Developing Nickel–Zirconia Co-Precipitated Catalysts for Production of Green Diesel

 , ,
, , 
 and
and
Abstract
1. Introduction
2. Results and Discussion
2.1. Catalyst Preparation
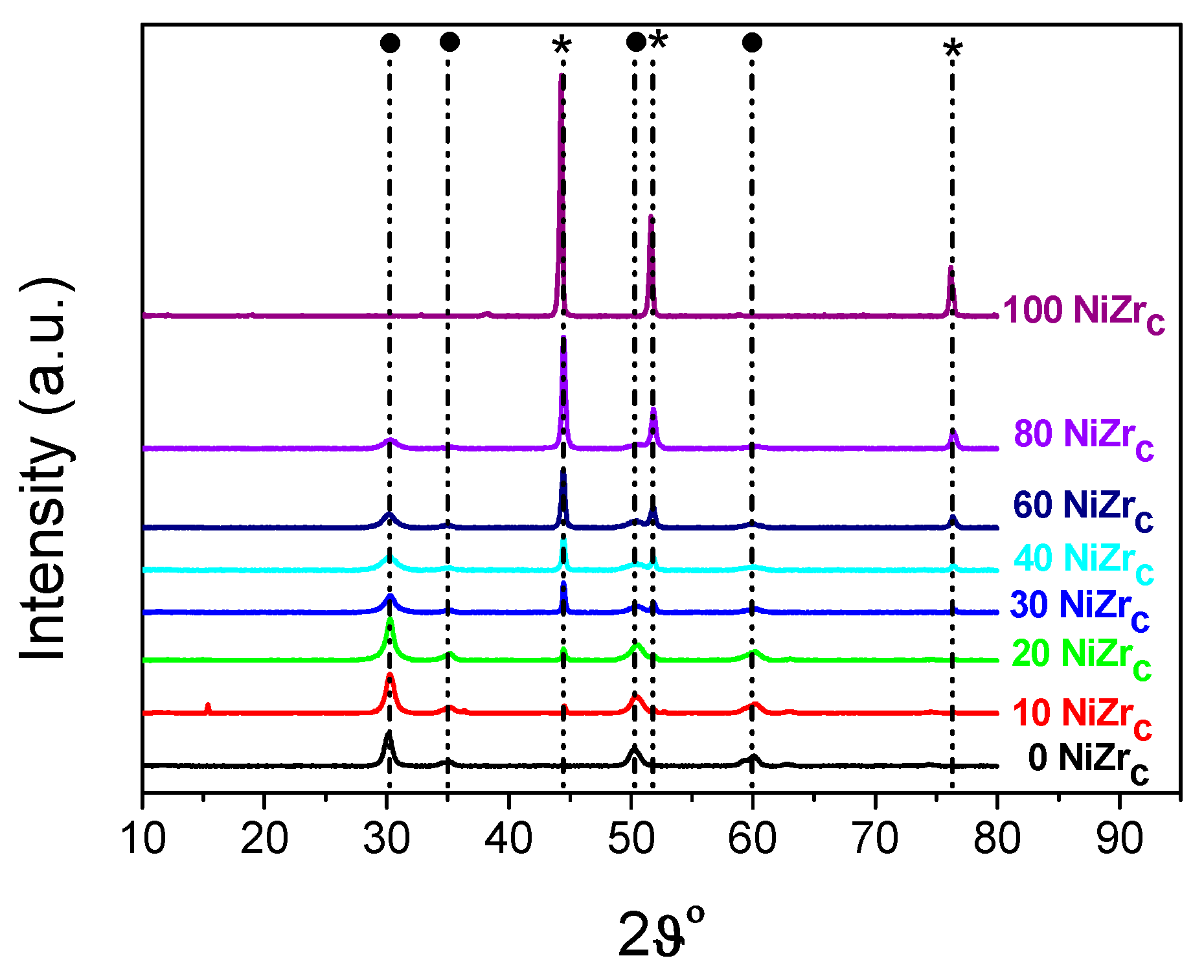
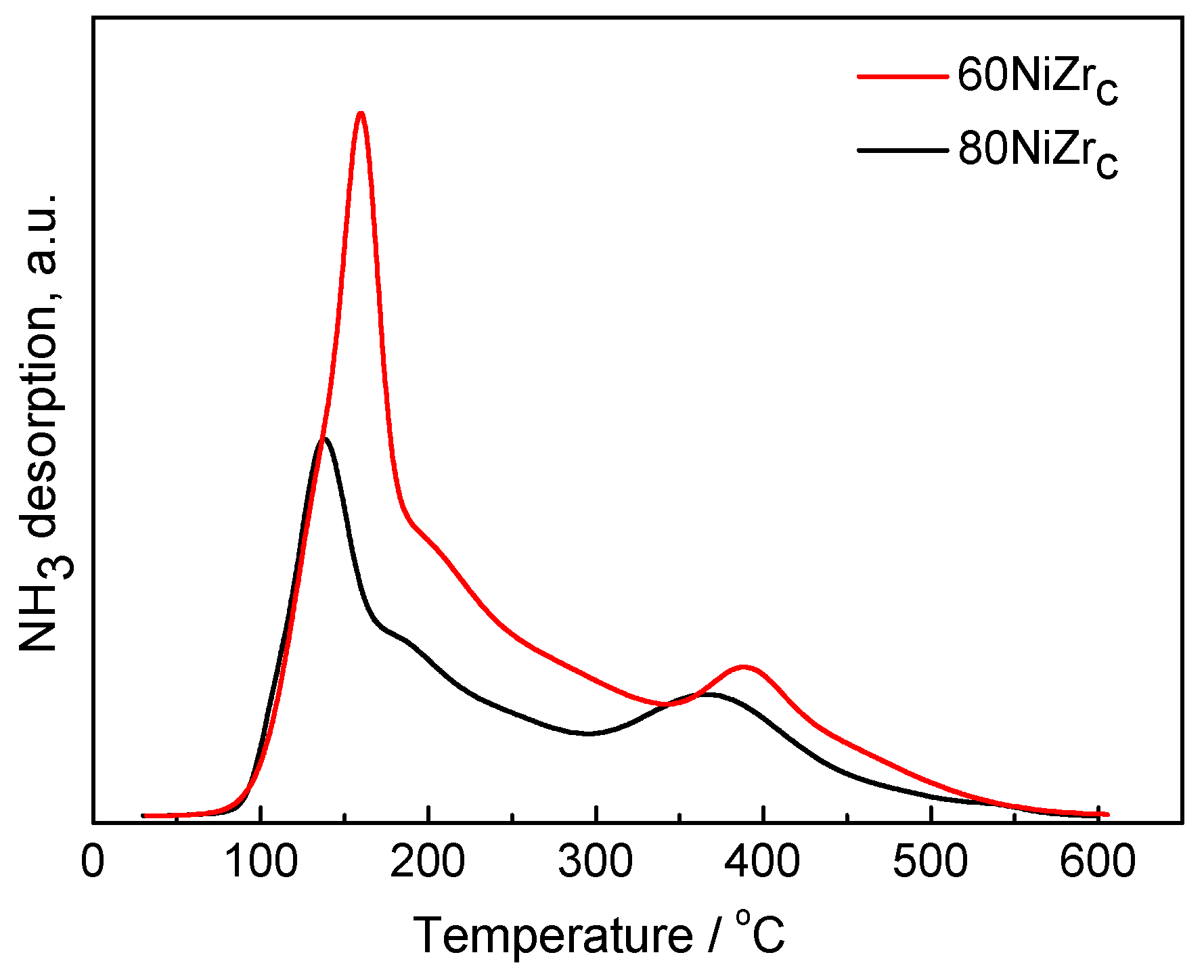
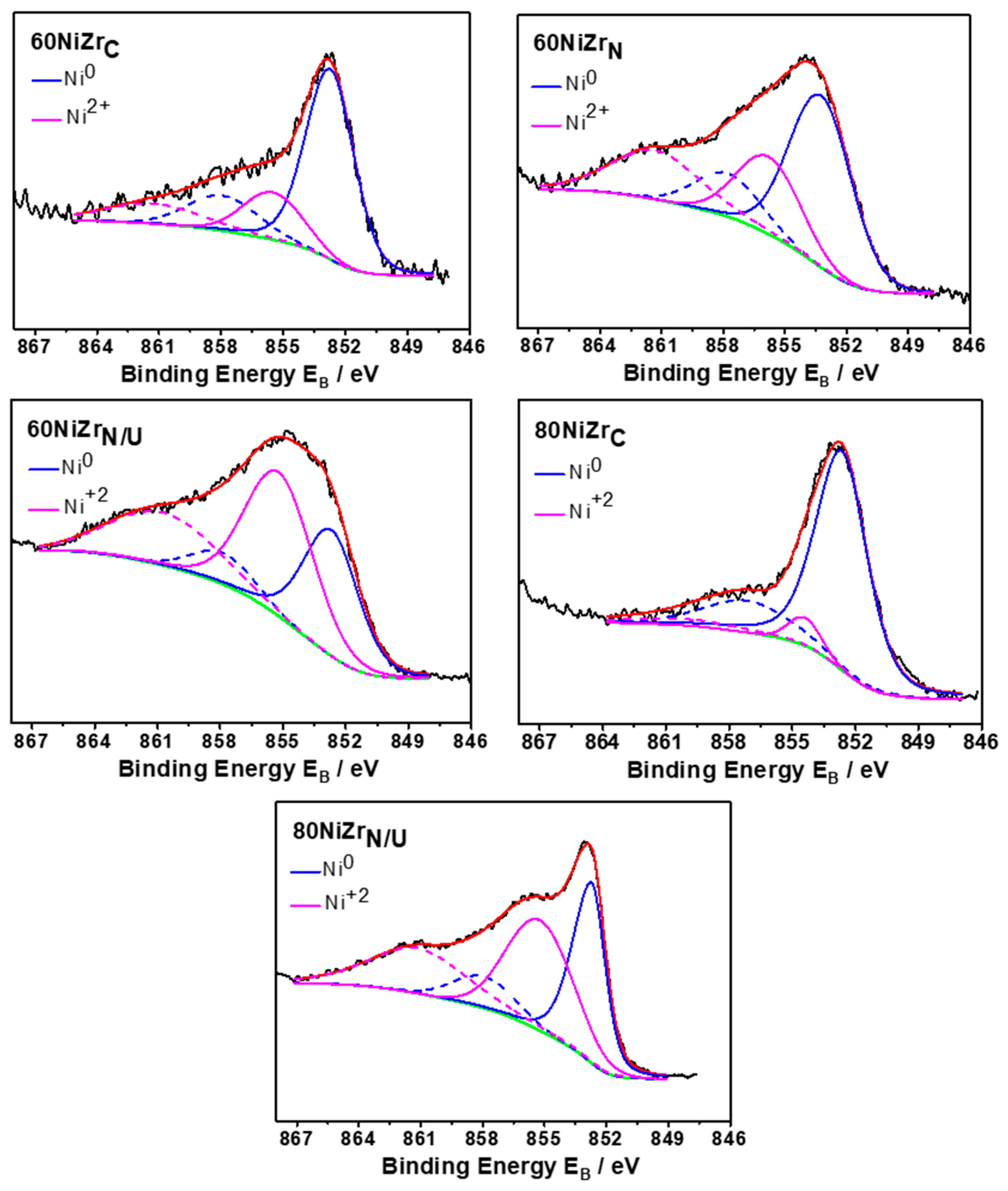
2.2. First Series of Catalysts: Effect of Loading–The Poisoning Action of Chlorine
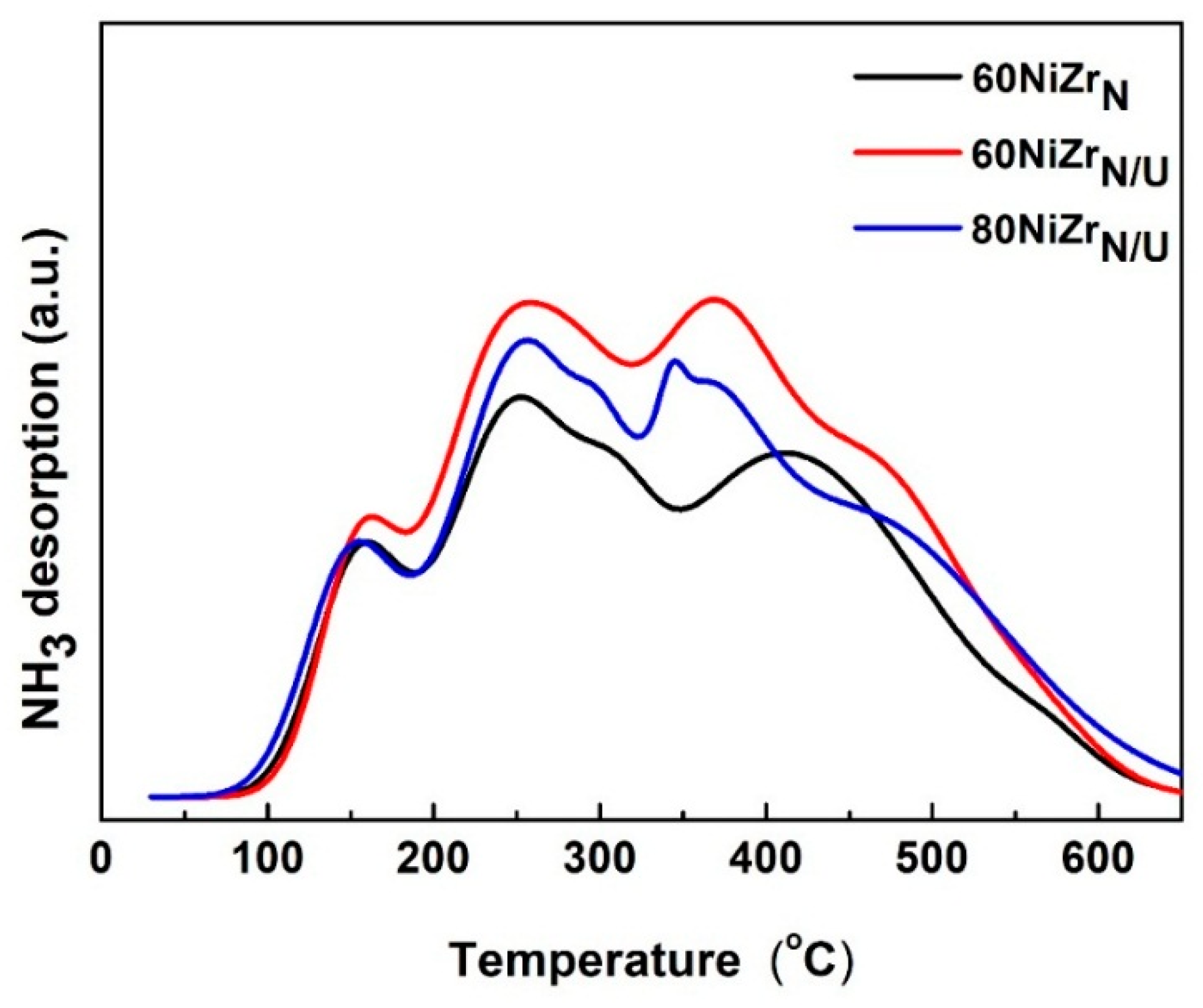
2.3. Second Series of Catalysts: Effect of the Preparation Parameters
3. Experimental Section
3.1. Feedstock
3.2. Preparation of the Catalysts
3.2.1. Materials
3.2.2. Catalyst Preparation
3.2.3. Activation
3.3. Characterization of the Catalysts
3.3.1. Scanning Electron Microscopy
3.3.2. Determination of the Texture
3.3.3. X-ray Diffraction
3.3.4. X-ray Photoelectron Spectroscopy
3.3.5. Temperature Programmed Reduction
3.3.6. Acidity Measurements
3.4. Evaluation of the Catalysts
3.4.1. Catalytic Tests
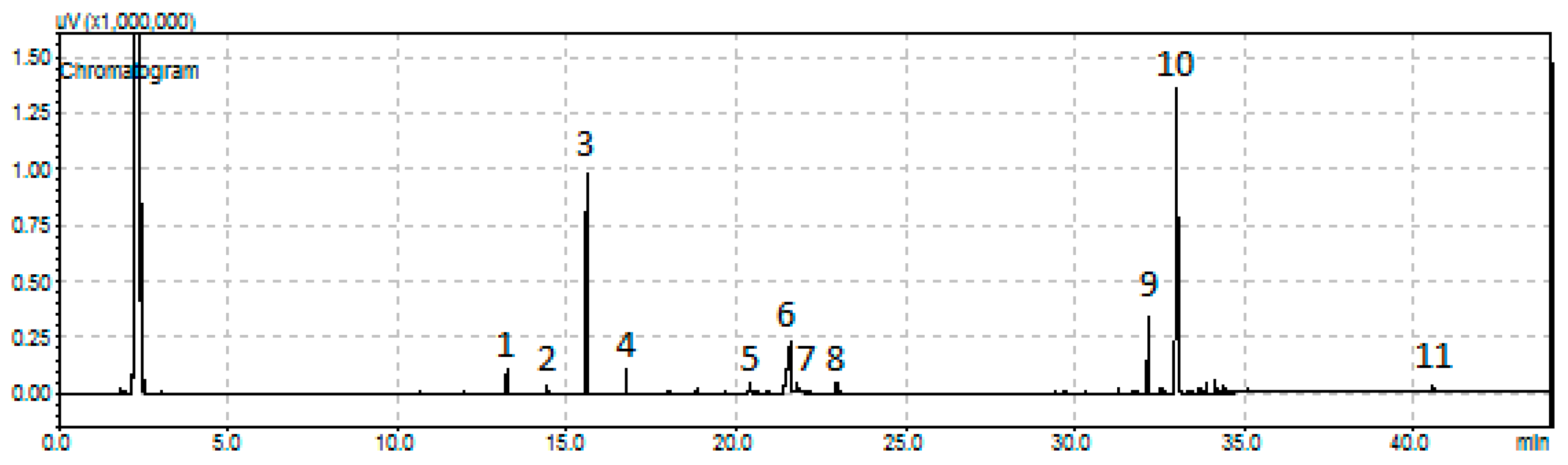
3.4.2. Chromatographic and GC-MS Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Armaroli, N.; Balzani, V. The Future of Energy Supply: Challenges and Opportunities. Angew. Chem. Int. Ed. 2006, 46, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.M.; Harris, K.D.M. Some of tomorrow’s catalysts for processing renewable and non-renewable feedstocks, diminishing anthropogenic carbon dioxide and increasing the production of energy. Energy Environ. Sci. 2016, 9, 687–708. [Google Scholar] [CrossRef]
- Armstrong, R.C.; Wolfram, C.; De Jong, K.P.; Gross, R.; Lewis, N.S.; Boardman, B.; Ragauskas, A.J.; Ehrhardt-Martinez, K.; Crabtree, G.; Ramana, M.V. The frontiers of energy. Nat. Energy 2016, 1, 15020. [Google Scholar] [CrossRef]
