Boosting the Characterization of Heterogeneous Catalysts for H2O2 Direct Synthesis by Infrared Spectroscopy


Abstract
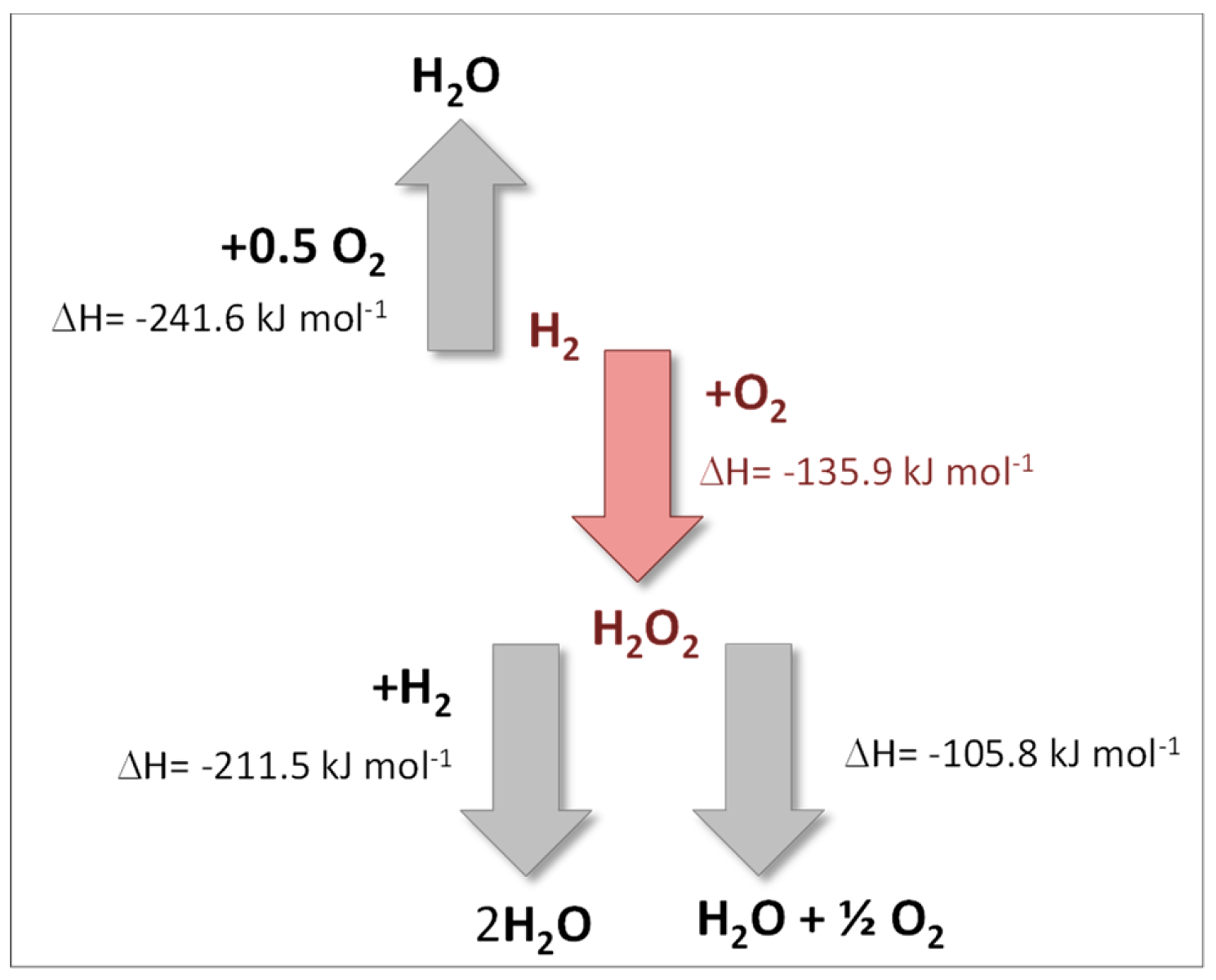
:1. The Direct Synthesis of Hydrogen Peroxide from Molecular Hydrogen and Oxygen
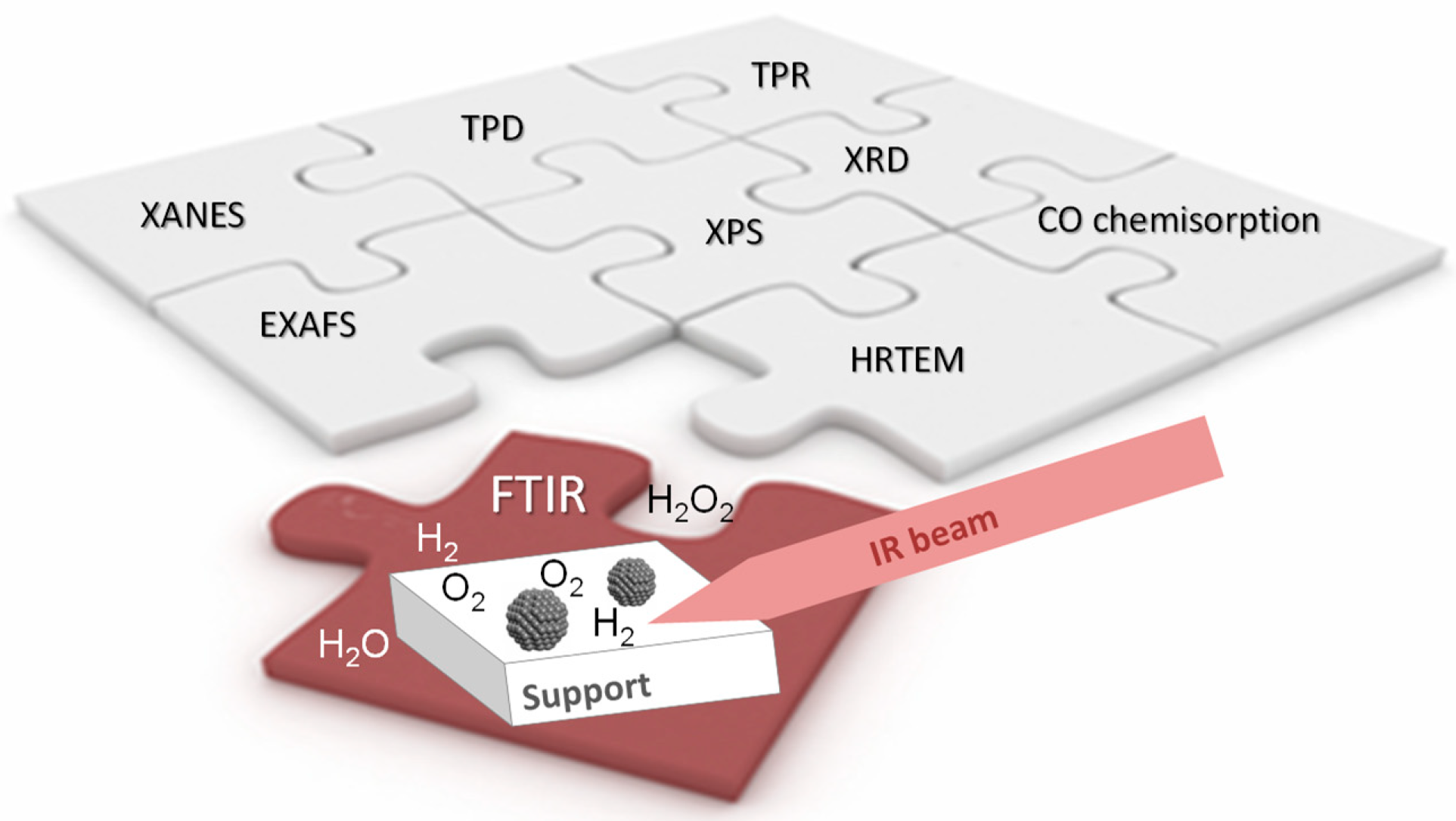
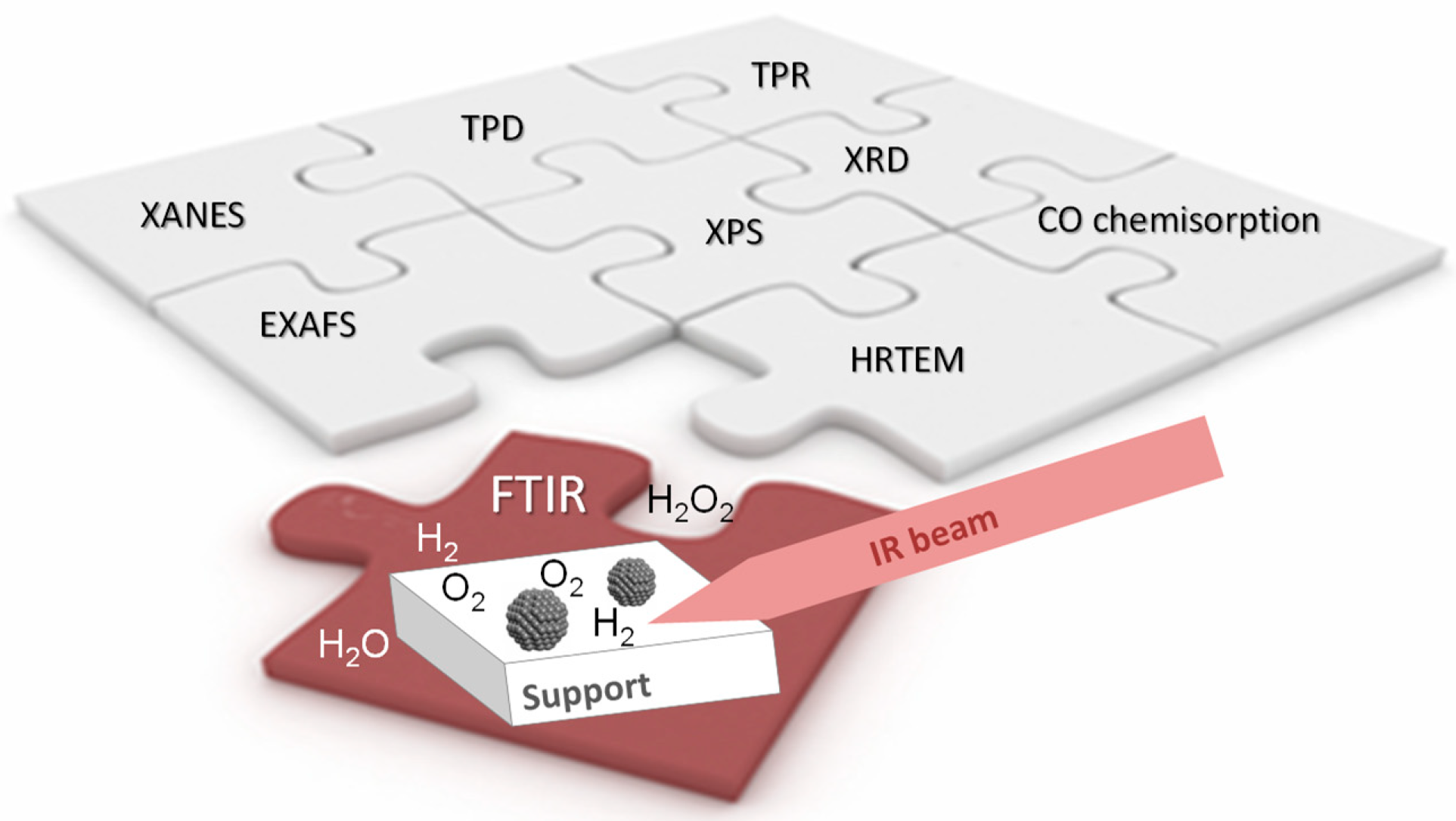
2. Infrared Absorption Spectroscopy as a Tool for Catalyst Characterization
2.1. FTIR Spectroscopy of Adsorbed Probe Molecules
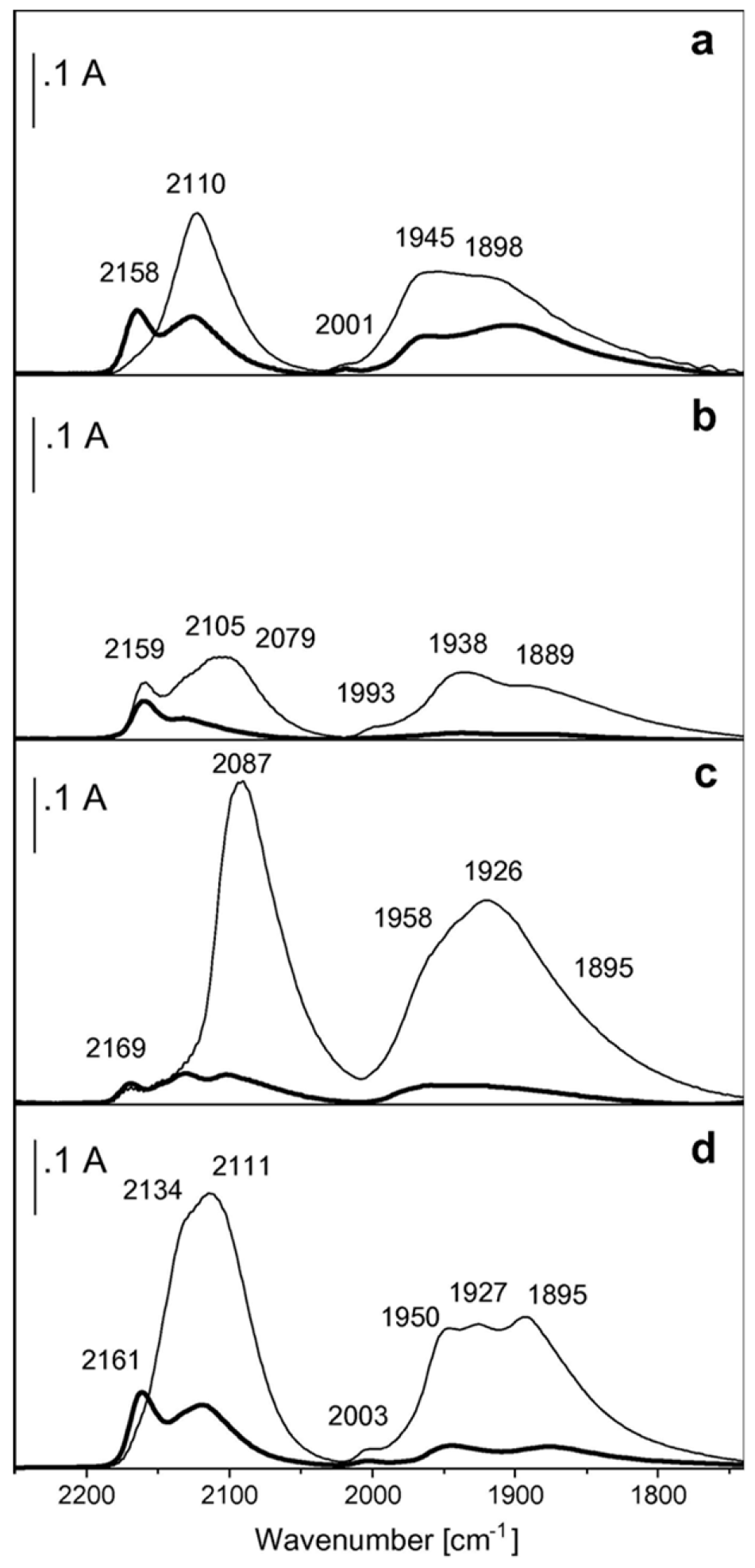
Carbon Monoxide
2.2. Experimental Setup
2.2.1. Transmission FTIR Mode
2.2.2. Diffuse-Reflectance (DRIFTS) Mode
3. FTIR Spectroscopic Characterization: What Can Be Learned?
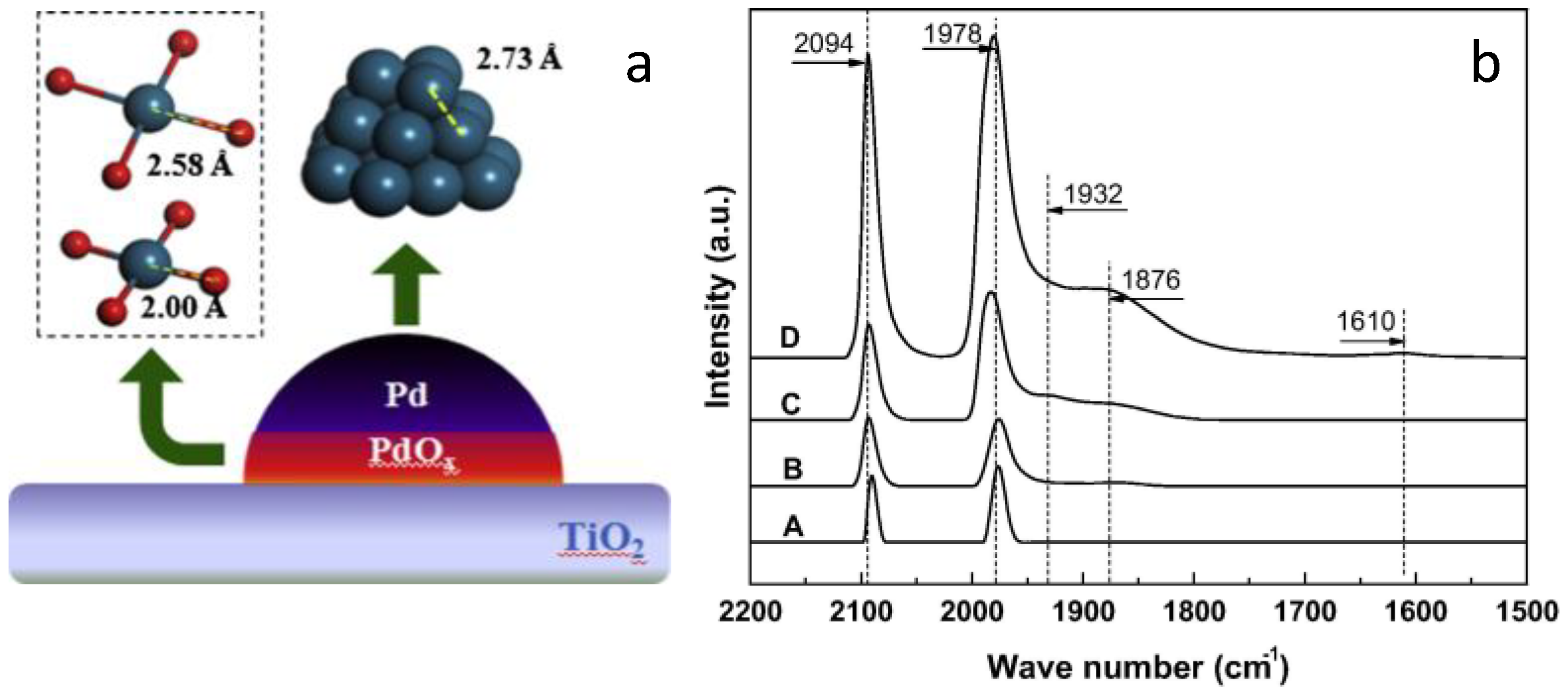
3.1. Nature of the Active Sites on Pd Catalysts
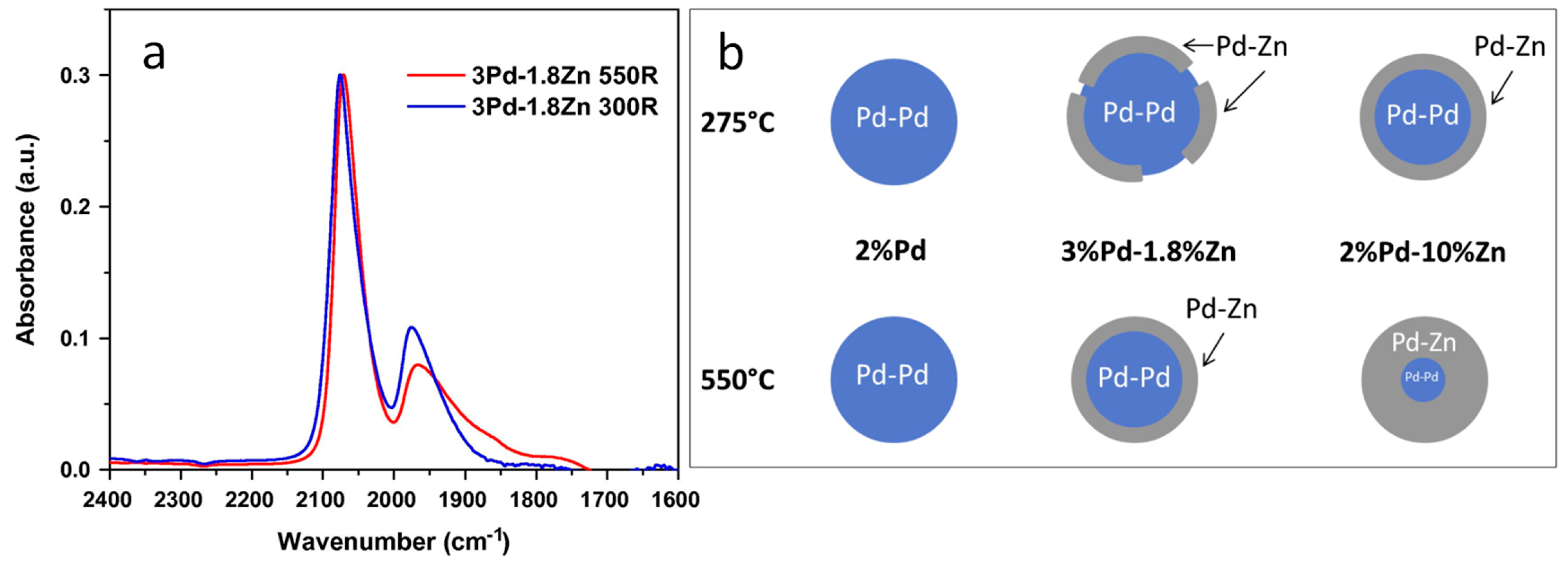
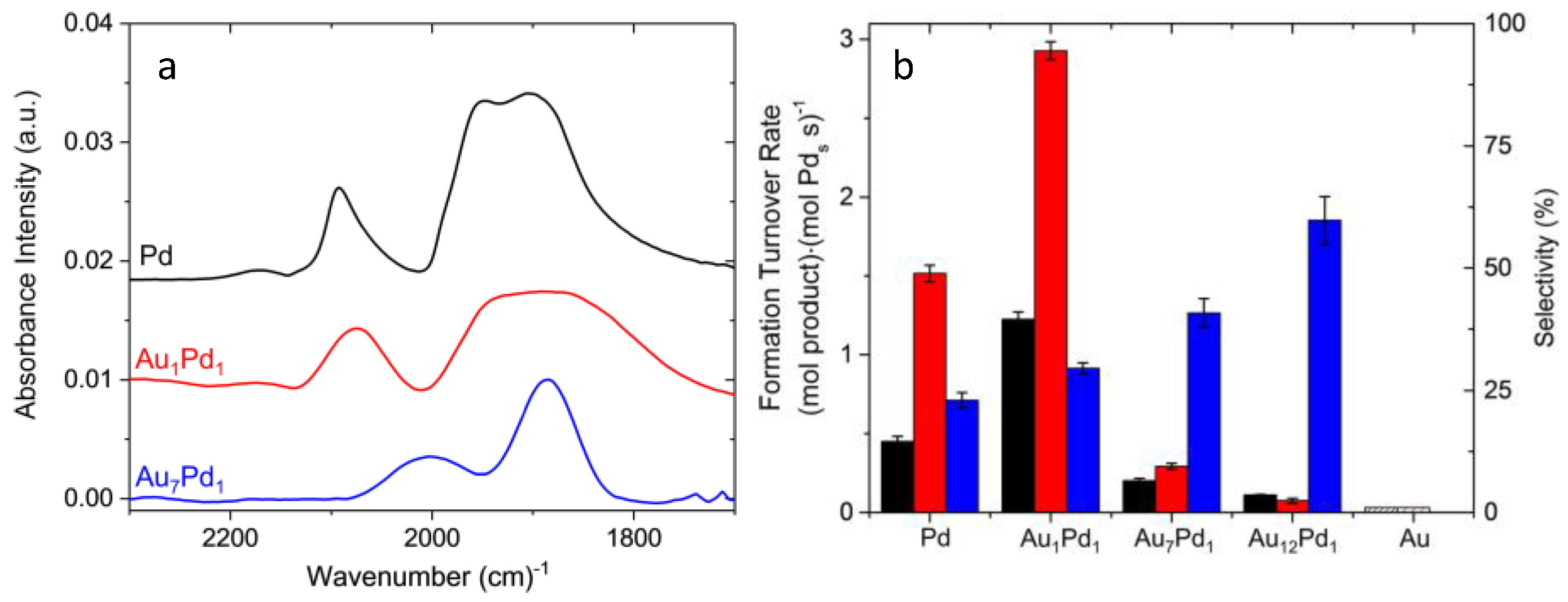
3.2. Site Isolation and Electronic Effects
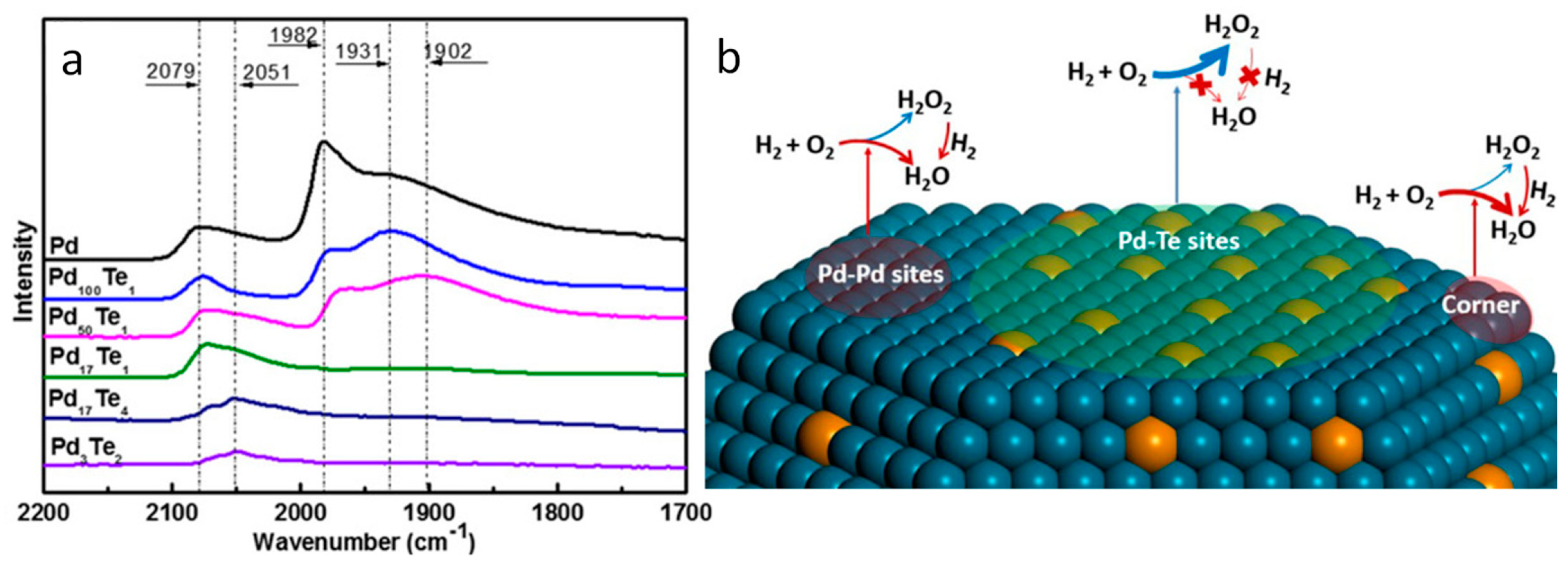
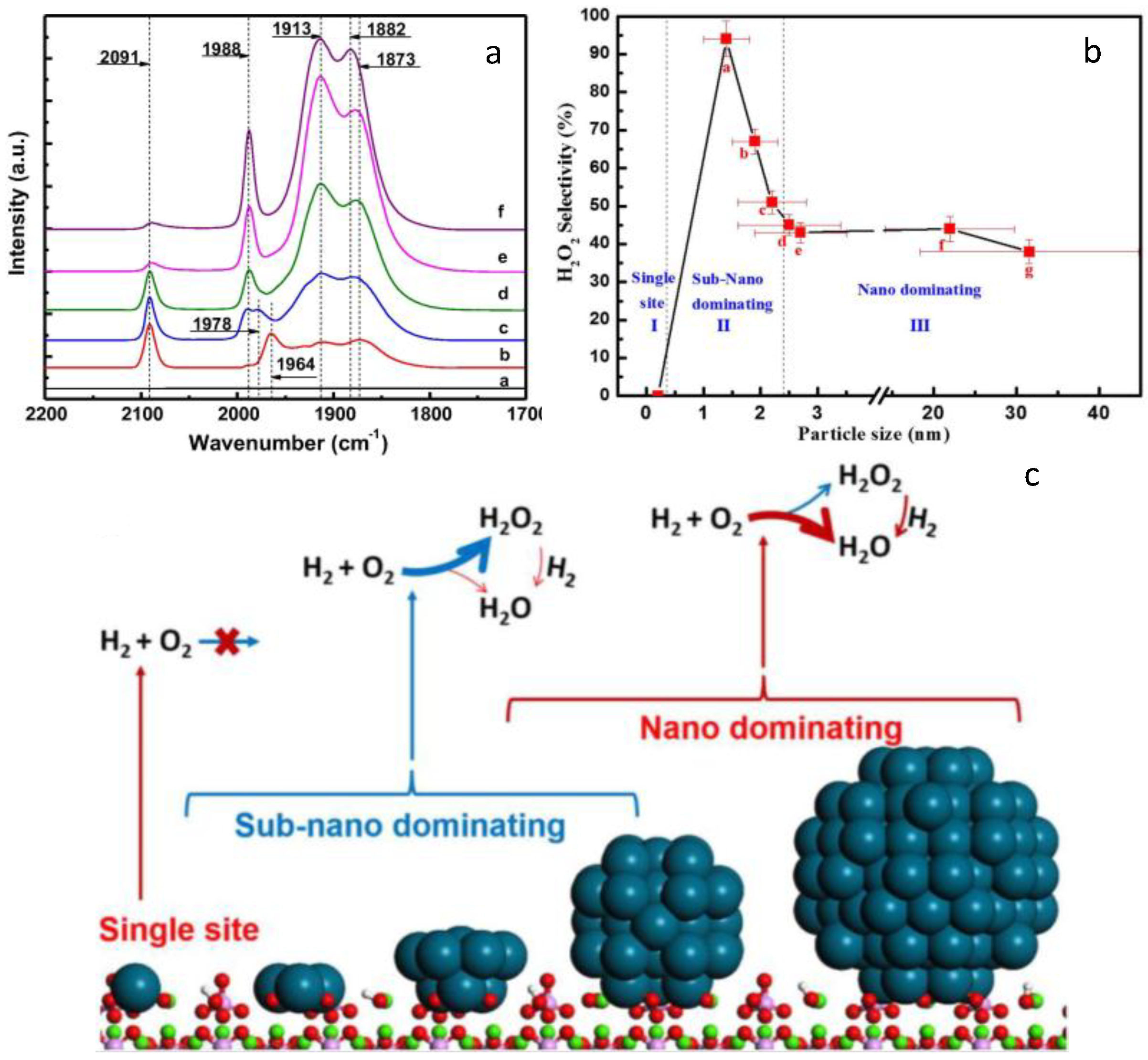
3.3. Reactivity of the Exposed Metal Sites
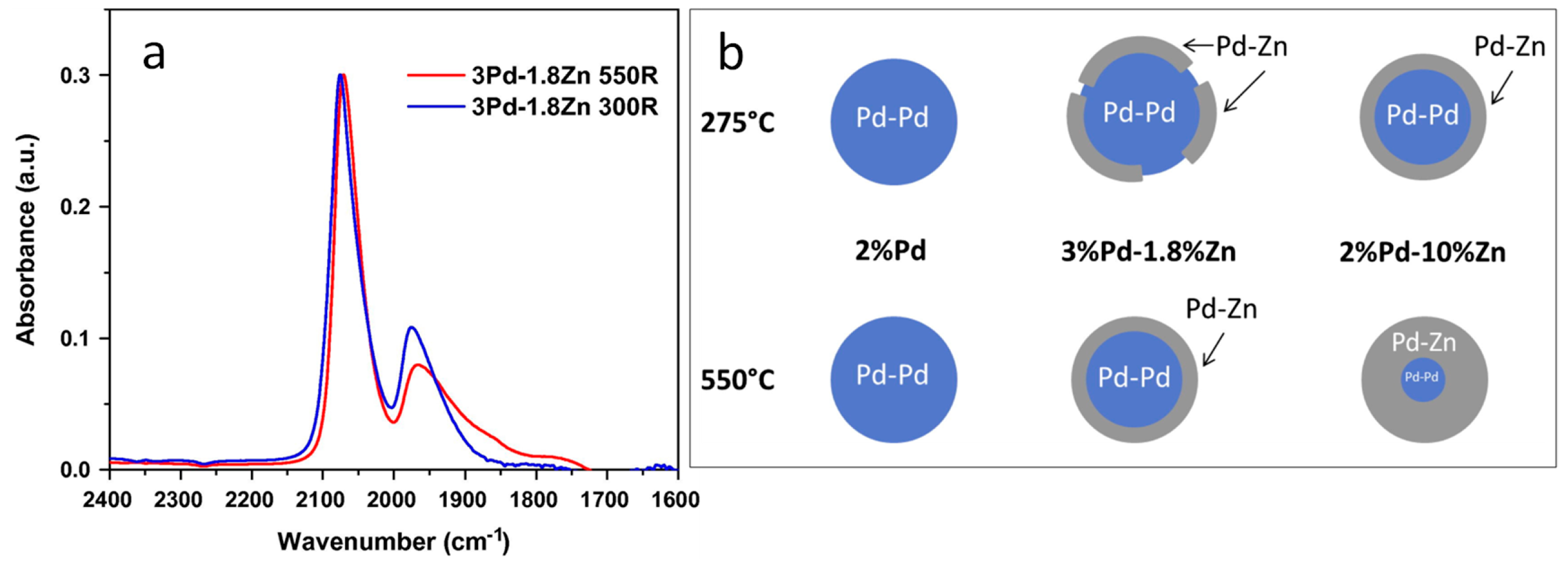
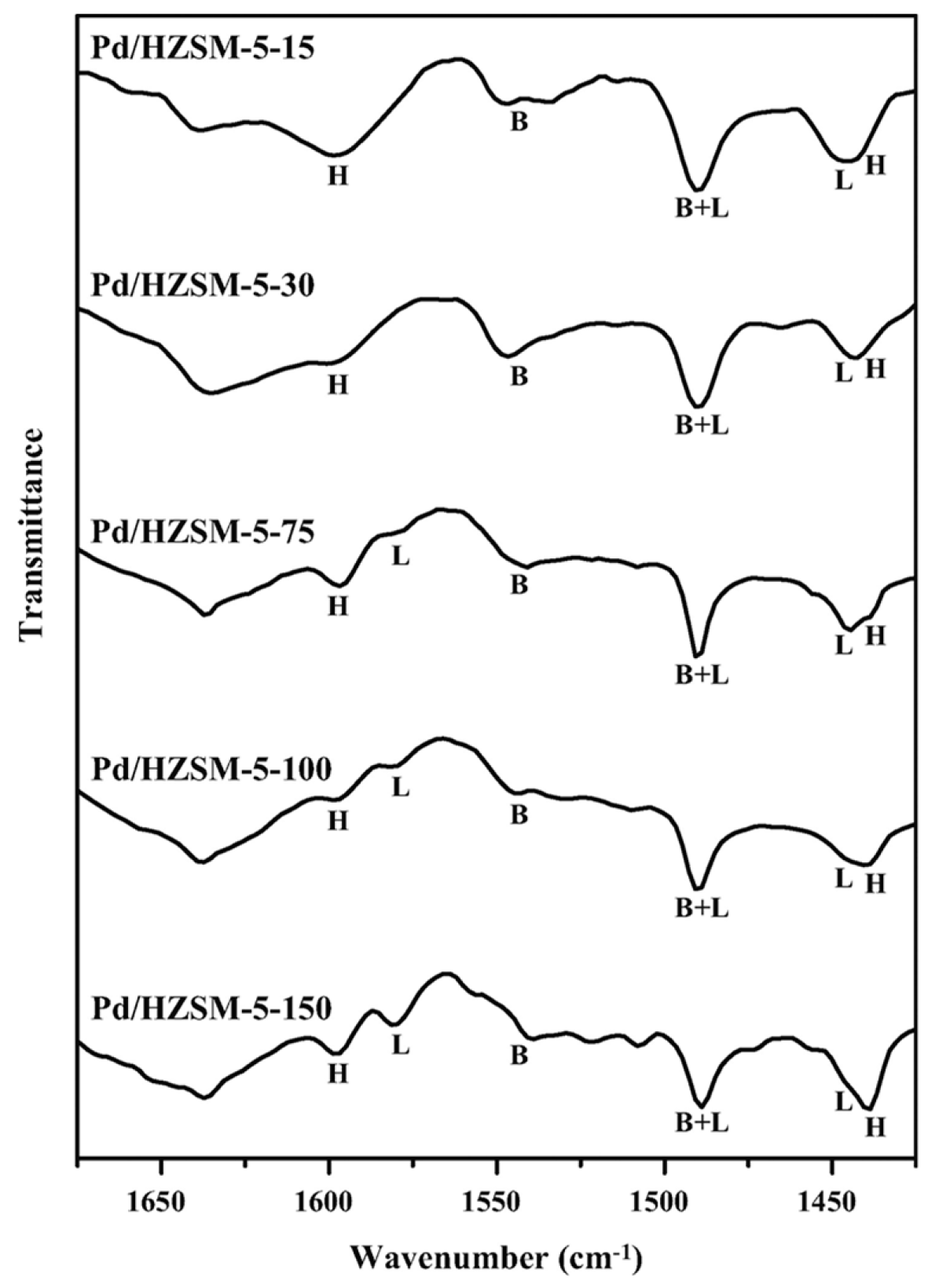
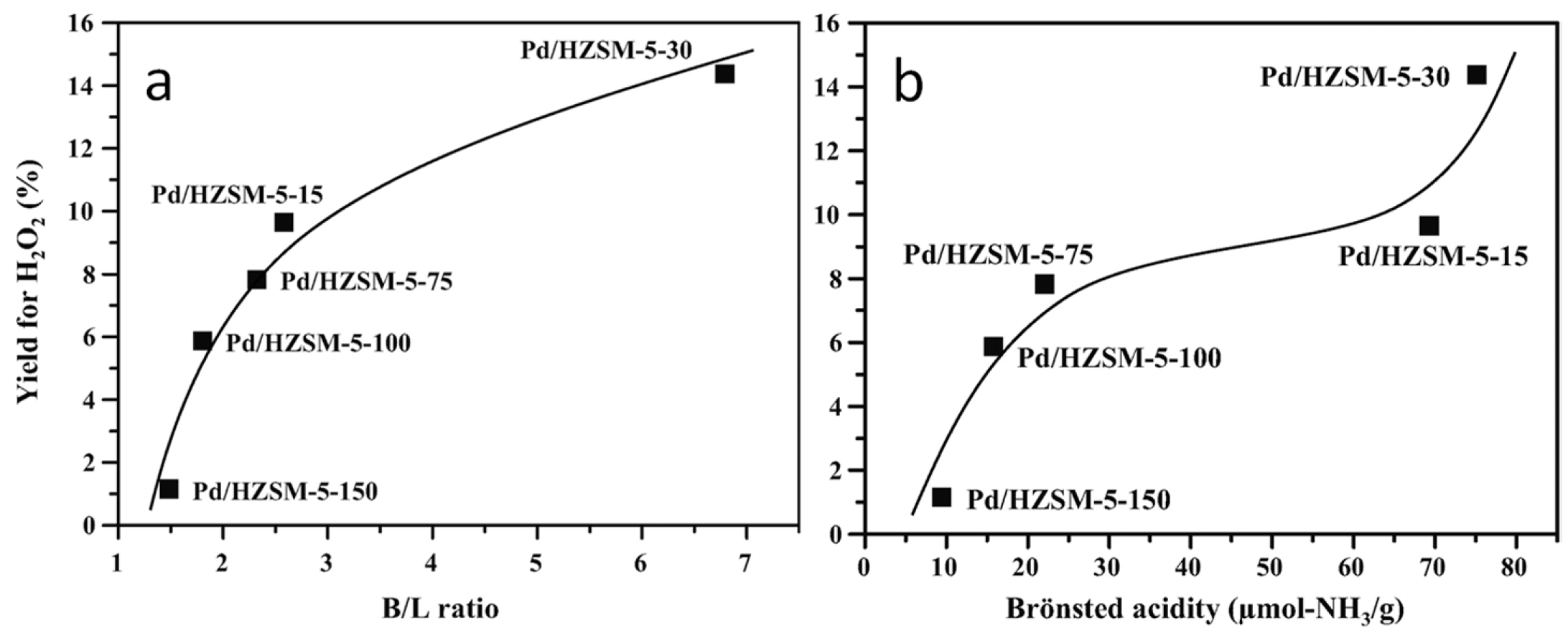
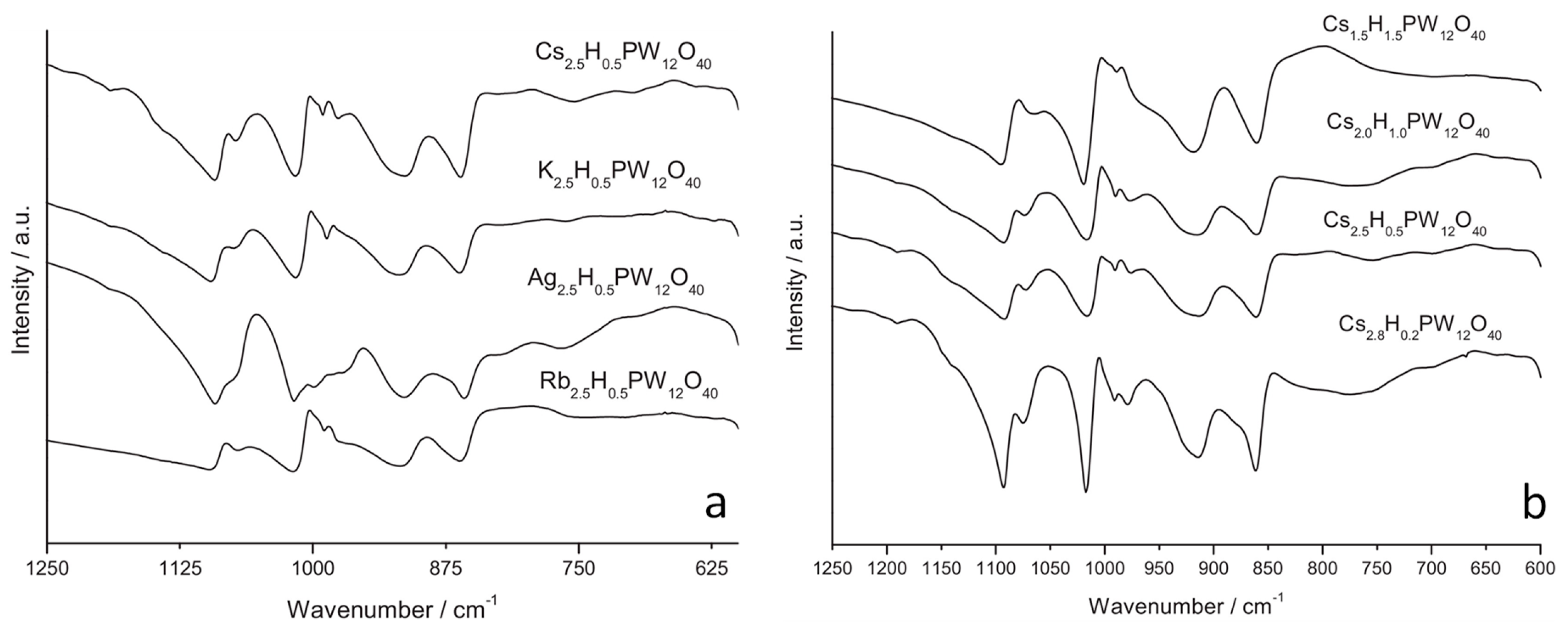
3.4. Insights on the Exposed Sites and on the Properties of the Support
4. Final Remarks and Open Issues
Acknowledgments
Conflicts of Interest
References
- Campos-Martin, J.M.; Blanco-Brieva, G.; Fierro, J.L.G. Hydrogen Peroxide Synthesis: An Outlook beyond the Anthraquinone Process. Angew. Chem. Int. Ed. 2006, 45, 6962–6984. [Google Scholar] [CrossRef] [PubMed]
- Riedl, H.-J.; Pfleiderer, G. Production of Hydrogen Peroxide. U.S. Patent No. 2215883, 24 September 1940. [Google Scholar]
- Wilson, N.M.; Flaherty, D.W. Mechanism for the Direct Synthesis of H2O2 on Pd Clusters: Heterolytic Reaction Pathways at the Liquid−Solid Interface. J. Am. Chem. Soc. 2015, 138, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, D.W. Direct Synthesis of H2O2 from H2 and O2 on Pd Catalysts: Current Understanding, Outstanding Questions, and Research Needs Motivation and Challenges for Direct Synthesis of H2O2. ACS Catal. 2018, 8, 1520–1527. [Google Scholar] [CrossRef]
- Wilson, N.M.; Bregante, D.T.; Priyadarshini, P.; Flaherty, D.W. Production and use of H2O2 for atom-efficient functionalization of hydrocarbons and small molecules. In Catalysis; Spivey, J., Han, Y.-F., Eds.; Royal Society of Chemistry: London, UK, 2017; Volume 29, pp. 122–212. [Google Scholar]
- Yang, S.; Verdaguer-Casadevall, A.; Arnarson, L.; Silvioli, L.; Frydendal, R.; Rossmeisl, J.; Chorkendorff, I.L.; Stephens, I.E. Toward the Decentralized Electrochemical Production of H2O2: A Focus on the Catalysis. ACS Catal. 2018, 8, 4064–4081. [Google Scholar] [CrossRef]
- Gosser, L.W. Catalytic Process for Making H2O2 from Hydrogen and Oxygen. U.S. Patent No. 4681751, 21 July 1987. [Google Scholar]
- Gosser, L.W.; Schwartz, J.-A.T. Catalytic Process for Making Hydrogen Peroxide from Hydrogen and Oxygen Employing a Bromide Promoter. U.S. Patent No. 4772458, 20 September 1988. [Google Scholar]
- Colery, J.-C.; Van Weynbergh, J.; Schoebrechts, J.-P. Direct Synthesis of Hydrogen Peroxide by Heterogeneous Catalysis, Catalyst for the Said Synthesis and Method of Preparation of the Said Catalyst. U.S. Patent No. 5447706, 5 September 1995. [Google Scholar]
- Bertsch-Frank, B.; Balduf, T.; Becker-Balfanz, C.; Hemme, I.; Rollmann, J.; Schutte, R.; Wildner, W. Process for Producing Hydrogen Peroxide by Direct Synthesis. U.S. Patent No. 6,387,346B1, 14 May 2002. [Google Scholar]
- Vanden Bussche, K.M.; Abdo, S.F.; Oroskar, A.R. Process for Mixing and Reacting Two or More Fluids. U.S. Patent No. 6713036 B1, 30 March 2004. [Google Scholar]
- Samanta, C.; Choudhary, V.R. Direct formation of H2O2 from H2 and O2 and decomposition/hydrogenation of H2O2 in aqueous acidic reaction medium over halide-containing Pd/SiO2 catalytic system. Catal. Commun. 2007, 8, 2222–2228. [Google Scholar] [CrossRef]
