Landon, P.; Collier, P.J.; Carley, A.F.; Chadwick, D.; Papworth, A.J.; Burrows, A.; Kiely, C.J.; Hutchings, G.J. Direct synthesis of hydrogen peroxide from H2 and O2 using Pd and Au catalysts. Phys. Chem. Chem. Phys. 2003, 5, 1917–1923. [Google Scholar] [CrossRef]
- Freakley, S.J.; He, Q.; Harrhy, J.H.; Lu, L.; Crole, D.A.; Morgan, D.J.; Ntainjua, E.N.; Edwards, J.K.; Carley, A.F.; Borisevich, A.Y. Palladium-tin catalysts for the direct synthesis of H2O2 with high selectivity. Science 2016, 351, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Tian, P.; Xu, X.; Ao, C.; Ding, D.; Li, W.; Si, R.; Tu, W.; Xu, J.; Han, Y.-F. Direct and selective synthesis of hydrogen peroxide over palladium–tellurium catalysts at ambient pressure. ChemSusChem 2017, 10, 3342–3346. [Google Scholar] [CrossRef] [PubMed]
- Biasi, P.; Mikkola, J.-P.; Sterchele, S.; Salmi, T.; Gemo, N.; Shchukarev, A.; Centomo, P.; Zecca, M.; Canu, P.; Rautio, A.-R. Revealing the role of bromide in the H2O2 direct synthesis with the catalyst wet pretreatment method (CWPM). AIChE J. 2017, 63, 32–42. [Google Scholar] [CrossRef]
- Ouyang, L.; Tian, P.; Da, G.; Xu, X.-C.; Ao, C.; Chen, T.; Si, R.; Xu, J.; Han, Y.-F. The origin of active sites for direct synthesis of H2O2 on Pd/TiO2 catalysts: Interfaces of Pd and PdO domains. J. Catal. 2015, 321, 70–80. [Google Scholar] [CrossRef]
- Ouyang, L.; Da, G.; Tian, P.; Chen, T.; Xu, J.; Han, Y.-F. Insight into active sites of Pd–Au/TiO2 catalysts in hydrogen peroxide synthesis directly from H2 and O2. J. Catal. 2014, 311, 129–136. [Google Scholar] [CrossRef]
- Arrigo, R.; Schuster, M.E.; Xie, Z.; Yi, Y.; Wowsnick, G.; Sun, L.L.; Hermann, K.E.; Friedrich, M.; Kast, P.; Hävecker, M. Nature of the N–Pd interaction in nitrogen-doped carbon nanotube catalysts. ACS Catal. 2015, 5, 2740–2753. [Google Scholar] [CrossRef]
- Arrigo, R.; Schuster, M.E.; Abate, S.; Giorgianni, G.; Centi, G.; Perathoner, S.; Wrabetz, S.; Pfeifer, V.; Antonietti, M.; Schlögl, R. Pd supported on carbon nitride boosts the direct hydrogen peroxide synthesis. ACS Catal. 2016, 6, 6959–6966. [Google Scholar] [CrossRef]
- Plauck, A.; Stangland, E.E.; Dumesic, J.A.; Mavrikakis, M. Active sites and mechanisms for H2O2 decomposition over Pd catalysts. Proc. Natl. Acad. Sci. USA 2016, 113, E1973–E1982. [Google Scholar] [CrossRef] [PubMed]
- Selinsek, M.; Deschner, B.J.; Doronkin, D.E.; Sheppard, T.L.; Grunwaldt, J.-D.; Dittmeyer, R. Revealing the Structure and Mechanism of Palladium during Direct Synthesis of Hydrogen Peroxide in Continuous Flow Using Operando Spectroscopy. ACS Catal. 2018, 8, 2546–2557. [Google Scholar] [CrossRef]
- Tian, P.; Ouyang, L.; Xu, X.; Ao, C.; Xu, X.; Si, R.; Shen, X.; Lin, M.; Xu, J.; Han, Y.-F. The origin of palladium particle size effects in the direct synthesis of H2O2: Is smaller better? J. Catal. 2017, 349, 30–40. [Google Scholar] [CrossRef]