- Lourinho, G.; Brito, P. Advanced biodiesel production technologies: Novel developments. Rev. Environ. Sci. Biotechnol. 2015, 14, 287–316. [Google Scholar] [CrossRef]
- Veljković, V.B.; Banković-Ilić, I.B.; Stamenković, O.S. Purification of crude biodiesel obtained by heterogeneously-catalyzed transesterification. Renew. Sustain. Energy Rev. 2015, 49, 500–516. [Google Scholar] [CrossRef]
- Mansir, N.; Taufiq-Yap, Y.H.; Rashid, U.; Lokman, I.M. Investigation of heterogeneous solid acid catalyst performance on low grade feedstocks for biodiesel production: A review. Energy Convers. Manag. 2017, 141, 171–182. [Google Scholar] [CrossRef]
- Abdullah, S.H.Y.S.; Hanapi, N.H.M.; Azid, A.; Umar, R.; Juahir, H.; Khatoon, H.; Endut, A. A review of biomass-derived heterogeneous catalyst for a sustainable biodiesel production. Renew. Sustain. Energy Rev. 2017, 70, 1040–1051. [Google Scholar] [CrossRef]
- Tran, D.T.; Chang, J.S.; Lee, D.J. Recent insights into continuous-flow biodiesel production via catalytic and non-catalytic transesterification processes. Appl. Energy 2017, 185, 376–409. [Google Scholar] [CrossRef]
- Kubickova, I.; Kubicka, D. Utilization of Triglycerides and Related Feedstocks for Production of Clean Hydrocarbon Fuels and Petrochemicals: A Review. Waste Biomass Valoriz. 2010, 1, 293–308. [Google Scholar] [CrossRef]
- Gosselink, R.W.; Hollak, S.A.W.; Chang, S.-W.; van Haveren, J.; de Jong, K.P.; Bitter, J.H.; van Es, D.S. Reaction Pathways for the Deoxygenation of Vegetable Oils and Related Model Compounds. Chem. Sustain. Chem. 2013, 6, 1576–1594. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Brück, T.; Lercher, J.A. Catalytic deoxygenation of microalgae oil to green hydrocarbons. Green Chem. 2013, 15, 1720–1739. [Google Scholar] [CrossRef]
