Abstract
Three new tetracopper(II) coordination compounds were easily generated from Cu(NO3)2, a trifunctional aminoalcohol sulfonic acid (H3bes, N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid) as a principal building block, and a benzene carboxylic acid as a supporting ligand (i.e., benzoic (Hba), 4-hydroxybenzoic (Hfba), or 3-hydroxybenzoic (Hthba) acid). The obtained microcrystalline products, [Cu4(µ-Hbes)3(µ-H2bes)(µ-L)]·2H2O (L = ba− (1), fhba− (2), and thba− (3)), were fully characterized by FTIR (Fourier-transform infrared spectroscopy), elemental analysis, ESI-MS (Electrospray Ionisation Mass Spectrometry), and single-crystal X-ray diffraction methods. Compounds 1–3 were applied as effective homogeneous catalysts in the oxidative C−H functionalization of alkanes (cycloalkanes and propane). Two different model reactions were explored: (1) mild oxidation of alkanes with hydrogen peroxide to give alcohols and ketones, and (2) mild carboxylation of alkanes with carbon monoxide, water, and potassium peroxodisulfate to give carboxylic acids. For these reactions, effects of different parameters, as well as mechanistic and selectivity characteristics, were studied.
1. Introduction
The search for active catalytic systems and novel protocols toward mild C–H functionalization of saturated hydrocarbons represents a challenging research direction with relevance to areas of homogeneous and heterogeneous catalysis, as well as coordination and organic chemistry [1,2,3,4,5,6,7,8,9,10,11,12]. Alkanes represent a class of compounds that are particularly attractive for the synthesis of industrially valuable products [1,2,3]. However, these naturally abundant carbon raw materials have limitations on their synthetic use due to the high inertness. Thus, saturated hydrocarbons are mainly converted, via energy-demanding processes, into more reactive intermediates (e.g., olefins, syngas) for subsequent large-scale organic synthesis [1,2,3].
Considering these factors, there is a growing interest for the development of effective and single-step protocols for the oxidative transformation of saturated hydrocarbons, including the tuning of reaction conditions and the selection of appropriate oxidizing agents and metal catalysts [4,5,6,7,8,9,10,11,12,13,14,15,16,17,18]. In particular, Cu-based coordination compounds represent an interesting class of catalysts capable of functionalizing diverse C–H bonds [19,20,21,22], including those in rather inert substrates such as alkanes [1,8,20,21,22,23,24,25,26,27,28]. In fact, various copper enzymes are known to act as biocatalysts for the oxidative functionalization of organic substrates [20,21,22]. Although a plethora of bioinspired copper systems were tested for the oxidation of hydrocarbons, there are still a few drawbacks concerning their catalytic application, including the design and preparation of complex and costly ligands, among others [8,20,21,22].
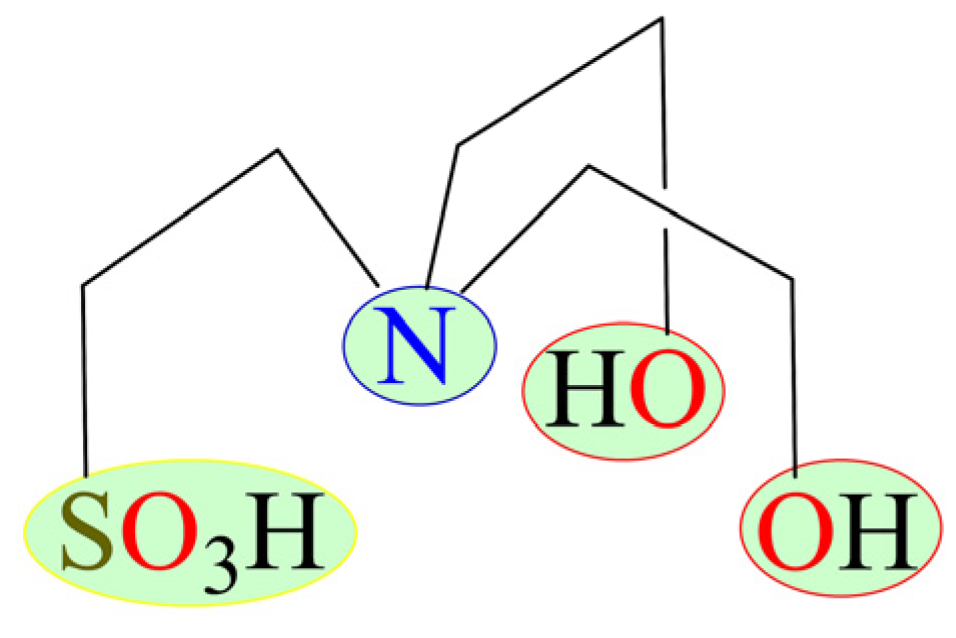
An interesting approach regarding the preparation of Cu-complex catalysts consists of the usage of commercially available, water-soluble, nontoxic, and multifunctional ligands. One very interesting example of such ligands concerns a trifunctional aminoalcohol sulfonic acid, namely N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid (H3bes, Scheme 1). Although H3bes is a very common biobuffer in biochemical research [29,30], its use in catalysis and coordination chemistry remains little explored [31,32,33]. Due to its structure with three different functionalities (tertiary amine, alcohol, and sulfonic acid groups), versatile multidentate nature, stability, and aqueous solubility, we selected H3bes as a main building block in the present work.
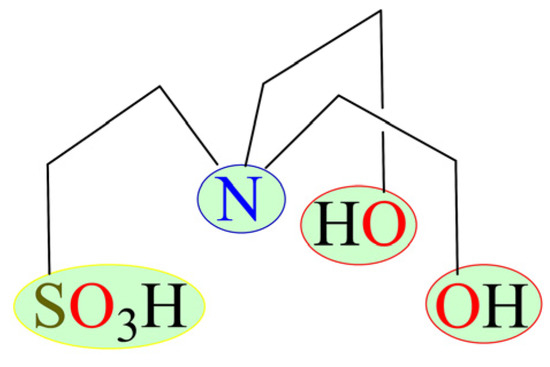
Scheme 1.
Formula of N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid (H3bes) highlighting three types of functional groups (−N, −OH, −SO3H).
Hence, the principal goals of this study consisted of (A) the preparation and characterization of new multicopper(II) coordination compounds using H3bes as a primary N,O-ligand source, and (B) the catalytic application of the obtained compounds in oxidation and carboxylation of alkanes (cycloalkanes and propane) to form value-added products (alcohols, ketones, carboxylic acids). Herein we report self-assembly generation, full characterization, X-ray crystal structures, and application in homogeneous catalysis of three novel tetracopper coordination compounds, namely [Cu4(µ-Hbes)3(µ-H2bes)(µ-ba)]·2H2O (1), [Cu4(µ-Hbes)3(µ-H2bes)(µ-fhba)]·2H2O (2), and [Cu4(µ-Hbes)3(µ-H2bes)(µ-thba)]·2H2O (3), derived from H3bes and various benzene carboxylic acids (benzoic (Hba), 4-hydroxybenzoic (Hfba), or 3-hydroxybenzoic (Hthba) acid). The effects of different reaction parameters, as well as selectivity and mechanistic features, were studied and discussed in detail.
