Enhancing Light-Driven Production of Hydrogen Peroxide by Anchoring Au onto C3N4 Catalysts




Abstract
:
1. Introduction
2. Results
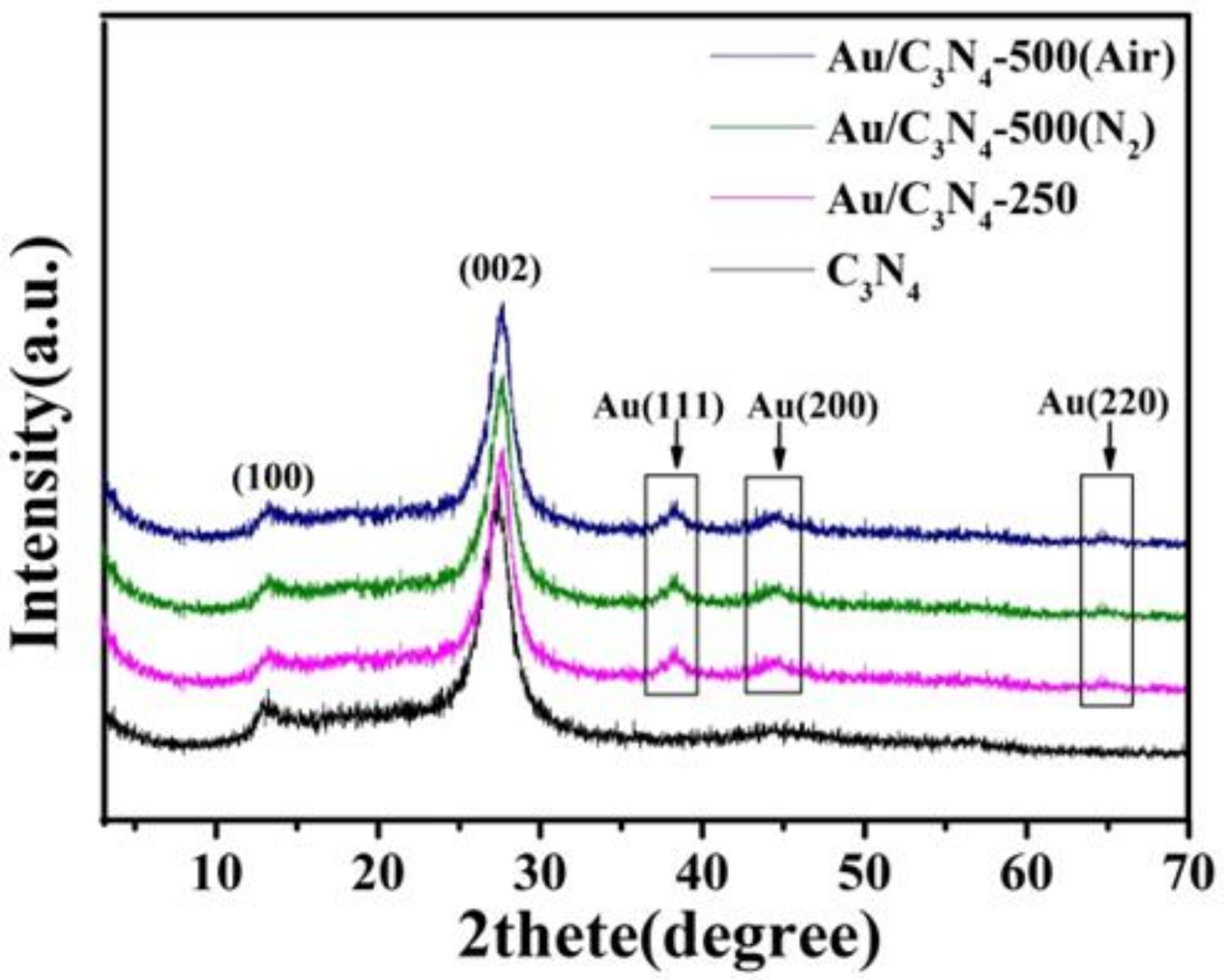
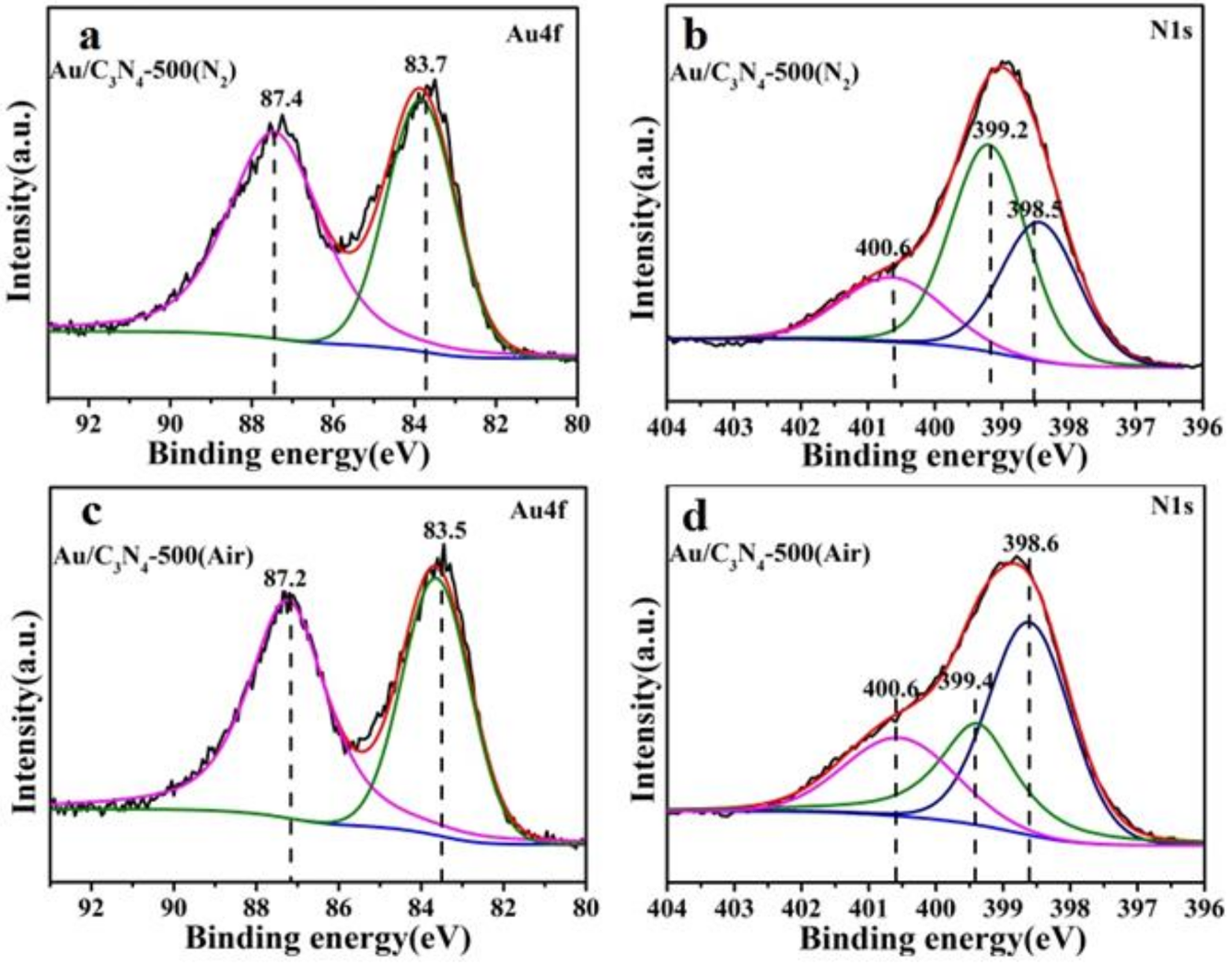
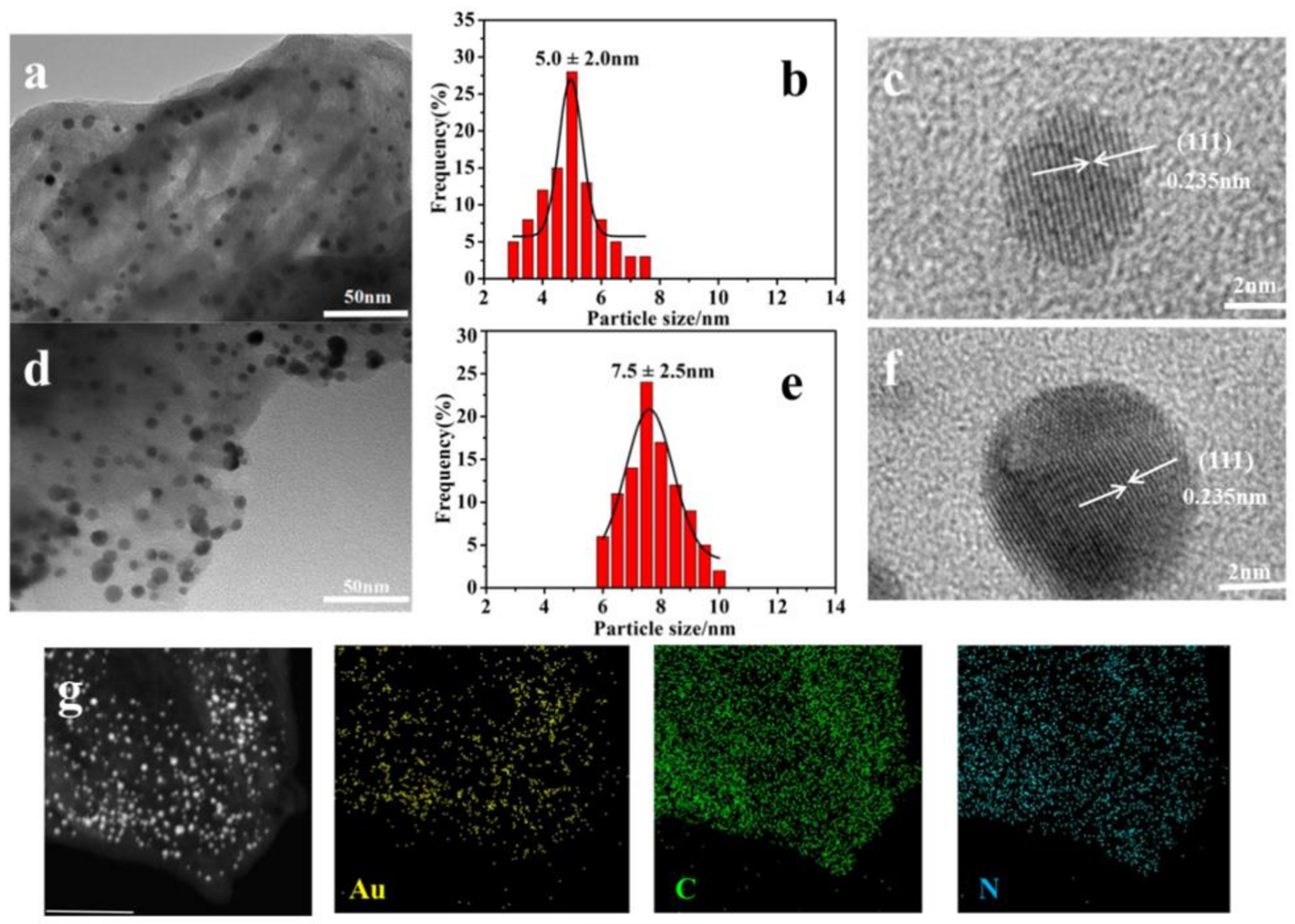
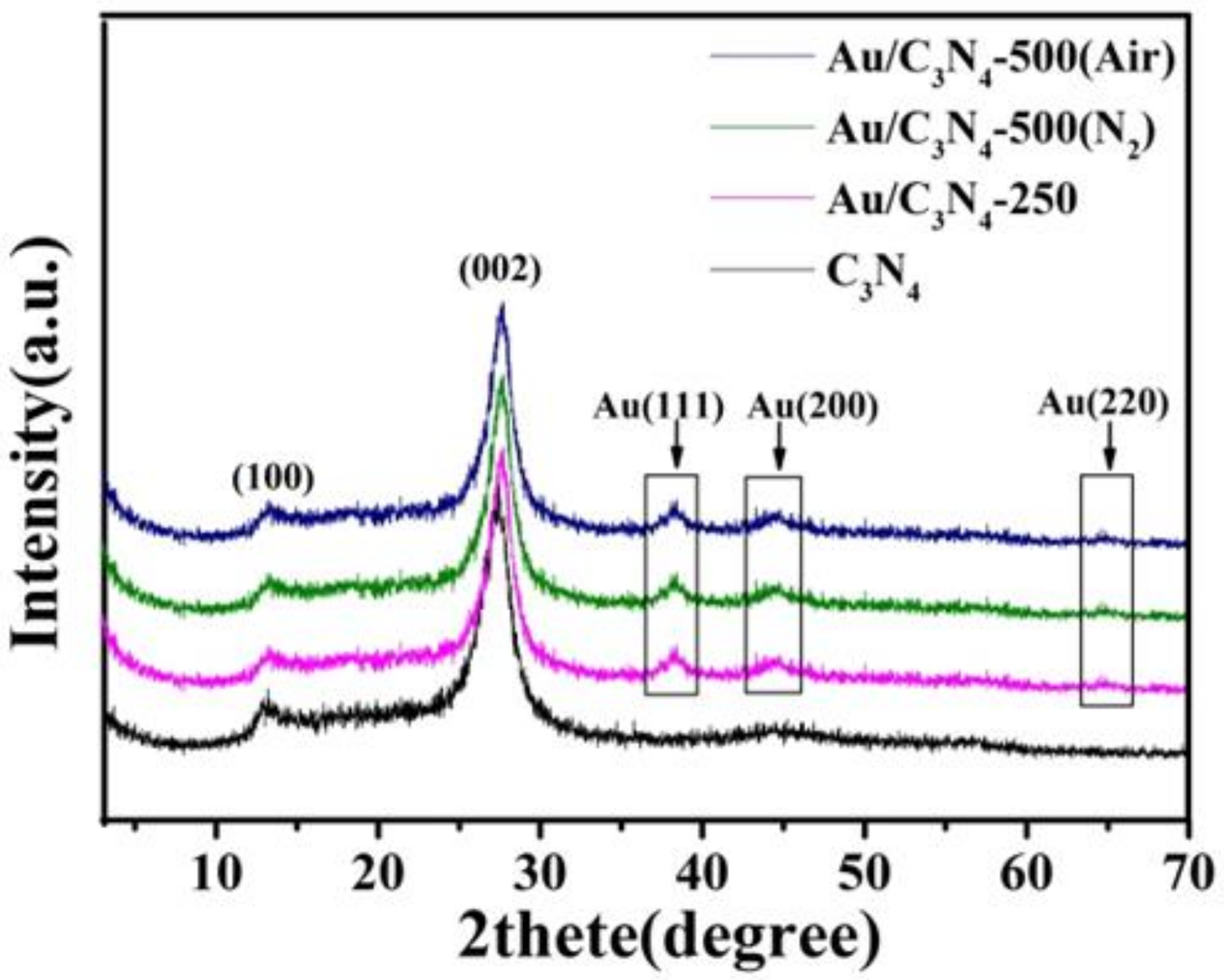
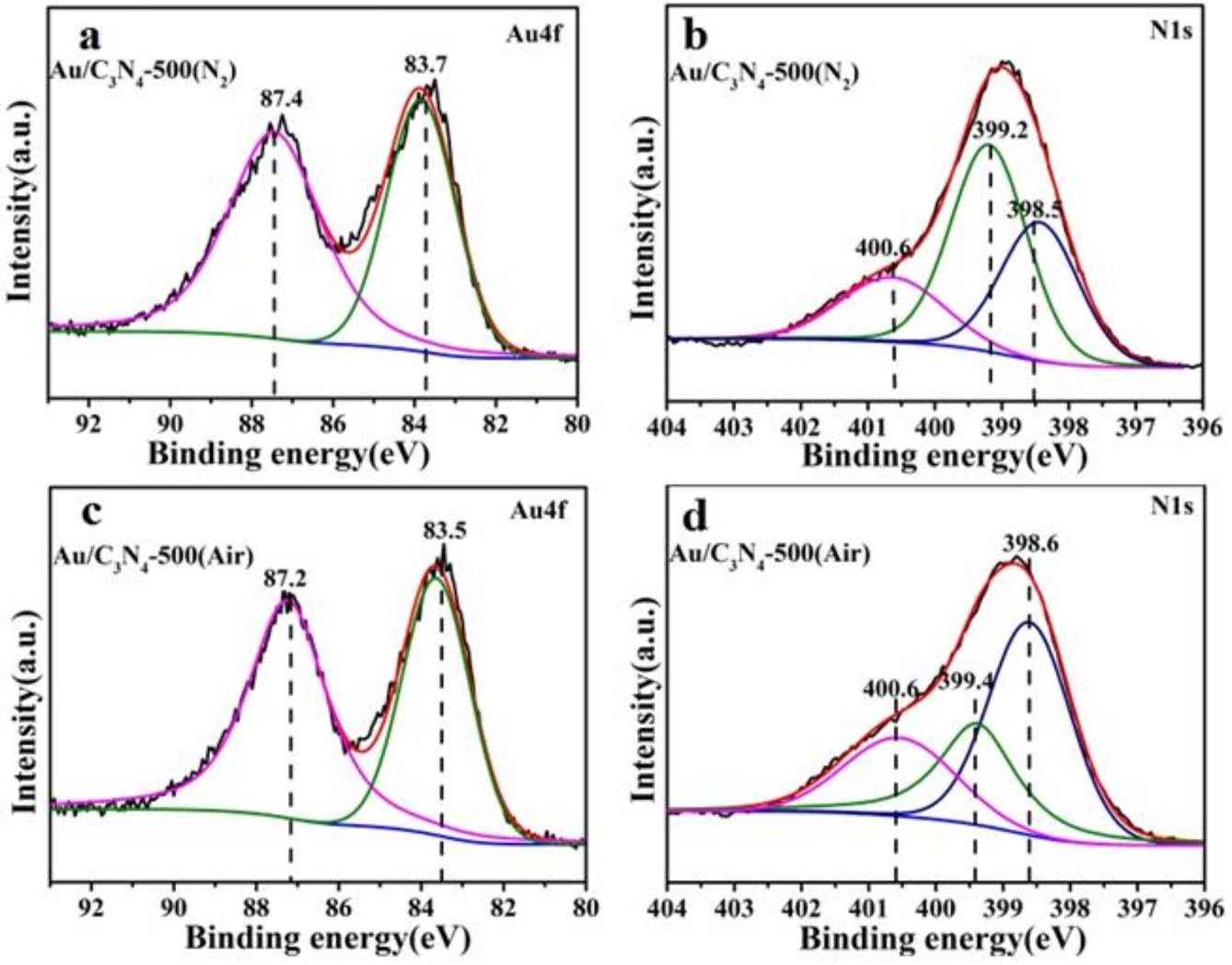
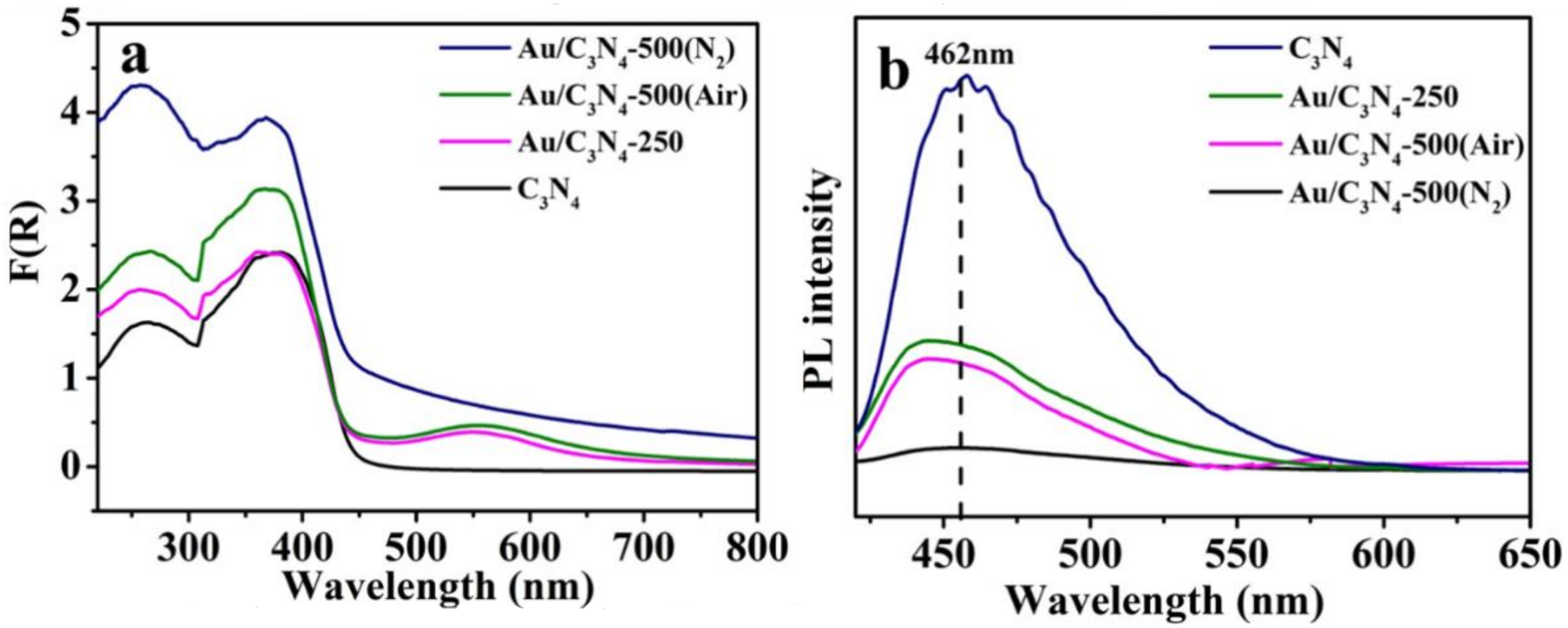
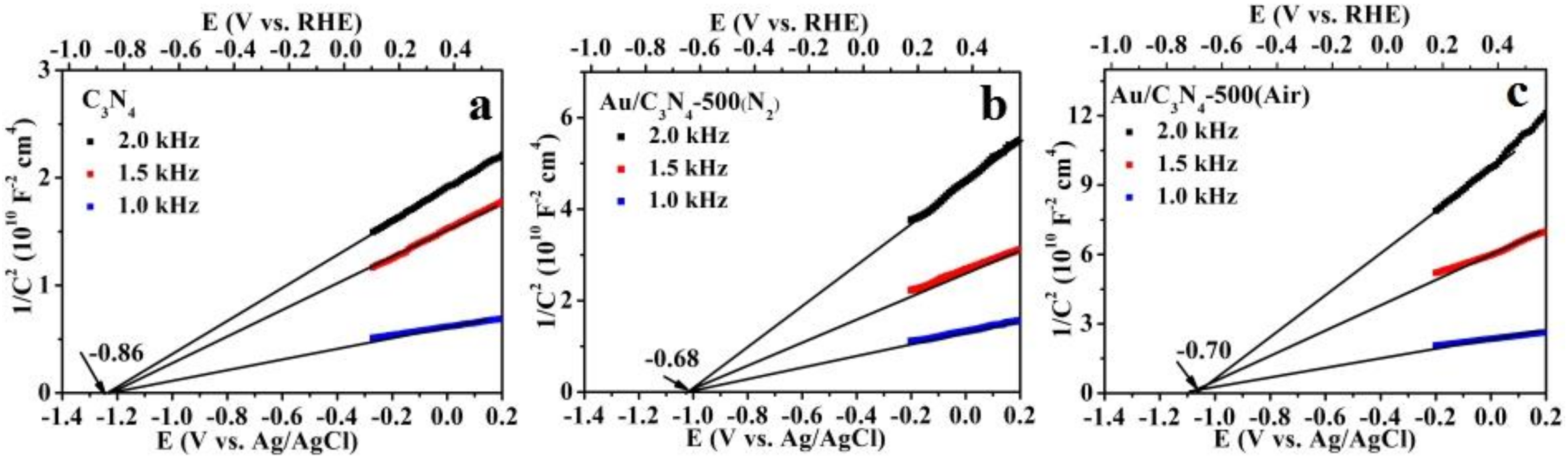
2.1. Catalyst Characterization
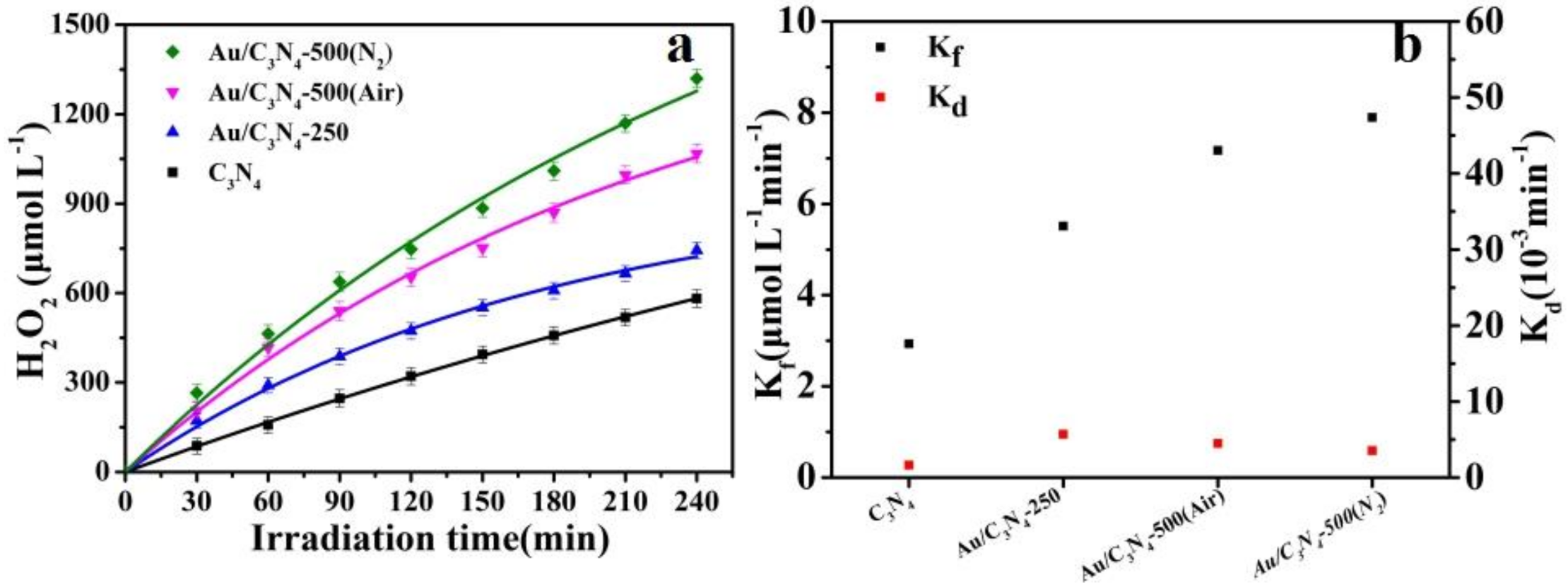
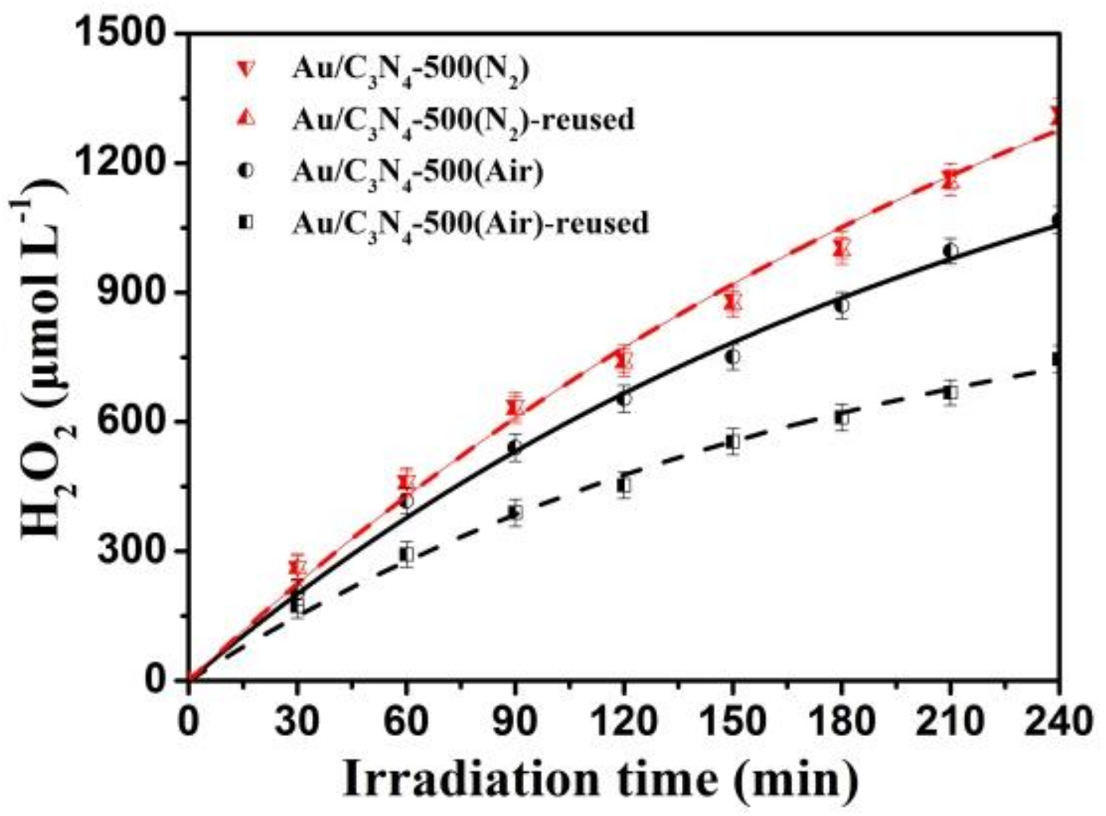
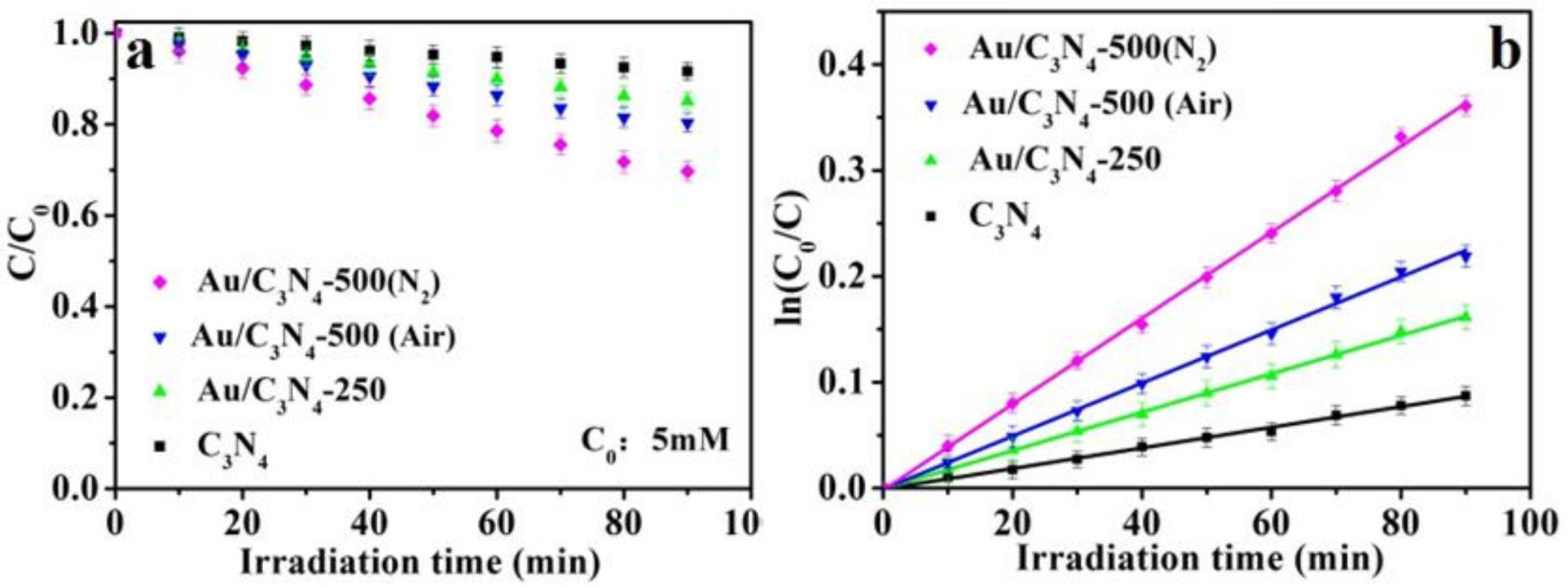
2.2. Photosynthesis of H2O2
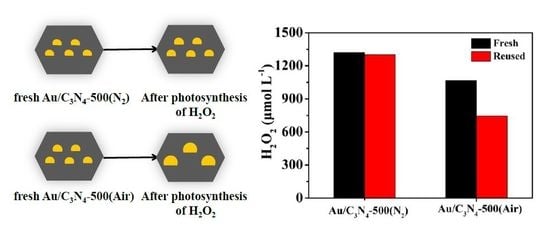
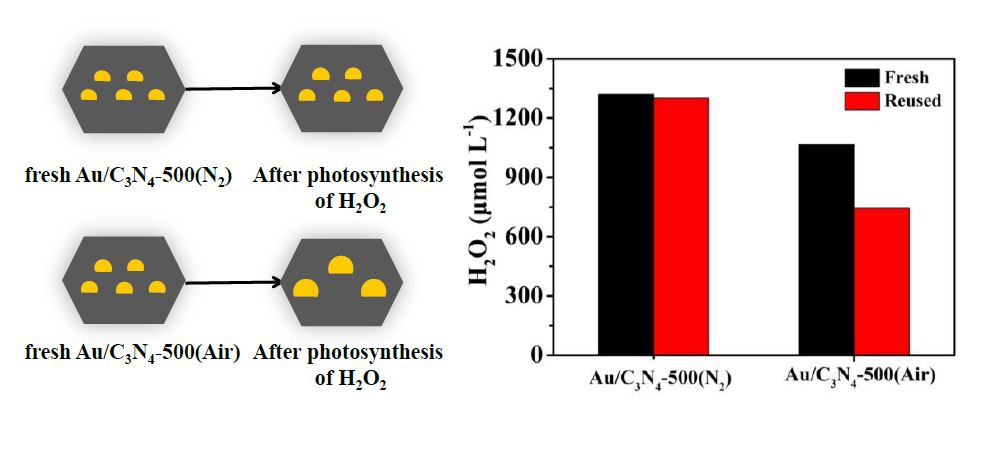
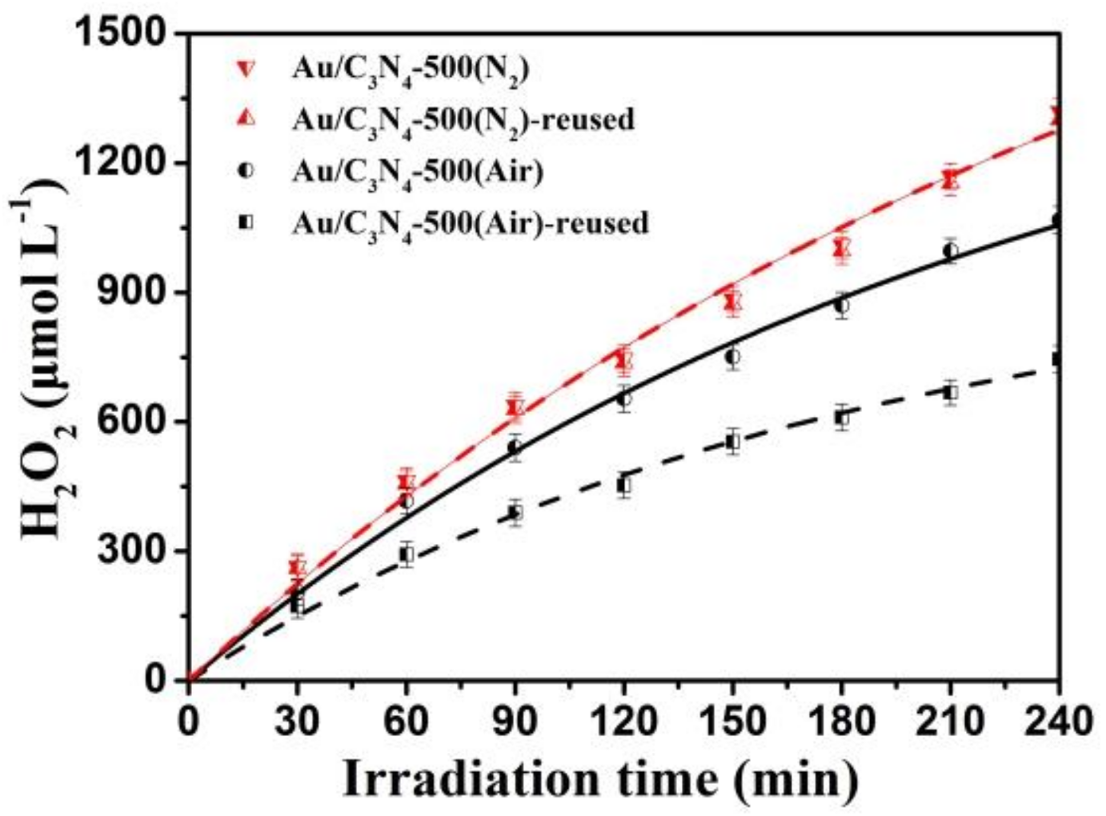
2.3. Stability of the Photocatalysts
3. Materials and Methods
3.1. Materials
3.2. Catalyst Preparation
3.2.1. Preparation of g-C3N4
3.2.2. Synthesis of Au-Supported C3N4 Catalysts
Au/C3N4-250 Catalyst
Au/C3N4-500(N2) Catalyst
Au/C3N4-500(Air) Catalyst
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Campos-Martin, J.M.; Blanco-Brieva, G.; Fierro, J.L.G. Hydrogen peroxide synthesis: An outlook beyond the anthraquinone process. Angew. Chem. Int. Ed. 2006, 45, 6962–6984. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Jung, J.; Suenobua, T.; Fukuzumi, S. Production of hydrogen peroxide as a sustainable solar fuel from water and dioxygen. Energy Environ. Sci. 2013, 6, 3756–3764. [Google Scholar] [CrossRef]
- Isaka, Y.; Kato, S.; Hong, D.; Suenobu, T.; Yamada, Y.; Fukuzumi, S. Bottom-up and top-down methods to improve catalytic reactivity for photocatalytic production of hydrogen peroxide using a Ru-complex and water oxidation catalysts. J. Mater. Chem. A 2015, 3, 12404–12412. [Google Scholar] [CrossRef]
