Chemical and Electronic Changes of the CeO2 Support during CO Oxidation on Au/CeO2 Catalysts: Time-Resolved Operando XAS at the Ce LIII Edge

 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. State of the Au/CeO2 Catalysts after Different Pretreatment Procedures
2.2. Operando X-Ray Absorption Spectroscopy during CO Oxidation
2.3. Role of Ce3+ Species/O-Vacancies in the CO Oxidation Reaction
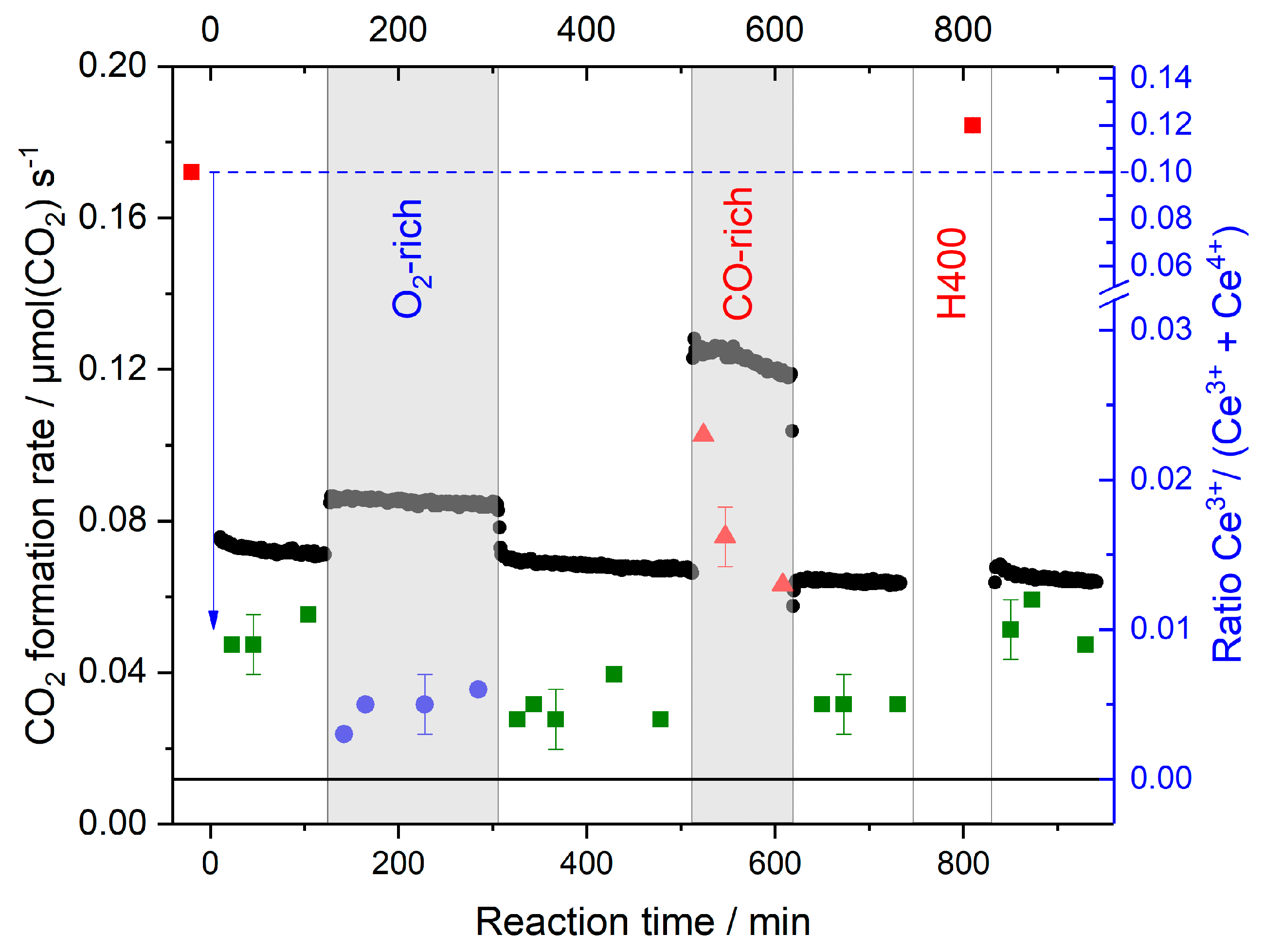
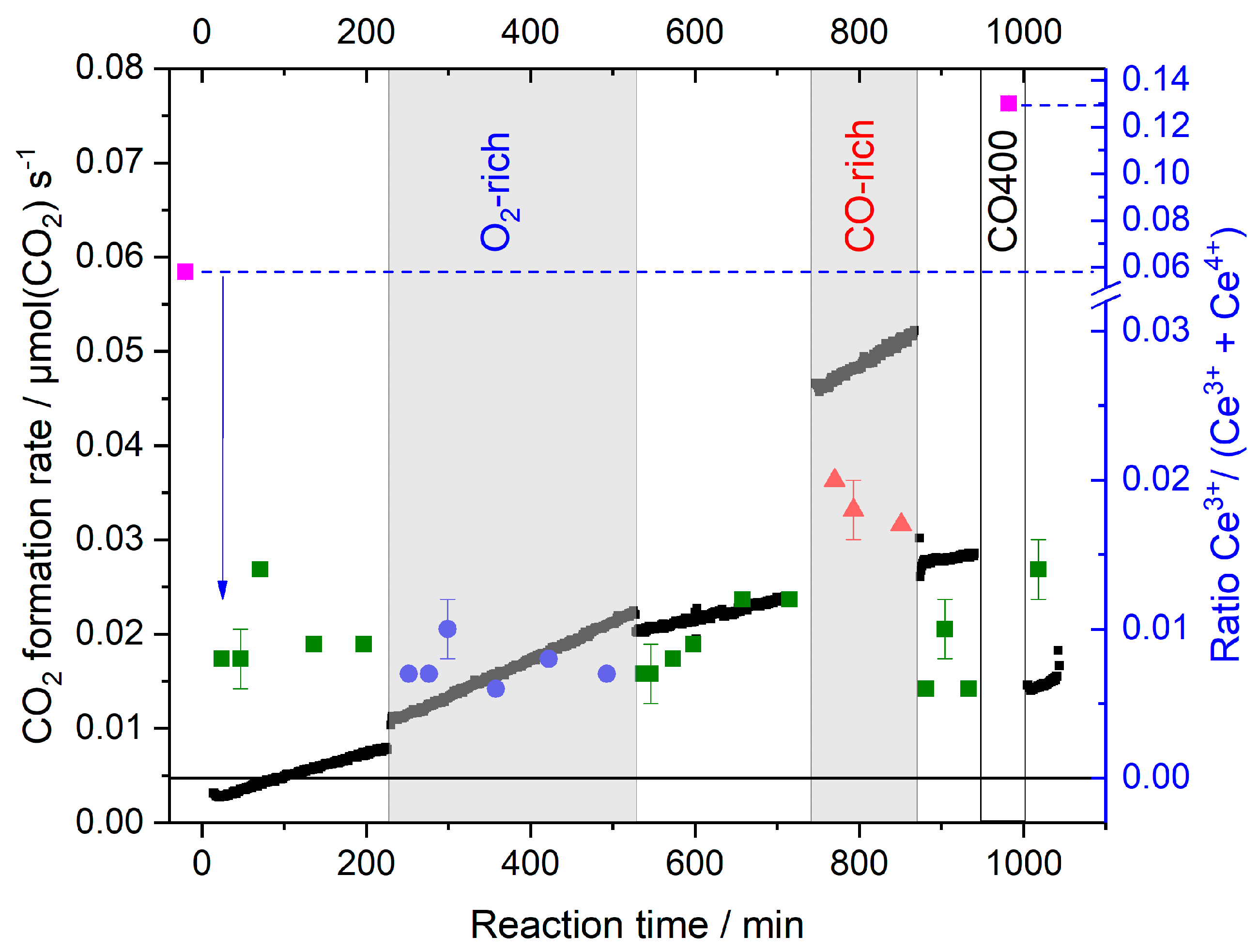
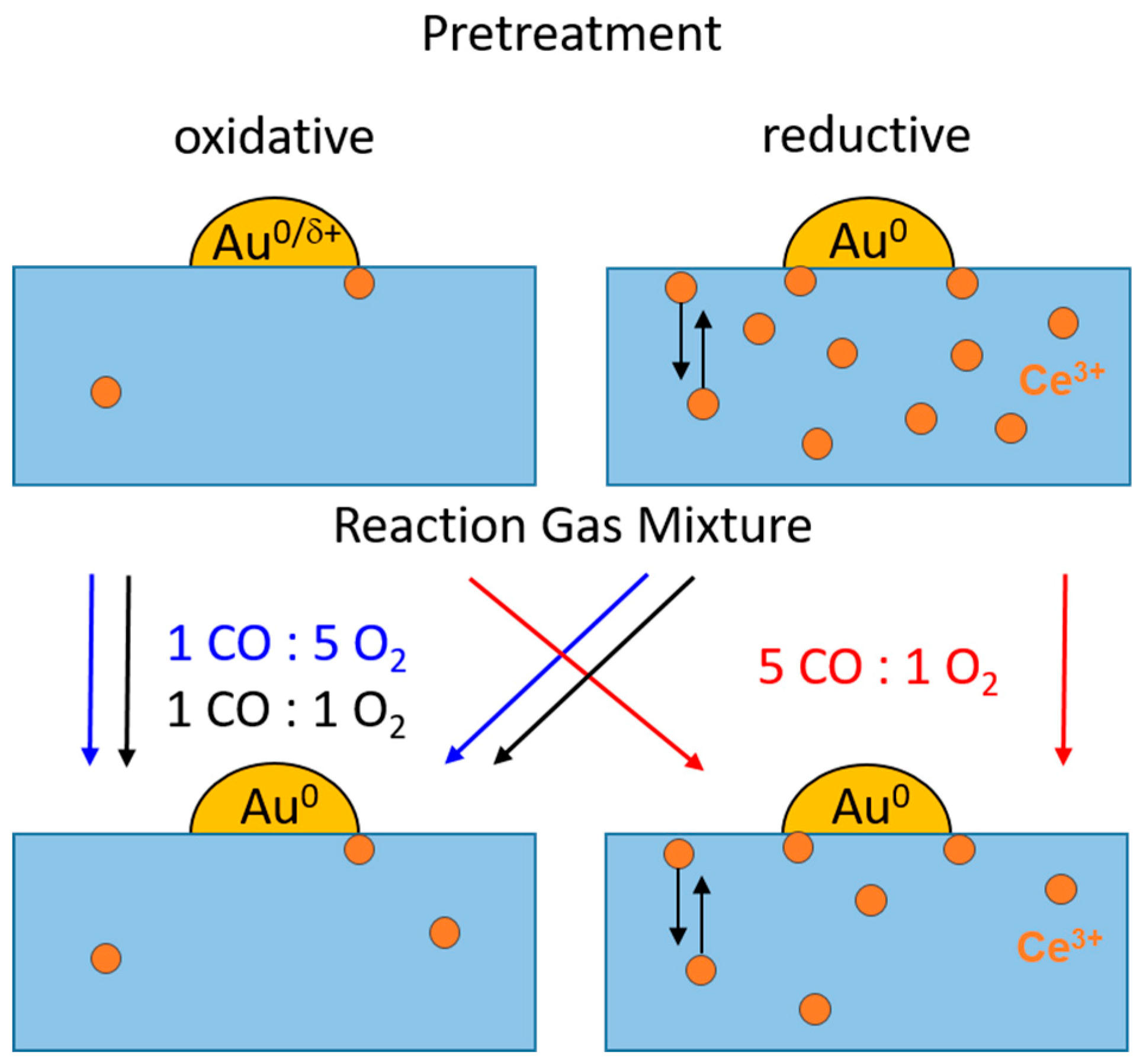
- While oxidative O400 pretreatment results in a fully oxidized CeO2 support with negligible Ce3+ contents, reductive H400 or CO400 pretreatment leads to the significant formation of Ce3+ species, between 6% and 10% in the catalyst region accessible by XANES under present conditions, and hence formation of O-vacancies (cf. upper panel of Figure 8). At the same time, Au species are fully reduced after reductive pretreatment, while the formation of oxidic Au species was reported after oxidative O400 pretreatment [30,46];
- The concentration of Ce3+ species on the pure CeO2 support (without Au NPs) seems to be slightly higher than on the Au/CeO2 catalyst, with 1.7% and 17% Ce3+ species after O400 and CO400 treatment, respectively. The higher Ce3+ content compared to those obtained on the Au/CeO2 catalysts seems to contradict earlier claims that the presence of the Au NPs enhances the formation of O vacancies [19,27,48]. This apparent contradiction may arise from the rather high temperature of the reductive pretreatment, where ceria reduction is facile also without additional enhancement by Au NPs;
- During the reaction in formally oxidative gas mixtures (standard gas mixture and O2-rich gas mixture), most of the Ce3+ species are rapidly re-oxidized. Due to the high mobility of O-vacancies, the bulk O-vacancies are essentially in equilibrium with those in the surface near region. In the O400 pretreated sample, this leads to complete removal of Ce3+ species/O-vacancies, while on the reductively pretreated samples a small fraction of Ce3+ species/O-vacancies (~0.5%) remains, which cannot be re-oxidized under present reaction conditions. They are attributed to deeper lying defects, which can be created during pretreatment at 400 °C, but are metastable during reaction at 80 °C due to the limited mobility of O-vacancy defects under these conditions. This agrees fully with findings from operando Raman studies, which also identified surface and bulk type defects and could discriminate between them [22,33,39,43];