- Lee, J.W.; Kim, J.K.; Kang, T.H.; Lee, E.J.; Song, I.K. Direct synthesis of hydrogen peroxide from hydrogen and oxygen over palladium catalyst supported on heteropolyacid-containing ordered mesoporous carbon. Catal. Today 2017, 293, 49–55. [Google Scholar] [CrossRef]
- Blanco-Brieva, G.; Montiel-Argaiz, M.; Desmedt, F.; Miquel, P.; Campos-Martin, J.M.; Fierro, J.L.G. Effect of the Acidity of the Groups of Functionalized Silicas on the Direct Synthesis of H2O2. Top. Catal. 2017, 60, 1151–1155. [Google Scholar] [CrossRef]
- Tu, R.; Li, L.; Zhang, S.; Chen, S.; Li, J.; Lu, X. Carbon-modified mesoporous anatase/TiO2 (B) whisker for enhanced activity in direct synthesis of hydrogen peroxide by palladium. Catalysts 2017, 7, 175. [Google Scholar] [CrossRef]
- Seo, M.; Lee, D.-W.; Han, S.S.; Lee, K.-Y. Direct synthesis of hydrogen peroxide from hydrogen and oxygen over mesoporous silica-shell-coated, palladium-nanocrystal-grafted SiO2 nanobeads. ACS Catal. 2017, 7, 3039–3048. [Google Scholar] [CrossRef]
- Grabow, L.C.; Hvolbæk, B.; Falsig, H.; Nørskov, J.K. Search directions for direct H2O2 synthesis catalysts starting from Au 12 nanoclusters. Top. Catal. 2012, 55, 336–344. [Google Scholar] [CrossRef]
- Deguchi, T.; Iwamoto, M. Catalytic properties of surface sites on Pd clusters for direct H2O2 synthesis from H2 and O2: A DFT study. J. Phys. Chem. C 2013, 117, 18540–18548. [Google Scholar] [CrossRef]
- Staykov, A.; Kamachi, T.; Ishihara, T.; Yoshizawa, K. Theoretical study of the direct synthesis of H2O2 on Pd and Pd/Au surfaces. J. Phys. Chem. C 2008, 112, 19501–19505. [Google Scholar] [CrossRef]
- Wilson, N.M.; Flaherty, D.W. Mechanism for the direct synthesis of H2O2 on Pd clusters: Heterolytic reaction pathways at the liquid–solid interface. J. Am. Chem. Soc. 2015, 138, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Wang, L.; Li, G.; Guo, H. A review on research progress in the direct synthesis of hydrogen peroxide from hydrogen and oxygen: Noble-metal catalytic method, fuel-cell method and plasma method. Catal. Sci. Technol. 2016, 6, 1593–1610. [Google Scholar] [CrossRef]
- Biasi, P.; Menegazzo, F.; Pinna, F.; Eränen, K.; Salmi, T.O.; Canu, P. Continuous H2O2 direct synthesis over PdAu catalysts. Chem. Eng. J. 2011, 176, 172–177. [Google Scholar] [CrossRef]
- Biasi, P.; Canu, P.; Menegazzo, F.; Pinna, F.; Salmi, T.O. Direct synthesis of hydrogen peroxide in a trickle bed reactor: Comparison of Pd-based catalysts. Ind. Eng. Chem. Res. 2012, 51, 8883–8890. [Google Scholar] [CrossRef] [Green Version]
- Biasi, P.; García-Serna, J.; Bittante, A.; Salmi, T. Direct synthesis of hydrogen peroxide in water in a continuous trickle bed reactor optimized to maximize productivity. Green Chem. 2013, 15, 2502–2513. [Google Scholar] [CrossRef]