2. Results and Discussion
2.1. Synthesis of Compounds 1–3
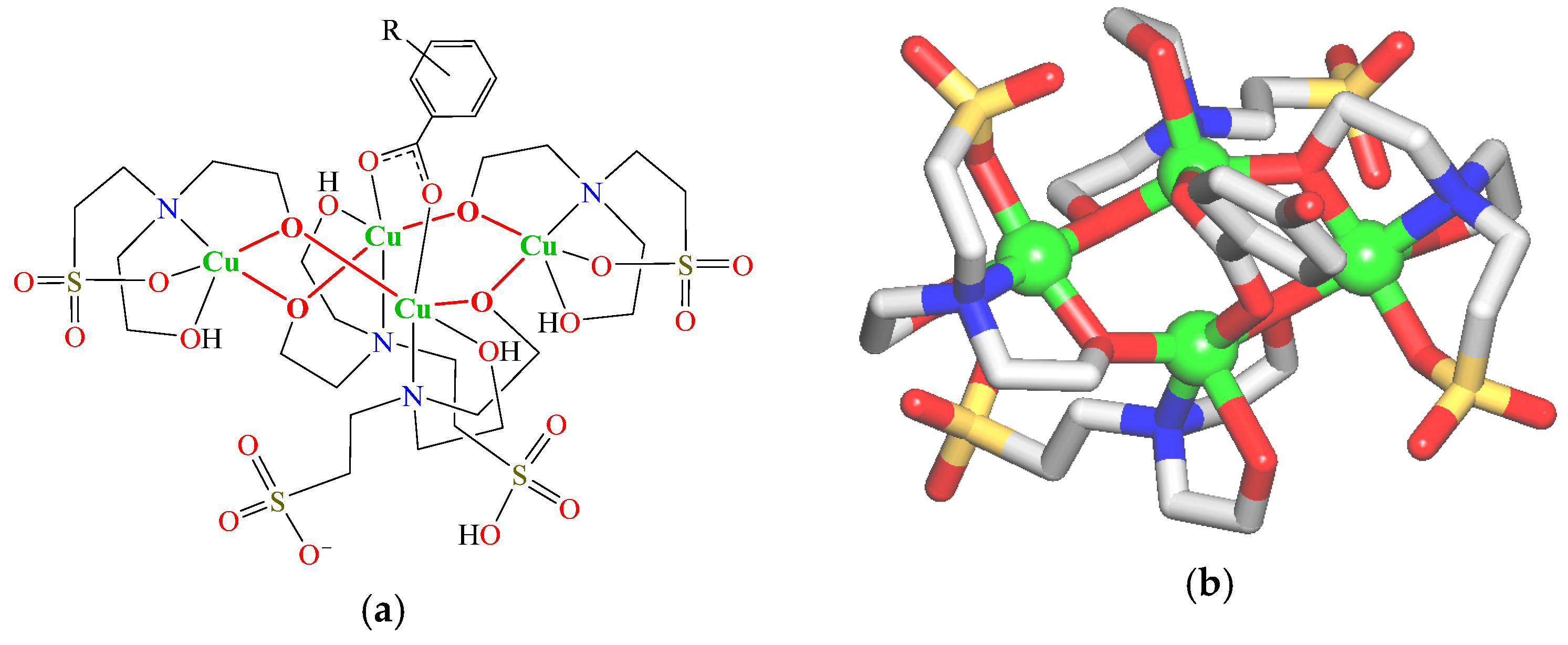
Tetracopper(II) complexes 1–3 were obtained using a self-assembly synthetic procedure, in H2O/MeCN at ~25 °C and under aerobic conditions. This method consists of the combination of copper nitrate(II) with H3bes as a principal building block and benzoic acid (Hba, Hfhba, or Hthba for 1–3, respectively) as a supporting ligand, and ammonia for alkalization of the obtained reaction mixture. This led to a generation, via a self-assembly process, of discrete complexes [Cu4(µ-Hbes)3(µ-H2bes)(µ-ba)]·2H2O (1), [Cu4(µ-Hbes)3(µ-H2bes)(µ-fhba)]·2H2O (2), and [Cu4(µ-Hbes)3(µ-H2bes)(µ-thba)]·2H2O (3), which slowly crystallized from the reaction mixture. These compounds were obtained as green microcrystals (including those of X-ray quality) and then analyzed by elemental analysis, ESI-MS, FTIR spectroscopy, and single-crystal X-ray diffraction (Figure 1 and Figure S1, Supplementary Materials). Discussion of IR and ESI-MS data is given in the Supplementary Materials (Figures S2−S7).
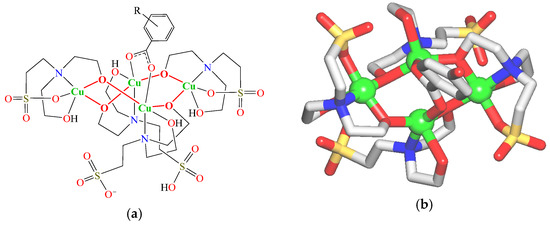
Figure 1.
(a) General formula of 1–3; R = H (1), 4-OH (2), and 3-OH (3). (b) Crystal structure of 2 (representative example); H atoms are omitted for clarity; Cu (green balls), O (red), N (blue), S (yellow), C (gray).
2.2. Structural Description
Crystal structures of 1–3 are essentially similar and are composed of the neutral discrete tetracopper(II) molecular units with a general formula [Cu4(µ-Hbes)3(µ-H2bes)(µ-L)] (L = ba− (1), fhba− (2), and thba− (3)) (Figure 1a and Figure S1, Supplementary Materials). As an example, the structure of compound 2 is described in detail (Figure 1b).
In 2, a tetracopper(II) unit is composed of two “central” Cu1/Cu3 and two “side” Cu2/Cu4 atoms, four bridging aminoalcoholate sulfonate ligands (three µ-Hbes2− and one µ-H2bes−), and one µ-4-hydroxybenzoate− linker. The “central” Cu1/Cu3 atoms are five-coordinate atoms and possess a distorted CuNO4 square-pyramidal environment, which is taken by three oxygen and one nitrogen donors of the µ-Hbes2−/µ-H2bes− moieties (Cu–O 1.931(7)–2.333(9), Cu–N 2.063(9)–2.098(11) Å) and an O atom of µ-fhba− linker (Cu–O 2.001(9)–2.029(9) Å). The “side” Cu2/Cu4 centers are also five-coordinate atoms and feature a distorted CuNO4 square-pyramidal geometry. It is formed by four oxygen and one nitrogen donors of µ-Hbes2− ligands (Cu–O 1.899(7)–2.336(8), Cu–N 2.003(9)–2.062(9) Å). These distances are comparable to those reported for related types of Cu derivatives [31,32,33]. The carboxylate group of the µ-fhba− linker adopts a bridging bidentate mode and interconnects the Cu1/Cu3 centers with a Cu1∙∙∙Cu3 separation of 3.054(2) Å. The µ-Hbes2−/µ-H2bes− moieties that act as chelating ligands to the “central” Cu1/Cu3 atoms have a sulfonic acid group uncoordinated, whereas the µ-Hbes2− ligands that chelate to the “side” Cu2 atoms have all “arms” coordinated.
Within the Cu4(µ-Hbes)3(µ-H2bes)(µ-fhba) molecular unit, the Cu centers are arranged into a cyclic Cu4(µ-O)4(µ-COO) core with a longest Cu2∙∙∙Cu4 separation of 5.876(2) Å. The Cu4 unit is additionally stabilized by means of the O–H∙∙∙O intramolecular hydrogen bonds between –SO3H or –OH functionalities of µ-Hbes2−/µ-H2bes− and the O atoms of adjacent aminoalcoholate moieties. Furthermore, the intermolecular H-bonds between the Cu4(µ-Hbes)3(µ-H2bes)(µ-fhba) units and water molecules of crystallization give rise to a structure extension (zero dimensions to three dimensions (0D→3D)) forming a 3D H-bonded network.