- Patil, S.J.; Vaidya, P.D. On the production of bio-hydrogenated diesel over hydrotalcite-like supported palladium and ruthenium catalysts. Fuel Proc. Technol. 2018, 169, 142–149. [Google Scholar] [CrossRef]
- Li, X.; Luo, X.Y.; Jin, Y.B.; Li, J.Y.; Zhang, H.D.; Zhang, A.P.; Xie, J. Heterogeneous sulfur-free hydrodeoxygenation catalysts for selectively upgrading the renewable bio-oils to second generation biofuels. Renew. Sustain. Energy 2018, 82, 3762–3797. [Google Scholar] [CrossRef]
- Kumar, R.; Farooqui, S.A.; Anand, M.; Kumar, R.; Joshi, R.; Khan, A.; Sinha, A.K. Hydrotreatment of jatropha oil over NiMoS catalyst supported on thermostable mesoporous silica doped titania for the production of renewable drop-in diesel. Catal. Commun. 2017, 98, 102–106. [Google Scholar] [CrossRef]
- Nepomnyashchiy, A.A.; Buluchevskiy, E.A.; Lavrenov, A.V.; Yurpalov, V.L.; Gulyaeva, T.I.; Leont’eva, N.N.; Talzi, V.P. Hydrodeoxygenation of Vegetable Oil on NiMoS/WO3-Al2O3 Catalysts. Russ. J. Appl. Chem. 2017, 90, 1944–1952. [Google Scholar] [CrossRef]
- Coumans, A.E.; Hensen, E.J.M. A model compound (methyl oleate, oleic acid, triolein) study of triglycerides hydrodeoxygenation over alumina-supported NiMo sulfide. Appl. Catal. B Environ. 2017, 201, 290–301. [Google Scholar] [CrossRef]
- Chen, S.Y.; Nishi, M.; Mochizuki, T.; Takagi, H.; Takatsuki, A.; Roschat, W.; Toba, M.; Yoshimura, Y. Co-Processing of Jatropha-Derived Bio-Oil with Petroleum Distillates over Mesoporous CoMo and NiMo Sulfide Catalysts. Catalysts 2018, 8, 59. [Google Scholar] [CrossRef]
- Patil, S.J.; Vaidya, P.D. Production of Hydrotreated Jatropha Oil Using Co-Mo and Ni-Mo Catalysts and Its Blending with Petroleum Diesel. Energy Fuels 2018, 32, 1812–1821. [Google Scholar] [CrossRef]