- Isaka, Y.; Oyama, K.; Yamada, Y.; Suenobu, T.; Fukuzumi, S. Photocatalytic production of hydrogen peroxide from water and dioxygen using cyano-bridged polynuclear transition metal complexes as water oxidation catalysts. Catal. Sci. Technol. 2016, 6, 681–684. [Google Scholar] [CrossRef]
- Thakur, S.; Kshetri, T.; Kim, N.H.; Lee, J.H. Sunlight-driven sustainable production of hydrogen peroxide using a CdS–graphene hybrid photocatalyst. J. Catal. 2017, 345, 78–86. [Google Scholar] [CrossRef]
- Fukushima, M.; Tatsumi, K.; Tanaka, S.; Nakamura, H. Photocatalytic production of hydrogen peroxide by tris(2,2′-bipyridine) ruthenium(II) using humic acids as electron donor. Environ. Sci. Technol. 1998, 32, 3948–3953. [Google Scholar] [CrossRef]
- Zhuang, H.; Yang, L.; Xu, J.; Li, F.; Zhang, Z.; Lin, H.; Long, J.; Wang, X. Robust photocatalytic H2O2 production by octahedral Cd3(C3N3S3)2 coordination polymer under visible light. Sci. Rep. 2015, 5, 16947. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhao, X.; Zhang, H.; Li, J.; Zhu, Y. Covalent combination of polyoxometalate and graphitic carbon nitride for light-driven hydrogen peroxide production. Nano Energy 2017, 35, 405–414. [Google Scholar] [CrossRef]
- Yamada, Y.; Nomura, A.; Miyahigashi, T.; Ohkubo, K.; Fukuzumi, S. Acetate induced enhancement of photocatalytic hydrogen peroxide production from qxalic acid and dioxygen. J. Phys. Chem. A 2013, 117, 3751–3760. [Google Scholar] [CrossRef] [PubMed]
- Aratani, Y.; Suenobu, T.; Ohkubo, K.; Yamada, Y.; Fukuzumi, S. Dual function photocatalysis of cyano-bridged heteronuclear metal complexes for water oxidation and two-electron reduction of dioxygen to produce hydrogen peroxide as a solar fuel. Chem. Commun. 2017, 53, 3473–3476. [Google Scholar] [CrossRef] [PubMed]