2.3. Mild Catalytic Oxidation of Cycloalkanes
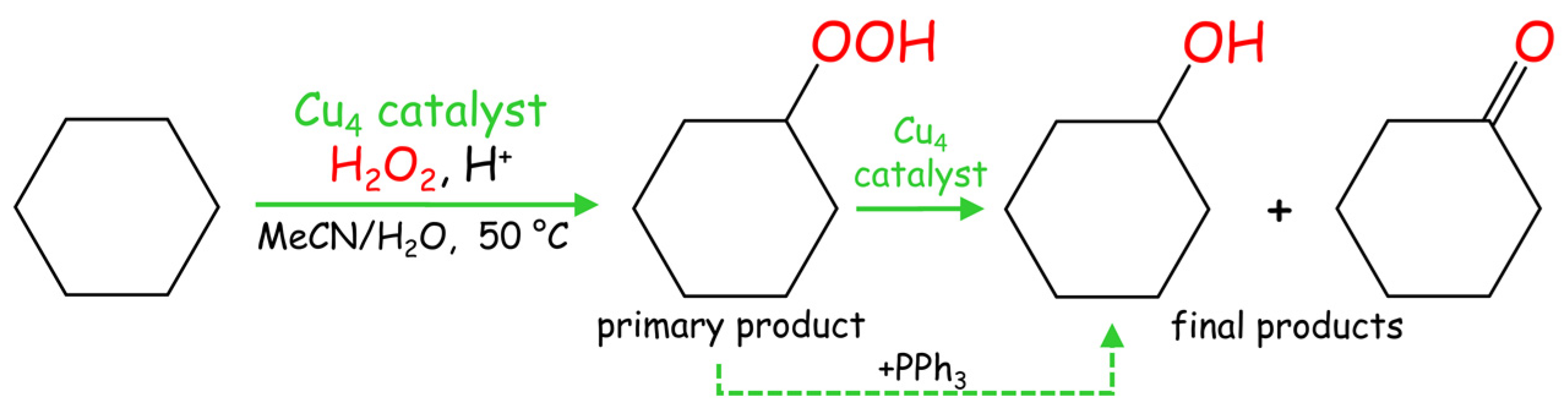
In pursuit of our prior research on alkane functionalization [23,24,25,26,32,33,34,35,36,37], we tested the obtained tetracopper(II) complexes 1–3 as catalysts in the homogeneous oxidation of cyclohexane and related cycloalkanes to generate cycloalkyl hydroperoxides as primary products and then alcohols and ketones as final products (Scheme 2).
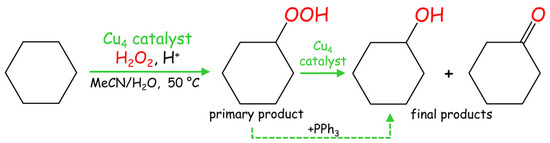
Scheme 2.
Mild Cu-catalyzed oxidation of cyclohexane (model cycloalkane substrate).
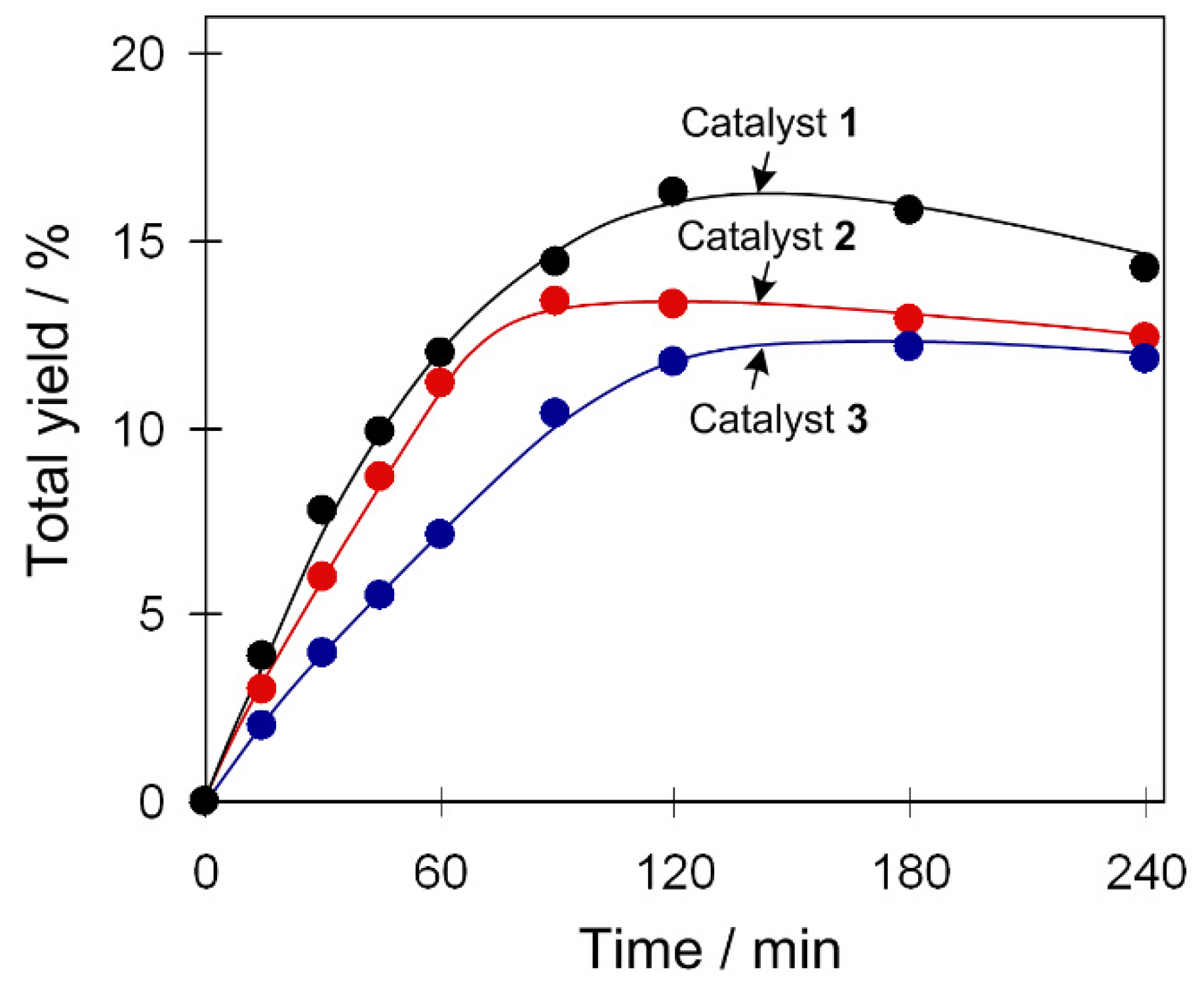
The oxidation of C6H12 was studied as a model reaction due to the relevance of cyclohexanol and cyclohexanone to nylon production [38,39,40,41]. Typical reactions were performed in acetonitrile/water medium at 50 °C and under atmospheric pressure, using hydrogen peroxide (50% in water) as a green oxidant and a small quantity of trifluoroacetic acid (TFA) as a promoter [13,14,15]. Compounds 1–3 catalyze the cyclohexane oxidation with hydrogen peroxide, resulting in the 12–16% total yields of cyclohexanol and cyclohexanone as final products (Figure 2). Hereinafter, the product yields are relative to substrate ((moles of alcohol and ketone per mol of substrate) × 100%), and were obtained after the treatment of the reaction mixtures with triphenylphosphine for reduction of peroxides (cyclohexyl hydroperoxide to cyclohexanol and H2O2 to water). The formation of cyclohexyl hydroperoxide as a primary (intermediate) product was confirmed following a method developed by Shul’pin [5,15], which is based on the GC (gas chromatography) analysis of the reaction mixtures before and after the treatment with solid PPh3. The maximum yield is attained after 90–120 min of the reaction. In some cases, a slight yield drop was observed due to an overoxidation that might occur at a prolonged reaction time. Influence of different reaction parameters was studied and is described below.
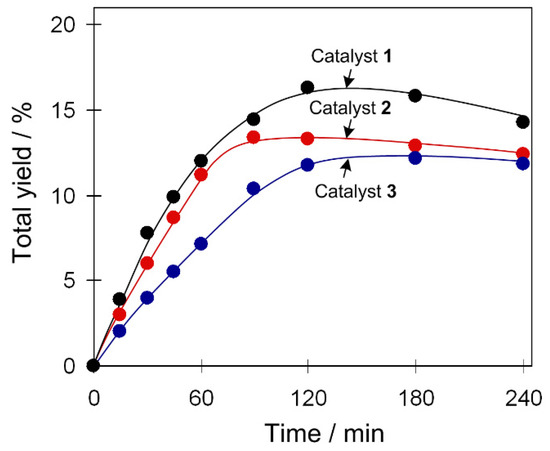
Figure 2.
Accumulation of products (cyclohexanol and cyclohexanone total yield) in Cu-catalyzed C6H12 oxidation with H2O2. Conditions: catalyst 1–3 (2.5 µmol), C6H12 (1 mmol), trifluoroacetic acid (TFA; 50 µmol), H2O2 (5 mmol), CH3CN (up to 2.5 mL of total volume), 50 °C.