- Pizzutilo, E.; Freakley, S.J.; Cherevko, S.; Venkatesan, S.; Hutchings, G.J.; Liebscher, C.H.; Dehm, G.; Mayrhofer, K.J.J. Gold−Palladium Bimetallic Catalyst Stability: Consequences for Hydrogen Peroxide Selectivity. ACS Catal. 2017, 7, 5699–5705. [Google Scholar] [CrossRef]
- Biasi, P.; Menegazzo, F.; Pinna, F.; Eränen, K.; Salmi, T.O.; Canu, P. Continuous H2O2 direct synthesis over PdAu catalysts. Chem. Eng. J. 2011, 176–177, 172–177. [Google Scholar] [CrossRef]
- Verdaguer-Casadevall, A.; Deiana, D.; Karamad, M.; Siahrostami, S.; Malacrida, P.; Hansen, T.W.; Rossmeisl, J.; Chorkendorff, I.; Stephens, I.E.L. Trends in the Electrochemical Synthesis of H2O2: Enhancing Activity and Selectivity by Electrocatalytic Site Engineering. Nano Lett. 2014, 14, 1603–1608. [Google Scholar] [CrossRef]
- Lunsford, J.H. The direct formation of H2O2 from H2 and O2 over palladium catalysts. J. Catal. 2003, 216, 455–460. [Google Scholar] [CrossRef]
- Yi, Y.; Wang, L.; Li, G.; Guo, H. A review on research progress in the direct synthesis of hydrogen peroxide from hydrogen and oxygen: Noble-metal catalytic method, fuel-cell method and plasma method. Catal. Sci. Technol. 2016, 6, 1593–1610. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Samanta, C. Role of chloride or bromide anions and protons for promoting the selective oxidation of H2 by O2 to H2O2 over supported Pd catalysts in an aqueous medium. J. Catal. 2006, 238, 28–38. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Samanta, C.; Choudhary, T.V. Direct oxidation of H2 to H2O2 over Pd-based catalysts: Influence of oxidation state, support and metal additives. Appl. Catal. A Gen. 2006, 308, 128–133. [Google Scholar] [CrossRef]
- Burch, R.; Ellis, P.R. An investigation of alternative catalytic approaches for the direct synthesis of hydrogen peroxide from hydrogen and oxygen. Appl. Catal. B Environ. 2003, 42, 203–211. [Google Scholar] [CrossRef]
- Edwards, J.K.; Solsona, B.; Ntainjua, E.; Carley, A.F.; Herzing, A.A.; Kiely, C.J.; Hutchings, G.J. Switching Off Hydrogen Peroxide Hydrogenation in the Direct Synthesis Process. Science 2009, 323, 1037–1041. [Google Scholar] [CrossRef]
- Akram, A.; Freakley, S.J.; Reece, C.; Piccinini, M.; Shaw, G.; Edwards, J.K.; Desmedt, F.; Miquel, P.; Seuna, E.; Willock, D.J.; et al. Gas phase stabiliser-free production of hydrogen peroxide using supported gold–palladium catalysts. Chem. Sci. 2016, 7, 5833–5837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crole, D.A.; Freakley, S.J.; Edwards, J.K.; Hutchings, G.J. Direct synthesis of hydrogen peroxide in water at ambient temperature. Proc. R. Soc. A Math. Phys. Eng. Sci. 2016, 472, 20160156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Baeck, S.-H.; Kim, T.J.; Chung, Y.-M.; Oh, S.-H.; Song, I.K. Direct synthesis of hydrogen peroxide from hydrogen and oxygen over palladium catalyst supported on SO3H-functionalized mesoporous silica. J. Mol. Catal. A Chem. 2010, 319, 98–107. [Google Scholar] [CrossRef]
- Edwards, J.K.; Freakley, S.J.; Carley, A.F.; Kiely, C.J.; Hutchings, G.J. Strategies for Designing Supported Gold-Palladium Bimetallic Catalysts for the Direct Synthesis of Hydrogen Peroxide. Acc. Chem. Res. 2013, 47, 845–854. [Google Scholar] [CrossRef]
- Solsona, B.E.; Edwards, J.K.; Landon, P.; Carley, A.F.; Herzing, A.; Kiely, C.J.; Hutchings, G.J. Direct Synthesis of Hydrogen Peroxide from H2 and O2 Using Al2O3 Supported Au–Pd Catalysts. Chem. Mater. 2006, 18, 2689–2695. [Google Scholar] [CrossRef]
- Edwards, J.K.; Solsona, B.E.; Landon, P.; Carley, A.F.; Herzing, A.; Kiely, C.J.; Hutchings, G.J. Direct synthesis of hydrogen peroxide from H2 and O2 using TiO2-supported Au–Pd catalysts. J. Catal. 2005, 236, 69–79. [Google Scholar] [CrossRef]
- Jeong, H.E.; Kim, S.; Seo, M.; Lee, D.-W.; Lee, K.-Y. Catalytic activity of Pd octahedrons/SiO2 for the direct synthesis of hydrogen peroxide from hydrogen and oxygen. J. Mol. Catal. A Chem. 2016, 420, 88–95. [Google Scholar] [CrossRef]
- Selinsek, M.; Deschner, B.J.; Doronkin, D.E.; Sheppard, T.L.; Grunwaldt, J.-D.; Dittmeyer, R. Revealing the Structure and Mechanism of Palladium during Direct Synthesis of Hydrogen Peroxide in Continuous Flow Using Operando Spectroscopy. ACS Catal. 2018, 8, 2546–2557. [Google Scholar] [CrossRef]