- Isaka, Y.; Yamada, Y.; Suenobu, T.; Nakagawa, T.; Fukuzumi, S. Production of hydrogen peroxide by combination of semiconductor-photocatalysed oxidation of water and photocatalytic two-electron reduction of dioxygen. RSC Adv. 2016, 6, 42041–42044. [Google Scholar] [CrossRef]
- Cai, R.; Kubota, Y.; Fujishima, A. Effect of copper ions on the formation of hydrogen peroxide from photocatalytic titanium dioxide particles. J. Catal. 2003, 219, 214–218. [Google Scholar] [CrossRef]
- Maurino, V.; Minero, C.; Mariella, G.; Pelizzetti, E. Sustained production of H2O2 on irradiated TiO2-fluoride systems. Chem. Commun. 2005, 20, 2627–2629. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, D.; Shiro, A.; Shiraishi, Y.; Shiraishi, Y.; Sugano, Y.; Ichikawa, S.; Tanaka, S.; Hirai, T. Photocatalytic H2O2 production from ethanol/O2 system using TiO2 loaded with Au-Ag bimetallic alloy nanoparticles. ACS Catal. 2012, 2, 599–603. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Kanazawa, S.; Tsukamoto, D.; Shiro, A.; Sugano, Y.; Hirai, T. Selective hydrogen peroxide formation by titanium dioxide photocatalysis with benzylic alcohols and molecular oxygen in water. ACS Catal. 2013, 3, 2222–2227. [Google Scholar] [CrossRef]
- Teranishi, M.; Naya, S.; Tada, H. In situ liquid phase synthesis of hydrogen peroxide from molecular oxygen using gold nanoparticle-loaded titanium(IV) dioxide photocatalyst. J. Am. Chem. Soc. 2010, 132, 7850–7851. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, C.; Zhao, J. Mechanism of photodecomposition of H2O2 on TiO2 surfaces under visible light irradiation. Langmuir 2001, 17, 4118–4122. [Google Scholar] [CrossRef]
- Wang, X.; Maeda, K.; Thomas, A.; Takanabe, K.; Xin, G.; Carlsson, J.M.; Domen, K.; Antonietti, M. A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat. Mater. 