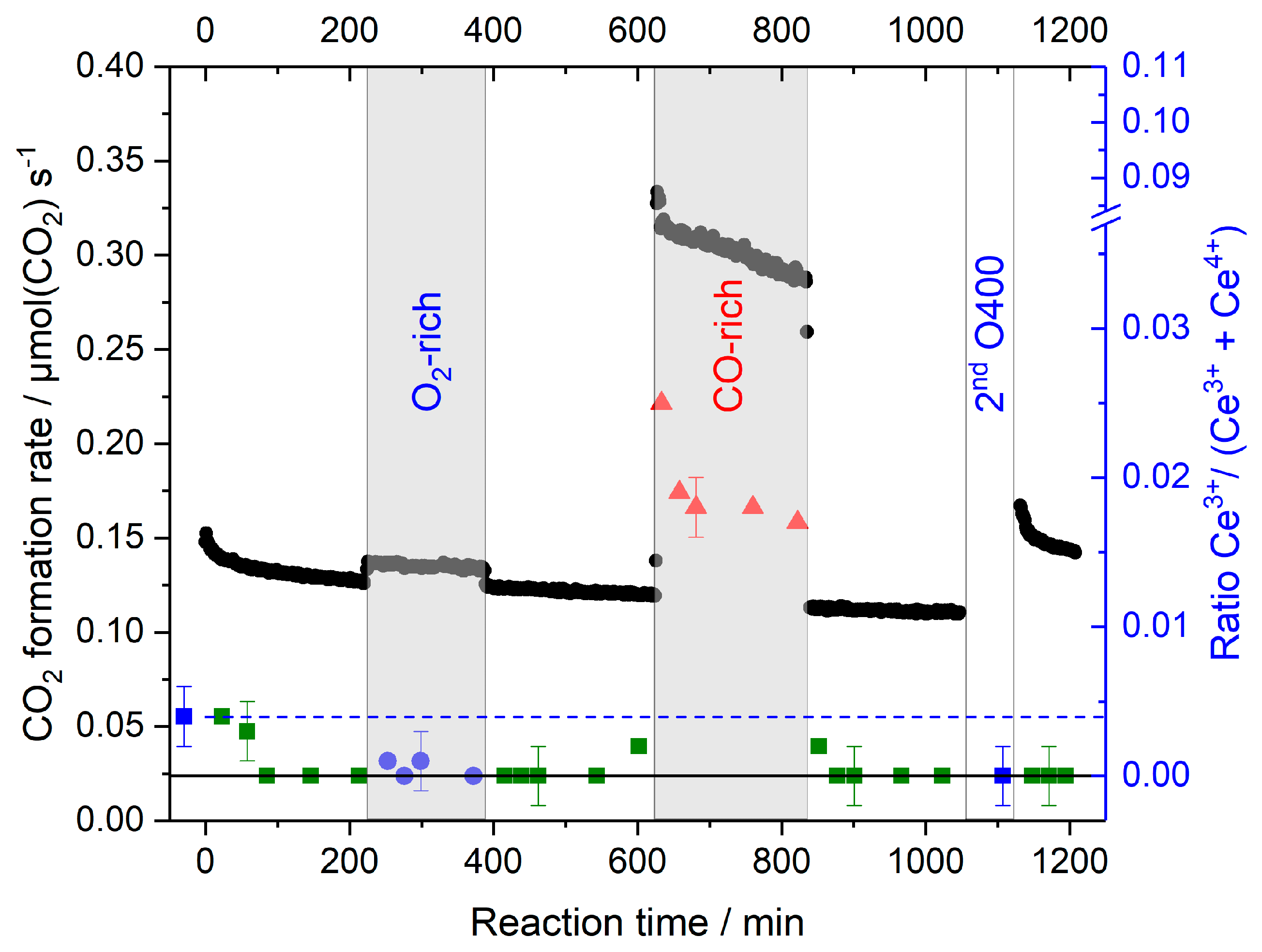
- On the Au/CeO2 catalysts the steady-state concentration of Ce3+ species/O-vacancies in the surface region is determined by the reaction gas composition, it is negligible in the standard and O2-rich gas mixtures, and in the range of 1%–2% in the CO-rich mixture, independent of the pretreatment (cf. lower panel in Figure 8);
- Based on the rapid re-oxidation of Ce3+ species/surface vacancies during reaction at 80 °C in oxidative gas mixtures and their stable concentration at longer reaction times we can exclude that the slow deactivation of the catalysts under these conditions is mainly related to an over-reduction of the catalyst support, in the sense that a reduced catalyst state is generated which is less active. Considering also previous reports this seems to be related to the slow build-up of site blocking surface species, mainly surface carbonates;
- The build-up of such surface species during pretreatment in CO rather than over-reduction is also proposed as main reason for the very low initial activity of the CO400 pretreated sample and the very slow increase in activity during time on stream, considering that the concentration of Ce3+ species/O vacancies is similarly high after pretreatment in H2 (H400), where such effects were not observed, as in CO (CO400). This is supported also by the fact that the concentration of Ce3+ species/O vacancies decreases rapidly during the time on stream, while the increase in activity during the reaction is very slow. We attribute this to a slow, possibly oxygen-induced decomposition of site blocking surface species;
- Considering that reduction/re-oxidation of the surface region is rather fast on the time scale of these measurements, it is not possible to decide from these measurements whether changes in the CO2 formation rate upon changing the composition of the reaction gas atmosphere are (mainly) caused by changes in the support oxidation state, leading to a modification of the rate constant, or by changes in the adsorbate coverages. This is mainly relevant when going to CO-rich gas mixtures with their higher CO2 formation rate, while increasing the O2 content has little effect on the CO2 formation rate.