- Srifa, A.; Viriya-empikul, N.; Assabumrungrat, S.; Faungnawakij, K. Catalytic behaviors of Ni/γ-Al2O3 and Co/γ-Al2O3 during the hydrodeoxygenation of palm oil. Catal. Sci. Technol. 2015, 5, 3693–3705. [Google Scholar] [CrossRef]
- Kordulis, C.; Bourikas, K.; Gousi, M.; Kordouli, E.; Lycourghiotis, A. Development of nickel based catalysts for the transformation of natural triglycerides and related compounds into green diesel: A critical review. Appl. Catal. B Environ. 2016, 181, 156–196. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, G.; Miao, C. Green and renewable bio-diesel produce from oil hydrodeoxygenation: Strategies for catalyst development and mechanism. Renew. Sustain. Energy Rev. 2019, 101, 568–589. [Google Scholar] [CrossRef]
- Gousi, M.; Andriopoulou, C.; Bourikas, K.; Ladas, S.; Sotiriou, M.; Kordulis, C.; Lycourghiotis, A. Green diesel production over nickel-alumina co-precipitated catalysts. Appl. Catal. A Gen. 2017, 536, 45–56. [Google Scholar] [CrossRef]
- Munnik, P.; de Jongh, P.E.; de Jong, K.P. Recent Developments in the Synthesis of Supported Catalysts. Chem. Rev. 2015, 115, 6687–6718. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Yuan, X.; Zhao, C.; Lercher, J.A. Stabilizing Catalytic Pathways via Redundancy: Selective Reduction of Microalgae Oil to Alkanes. J. Am. Chem. Soc. 2012, 134, 9400–9405. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Zhao, C.; Kasakov, S.; Foraita, S.; Lercher, J.A. Manipulating Catalytic Pathways: Deoxygenation of Palmitic Acid on Multifunctional Catalysts. Chem. Eur. J. 2013, 19, 4732–4741. [Google Scholar] [CrossRef] [PubMed]