2009, 8, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Shen, J.; Feng, Y.; Shen, Q.; Yang, H. Template-free preparation and characterization of nanoporous g-C3N4 with enhanced visible photocatalytic activity. J. Alloys Compd. 2015, 628, 372–378. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Kanazawa, S.; Sugano, Y.; Tsukamoto, D.; Sakamoto, H.; Ichikawa, S.; Hirai, T. Highly selective production of hydrogen peroxide on graphitic carbon nitride (g-C3N4) photocatalyst activated by visible light. ACS Catal. 2014, 4, 774–780. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Kanazawa, S.; Kofuji, Y.; Sakamoto, H.; Ichikawa, S.; Tanaka, S.; Hirai, T. Sunlight-driven hydrogen peroxide production from water and molecular oxygen by metal-free photocatalysts. Angew. Chem. Int. Ed. 2014, 53, 13454–13459. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, Y.; Kofuji, Y.; Sakamoto, H.; Tanaka, S.; Ichikawa, S.; Hirai, T. Effects of surface defects on photocatalytic H2O2 production by mesoporous graphitic carbon nitride under visible light irradiation. ACS Catal. 2015, 5, 3058–3066. [Google Scholar] [CrossRef]
- Kofuji, Y.; Ohkita, S.; Shiraishi, Y.; Sakamoto, H.; Tanaka, S.; Ichikawa, S.; Hirai, T. Graphitic carbon nitride doped with biphenyl diimide: Efficient photocatalyst for hydrogen peroxide production from water and molecular oxygen by sunlight. ACS Catal. 2016, 6, 7021–7029. [Google Scholar] [CrossRef]
- Kofuji, Y.; Isobe, Y.; Shiraishi, Y.; Sakamoto, H.; Tanaka, S.; Ichikawa, S.; Hirai, T. Carbon nitride-aromatic diimide-graphene nanohybrids: MetalFree photocatalysts for solar-to-hydrogen peroxide energy conversion with 0.2% efficiency. J. Am. Chem. Soc. 2016, 138, 10019–10025. [Google Scholar] [CrossRef] [PubMed]
- Moon, G.-H.; Fujitsuka, M.; Kim, S.; Majima, T.; Wang, X.; Choi, W. Eco-Friendly photochemical production of H2O2 through O2 reduction over carbon nitride frameworks incorporated with multiple heteroelements. ACS Catal. 2017, 7, 2886–2895. [Google Scholar] [CrossRef]