3. Experimental
3.1. Catalyst Preparation and Characterization
3.2. Operando Catalyst Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hutchings, G.J.; Joffe, R. A Novel Process for the Co-Synthesis of Vinyl Chloride Monomer and Sodium Carbonate Using a Gold Catalyst. Appl. Catal. 1986, 20, 215–218. [Google Scholar] [CrossRef]
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel Gold Catalysts for the Oxidation of Carbon Monoxide. Chem. Lett. 1987, 16, 405–408. [Google Scholar] [CrossRef]
- Bond, G.C.; Thompson, D.T. Catalysis by Gold. Catal. Rev. Sci. Eng. 1999, 41, 319–388. [Google Scholar] [CrossRef]
- Schubert, M.M.; Hackenberg, S.; van Veen, A.C.; Muhler, M.; Plzak, V.; Behm, R.J. CO Oxidation over Supported Gold Catalysts—“Inert” and “Active” Support Materials and their Role for the Oxygen Supply during Reaction. J. Catal. 2001, 197, 113–122. [Google Scholar] [CrossRef]
- Ab Rahim, M.H.; Forde, M.M.; Hammond, C.; Jenkins, R.L.; Dimitratos, N.; Lopez-Sanchez, J.A.; Carley, A.F.; Taylor, S.H.; Willock, D.J.; Hutchings, G.J. Systematic Study of the Oxidation of Methane Using Supported Gold Palladium Nanoparticles Under Mild Aqueous Conditions. Top. Catal. 2013, 56, 1843–1857. [Google Scholar] [CrossRef]
- Burch, R. Gold Catalysts for Pure Hydrogen Production in the Water–Gas Shift Reaction: Activity, Structure and Reaction Mechanism. Phys. Chem. Chem. Phys. 2006, 8, 5483–5500. [Google Scholar] [CrossRef]
- Flytzani-Stephanopoulos, M. Gold Atoms Stabilized on Various Supports Catalyze the Water-Gas Shift Reaction. Acc. Chem. Res. 2014, 47, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Enache, D.I.; Edwards, J.K.; Landon, P.; Solsona-Espriu, B.; Carley, A.F.; Herzing, A.A.; Watanabe, M.; Kiely, C.; Knight, D.W.; Hutchings, G.J. Solvent-Free Oxidation of Primary Alcohols to Aldehydes Using Au-Pd/TiO2 Catalysts. Science 2011, 311, 362–365. [Google Scholar] [CrossRef]