- Biasi, P.; Sterchele, S.; Bizzotto, F.; Manzoli, M.; Lindholm, S.; Ek, P.; Bobacka, J.; Mikkola, J.-P.; Salmi, T. Application of the Catalyst Wet Pretreatment Method (CWPM) for catalytic direct synthesis of H2O2. Catal. Today 2015, 246, 207–215. [Google Scholar] [CrossRef]
- Menegazzo, F.; Signoretto, M.; Frison, G.; Pinna, F.; Strukul, G.; Manzoli, M.; Boccuzzi, F. When high metal dispersion has a detrimental effect: Hydrogen peroxide direct synthesis under very mild and nonexplosive conditions catalyzed by Pd supported on silica. J. Catal. 2012, 290. [Google Scholar] [CrossRef]
- Abate, S.; Arrigo, R.; Schuster, M.E.; Perathoner, S.; Centi, G.; Villa, A.; Su, D.; Schlögl, R. Pd nanoparticles supported on N-doped nanocarbon for the direct synthesis of H2O2 from H2 and O2. Catal. Today 2010, 157, 280–285. [Google Scholar] [CrossRef]
- Kim, J.; Chung, Y.-M.; Kang, S.-M.; Choi, C.-H.; Kim, B.-Y.; Kwon, Y.-T.; Jin Kim, T.; Oh, S.-H.; Lee, C.-S. Palladium Nanocatalysts Immobilized on Functionalized Resin for the Direct Synthesis of Hydrogen Peroxide from Hydrogen and Oxygen. ACS Catal. 2012, 2, 1042–1048. [Google Scholar] [CrossRef]
- Seo, M.-G.; Lee, D.-W.; Han, S.S.; Lee, K.-Y. Direct Synthesis of Hydrogen Peroxide from Hydrogen and Oxygen over Mesoporous Silica-Shell-Coated, Palladium-Nanocrystal-Grafted SiO2 Nanobeads. ACS Catal. 2017, 7, 3039–3048. [Google Scholar] [CrossRef]
- Gervasini, A.; Carniti, P.; Frédé, F.; Desmedt, F.; Miquel, P. Liquid Phase Direct Synthesis of H2O2: Activity and Selectivity of Pd-Dispersed Phase on Acidic Niobia-Silica Supports. ACS Catal. 2017, 7, 3039–3048. [Google Scholar] [CrossRef]
- Krishnankutty, N.; Li, J.; Albert Vannice, M. The effect of Pd precursor and pretreatment on the adsorption and absorption behavior of supported Pd catalysts. Appl. Catal. A Gen. 1998, 173, 137–144. [Google Scholar] [CrossRef]
- Seo, M.; Kim, S.; Jeong, H.E.; Lee, D.-W.; Lee, K.-Y. A yolk–shell structured Pd@void@ZrO2 catalyst for direct synthesis of hydrogen peroxide from hydrogen and oxygen. J. Mol. Catal. A Chem. 2016, 413, 1–6. [Google Scholar] [CrossRef]
- Yook, S.; Kwon, H.C.; Kim, Y.-G.; Choi, W.; Choi, M. Significant Roles of Carbon Pore and Surface Structure in AuPd/C Catalyst for Achieving High Chemoselectivity in Direct Hydrogen Peroxide Synthesis. ACS Sustain. Chem. Eng. 2016, 5, 1208–1216. [Google Scholar] [CrossRef]
- Menegazzo, F.; Signoretto, M.; Manzoli, M.; Boccuzzi, F.; Cruciani, G.; Pinna, F.; Strukul, G. Influence of the preparation method on the morphological and composition properties of Pd–Au/ZrO2 catalysts and their effect on the direct synthesis of hydrogen peroxide from hydrogen and oxygen. J. Catal. 2009, 268, 122–130. [Google Scholar] [CrossRef]
- Menegazzo, F.; Manzoli, M.; Signoretto, M.; Pinna, F.; Strukul, G. H2O2 direct synthesis under mild conditions on Pd-Au samples: Effect of the morphology and of the composition of the metallic phase. Catal. Today 2014, 248, 18–27. [Google Scholar] [CrossRef]
- Edwards, J.K.; Hutchings, G.J. Palladium and Gold-Palladium Catalysts for the Direct Synthesis of Hydrogen Peroxide. Angew. Chem. Int. Ed. 2008, 47, 9192–9198. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Cheng, D.; Gao, Y. Design of High-Performance Pd-Based Alloy Nanocatalysts for Direct Synthesis of H2O2. ACS Catal. 2017, 7, 2164–2170. [Google Scholar] [CrossRef]
- García, T.; Agouram, S.; Dejoz, A.; Sánchez-Royo, J.F.; Torrente-Murciano, L.; Solsona, B. Enhanced H2O2 production over Au-rich bimetallic Au–Pd nanoparticles on ordered mesoporous carbons. Catal. Today 2015, 248, 48–57. [Google Scholar] [CrossRef]
- Arrigo, R.; Schuster, M.E.; Abate, S.; Wrabetz, S.; Amakawa, K.; Teschner, D.; Freni, M.; Centi, G.; Perathoner, S.; Hävecker, M.; Schlögl, R. Dynamics of Palladium on Nanocarbon in the Direct Synthesis of H2O2. ChemSusChem 2014, 7, 179–194. [Google Scholar] [CrossRef]