- Paunovic, V.; Ordomsky, V.; D’angelo, M.F.N.; Schouten, J.C.; Nijhuis, T.A. Direct synthesis of hydrogen peroxide over Au–Pd catalyst in a wall-coated microchannel. J. Catal. 2014, 309, 325–332. [Google Scholar] [CrossRef]
- Paunovic, V.; Schouten, J.C.; Nijhuis, T.A. Direct synthesis of hydrogen peroxide using concentrated H2 and O2 mixtures in a wall-coated microchannel–kinetic study. Appl. Catal. A Gen. 2015, 505, 249–259. [Google Scholar] [CrossRef]
- Kanungo, S.; Paunovic, V.; Schouten, J.C.; Neira D’Angelo, M.F. Facile synthesis of catalytic AuPd nanoparticles within capillary microreactors using polyelectrolyte multilayers for the direct synthesis of H2O2. Nano Lett. 2017, 17, 6481–6486. [Google Scholar] [CrossRef] [PubMed]
- Freakley, S.J.; Piccinini, M.; Edwards, J.K.; Ntainjua, E.N.; Moulijn, J.A.; Hutchings, G.J. Effect of reaction conditions on the direct synthesis of hydrogen peroxide with a AuPd/TiO2 catalyst in a flow reactor. ACS Catal. 2013, 3, 487–501. [Google Scholar] [CrossRef]
- Voloshin, Y.; Halder, R.; Lawal, A. Kinetics of hydrogen peroxide synthesis by direct combination of H2 and O2 in a microreactor. Catal. Today 2007, 125, 40–47. [Google Scholar] [CrossRef]
- Voloshin, Y.; Lawal, A. Overall kinetics of hydrogen peroxide formation by direct combination of H2 and O2 in a microreactor. Chem. Eng. Sci. 2010, 65, 1028–1036. [Google Scholar] [CrossRef]
- Ratchananusorn, W.; Gudarzi, D.; Turunen, I. Catalytic direct synthesis of hydrogen peroxide in a novel microstructured reactor. Chem. Eng. Process. Process. Intensif. 2014, 84, 24–30. [Google Scholar] [CrossRef]
- Inoue, T.; Schmidt, M.A.; Jensen, K.F. Microfabricated multiphase reactors for the direct synthesis of hydrogen peroxide from hydrogen and oxygen. Ind. Eng. Chem. Res. 2007, 46, 1153–1160. [Google Scholar] [CrossRef]
- Inoue, T.; Kikutani, Y.; Hamakawa, S.; Mawatari, K.; Mizukami, F.; Kitamori, T. Reactor design optimization for direct synthesis of hydrogen peroxide. Chem. Eng. J. 2010, 160, 909–914. [Google Scholar] [CrossRef]
- Inoue, T.; Adachi, J.; Ohtaki, K.; Lu, M.; Murakami, S.; Sun, X.; Wang, D.F. Direct hydrogen peroxide synthesis using glass microfabricated reactor–Paralleled packed bed operation. Chem. Eng. J. 2015, 278, 517–526. [Google Scholar] [CrossRef]
- Pashkova, A.; Svajda, K.; Dittmeyer, R. Direct synthesis of hydrogen peroxide in a catalytic membrane contactor. Chem. Eng. J. 2008, 139, 165–171. [Google Scholar] [CrossRef]
- Pashkova, A.; Dittmeyer, R.; Kaltenborn, N.; Richter, H. Experimental study of porous tubular catalytic membranes for direct synthesis of hydrogen peroxide. Chem. Eng. J. 2010, 165, 924–933. [Google Scholar] [CrossRef]
- Pashkova, A.; Greiner, L.; Krtschil, U.; Hofmann, C.; Zapf, R. Direct synthesis of hydrogen peroxide over supported Pd catalysts: Turning to dense CO2 as an alternative solvent. Appl. Catal. A Gen. 2013, 464, 281–287. [Google Scholar] [CrossRef]