- Wang, R.; Pan, K.; Han, D.; Jiang, J.; Xiang, C.; Huang, Z.; Zhang, L.; Xiang, X. Solar-driven H2O2 generation from H2O and O2 using earth-abundant mixed-metal oxide@carbon nitride photocatalysts. ChemSusChem 2016, 9, 2470–2479. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, L.; Xiang, X.; Yan, D.; Li, F. Engineering of ZnCo-layered double hydroxide nanowalls toward high-efficiency electrochemical water oxidation. J. Mater. Chem. A 2014, 2, 13250–13258. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, R.; Yang, Y.; Yan, D.; Xiang, X. Highly enhanced photoelectrochemical water oxidation efficiency based on triadic quantum dot/layered double hydroxide/BiVO4 photoanodes. ACS Appl. Mater. Interfaces 2016, 8, 19446–19455. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Liu, J.; Yang, J.; Xiang, X. In-situ conversion and catalytic properties of mixed-metal oxide catalysts for photosynthesis of hydrogen peroxide. Sci. China-Chem. 2017, 47, 465–473. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, X.; Li, F.; Cao, D.; Pu, M.; Han, D.; Yang, J.; Xiang, X. Energy-level dependent H2O2 production on metal-free, carbon-content tunable carbon nitride photocatalysts. J. Energy Chem. 2018, 27, 343–350. [Google Scholar] [CrossRef]
- Tan, Y.; Liu, X.Y.; Zhang, L.; Wang, A.; Li, L.; Pan, X.; Miao, S.; Haruta, M.; Wei, H.; Wang, H.; et al. ZnAl-hydrotalcite-supported Au25 nanoclusters as precatalysts for chemoselective hydrogenation of 3-nitrostyrene. Angew. Chem. Int. Ed. 2017, 56, 2709–2713. [Google Scholar] [CrossRef] [PubMed]