2.3.1. Acid Promoter Effect
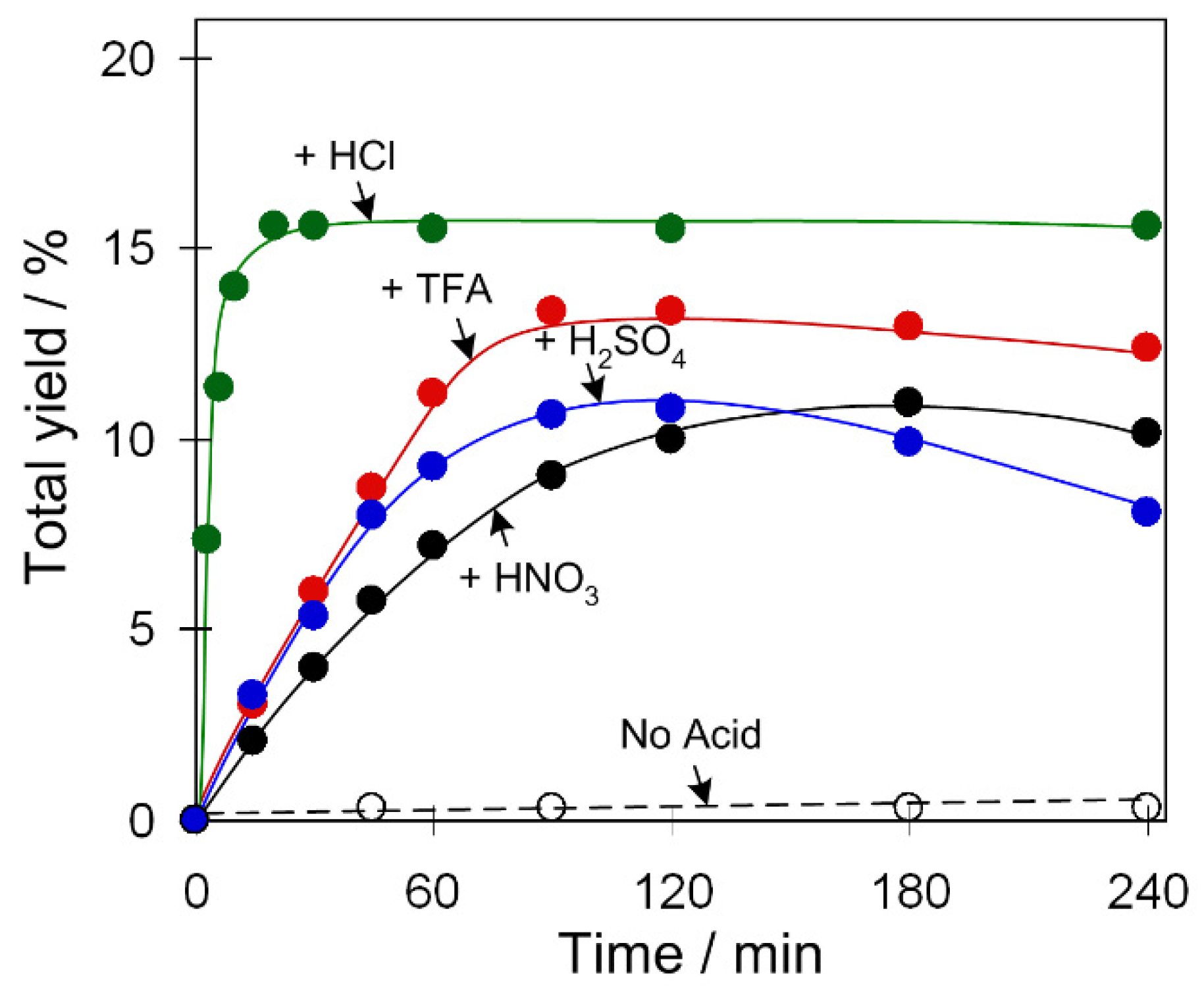
Since the Cu-catalyzed oxidation reactions require a minor quantity of an acid promoter, we tested the promoting effect of various acids, namely trifluoroacetic (TFA), nitric, sulfuric, or hydrochloric acids (Figure 3 and Figure S8, Supplementary Materials). According to a literature background [15,40], the function of an acid promoter might consist of the following: (A) activation of catalyst via unsaturation of copper centers by protonation of ligands; (B) facilitation of proton transfer steps; (C) increase of the reaction rate of oxidation; (D) facilitation of the formation of peroxo complexes; (E) increase of hydrogen peroxide reactivity; (F) suppressing a decomposition of H2O2 to H2O and O2 (eventual catalase activity of catalyst can be suppressed in the presence of acid).
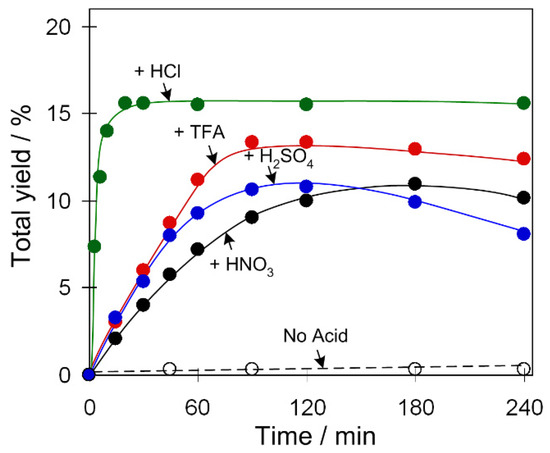
Figure 3.
Effect of acid promoter type on the total yield of products (cyclohexanol and cyclohexanone) in C6H12 oxidation with H2O2. Conditions: catalyst 2 (2.5 µmol), C6H12 (1 mmol), acid (50 µmol), H2O2 (5 mmol), CH3CN solvent (up to 2.5 mL of the total reaction volume), 50 °C.
Kinetic curves of product accumulation in the cyclohexane oxidation catalyzed by 2 in the systems containing various acid promoters are shown in Figure 3. The oxidation is exceptionally fast when using HCl as a promoter, leading to a 16% yield of products already after 15 min of reaction (turnover frequency: TOF ~250 h−1). Such a remarkable effect of hydrochloric acid on the reaction rate of cyclohexane oxidation was previously observed in other systems and can be associated with the participation of Cl− ions in the generation of catalytically active species bearing bridging or terminal chloride ligands [8]. TFA, HNO3, and H2SO4 exhibit a slightly weaker promoting behavior with total yields of 14–11%, which are achieved at more prolonged reaction times relative to a system operating with HCl. We also studied the promoting effects of different acids for catalysts 1 and 3 which reveal a behavior similar to 2 (Figure S8, Supplementary Materials).