- Selinsek, M.; Bohrer, M.; Vankayala, B.K.; Haas-Santo, K.; Kraut, M.; Dittmeyer, R. Towards a new membrane micro reactor system for direct synthesis of hydrogen peroxide. Catal. Today 2016, 268, 85–94. [Google Scholar] [CrossRef]
- Moreno, T.; García-Serna, J.; Plucinski, P.; Sánchez-Montero, M.J.; Cocero, M.J. Direct synthesis of H2O2 in methanol at low pressures over Pd/C catalyst: Semi-continuous process. Appl. Catal. A Gen. 2010, 386, 28–33. [Google Scholar] [CrossRef]
- Broka, K.; Ekdunge, P. Oxygen and hydrogen permeation properties and water uptake of Nafion® 117 membrane and recast film for PEM fuel cell. J. Appl. Electrochem. 1997, 27, 117–123. [Google Scholar] [CrossRef]
- Chiou, J.S.; Paul, D.R. Gas permeation in a dry Nafion membrane. Ind. Eng. Chem. Res. 1988, 27, 2161–2164. [Google Scholar] [CrossRef]
- Mohamed, H.F.; Ito, K.; Kobayashi, Y.; Takimoto, N.; Takeoka, Y.; Ohira, A. Free volume and permeabilities of O2 and H2 in Nafion membranes for polymer electrolyte fuel cells. Polymer 2008, 49, 3091–3097. [Google Scholar] [CrossRef]
- Kim, J.; Chung, Y.-M.; Kang, S.-M.; Choi, C.-H.; Kim, B.-Y.; Kwon, Y.-T.; Kim, T.J.; Oh, S.-H.; Lee, C.-S. Palladium nanocatalysts immobilized on functionalized resin for the direct synthesis of hydrogen peroxide from hydrogen and oxygen. ACS Catal. 2012, 2, 1042–1048. [Google Scholar] [CrossRef]
- Pashkova, A.; Svajda, K.; Black, G.; Dittmeyer, R. Automated system for spectrophotometric detection of liquid phase hydrogen peroxide for concentrations up to 5% w/w. Rev. Sci. Instrum. 2009, 80, 055104. [Google Scholar] [CrossRef] [PubMed]
- DECHEMA Web Page. Available online: http://i-systems.dechema.de/detherm (accessed on 25 October 2018).
- Edwards, J.K.; Solsona, B.E.; Landon, P.; Carley, A.F.; Herzing, A.; Kiely, C.J.; Hutchings, G.J. Direct synthesis of hydrogen peroxide from H2 and O2 using TiO2-supported Au–Pd catalysts. J. Catal. 2005, 236, 69–79. [Google Scholar] [CrossRef]
- Solvay Chemicals Web Page. Available online: https://www.solvay.us/en/binaries/HH-056_Passivation-236794.pdf (accessed on 25 October 2018).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exp.-Nr. | Micro Channel | Cat. | mcat, mg | Config. 1 | Membrane | Solvent Flow Rate, mL/min | p, bar | pO2/pH2 | Additives |
|---|---|---|---|---|---|---|---|---|---|
| 1–4 | 0.5 mm | Pd/C | 200 | mfb | 180 μm | 3/2/1/0.5 | 20 | 1 | 0.15 mM NaBr, 0.15 mM H2SO4 |
| 5–9 | 0.5 mm | Pd/TiO2 | 200 | mfb | 180 μm | 1/0.75/0.5/0.4/0.25 | 20 | 1 | 0.15 mM NaBr, 0.15 mM H2SO4 |
| 10–12 | 0.5 mm | Pd/TiO2 | 200 | mfb | 180 μm | 1/0.5/0.25 | 20 | 1 | 4 mM NaBr, 0.15 mM H2SO4 |
| 13–17 | 0.5 mm | Pd/TiO2 | 200 | mfb | 180 μm | 2/1/0.5/0.25/0.1 | 10 | 1 | 4 mM NaBr, 0.15 mM H2SO4 |
| 18–24 | 0.5 mm | Pd/TiO2 | 200 | mfb | 180 μm | 1 | 10 | 1/1.5/2/3/5/9/16 | 4 mM NaBr, 0.15 mM H2SO4 |