- Wilson, N.M.; Pan, Y.-T.; Shao, Y.-T.; Zuo, J.-M.; Yang, H.; Flaherty, D.W. Direct Synthesis of H2O2 on AgPt Octahedra: The Importance of Ag–Pt Coordination for High H2O2 Selectivity. ACS Catal. 2018, 8, 2880–2889. [Google Scholar] [CrossRef]
- Prati, L.; Villa, A.; Campisi, S.; Vindigni, F.; Manzoli, M.; Dimitratos, N. Support acid-base properties as a tool for directing selectivity in the Au-Pt catalyzed base-free glycerol oxidation. In Proceedings of the Engineering Sciences and Fundamentals 2013—Core Programming Area at the 2013 AIChE Annual Meeting: Global Challenges for Engineering a Sustainable Future, San Francisco, CA, USA, 3–8 November 2013; Volume 2. [Google Scholar]
- Bernardotto, G.; Menegazzo, F.; Pinna, F.; Signoretto, M.; Cruciani, G.; Strukul, G. New Pd–Pt and Pd–Au catalysts for an efficient synthesis of H2O2 from H2 and O2 under very mild conditions. Appl. Catal. A Gen. 2009, 358, 129–135. [Google Scholar] [CrossRef]
- Liu, Q.; Bauer, J.C.; Schaak, R.E.; Lunsford, J.H. Direct synthesis of H2O2 from H2 and O2 over Pd–Pt/SiO2 bimetallic catalysts in a H2SO4/ethanol system. Appl. Catal. A Gen. 2008, 339, 130–136. [Google Scholar] [CrossRef]
- Freakley, S.J.; He, Q.; Harrhy, J.H.; Lu, L.; Crole, D.A.; Morgan, D.J.; Ntainjua, E.N.; Edwards, J.K.; Carley, A.F.; Borisevich, A.Y.; et al. Palladium-tin catalysts for the direct synthesis of H2O2 with high selectivity. Science 2016, 351, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Shao, Q.; Hu, M.; Chen, Y.; Huang, X. Hollow Pd−Sn Nanocrystals for Efficient Direct H2O2 Synthesis: The Critical Role of Sn on Structure Evolution and Catalytic Performance. ACS Catal. 2018, 8, 3418–3423. [Google Scholar] [CrossRef]
- Maity, S.; Eswaramoorthy, M. Ni–Pd bimetallic catalysts for the direct synthesis of H2O2—Unusual enhancement of Pd activity in the presence of Ni. J. Mater. Chem. A 2016, 4, 3233–3237. [Google Scholar] [CrossRef]
- Tian, P.; Xu, X.; Ao, C.; Ding, D.; Li, W.; Si, R.; Tu, W.; Xu, J.; Han, Y.-F. Direct and Selective Synthesis of Hydrogen Peroxide over Palladium-Tellurium Catalysts at Ambient Pressure. ChemSusChem 2017, 10, 3342–3346. [Google Scholar] [CrossRef]
- Childers, D.J.; Schweitzer, N.M.; Shahari, S.M.K.; Rioux, R.M.; Miller, J.T.; Meyer, R.J. Modifying structure-sensitive reactions by addition of Zn to Pd. J. Catal. 2014, 318, 75–84. [Google Scholar] [CrossRef]
- Wang, S.; Gao, K.; Li, W.; Zhang, J. Effect of Zn addition on the direct synthesis of hydrogen peroxide over supported palladium catalysts. Appl. Catal. A Gen. 2017, 531, 89–95. [Google Scholar] [CrossRef]
- Li, G.; Edwards, J.; Carley, A.F.; Hutchings, G.J. Direct synthesis of hydrogen peroxide from H2 and O2 and in situ oxidation using zeolite-supported catalysts. Catal. Commun. 2007, 8, 247–250. [Google Scholar] [CrossRef]
- Hu, B.; Deng, W.; Li, R.; Zhang, Q.; Wang, Y.; Delplanque-Janssens, F.; Paul, D.; Desmedt, F.; Miquel, P. Carbon-supported palladium catalysts for the direct synthesis of hydrogen peroxide from hydrogen and oxygen. J. Catal. 2014, 319, 15–26. [Google Scholar] [CrossRef]
- Tian, P.; Ouyang, L.; Xu, X.; Ao, C.; Xu, X.; Si, R.; Shen, X.; Lin, M.; Xu, J.; Han, Y.-F. The origin of palladium particle size effects in the direct synthesis of H2O2: Is smaller better? J. Catal. 2017, 349, 30–40. [Google Scholar] [CrossRef]
- Han, Y.-F.; Lunsford, J.H. Direct formation of H2O2 from H2 and O2 over a Pd/SiO2 catalyst: The roles of the acid and the liquid phase. J. Catal. 2005, 230, 313–316. [Google Scholar] [CrossRef]
- Ouyang, L.; Tian, P.; Da, G.; Xu, X.-C.; Ao, C.; Chen, T.; Si, R.; Xu, J.; Han, Y.-F. The origin of active sites for direct synthesis of H2O2 on Pd/TiO2 catalysts: Interfaces of Pd and PdO domains. J. Catal. 2015, 321, 70–80. [Google Scholar] [CrossRef]
- Lari, G.M.; Puértolas, B.; Shahrokhi, M.; López, N.; Pérez-Ramírez, J. Hybrid Palladium Nanoparticles for Direct Hydrogen Peroxide Synthesis: The Key Role of the Ligand. Angew. Chem. Int. Ed. 2017, 56, 1775–1779. [Google Scholar] [CrossRef] [PubMed]