- Min, B.K.; Friend, C.M. Heterogeneous Gold-Based Catalysis for Green Chemistry: Low-Temperature CO Oxidation and Propene Oxidation. Chem. Rev. 2007, 107, 2709–2724. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, B.; Su, D.; Huang, W. Titania Morphology-Dependent Gold-Titania Interaction, Structure, and Catalytic Performance of Gold/Titania Catalysts. ChemCatChem 2015, 7, 3290–3298. [Google Scholar] [CrossRef]
- Sakurai, H.; Haruta, M. Carbon Dioxide and Carbon Monoxide Hydrogenation Over Gold Supported on Titanium, Iron and Zinc Oxides. Appl. Catal. A 1995, 127, 93–105. [Google Scholar] [CrossRef]
- Sakurai, H.; Haruta, M. Synergism in Methanol Synthesis from Carbon Dioxide over Gold Catalysts Supported on Metal Oxides. Catal. Today 1996, 29, 361–365. [Google Scholar] [CrossRef]
- Hartadi, Y.; Widmann, D.; Behm, R.J. CO2 Hydrogenation to Methanol on Supported Au Catalysts under Moderate Reaction Conditions: Support and Particle Size Effects. ChemSusChem 2015, 8, 456–465. [Google Scholar] [CrossRef]
- Abdel-Mageed, A.M.; Klyushin, A.Y.; Knop-Gericke, A.; Schlögl, R.; Behm, R.J. Negative Charging of Au Nanoparticles during Methanol Synthesis from CO2/H2 on a Au/ZnO Catalyst: Insights from Operando Infrared and Near-ambient Pressure XPS and XAS. Angew. Chem. Int. Ed. 2018, 58, 10325–10329. [Google Scholar] [CrossRef]
- Trovarelli, A. Catalytic Properties of Ceria and CeO2-Containing Materials. Catal. Rev. Sci. Eng. 1996, 38, 439–520. [Google Scholar] [CrossRef]
- Dutta, P.; Pal, S.; Seehra, M.S.; Shi, Y.; Eyring, E.M.; Ernst, R.D. Concentration of Ce3+ and Oxygen Vacancies in Cerium Oxide Nanoparticles. Chem. Mater. 2006, 18, 5144–5146. [Google Scholar] [CrossRef]
- Zhang, C.; Michaelides, A.; King, D.A.; Jenkins, S.J. Oxygen Vacancy Clusters on Ceria: Decisive Role of Cerium f Electrons. Phys. Rev. B 2009, 79, 075433. [Google Scholar] [CrossRef]
- Mullins, D.R. The Surface Chemistry of Cerium Oxide. Surf. Sci. Rep. 2015, 70, 42–85. [Google Scholar] [CrossRef]
- Tabakova, T.; Boccuzzi, F.; Manzoli, M.; Andreeva, D. FTIR Study of Low-Temperature Water-Gas Shift Reaction on Gold/Ceria Catalyst. Appl. Catal. A 2003, 252, 385–397. [Google Scholar] [CrossRef]
- Wang, L.C.; Tahvildar Khazaneh, M.; Widmann, D.; Behm, R.J. Dynamic TAP Reactor Studies of the Oxidizing Capability of CO2 on a Au/CeO2 Catalyst—A First Step Toward Identifying a Redox Mechanism in the Reverse Water-Gas Shift Reaction. J. Catal. 2013, 302, 20–30. [Google Scholar] [CrossRef]