- Foraita, S.; Fulton, J.L.; Chase, Z.A.; Vjunov, A.; Xu, P.; Barath, E.; Camaioni, D.M.; Zhao, C.; Lercher, J.A. Impact of the Oxygen Defects and the Hydrogen Concentration on the Surface of Tetragonal and Monoclinic ZrO2 on the Reduction Rates of Stearic Acid on Ni/ZrO2. Chem. Eur. J. 2015, 21, 2423–2434. [Google Scholar] [CrossRef] [PubMed]
- Foraita, S.; Liu, Y.; Haller, G.L.; Barath, E.; Zhao, C.; Lercher, J.A. Controlling Hydrodeoxygenation of Stearic Acid to n-Heptadecane and n-Octadecane by Adjusting the Chemical Properties of Ni/SiO2–ZrO2 Catalyst. ChemCatChem 2017, 9, 195–203. [Google Scholar] [CrossRef]
- Yakovlev, V.A.; Khromova, S.A.; Sherstyuk, O.V.; Dundich, V.O.; Ermakov, D.Y.; Novopashina, V.M.; Lebedev, M.Y.; Bulavchenko, O.; Parmon, V.N. Development of new catalytic systems for upgraded bio-fuels production from bio-crude-oil and biodiesel. Catal. Today 2009, 144, 362–366. [Google Scholar] [CrossRef]
- Dundich, V.O.; Khromova, S.A.; Ermakov, D.Y.; Lebedev, M.Y.; Novopashina, V.M.; Sister, V.G.; Yakimchuk, A.I.; Yakovlev, V.A. Nickel catalysts for the hydrodeoxygenation of biodiesel. Kinet. Catal. 2010, 51, 704–709. [Google Scholar] [CrossRef]
- Goula, M.A.; Charisiou, N.D.; Siakavelas, G.; Tzounis, L.; Tsiaoussis, I.; Panagiotopoulou, P.; Goula, G.; Yentekakis, I.V. Syngas production via the biogas dry reforming reaction over Ni supported on zirconia modified with CeO2 or La2O3 catalysts. Int. J. Hydrogen Energy 2017, 42, 13724–13740. [Google Scholar] [CrossRef]
- Basahel, S.N.; Ali, T.T.; Mokhtar, M.; Narasimharao, K. Influence of crystal structure of nanosized ZrO2 on photocatalytic degradation of methyl orange. Nanoscale Res. Lett. 2015, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Liu, W.-Q.; Liu, Y.-A.; Etim, U.J.; Liu, X.-M.; Yan, Z.-F. Pore confinement effect of MoO3/Al2O3 catalyst for deep hydrodesulfurization. Chem. Eng. J. 2017, 330, 706–717. [Google Scholar] [CrossRef]