- Samanta, C. Direct synthesis of hydrogen peroxide from hydrogen and oxygen: An overview of recent developments in the process. Appl. Catal. A Gen. 2008, 350, 133–149. [Google Scholar] [CrossRef]
- Chinta, S.; Lunsford, J.H. A mechanistic study of H2O2 and H2O formation from H2 and O2 catalyzed by palladium in an aqueous medium. J. Catal. 2004, 225, 249–255. [Google Scholar] [CrossRef]
- Melada, S.; Rioda, R.; Menegazzo, F.; Pinna, F.; Strukul, G. Direct synthesis of hydrogen peroxide on zirconia-supported catalysts under mild conditions. J. Catal. 2006, 239, 422–430. [Google Scholar] [CrossRef]
- Park, S.; Lee, S.H.; Song, S.H.; Park, D.R.; Baeck, S.-H.; Kim, T.J.; Chung, Y.-M.; Oh, S.-H.; Song, I.K. Direct synthesis of hydrogen peroxide from hydrogen and oxygen over palladium-exchanged insoluble heteropolyacid catalysts. Catal. Commun. 2009, 10, 391–394. [Google Scholar] [CrossRef]
- Freakley, S.J.; Lewis, R.J.; Morgan, D.J.; Edwards, J.K.; Hutchings, G.J. Direct synthesis of hydrogen peroxide using Au–Pd supported and ion-exchanged heteropolyacids precipitated with various metal ions. Catal. Today 2015, 248, 10–17. [Google Scholar] [CrossRef]
- Park, S.; Park, D.R.; Choi, J.H.; Kim, T.J.; Chung, Y.-M.; Oh, S.-H.; Song, I.K. Direct synthesis of hydrogen peroxide from hydrogen and oxygen over palladium catalyst supported on H3PW12O40-incorporated MCF silica. J. Mol. Catal. A Chem. 2011, 336, 78–86. [Google Scholar] [CrossRef]
- Park, S.; Lee, J.; Song, J.H.; Kim, T.J.; Chung, Y.-M.; Oh, S.-H.; Song, I.K. Direct synthesis of hydrogen peroxide from hydrogen and oxygen over Pd/HZSM-5 catalysts: Effect of Brönsted acidity. J. Mol. Catal. A Chem. 2012, 363–364, 230–236. [Google Scholar] [CrossRef]
- Park, S.; Choi, J.H.; Kim, T.J.; Chung, Y.-M.; Oh, S.-H.; Song, I.K. Direct synthesis of hydrogen peroxide from hydrogen and oxygen over Pd/CsXH3−XPW12O40/MCF (X = 1.7, 2.0, 2.2, 2.5, and 2.7) catalysts. J. Mol. Catal. A Chem. 2012, 353, 37–43. [Google Scholar] [CrossRef]
- Park, S.; Park, D.R.; Choi, J.H.; Kim, T.J.; Chung, Y.-M.; Oh, S.-H.; Song, I.K. Direct synthesis of hydrogen peroxide from hydrogen and oxygen over insoluble Cs2.5H0.5PW12O40 heteropolyacid supported on Pd/MCF. J. Mol. Catal. A Chem. 2010, 332, 76–83. [Google Scholar] [CrossRef]
- Lewis, R.J.; Edwards, J.K.; Freakley, S.J.; Hutchings, G.J. Solid Acid Additives as Recoverable Promoters for the Direct Synthesis of Hydrogen Peroxide. Ind. Eng. Chem. Res. 2017, 56, 13287–13293. [Google Scholar] [CrossRef]
- Zaera, F. New advances in the use of infrared absorption spectroscopy for the characterization of heterogeneous catalytic reactions. Chem. Soc. Rev. 2014, 43, 7624–7663. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.R.; De Haseth, J.A. Fourier Transform Infrared Spectrometry; Wiley-Interscience: New York, NY, USA, 2007; ISBN 978-0-47-119404-0. [Google Scholar]
- Blyholder, G. Molecular Orbital View of Chemisorbed Carbon Monoxide. J. Phys. Chem. 1964, 68, 2772–2777. [Google Scholar] [CrossRef]
- Lavalley, J.C. Infrared spectrometric studies of the surface basicity of metal oxides and zeolites using adsorbed probe molecules. Catal. Today 1996, 27, 377–401. [Google Scholar] [CrossRef]
- Busca, G. Spectroscopic characterization of the acid properties of metal oxide catalysts. Catal. Today 1998, 41, 191–206. [Google Scholar] [CrossRef]
- Corma, A. Inorganic Solid Acids and Their Use in Acid-Catalyzed Hydrocarbon Reactions. Chem. Rev. 1995, 95, 559–614. [Google Scholar] [CrossRef]
- Zecchina, A.; Scarano, D.; Bordiga, S.; Ricchiardi, G.; Spoto, G.; Geobaldo, F. IR studies of CO and NO adsorbed on well characterized oxide single microcrystals. Catal. Today 1996, 27, 403–435. [Google Scholar] [CrossRef]
- Bensitel, M.; Saur, O.; Lavalley, J.C. Use of methanol as a probe to study the adsorption sites of different MgO samples. Mater. Chem. Phys. 1991, 28, 309–320. [Google Scholar] [CrossRef]
- Zaera, F. Infrared Absorption Spectroscopy of Adsorbed CO: New Applications in Nanocatalysis for an Old Approach. ChemCatChem 2012, 4, 1525–1533. [Google Scholar] [CrossRef]