- Wang, F.; Li, C.; Zhang, X.; Wei, M.; Evans, D.G.; Duan, X. Catalytic Behavior of Supported Ru Nanoparticles on the {100},{110}, and {111} Facet of CeO2. J. Catal. 2015, 329, 177–186. [Google Scholar] [CrossRef]
- Vindigni, F.; Manzoli, M.; Damin, A.; Tabakova, T.; Zecchina, A. Surface and Inner Defects in Au/CeO2 WGS Catalysts: Relation between Raman Properties, Reactivity and Morphology. Chem. Eur. J. 2011, 17, 4356–4361. [Google Scholar] [CrossRef]
- Schilling, C.; Hess, C. Elucidating the Role of Support Oxygen in the Water-Gas Shift Reaction over Ceria-Supported Gold Catalysts Using Operando Spectroscopy. ACS Catal. 2019, 9, 1159–1171. [Google Scholar] [CrossRef]
- Abdel-Mageed, A.M.; Kucèrová, G.; Bansmann, J.; Behm, R.J. Active Au Species During the Low-Temperature Water Gas Shift Reaction on Au/CeO2: A Time Resolved Operando XAS and DRIFTS Study. ACS Catal. 2017, 17, 6471–6484. [Google Scholar] [CrossRef]
- Liu, W.; Flytzani-Stephanopoulos, M. Total Oxidation of Carbon Monoxide and Methane over Transition Metal-Fluorite Oxide Composite Catalysis. II. Catalyst Characterization and Reaction Kinetics. J. Catal. 1995, 153, 317–332. [Google Scholar] [CrossRef]
- Plzak, V.; Garche, J.; Behm, R.J. Au/MeOx Catalysts in Gas Processing for PEM Fuel Cells: An Overview of the Results at ZSW-Uni Ulm—Part 2: Low Temperature PROX Yeaction over New Highly Active Au/TiO2 and Au/CeO2 Catalysts. Eur. Fuel Cell News 2003, 10, 16–20. [Google Scholar]
- Widmann, D.; Leppelt, R.; Behm, R.J. Activation of Au/CeO2 Catalyst for the CO Oxidation Reaction by Surface Oxygen Removal/Oxygen Vacancy Formation. J. Catal. 2007, 251, 437–442. [Google Scholar] [CrossRef]
- Camellone, M.F.; Fabris, S. Reaction Mechanisms for the CO Oxidation on Au/CeO2 Catalysts: Activity of Substitutional Au3+/Au+ Cations and Deactivation of Supported Au+ Adatoms. J. Am. Chem. Soc. 2009, 131, 10473–10483. [Google Scholar] [CrossRef]
- Zhang, R.-R.; Ren, L.-H.; Lu, A.-H.; Li, W.-C. Influence of Pretreatment Atmospheres on the Activity of Au/CeO2 Catalyst for Low-Temperature CO Oxidation. Catal. Commun. 2011, 13, 18–21. [Google Scholar] [CrossRef]