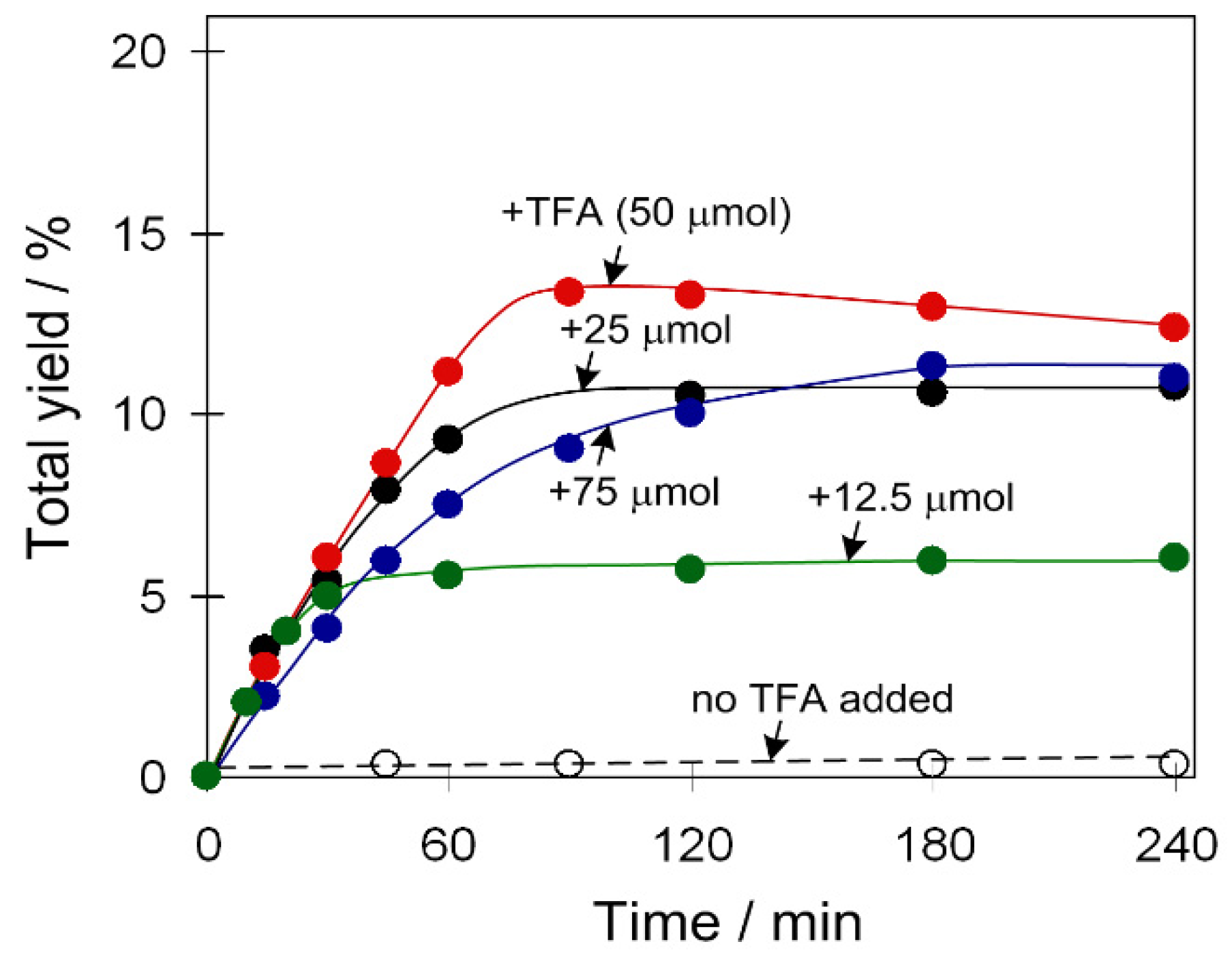
An influence of the TFA amount was also investigated for catalyst 2 (Figure 4) that is barely active without an acid promoter. However, total yield of products gradually grew upon elevating the amount of acid promoter up to 20 equivalents relative to the catalyst.
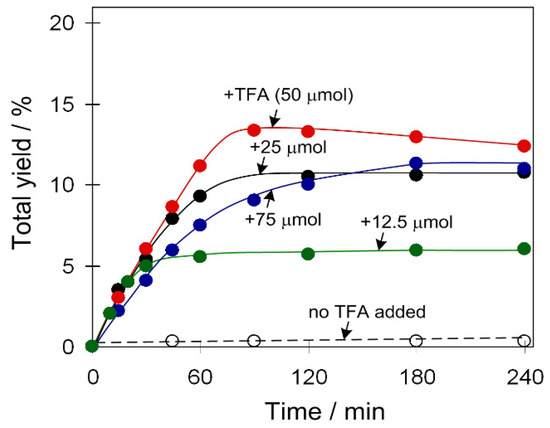
Figure 4.
Effect of TFA amount on total product yield (cyclohexanol and cyclohexanone) in the C6H12 oxidation with H2O2. Conditions: catalyst 2 (2.5 µmol), C6H12 (1 mmol), TFA (0–75 µmol), H2O2 (5 mmol), CH3CN (up to 2.5 mL of the total reaction volume), 50 °C.
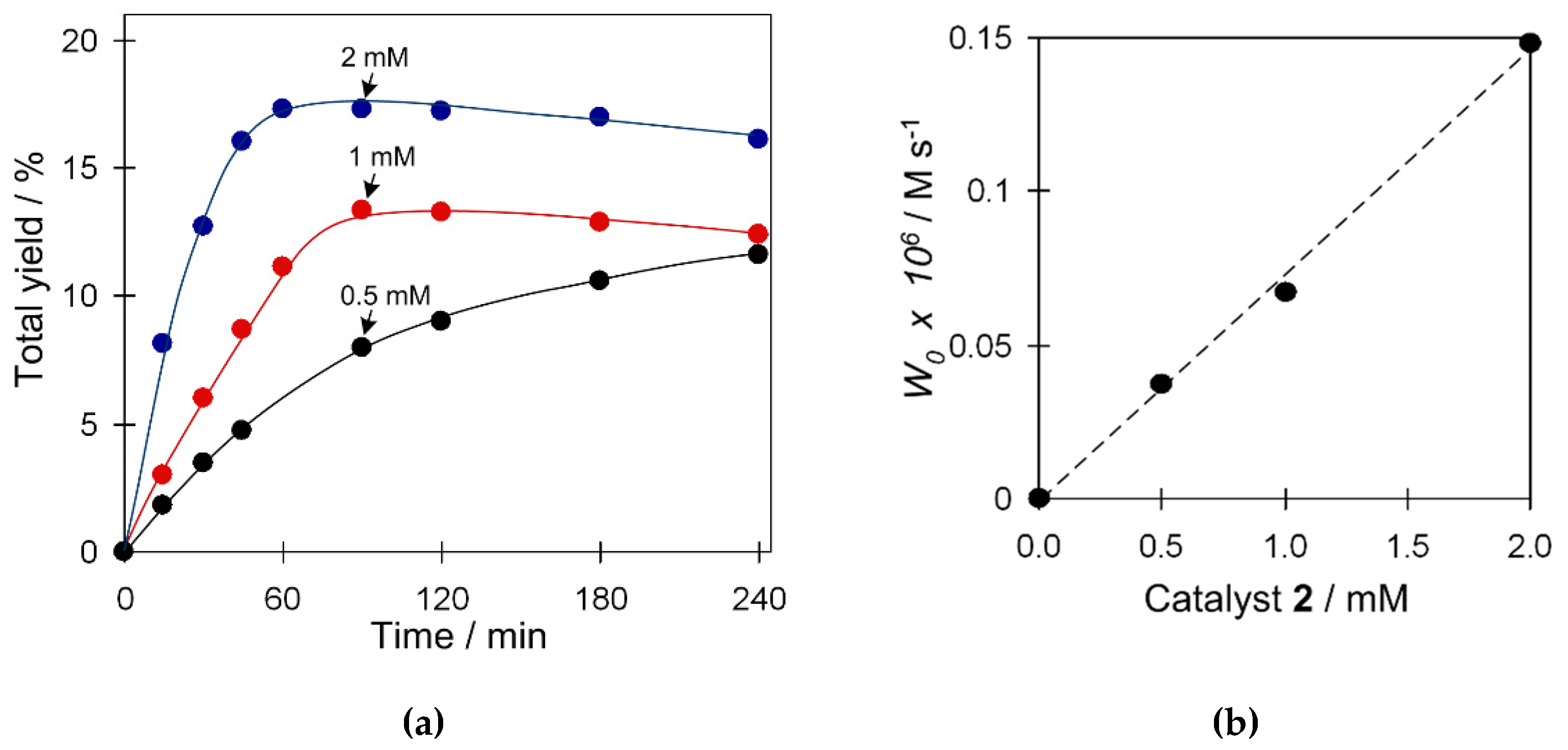
2.3.2. Effect of Catalyst Amount
We also studied the influence of the amount of catalyst 2 on the yield and W0 (maximum initial reaction rate) in the oxidation of cyclohexane (Figure 5). Blank experiments revealed that the cyclohexane oxidation products are not formed unless copper catalyst is added. An increase of the catalyst amount from 1.25 to 5 µmol results in an increase of both W0 and total yield of products (Figure 5a). The highest yield of 17% was obtained when using 5 µmol of catalyst. A linear trend between W0 and the amount of catalyst was observed (Figure 5b), suggesting kinetics of the first order and supporting an eventual participation of a single type of copper species in a rate-limiting step of the oxidation reaction.