- Zhan, W.; He, Q.; Liu, X.; Guo, Y.; Wang, Y.; Wang, L.; Guo, Y.; Borisevich, A.Y.; Zhang, J.; Lu, G.; et al. A sacrificial coating strategy toward enhancement of metal-support interaction for ultrastable Au nanocatalysts. J. Am. Chem. Soc. 2016, 138, 16130–16139. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Shen, S.; Guo, P.; Wu, P.; Guo, L. Spatial engineering of photo-active sites on g-C3N4 for efficient solar hydrogen generation. J. Mater. Chem. A 2014, 2, 4605–4612. [Google Scholar] [CrossRef]
- Sun, X.; Dong, S.; Wang, E. Large-Scale synthesis of micrometer-scale single-crystalline Au plates of nanometer thickness by a wet-chemical route. Angew. Chem. Int. Ed. 2004, 116, 6520–6523. [Google Scholar] [CrossRef]
- Leff, D.V.; Brandt, L.; Heath, J.R. Synthesis and characterization of hydrophobic, organically-soluble gold nanocrystals functionalized with primary amines. Langmuir 1996, 12, 4723–4730. [Google Scholar] [CrossRef]
- Park, M.; Shin, K.S.; Lee, J.W.; Kim, K. Novel fabrication of Au nanoparticle film on a polyelectrolyte-coated glass for efficient surface-enhanced raman scattering. Bull. Korean Chem. Soc. 2015, 36, 743–747. [Google Scholar]
- Singh, J.A.; Overbury, S.H.; Dudney, N.J.; Li, M.; Veith, G.M. Gold nanoparticles supported on carbon nitride: Influence of surface hydroxyls on low temperature carbon monoxide oxidation. ACS Catal. 2012, 2, 1138–1146. [Google Scholar] [CrossRef]
- Kartusch, C.; Krumeich, F.; Safonova, O.; Hartfelder, U.; Makosch, M.; Sa, J.; Bokhoven, J.A. Redispersion of gold multiple-twinned particles during liquid-phase hydrogenation. ACS Catal. 2012, 2, 1394–1403. [Google Scholar] [CrossRef]
- Gao, L.F.; Wen, T.; Xu, J.Y.; Zhai, X.P.; Zhao, M.; Hu, G.W.; Chen, P.; Wang, Q.; Zhang, H.L. Iron-doped carbon nitride-type polymers as homogeneous organocatalysts for visible light-driven hydrogen evolution. ACS Appl. Mater. Interfaces 2016, 8, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Khabashesku, V.N.; Zimmerman, J.L.; Margrave, J.L. Powder synthesis and characterization of amorphous carbon nitride. Chem. Mater. 2000, 12, 3264–3270. [Google Scholar] [CrossRef]