| 25–28 | 0.5 mm | Pd/TiO2 | 200 | mfb | 180 μm | 1/0.5/0.25/0.1 | 10 | 9 | 4 mM NaBr, 0.15 mM H2SO4 |
| 29–33 | 0.5 mm | Pd/TiO2 | 100 | coating | 180 μm | 0.5 | 10/20/30/40/50 | 1 | 4 mM NaBr, 0.15 mM H2SO4 |
| 34–37 | 0.5 mm | Pd/TiO2 | 100 | coating | 180 μm | 0.5 | 10/20/30/40 | 1 | no Additives |
| 38–40 | 0.5 mm | Pd/TiO2 | 100 | fb | 180 μm | 0.5/0.25/0.1 | 10 | 1 | 5 mM NaBr |
| 41–43 | 0.5 mm | Pd/TiO2 | 200 | fb | 180 μm | 0.5/0.25/0.1 | 10 | 1 | 5 mM NaBr |
| 44–45 | 0.5 mm | Pd/TiO2 | 50 | fb | 180 μm | 0.5/0.1 | 10 | 1 | 5 mM NaBr |
| 46–48 | 3 × 0.125 mm | Pd/TiO2 | 100 | fb | 180 μm | 1/0.4/0.2 | 10 | 1 | 5 mM NaBr |
| 49–50 | 3 × 0.125 mm | Pd/TiO2 | 100 | fb | 180 μm | 1/0.4/0.2 | 10 | 1 | 5 mM NaBr, Sodium acetate buffer, pH 3 |
| 51–53 | 3 × 0.125 mm | Pd/TiO2 | 100 | fb | 180 μm | 1/0.4/0.2 | 10 | 1 | 5 mM NaBr |
| 54–57 | 0.5 mm | Pd/TiO2 | 100 | fb | 15 μm | 2/1/0.4/0.2 | 10 | 1 | 5 mM NaBr |
| 58–59 | 0.5 mm | Pd/TiO2 | 100 | coating | 30 μm | 1 | 10/40 | 1 | 5 mM NaBr |
| 60–62 | 0.5 mm | Pd/TiO2 | 100 | coating | 30 μm | 1/0.4/0.2 | 40 | 1 | 5 mM NaBr |
| 63–64 | 0.5 mm | Pd/TiO2 | 100 | fb | 30 μm | 1/0.4 | 10 | 1 | 5 mM NaBr |
| 65–66 | 0.5 mm | Pd/TiO2 | 100 | fb | 30 μm | 1/0.4 | 10 | 1 | 5 mM NaBr, 4 mM H3PO4, 25 mM H2SO4 |
| 67–69 | 0.5 mm | Pd/TiO2 | 100 | fb | 30 μm | 1/0.4/0.2 | 40 | 1 | 5 mM NaBr, 4 mM H3PO4, 25 mM H2SO4 |
| 70 | 0.5 mm | Pd/TiO2 | 100 | fb | 30 μm | 0.1 | 40 | 4 | 5 mM NaBr, 4 mM H3PO4, 25 mM H2SO4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Selinsek, M.; Kraut, M.; Dittmeyer, R. Experimental Evaluation of a Membrane Micro Channel Reactor for Liquid Phase Direct Synthesis of Hydrogen Peroxide in Continuous Flow Using Nafion® Membranes for Safe Utilization of Undiluted Reactants. Catalysts 2018, 8, 556. https://doi.org/10.3390/catal8110556
Selinsek M, Kraut M, Dittmeyer R. Experimental Evaluation of a Membrane Micro Channel Reactor for Liquid Phase Direct Synthesis of Hydrogen Peroxide in Continuous Flow Using Nafion® Membranes for Safe Utilization of Undiluted Reactants. Catalysts. 2018; 8(11):556. https://doi.org/10.3390/catal8110556
Chicago/Turabian StyleSelinsek, Manuel, Manfred Kraut, and Roland Dittmeyer. 2018. "Experimental Evaluation of a Membrane Micro Channel Reactor for Liquid Phase Direct Synthesis of Hydrogen Peroxide in Continuous Flow Using Nafion® Membranes for Safe Utilization of Undiluted Reactants" Catalysts 8, no. 11: 556. https://doi.org/10.3390/catal8110556
APA StyleSelinsek, M., Kraut, M., & Dittmeyer, R. (2018). Experimental Evaluation of a Membrane Micro Channel Reactor for Liquid Phase Direct Synthesis of Hydrogen Peroxide in Continuous Flow Using Nafion® Membranes for Safe Utilization of Undiluted Reactants. Catalysts, 8(11), 556. https://doi.org/10.3390/catal8110556