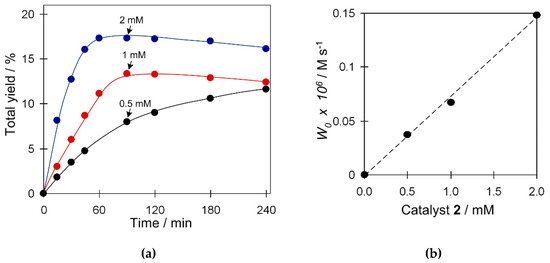
Figure 5.
Effect of catalyst loading on (a) total product yield (cyclohexanol and cyclohexanone), and (b) W0 (initial reaction rate) in the C6H12 oxidation with H2O2. Conditions: catalyst 2 (1.25–5 µmol), C6H12 (1 mmol), TFA (50 µmol), H2O2 (5 mmol), CH3CN (up to 2.5 mL of the total reaction volume), 50 °C.
2.3.3. Substrate Scope
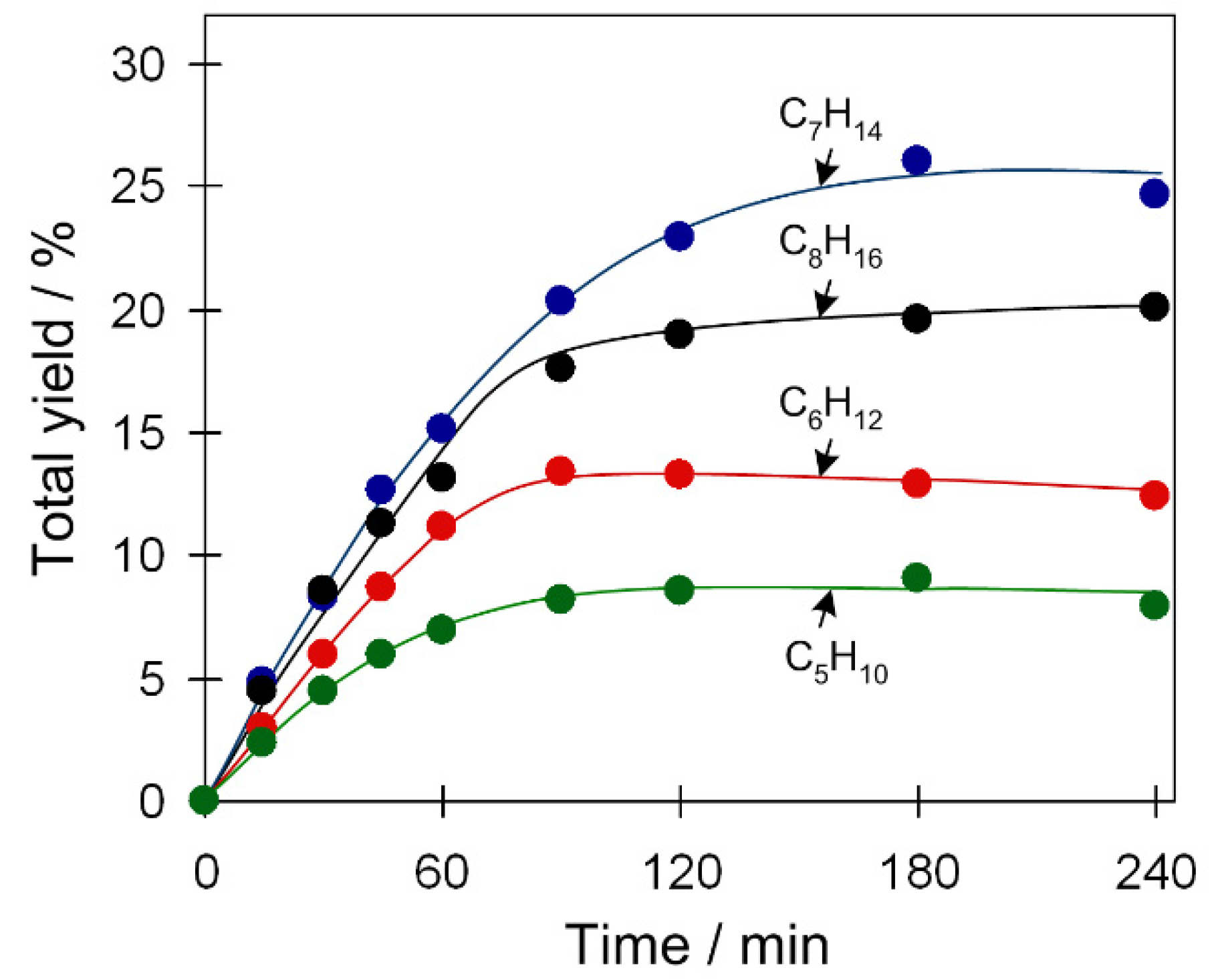
Apart from cyclohexane, oxidation of other cycloalkane substrates was also explored (Table 1, Figure 6 and Figure S9, Supplementary Materials). For catalyst 2, more reactive substrates are cycloheptane and cyclooctane, which lead to the maximum total yields of 26 and 20%, respectively. In comparison, the oxidation of cyclohexane and cyclopentane is less efficient (13–9% product yield). This might be influenced by the stability of formed radicals (cycloalkyl radicals) that increases with the ring size. Moreover, cyclopentane is rather volatile (boiling point (bp): 49 °C) and, at typical reaction conditions (50 °C), can partially be present in the gas phase; this may explain its lower reactivity. Such a trend of substrate reactivity is also observed for catalytic systems based on 1 or 3 (Table 1, Figure S9, Supplementary Materials).

Table 1.
Substrate scope in cycloalkane oxidation with H2O2 catalyzed by 1–3 a.
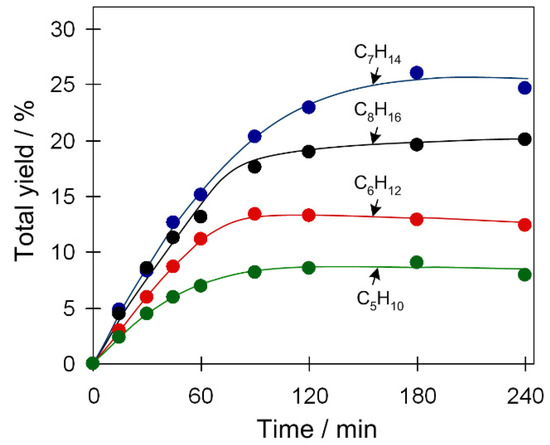
Figure 6.
Substrate scope in the cycloalkane oxidation to cyclic alcohols and ketones (total product yield vs. time) with H2O2. Conditions: catalyst 2 (2.5 µmol), cycloalkane (1 mmol), TFA (50 µmol), H2O2 (5 mmol), CH3CN (up to 2.5 mL of the total reaction volume), 50 °C.