- Abd El-Moemen, A.; Abdel-Mageed, A.M.; Bansmann, J.; Parlinska-Wojtan, M.; Behm, R.J.; Kucèrová, G. Deactivation of Au/CeO2 Catalysts in the CO Oxidation Reaction: Influence of Pretreatment and Reaction Conditions. J. Catal. 2016, 341, 160–179. [Google Scholar] [CrossRef]
- Longo, A.; Liotta, L.F.; Pantaleo, G.; Giannici, F.; Venezia, A.M.; Martorana, A. Structure of the Metal-Support Interface and Oxidation State of Gold Nanoparticles Supported on Ceria. J. Phys. Chem. C 2012, 116, 2960–2966. [Google Scholar] [CrossRef]
- Wang, Y.-G.; Mei, D.; Glezakou, V.A.; Li, J.; Rousseau, R. Dynamic Formation of Single-atom Catalytic Active sites on Ceria-supported Gold Nanoparticles. Nat. Commun. 2015, 6, 1–7. [Google Scholar] [CrossRef]
- Schilling, C.; Hess, C. Real-Time Observation of the Defect Dynamics in Working Au/CeO2 Catalysts by Combined Operando Raman/UV-Vis Spectroscopy. J. Phys. Chem. C 2018, 122, 2909–2917. [Google Scholar] [CrossRef]
- Guzman, J.; Carrettin, S.; Corma, A. Spectroscopic Evidence for the Supply of Reactive Oxygen during CO Oxidation Catalyzed by Gold Supported on Nanocrystalline CeO2. J. Am. Chem. Soc. 2005, 127, 3286–3287. [Google Scholar] [CrossRef]
- Abdel-Mageed, A.M.; Kucèrová, G.; Abd El-Moemen, A.; Bansmann, J.; Widmann, D.; Behm, R.J. Geometric and Electronic Structure of Au on Au/CeO2 Catalysts During the CO Oxidation: Deactivation by Reaction Induced Particle Growth. J. Phys. Conf. Ser. 2015, 712, 012044. [Google Scholar] [CrossRef]
- Romero-Sarria, F.; Martínez, T.; Centeno, M.A.; Odriozola, J.A. Surface Dynamics of Au/CeO2 Catalysts during CO Oxidation. J. Phys. Chem. C 2007, 111, 14469–14475. [Google Scholar] [CrossRef]
- Chen, S.; Luo, L.; Jiang, Z.; Huang, W. Size-Dependent Reaction Pathways of Low-Temperature CO Oxidation on Au/CeO2 Catalysts. ACS Catal. 2015, 5, 1653–1662. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, W.; Tian, M.; Wang, L.; Wang, Z. A Rational Design of the Sintering-Resistant Au-CeO2 Nanoparticles Catalysts for CO Oxidation: The Influence of H2 Pretreatments. Materials 2018, 11, 1952. [Google Scholar] [CrossRef]
- Schilling, C.; Hess, C. CO Oxidation on Ceria Supported Gold Catalysts Studied by Combined Operando Raman/UV-Vis and IR Spectroscopy. Top. Catal. 2017, 60, 131–140. [Google Scholar] [CrossRef]