- Hadjiivanov, K.; Knözinger, H. Characterization of vacant coordination sites of cations on the surfaces of oxides and zeolites using infrared spectroscopy of adsorbed probe molecules. Surf. Sci. 2009, 603, 1629–1636. [Google Scholar] [CrossRef]
- Hadjiivanov, K.I.; Vayssilov, G.N. Characterization of oxide surfaces and zeolites by carbon monoxide as an IR probe molecule. Adv. Catal. 2002, 47, 307–511. [Google Scholar] [CrossRef]
- Hollins, P. The influence of surface defects on the infrared spectra of adsorbed species. Surf. Sci. Rep. 1992, 16, 51–94. [Google Scholar] [CrossRef]
- Gao, F.; Wang, Y.; Goodman, D.W. CO Oxidation over AuPd(100) from Ultrahigh Vacuum to Near-Atmospheric Pressures: The Critical Role of Contiguous Pd Atoms. J. Am. Chem. Soc. 2009, 131, 5734–5735. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, D.-W.; Lee, K.-Y. Shape-dependent catalytic activity of palladium nanoparticles for the direct synthesis of hydrogen peroxide from hydrogen and oxygen. J. Mol. Catal. A Chem. 2014, 391, 48–54. [Google Scholar] [CrossRef]
- Kim, S.; Lee, D.-W.; Lee, K.-Y. Direct synthesis of hydrogen peroxide from hydrogen and oxygen over single-crystal cubic palladium on silica catalysts. J. Mol. Catal. A Chem. 2014, 383, 64–69. [Google Scholar] [CrossRef]
- Ouyang, L.; Da, G.; Tian, P.; Chen, T.; Liang, G.; Xu, J.; Han, Y.-F. Insight into active sites of Pd–Au/TiO2 catalysts in hydrogen peroxide synthesis directly from H2 and O2. J. Catal. 2014, 311, 129–136. [Google Scholar] [CrossRef]
- Chul Ham, H.; Hwang, G.S.; Han, J.; Woo Nam, S.; Hoon Lim, T. On the Role of Pd Ensembles in Selective H2O2 Formation on PdAu Alloys. J. Phys. Chem. C 2009, 113, 12943–12945. [Google Scholar] [CrossRef]
- Yi, C.-W.; Luo, K.; Wei, T.; Goodman, D.W. The Composition and Structure of Pd-Au Surfaces. J. Phys. Chem. B 2005, 109, 18535–18540. [Google Scholar] [CrossRef]
- Han, Y.-F.; Zhong, Z.; Ramesh, K.; Chen, F.; Chen, L.; White, T.; Tay, Q.; Nurbaya Yaakub, S.; Wang, Z. Au Promotional Effects on the Synthesis of H2O2 Directly from H2 and O2 on Supported Pd-Au Alloy Catalysts. J. Phys. Chem. C 2007, 111, 8410–8413. [Google Scholar] [CrossRef]
- Wilson, N.M.; Priyadarshini, P.; Kunz, S.; Flaherty, D.W. Direct synthesis of H2O2 on Pd and AuxPd1 clusters: Understanding the effects of alloying Pd with Au. J. Catal. 2018, 357, 163–175. [Google Scholar] [CrossRef]
- Kunz, S.; Iglesia, E. Mechanistic Evidence for Sequential Displacement–Reduction Routes in the Synthesis of Pd–Au Clusters with Uniform Size and Clean Surfaces. J. Phys. Chem. C 2014, 118, 7468–7479. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.K.; Pritchard, J.; Piccinini, M.; Shaw, G.; He, Q.; Carley, A.F.; Kiely, C.J.; Hutchings, G.J. The effect of heat treatment on the performance and structure of carbon-supported Au–Pd catalysts for the direct synthesis of hydrogen peroxide. J. Catal. 2012, 292, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, J.B.; Schroeder, T.; Bäumer, M.; Freund, H.-J. Study of CO adsorption on crystalline-silica-supported palladium particles. Surf. Sci. 2002, 498, L71–L77. [Google Scholar] [CrossRef]
- Tew, M.W.; Emerich, H.; van Bokhoven, J.A. Formation and Characterization of PdZn Alloy: A Very Selective Catalyst for Alkyne Semihydrogenation. J. Phys. Chem. C 2011, 115, 8457–8465. [Google Scholar] [CrossRef]
- Zhang, L.; Mao, J.; Li, S.; Yin, J.; Sun, X.; Guo, X.; Song, C.; Zhou, J. Hydrogenation of levulinic acid into gamma-valerolactone over in situ reduced CuAg bimetallic catalyst: Strategy and mechanism of preventing Cu leaching. Appl. Catal. B Environ. 2018, 232, 1–10. [Google Scholar] [CrossRef]
- Kale, M.J.; Christopher, P. Utilizing Quantitative in Situ FTIR Spectroscopy to Identify Well-Coordinated Pt Atoms as the Active Site for CO Oxidation on Al2O3-Supported Pt Catalysts. ACS Catal 2016, 6, 21. [Google Scholar] [CrossRef]
- Menegazzo, F.; Burti, P.; Signoretto, M.; Manzoli, M.; Vankova, S.; Boccuzzi, F.; Pinna, F.; Strukul, G. Effect of the addition of Au in zirconia and ceria supported Pd catalysts for the direct synthesis of hydrogen peroxide. J. Catal. 2008, 257, 369–381. [Google Scholar] [CrossRef]