2.3.4. Effect of Substrate and Oxidant Amount
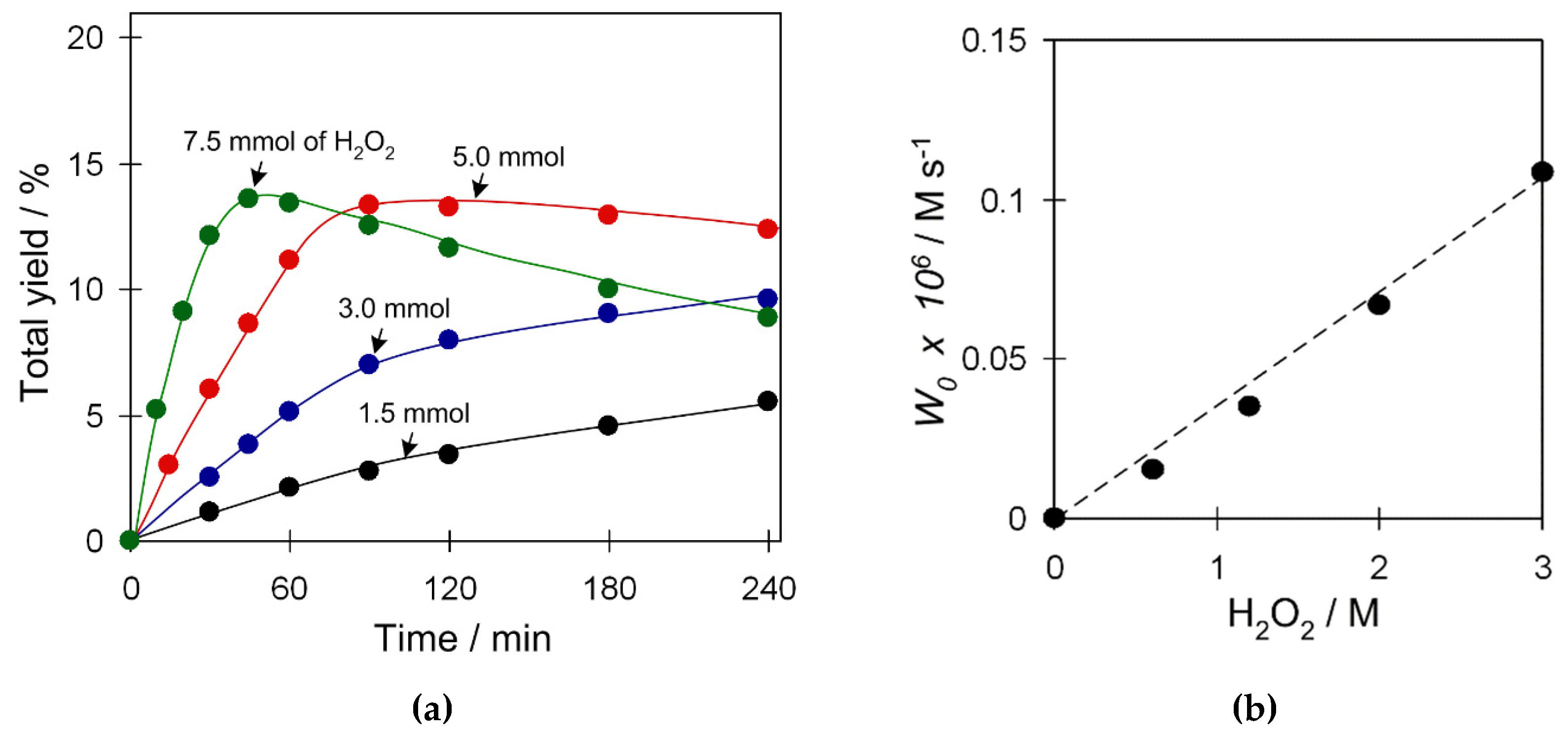
In cyclohexane oxidation catalyzed by 2 (Figure S10, Supplementary Materials), the reaction efficiency practically does not depend on the substrate amount within the studied range of loadings (0.5–2 mmol C6H12). In contrast, the amount of oxidant has a significant effect (Figure 7) and an excess of hydrogen peroxide is required for efficient oxidation of cyclohexane. An increase of the H2O2 amount from 1.5 to 7.5 mmol results in the growth of W0 (initial reaction rate) and product yield. Furthermore, higher loadings of H2O2 (up to 7.5 mmol) may lead to a yield drop due to the overoxidation at prolonged reaction times. The W0 linearly depends on the amount of hydrogen peroxide (Figure 7b), suggesting reaction kinetics of the first order for the oxidant.
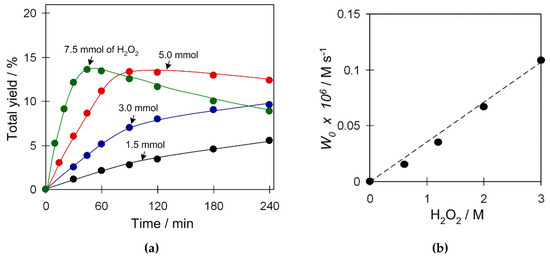
Figure 7.
Effect of the H2O2 amount on (a) total product yield (cyclohexanol and cyclohexanone), and (b) W0 in C6H12 oxidation with H2O2. Conditions: catalyst 2 (2.5 µmol), C6H12 (1 mmol), TFA (50 µmol), H2O2 (1.5–7.5 mmol), CH3CN (up to 2.5 mL of total reaction volume), 50 °C.
2.3.5. Effect of Water
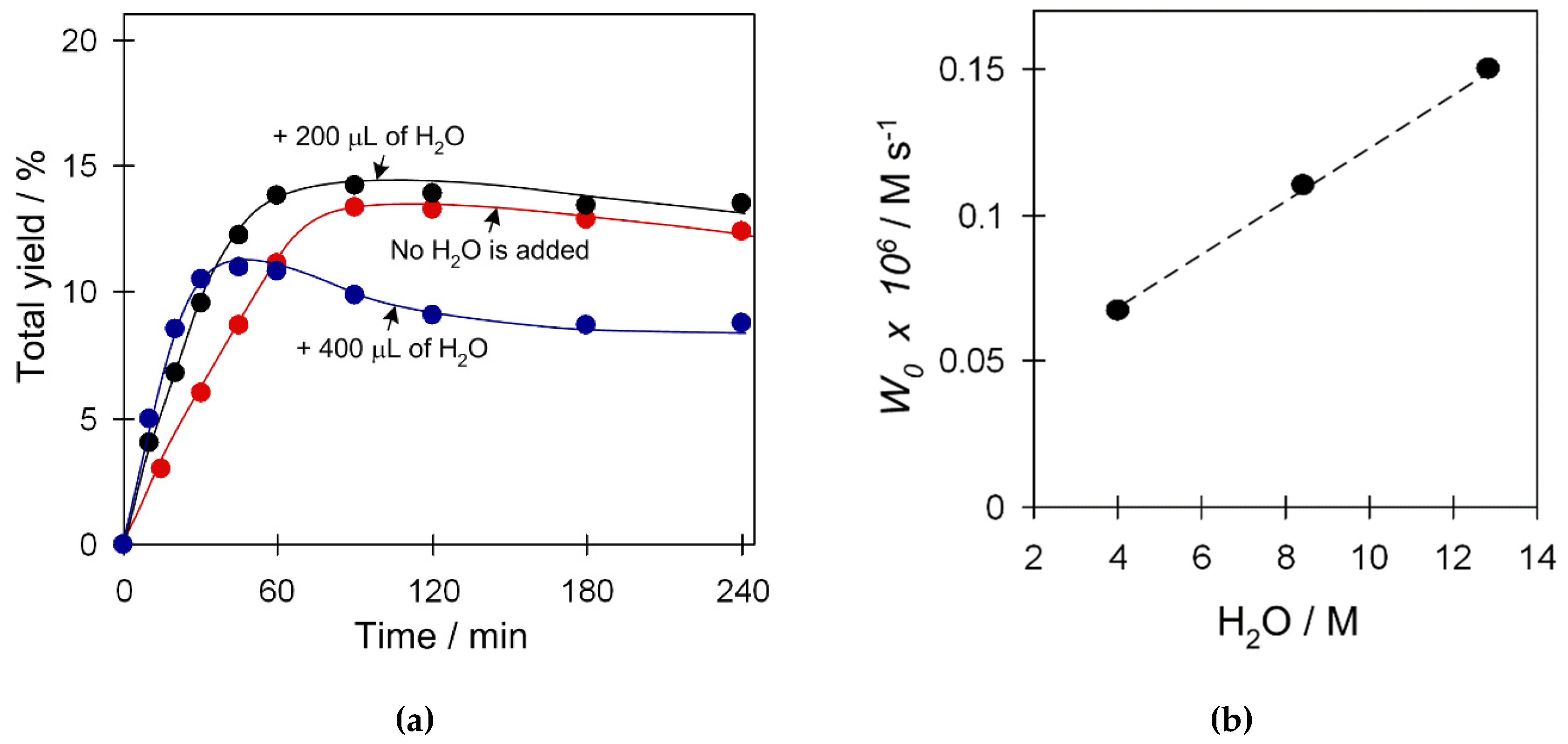
Recently, we reported an unexpected acceleration effect of H2O on cyclohexane oxidation catalyzed by other Cu catalytic systems [23,24]. Aiming at understanding whether H2O plays any role in the present Cu4 catalytic systems, we investigated the influence of water quantity in the system on both the W0 (maximum initial reaction rate) and product yield values in the C6H12 oxidation in the presence of catalyst 2 (Figure 8). Some water quantity (4.0 M) is already present in the reaction system that comes from the aqueous 50% H2O2. Addition of an extra portion of H2O (200 µL, 4.4 M; 8.4 M H2O total) into the reaction mixture results in an increase of the initial reaction rate W0 while not affecting the maximum yield of products. Addition of a double amount of water (400 µL, 8.4 M; 12.8 M H2O total) leads to further acceleration of W0 which, however, is accompanied by the decrease of total yield. The W0 values reveal a linear trend in the concentration of water within a studied range of concentrations (Figure 8b), thus suggesting an involvement of H2O in a rate-limiting step of the reaction. Such a promoting H2O behavior opens up a possibility of using the diluted H2O2 solutions (generated in situ) for cycloalkane oxidation.