- Guo, Q.; Xie, Y.; Wang, X.; Lv, S.; Hou, T.; Liu, X. Characterization of well crystallized graphitic carbon nitride nanocrystallites via a benzene thermal route at low temperatures. Chem. Phys. Lett. 2003, 380, 84–87. [Google Scholar] [CrossRef]
- Guo, Q.; Yang, Q.; Zhu, L.; Yi, C.; Zhang, S.; Xie, Y. A facile one pot solvothermal route to tubular forms of luminescent polymeric networks [(C3N3)2(NH)3]n. Solid State Commun. 2004, 132, 369–374. [Google Scholar] [CrossRef]
- Wang, S.; Li, C.; Wang, T.; Zhang, P.; Li, A.; Gong, J. Controllable synthesis of nanotube-type graphitic C3N4 and their visible-light photocatalytic and fluorescent properties. J. Mater. Chem. A 2014, 2, 2885–2890. [Google Scholar] [CrossRef]
- Zhan, G.; Huang, J.; Du, M.; Sun, D.; Abdul-Rauf, I.; Lin, W.; Hong, Y.; Li, Q. Liquid phase oxidation of benzyl alcohol to benzaldehyde with novel uncalcined bioreduction Au catalysts: High activity and durability. Chem. Eng. J. 2012, 187, 232–238. [Google Scholar] [CrossRef]
- Xu, Z.; Quintanilla, M.; Vetrone, F.; Govorov, A.O.; Chaker, M.; Ma, D. Harvesting lost photons: Plasmon and upconversion enhanced broadband photocatalytic activity in core@shell microspheres based on lanthanide-doped NaYF4, TiO2, and Au. Adv. Funct. Mater. 2015, 25, 2950–2960. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Y.; Liu, N.; Han, Y.; Zhang, X.; Huang, H.; Lifshitz, Y.; Lee, S.-T.; Zhong, J.; Kang, Z. Metal-free efficient photocatalyst for stable visible water splitting via a two-electron pathway. Science 2015, 347, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Moon, G.-H.; Kim, W.; Bokare, A.-D.; Sung, N.-H.; Choi, W. Solar production of H2O2 on reduced graphene oxide—TiO2 hybrid photocatalysts consisting of earth-abundant elements only. Energy Environ. Sci. 2014, 7, 4023–4028. [Google Scholar] [CrossRef]