- Groppo, M.E.; Bertarione, S.; Rotunno, F.; Agostini, G.; Scarano, D.; Pellegrini, R.; Leofanti, G.; Zecchina, A.; Lamberti, C. Role of the Support in Determining the Vibrational Properties of Carbonyls Formed on Pd Supported on SiO. J. Phys. Chem. C 2007, 111, 7021–7028. [Google Scholar] [CrossRef]
- Roques, J.; Lacaze-Dufaure, C.; Mijoule, C. Dissociative Adsorption of Hydrogen and Oxygen on Palladium Clusters: A Comparison with the (111) Infinite Surface. J. Chem. Theory Comput. 2007, 3, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Wang, J.; Goodman, D.W. Characterization and Chemical Properties of Pd−Au Alloy Surfaces. J. Phys. Chem. C 2007, 111, 8781–8788. [Google Scholar] [CrossRef]
- Schalow, T.; Brandt, B.; Starr, D.E.; Laurin, M.; Shaikhutdinov, S.K.; Schauermann, S.; Libuda, J.; Freund, H.-J. Particle size dependent adsorption and reaction kinetics on reduced and partially oxidized Pd nanoparticles. Phys. Chem. Chem. Phys. 2007, 9, 1347–1361. [Google Scholar] [CrossRef] [PubMed]
- Wolter, K.; Seiferth, O.; Libuda, J.; Kuhlenbeck, H.; Bäumer, M.; Freund, H.-J. IR spectroscopy of a Pd-carbonyl surface compound. Chem. Phys. Lett. 1997, 277, 513–520. [Google Scholar] [CrossRef]
- Abate, S.; Centi, G.; Melada, S.; Perathoner, S.; Pinna, F.; Strukul, G. Preparation, performances and reaction mechanism for the synthesis of H2O2 from H2 and O2 based on palladium membranes. Catal. Today 2005, 104, 323–328. [Google Scholar] [CrossRef]
- Edwards, J.K.; Solsona, B.; Landon, P.; Carley, A.F.; Herzing, A.; Watanabe, M.; Kiely, C.J.; Hutchings, G.J. Direct synthesis of hydrogen peroxide from H2 and O2 using Au–Pd/Fe2O3 catalysts. J. Mater. Chem. 2005, 15, 4595. [Google Scholar] [CrossRef]
- Enache, D.I.; Edwards, J.K.; Landon, P.; Solsona-Espriu, B.; Carley, A.F.; Herzing, A.A.; Watanabe, M.; Kiely, C.J.; Knight, D.W.; Hutchings, G.J. Solvent-free oxidation of primary alcohols to aldehydes using Au-Pd/TiO2 catalysts. Science 2006, 311, 362–365. [Google Scholar] [CrossRef]
- Herzing, A.A.; Carley, A.F.; Edwards, J.K.; Hutchings, G.J.; Kiely, C.J. Microstructural Development and Catalytic Performance of Au−Pd Nanoparticles on Al2O3 Supports: The Effect of Heat Treatment Temperature and Atmosphere. Chem. Mater. 2008, 20, 1492–1501. [Google Scholar] [CrossRef]
- Sales, E.A.; Jove, J.; de Jesus Mendes, M.; Bozon-Verduraz, F. Palladium, Palladium–Tin, and Palladium–Silver Catalysts in the Selective Hydrogenation of Hexadienes: TPR, Mössbauer, and Infrared Studies of Adsorbed CO. J. Catal. 2000, 195, 88–95. [Google Scholar] [CrossRef]
- Schalow, T.; Brandt, B.; Laurin, M.; Schauermann, S.; Guimond, S.; Kuhlenbeck, H.; Libuda, J.; Freund, H.-J. Formation of interface and surface oxides on supported Pd nanoparticles. Surf. Sci. 2006, 600, 2528–2542. [Google Scholar] [CrossRef]
- Hinojosa, J.A., Jr.; Kan, H.H.; Weaver, J.F. Molecular Chemisorption of O2 on a PdO(101) Thin Film on Pd(111). J. Phys. Chem. C 2008, 112, 8324–8331. [Google Scholar] [CrossRef]
- Ozensoy, E.; Wayne Goodman, D. Vibrational spectroscopic studies on CO adsorption, NO adsorption CO + NO reaction on Pd model catalysts. Phys. Chem. Chem. Phys. 2004, 6, 3765–3778. [Google Scholar] [CrossRef]
- Yuan, D.; Gong, X.; Wu, R. Peculiar distribution of Pd on Au nanoclusters: First-principles studies. Phys. Rev. B 2008, 78, 035441. [Google Scholar] [CrossRef]
- Lee, S.; Jeong, H.; Chung, Y.-M. Direct synthesis of hydrogen peroxide over Pd/C catalyst prepared by selective adsorption deposition method. J. Catal. 2018, 365, 125–137. [Google Scholar] [CrossRef]
- Fanning, P.E.; Vannice, M.A. A DRIFTS study of the formation of surface groups on carbon by oxidation. Carbon 1993, 31, 721–730. [Google Scholar] [CrossRef]
- Figueiredo, J.; Pereira, M.F.; Freitas, M.M.; Órfão, J.J. Modification of the surface chemistry of activated carbons. Carbon 1999, 37, 1379–1389. [Google Scholar] [CrossRef]