- Hernández, J.A.; Gómez, S.A.; Zepeda, T.A.; Fierro-González, J.C.; Fuentes, G.A. Insight into the Deactivation of Au/CeO2 Catalysts Studied by In Situ Spectroscopy during the CO-PROX Reaction. ACS Catal. 2015, 5, 4003–4012. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, P.; Koberstein, J.; Khalid, S.; Chan, S.-W. Cerium Oxidation State in Ceria Nanoparticles Studied with X-ray Photoelecton Spectroscopy and Absorption Near Edge Spectroscopy. Surf. Sci. 2004, 563, 74–82. [Google Scholar] [CrossRef]
- McBride, J.R.; Hass, K.C.; Poindexter, B.D.; Weber, W.H. Raman and X-ray Studies of Ce1-xRexO2-y, where RE=La, Pr, Nd, Eu, Gd, and Tb. J. Appl. Phys. 1994, 76, 2435–2441. [Google Scholar] [CrossRef]
- Lee, Y.; He, G.; Akey, A.J.; Si, R.; Flytzani-Stephanopoulos, M.; Herman, I.P. Raman Analysis of Mode Softening in Nanoparticle CeO2-d and Au-CeO2-d during CO Oxidation. J. Am. Chem. Soc. 2011, 133, 12952–12955. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data Analysis for X-ray Absorption Spectroscopy using IFEFFIT. J. Synchrotron Rad. 2005, 12, 537–541. [Google Scholar] [CrossRef]
- Yoshida, H.; Yuliati, L.; Hamajima, T.; Hattori, T. Valence of Highly Dispersed Cerium Oxide Species on Silica Quantitatively Estimated by Ce LIII-edge XANES. Mater. Transact. 2004, 45, 2062–2067. [Google Scholar] [CrossRef]
- Bansmann, J.; Kucèrová, G.; Abdel-Mageed, A.M.; Abd El-Moemen, A.; Behm, R.J. Influence of Re-Activation and Ongoing CO Oxidation Reaction on the Chemical and Electronic Properties of Au on a Au/CeO2 Catalyst: A XANES Study at the Au LIII edge. J. Electron Spectrosc. Relat. Phenom. 2017, 220, 86–90. [Google Scholar] [CrossRef]
- Widmann, D.; Liu, Y.; Schüth, F.; Behm, R.J. Support Effects in the Au Catalyzed CO Oxidation—Correlation Between Activity, Oxygen Storage capacity and Support Reducibility. J. Catal. 2010, 276, 292–305. [Google Scholar] [CrossRef]
- Widmann, D.; Behm, R.J. Activation of Molecular Oxygen and the Nature of theActive Oxygen Species for CO Oxidation on Oxide Supported Au Catalysts. Acc. Chem. Res. 2014, 47, 740–749. [Google Scholar] [CrossRef]
- Wang, Y.; Widmann, D.; Behm, R.J. Influence of TiO2 Bulk Defects on CO Adsorption and CO Oxidation on Au/TiO2: Electronic Metal-Support Interactions (EMSIs) in Supported Au Catalysts. ACS Catal. 2017, 7, 2339–2345. [Google Scholar] [CrossRef]