- Wijarnprecha, K.; Aryusuk, K.; Santiwattana, P.; Sonwai, S.; Rousseau, D. Structure and rheology of oleogels made from rice bran wax and rice bran oil. Food Res. Int. 2018, 112, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Das, U.K.; Ben-David, Y.; Leitus, G.; Diskin-Posner, Y.; Milstein, D. Dehydrogenative Cross-Coupling of Primary Alcohols to Form Cross-Esters Catalyzed by a Manganese Pincer Complex. ACS Catal. 2019, 9, 479–484. [Google Scholar] [CrossRef]
- Zhong, Y.; Deng, Q.; Zhang, P.; Wang, J.; Wang, R.; Zeng, Z.; Deng, S. Sulfonic acid functionalized hydrophobic mesoporous biochar: Design, preparation and acid-catalytic properties. Fuel 2019, 240, 270–277. [Google Scholar] [CrossRef]
- Park, B.-G.; Chung, K.-H. Catalytic properties of microporous zeolites in the catalytic cracking of m-diisopropylbenzene. Mol. Catal. 2019, 461, 80–85. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Papageridis, K.N.; Siakavelas, G.; Sebastian, V.; Hinder, S.J.; Baker, M.A.; Polychronopoulou, K.; Goula, M.A. The influence of SiO2 doping on the Ni/ZrO2 supported catalyst for hydrogen production through the glycerol steam reforming reaction. Catal. Today 2019, 319, 206–219. [Google Scholar] [CrossRef]
- Bespalov, I.; Datler, M.; Buhr, S.; Drachsel, W.; Rupprechter, G.; Suchorski, Y. Initial stages of oxide formation on the Zr surface at low oxygen pressure: An in situ FIM and XPS study. Ultramicroscopy 2015, 159, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.N. Characterization of NiO by XPS. Surf. Sci. Spectra 1994, 3, 231. [Google Scholar] [CrossRef]
- Mansour, A.N.; Melendres, C.A. Characterization of α-Ni(OH)2 by XPS. Surf. Sci. Spectra 1994, 3, 255. [Google Scholar] [CrossRef]