- Reddy, C.R.; Bhat, Y.S.; Nagendrappa, G.; Jai Prakash, B.S. Brønsted and Lewis acidity of modified montmorillonite clay catalysts determined by FT-IR spectroscopy. Catal. Today 2009, 141, 157–160. [Google Scholar] [CrossRef] [Green Version]
- Jin, F.; Li, Y. A FTIR and TPD examination of the distributive properties of acid sites on ZSM-5 zeolite with pyridine as a probe molecule. Catal. Today 2009, 145, 101–107. [Google Scholar] [CrossRef]
- Chakraborty, B.; Viswanathan, B. Surface acidity of MCM-41 by in situ IR studies of pyridine adsorption. Catal. Today 1999, 49, 253–260. [Google Scholar] [CrossRef]
- Emeis, C.A. Determination of Integrated Molar Extinction Coefficients for Infrared Absorption Bands of Pyridine Adsorbed on Solid Acid Catalysts. J. Catal. 1993, 141, 347–354. [Google Scholar] [CrossRef]
- Glazneva, T.S.; Kotsarenko, N.S.; Paukshtis, E.A. Surface acidity and basicity of oxide catalysts: From aqueous suspensions to in situ measurements. Kinet. Catal. 2008, 49, 859–867. [Google Scholar] [CrossRef]
- Sen, S.; Wusirika, R.R.; Youngman, R.E. High temperature thermal expansion behavior of H[Al]ZSM-5 zeolites: The role of Brønsted sites. Microporous Mesoporous Mater. 2006, 87, 217–223. [Google Scholar] [CrossRef]
- Essayem, N.; Holmqvist, A.; Gayraud, P.; Vedrine, J.; Ben Taarit, Y. In Situ FTIR Studies of the Protonic Sites of H3PW12O40 and Its Acidic Cesium Salts MxH3−xPW12O40. J. Catal. 2001, 197, 273–280. [Google Scholar] [CrossRef]
- Gemo, N.; Biasi, P.; Canu, P.; Menegazzo, F.; Pinna, F.; Samikannu, A.; Kordás, K.; Salmi, T.O.; Mikkola, J.-P. Reactivity Aspects of SBA15-Based Doped Supported Catalysts: H2O2 Direct Synthesis and Disproportionation Reactions. Top. Catal. 2013, 56, 540–549. [Google Scholar] [CrossRef]
- Gibson, E.K.; Beale, A.M.; Richard, C.; Catlow, A.; Chutia, A.; Gianolio, D.; Gould, A.; Kroner, A.; Mohammed, K.M.H.; Perdjon, M.; et al. Restructuring of AuPd Nanoparticles Studied by a Combined XAFS/ DRIFTS Approach. Chem. Mater. 2015, 27, 3714–3720. [Google Scholar] [CrossRef]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample 1 | Reduction Temperature (°C) | CO (Initial) Heat of Adsorption with Chemisorbed H (kJ/mol CO) 2 | Linear-to-Bridge Ratio |
|---|---|---|---|
| 2Pd/SiO2 3 | 225 | 92 | 0.2:1 |
| 2Pd–10Zn/SiO2 | 300 | 102 | 2.5:1 |
| 3Pd–1.8Zn/SiO2 | 300 | 99 | 2.1:1 |
| 550 | 93 | 1.8:1 |
| Catalyst | Productivity H2O2 (mol H2O2/molmetal h) | Selectivity (%) | H2 Conversion @ 15 min | Complete H2 Conversion | Acidity (μmol/g) | |||
|---|---|---|---|---|---|---|---|---|
| @ 15 min | @ Complete H2 Conversion | @ 15 min | @ Complete H2 Conversion | (%) | (min) | Lewis | Brønsted | |
| Pd/SBA15 | 359 | 311 | 20 | 20 | 99 | 20 | - | - |
| PdAu/SBA15 | 634 | 326 | 28 | 24 | 65 | 41 | - | - |
| Br-Pd/SBA15 | 39 | - | - | - | - | - | - | - |
| Br-PdAu/SBA15 | 192 | - | 24 | - | 13 | - | - | - |
| PdAu-Br/SBA15 | 0 | 0 | 0 | 0 | - | - | - | - |
| Pd/Si | 850 | 234 | 17 | 15 | 51 | 85 | - | - |
| PdAu/Si | 183 | 77 | 8 | 8 | 77 | 57 | - | - |
| PdAu/CeO2-SBA15 | 399 | 314 | 16 | 14 | 48 | 34 | 39 | 0 |
| PdAu/Ti-SBA15 | 538 | 463 | 13 | 12 | 77 | 18 | 16 | 0 |
| PdAu/Al-SBA15 | 870 | 422 | 30 | 24 | 67 | 37 | 22 | 8 |
| Br-PdAu/Al-SBA15 | 76 | - | 20 | - | 11 | - | 9 | 0 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manzoli, M. Boosting the Characterization of Heterogeneous Catalysts for H2O2 Direct Synthesis by Infrared Spectroscopy. Catalysts 2019, 9, 30. https://doi.org/10.3390/catal9010030
Manzoli M. Boosting the Characterization of Heterogeneous Catalysts for H2O2 Direct Synthesis by Infrared Spectroscopy. Catalysts. 2019; 9(1):30. https://doi.org/10.3390/catal9010030
Chicago/Turabian StyleManzoli, Maela. 2019. "Boosting the Characterization of Heterogeneous Catalysts for H2O2 Direct Synthesis by Infrared Spectroscopy" Catalysts 9, no. 1: 30. https://doi.org/10.3390/catal9010030
APA StyleManzoli, M. (2019). Boosting the Characterization of Heterogeneous Catalysts for H2O2 Direct Synthesis by Infrared Spectroscopy. Catalysts, 9(1), 30. https://doi.org/10.3390/catal9010030