- Di Cicco, A.; Aquilanti, G.; Minicucci, M.; Principi, E.; Novello, N.; Cognigni, A.; Olivi, L. Novel XAFS Capabilities at ELETTRA Synchrotron Light Source. J. Phys. Conf. Ser. 2009, 190, 012043. [Google Scholar] [CrossRef]
- Kokoric, V.; Widmann, D.; Wittmann, M.; Behm, R.J.; Mizaikoff, B. Infrared Spectroscopy via Substrate-Integrated Hollow Waveguides: A Powerful Tool in Catalysis Research. Analyst 2016, 141, 5990–5995. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
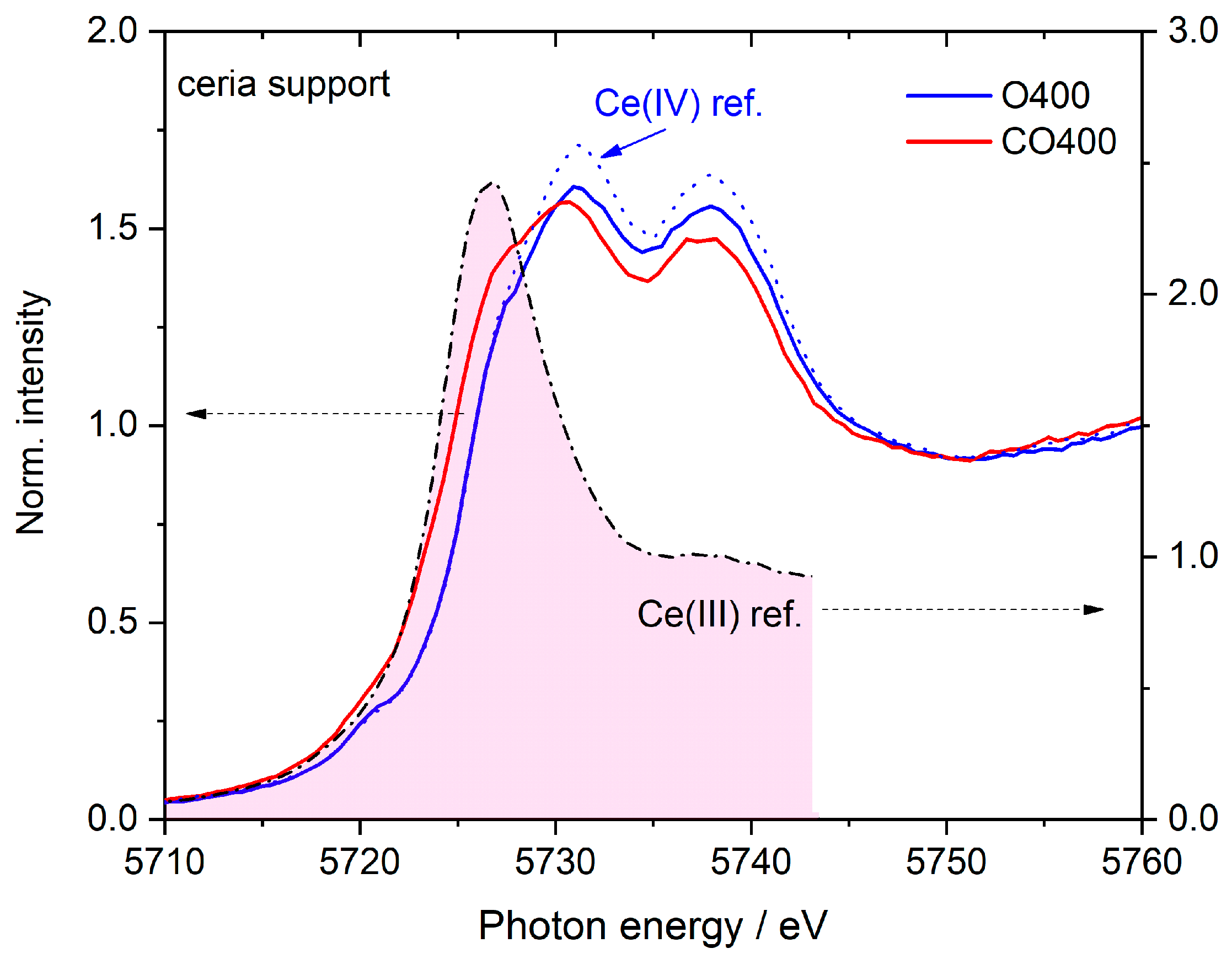
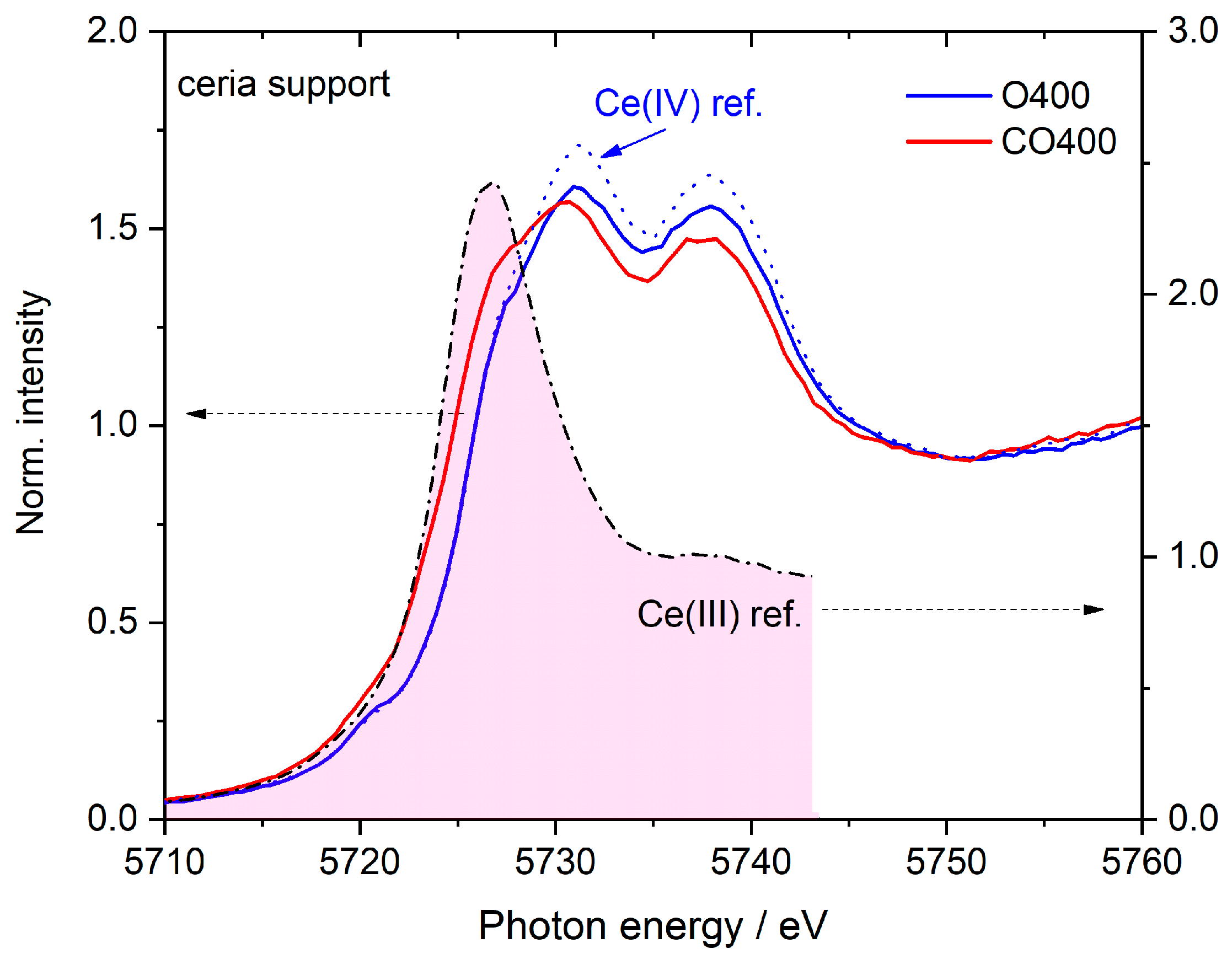
| Peak | A | B0 | B1 | B2 | C1 | C2 |
|---|---|---|---|---|---|---|
| Binding energy/eV | 5720.2 | 5726.1 | 5727.8 | 5730.2 | 5736.0 | 5738.1 |
| Ce(III) reference | 0 | 1.87 | 0 | 0 | 0 | 0 |
| Ce(IV) reference | 0.02 | 0.2 | 0.6 | 0.81 | 0.02 | 0.62 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bansmann, J.; Abdel-Mageed, A.M.; Chen, S.; Fauth, C.; Häring, T.; Kučerová, G.; Wang, Y.; Behm, R.J. Chemical and Electronic Changes of the CeO2 Support during CO Oxidation on Au/CeO2 Catalysts: Time-Resolved Operando XAS at the Ce LIII Edge. Catalysts 2019, 9, 785. https://doi.org/10.3390/catal9100785
Bansmann J, Abdel-Mageed AM, Chen S, Fauth C, Häring T, Kučerová G, Wang Y, Behm RJ. Chemical and Electronic Changes of the CeO2 Support during CO Oxidation on Au/CeO2 Catalysts: Time-Resolved Operando XAS at the Ce LIII Edge. Catalysts. 2019; 9(10):785. https://doi.org/10.3390/catal9100785
Chicago/Turabian StyleBansmann, Joachim, Ali M. Abdel-Mageed, Shilong Chen, Corinna Fauth, Thomas Häring, Gabriela Kučerová, Yuchen Wang, and R. Jürgen Behm. 2019. "Chemical and Electronic Changes of the CeO2 Support during CO Oxidation on Au/CeO2 Catalysts: Time-Resolved Operando XAS at the Ce LIII Edge" Catalysts 9, no. 10: 785. https://doi.org/10.3390/catal9100785
APA StyleBansmann, J., Abdel-Mageed, A. M., Chen, S., Fauth, C., Häring, T., Kučerová, G., Wang, Y., & Behm, R. J. (2019). Chemical and Electronic Changes of the CeO2 Support during CO Oxidation on Au/CeO2 Catalysts: Time-Resolved Operando XAS at the Ce LIII Edge. Catalysts, 9(10), 785. https://doi.org/10.3390/catal9100785