- Kordouli, E.; Sygellou, L.; Kordulis, C.; Bourikas, K.; Lycourghiotis, A. Probing the synergistic ratio of the NiMo/γ-Al2O3 reduced catalysts for the transformation of natural triglycerides into green diesel. Appl. Catal. B Environ. 2017, 209, 12–22. [Google Scholar] [CrossRef]
- Raikwar, D.; Munagala, M.; Majumdar, S.; Shee, D. Hydrodeoxygenation of guaiacol over Mo, W and Ta modified supported nickel catalysts. Catal. Today 2019, 325, 117–130. [Google Scholar] [CrossRef]
- Kordouli, E.; Pawelec, B.; Kordulis, C.; Lycourghiotis, A.; Fierro, J.L.G. Hydrodeoxygenation of phenol on bifunctional Ni-based catalysts: Effects of Mo promotion and support. Appl. Catal. B Environ. 2018, 238, 147–160. [Google Scholar] [CrossRef]
- Tribalis, A.; Panagiotou, G.D.; Bourikas, K.; Sygellou, L.; Kennou, S.; Ladas, S.; Lycourghiotis, A.; Kordulis, C. Ni Catalysts Supported on Modified Alumina for Diesel Steam Reforming. Catalysts 2016, 6, 11. [Google Scholar] [CrossRef]
- Kordouli, E.; Pawelec, B.; Bourikas, K.; Kordulis, C.; Fierro, J.L.G.; Lycourghiotis, A. Mo promoted Ni-Al2O3 co-precipitated catalysts for green diesel production. Appl. Catal. B Environ. 2018, 229, 139–154. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalysts | SSA (m2/g) | PV (cm3/g) | dp (nm) | Nid (nm) | ZrO2d (nm) | NH3 desorbed (a.u.) × 104 |
|---|---|---|---|---|---|---|---|
| 1 | 0NiZrC | 131 | 0.10 | 3.0 | - | 11.6 | - |
| 2 | 10NiZrC | 84 | 0.07 | 3.0 | 25.1 | 10.4 | - |
| 3 | 20NiZrC | 99 | 0.05 | 2.9 | 27.4 | 11.5 | - |
| 4 | 30NiZrC | 81 | 0.03 | 2.8 | 29.3 | 8.9 | - |
| 5 | 40NiZrC | 68 | 0.05 | 4.2 | 30.2 | 6.2 | - |
| 6 | 60NiZrC | 84 | 0.16 | 7.3 | 25.5 | 7.0 | 5.4 |
| 7 | 80NiZrC | 61 | 0.28 | 15.3 | 27.1 | 6.8 | 3.5 |