- Chao, C.; Liu, J.; Wang, J.; Zhang, Y.; Zhang, B.; Zhang, Y.; Xiang, X.; Chen, R. Surface Modification of Halloysite Nanotubes with Dopamine for Enzyme Immobilization. ACS Appl. Mater. Interfaces 2013, 5, 10559–10564. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Au4f/eV | N1s/eV | |||
|---|---|---|---|---|---|
| 4f5/2 | 4f7/2 | N-(C)3 | =N- | C2NH | |
| Au/C3N4-500(N2) | 87.4 | 83.7 | 400.6 | 399.2 | 398.5 |
| Au/C3N4-500(N2)-reused | 87.3 | 83.6 | 400.6 | 399.2 | 398.5 |
| Au/C3N4-500(Air) | 87.2 | 83.5 | 400.6 | 399.4 | 398.6 |
| Au/C3N4-500(Air)-reused | 87.0 | 83.3 | 400.6 | 399.4 | 398.6 |
| Sample | H2O2 Yield at 120 min (μmol L−1) | H2O2 Yield at 240 min (μmol L−1) | Kf (μmol L−1 min−1) | Kd (10−3 min−1) |
|---|---|---|---|---|
| C3N4 | 321 | 582 | 2.929 | 1.61 |
| Au/C3N4-250 | 473 | 743 | 5.514 | 5.68 |
| Au/C3N4-500(Air) | 653 | 1067 | 7.170 | 4.46 |
| Au/C3N4-500(N2) | 747 | 1320 | 7.893 | 3.54 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, X.; Yang, J.; Han, D.; Zhang, B.; Xiang, X.; He, J. Enhancing Light-Driven Production of Hydrogen Peroxide by Anchoring Au onto C3N4 Catalysts. Catalysts 2018, 8, 147. https://doi.org/10.3390/catal8040147
Chang X, Yang J, Han D, Zhang B, Xiang X, He J. Enhancing Light-Driven Production of Hydrogen Peroxide by Anchoring Au onto C3N4 Catalysts. Catalysts. 2018; 8(4):147. https://doi.org/10.3390/catal8040147
Chicago/Turabian StyleChang, Xiaoyu, Junjiao Yang, Dandan Han, Bing Zhang, Xu Xiang, and Jing He. 2018. "Enhancing Light-Driven Production of Hydrogen Peroxide by Anchoring Au onto C3N4 Catalysts" Catalysts 8, no. 4: 147. https://doi.org/10.3390/catal8040147