| 8 | 100NiZrC | 4 | 0.01 | 16.9 | 31.1 | - | - |
| 9 | 60NiZrN | 60 | 0.17 | 8.4 | 21.9 | 7.2 | 1.6 |
| 10 | 60NiZrN/U | 107 | 0.32 | 10.3 | 21.5 | 6.2 | 2.1 |
| 11 | 80NiZrN/U | 63 | 0.49 | 28.8 | 17.5 | - | 1.9 |
| Catalyst | % wt Cl Content |
|---|---|
| 0NiZrC | 1.91 |
| 10NiZrC | 1.81 |
| 20NiZrC | 1.70 |
| 60NiZrC | 1.44 |
| 80NiZrC | 1.05 |
| Catalyst | Surface Atomic Composition (atoms %) | ||
|---|---|---|---|
| Ni | Zr | O | |
| 60NiZrC | 8 | 30 | 62 |
| 60NiZrN | 16 | 24 | 60 |
| 60NiZrN/U | 26 | 21 | 53 |
| 80NiZrC | 25 | 27 | 48 |
| 80NiZrN/U | 36 | 16 | 48 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zafeiropoulos, G.; Nikolopoulos, N.; Kordouli, E.; Sygellou, L.; Bourikas, K.; Kordulis, C.; Lycourghiotis, A. Developing Nickel–Zirconia Co-Precipitated Catalysts for Production of Green Diesel. Catalysts 2019, 9, 210. https://doi.org/10.3390/catal9030210
Zafeiropoulos G, Nikolopoulos N, Kordouli E, Sygellou L, Bourikas K, Kordulis C, Lycourghiotis A. Developing Nickel–Zirconia Co-Precipitated Catalysts for Production of Green Diesel. Catalysts. 2019; 9(3):210. https://doi.org/10.3390/catal9030210
Chicago/Turabian StyleZafeiropoulos, Georgios, Nikolaos Nikolopoulos, Eleana Kordouli, Labrini Sygellou, Kyriakos Bourikas, Christos Kordulis, and Alexis Lycourghiotis. 2019. "Developing Nickel–Zirconia Co-Precipitated Catalysts for Production of Green Diesel" Catalysts 9, no. 3: 210. https://doi.org/10.3390/catal9030210
APA StyleZafeiropoulos, G., Nikolopoulos, N., Kordouli, E., Sygellou, L., Bourikas, K., Kordulis, C., & Lycourghiotis, A. (2019). Developing Nickel–Zirconia Co-Precipitated Catalysts for Production of Green Diesel. Catalysts, 9(3), 210. https://doi.org/10.3390/catal9030210