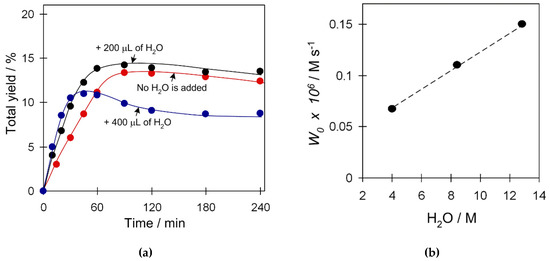
Figure 8.
Effect of H2O amount on (a) product yield (cyclohexanol and cyclohexanone), and (b) W0 in C6H12 oxidation with H2O2. Conditions: catalyst 2 (2.5 µmol), C6H12 (1 mmol), TFA (50 µmol), H2O2 (5 mmol), added H2O (200; 400 µL), CH3CN (up to 2.5 mL of total reaction volume), 50 °C.
2.4. Mild Cu-Catalyzed Oxidation of Propane
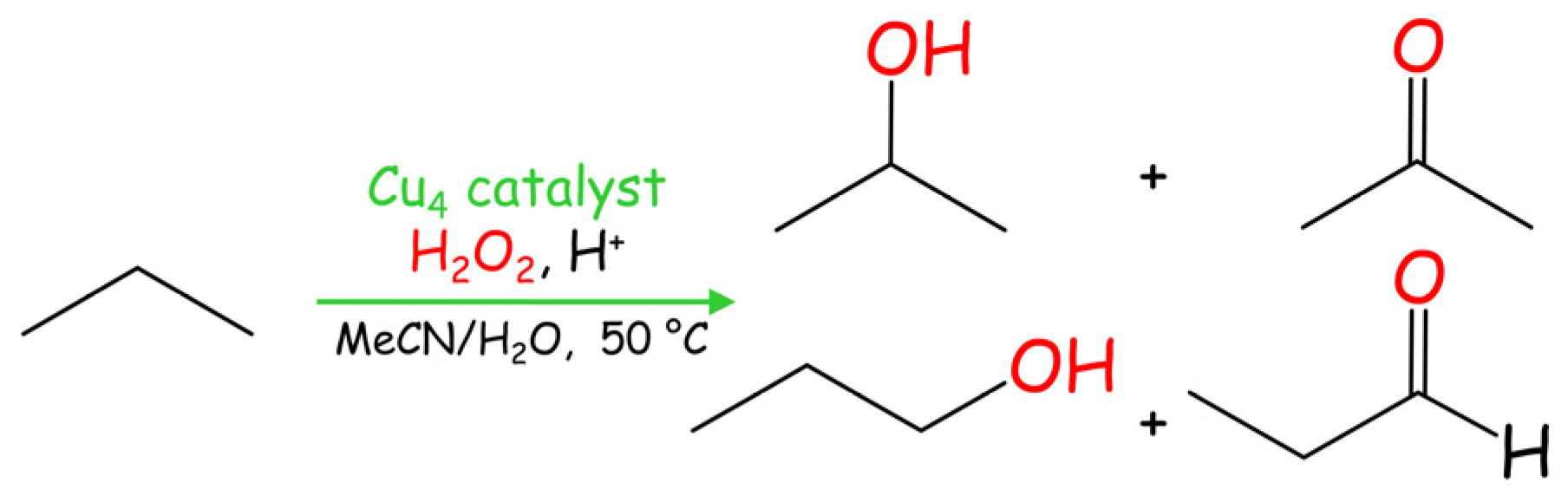
Development of the direct oxidation of propane constitutes a very promising approach in catalysis due to the abundance of this hydrocarbon as a C3 feedstock for the generation of value-added oxidation products [1,2]. Hence, catalytic performance of compounds 1–3 was also evaluated the in the propane oxidation with H2O2 under mild conditions, resulting in a mixture of isopropanol, acetone, n-propanol, and propanal (Scheme 3, Table 2).
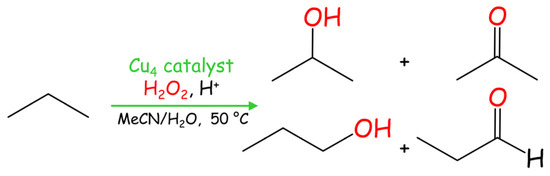
Scheme 3.
Mild Cu-catalyzed oxidation of propane.
Table 2.
Mild propane oxidation with hydrogen peroxide catalyzed by 1–3 a.
All catalysts 1–3 exhibit a comparable level of activity regarding the total product yield, which is in the 7–9% range (based on propane). As expected, the oxidation at the secondary C(2) atom is preferable, resulting in the generation of i-propanol and acetone as major products, due to an easier activation of the secondary carbon in propane in comparison with the two primary ones. Thus, n-propanol and propanal are formed in minor quantities (Table 2). Despite not being very high, such a level of total yields (up to 9%) in propane oxidation can be considered as significant given the high inertness of propane, its low solubility in the reaction medium, and the mild reaction conditions applied (50 °C, low-pressure reaction).
2.5. Selectivity Parameters and Mechanistic Considerations in Alkane Oxidation
Catalysts 1–3 were also tested in oxidation reactions of other alkanes, including a linear alkane (n-heptane), polycyclic alkane (adamantane), and cycloalkanes with methyl functionality (methylcyclohexane, cis-dimethylcyclohexane), aiming at establishing various selectivity parameters (Table 3) and getting additional mechanistic information. Hence, n-heptane oxidation undergoes with no regio-preference to a secondary C atom, showing the C(1):C(2):C(3):C(4) regioselectivity parameters of 1:4:5:8 (for 1), 1:5:5:7 (for 2), and 1:5:5:8 (for 3). Oxidation of methylcyclohexane proceeds with a moderate bond selectivity 1°:2°:3° of 1:5:19, 1:5:14, and 1:6:15 for catalysts 1–3, respectively. For the oxidation of adamantane, the 2°:3° bond selectivity parameter is close to 1:4 for all the catalysts (Table 3). Oxidation of cis-dimethylcyclohexane provides the information about stereoselectivity through the determination trans/cis ratio between the isomeric tertiary alcohol products. The observed trans/cis values are 0.9, 0.8, and 1.1 for 1–3, respectively; these suggest that the reactions are non-stereoselective.
Table 3.
Selectivity parameters in the oxidation of alkanes with H2O2 catalyzed by 1–3 a.
The obtained selectivity parameters, the generation of alkyl hydroperoxides as primary intermediate products (detected by Shul’pin’s method [5,15]), and the analysis of the relevant literature [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15] support a radical-type mechanism that involves indiscriminate oxidizing species such as hydroxyl radicals. Hence, we can propose a simplified mechanism for the Cu-catalyzed alkane oxidation with H2O2. It involves the following steps [8]: (1) formation of HO• radicals upon the interaction of H2O2 with Cu catalyst; (2) reaction of HO• radicals with alkane, abstracting H atom and forming R• (alkyl radicals); (3) formation of ROO• radicals in the reaction of alkyl radicals with O2; (4) transformation of ROO• radicals to ROOH (alkyl hydroperoxides) as a primary intermediate product; (5) decomposition of ROOH (via Cu-catalyzed processes), leading to alcohols (ROH) and ketones (R’=O) as final oxidation products.
2.6. Mild Cu-Catalyzed Carboxylation of C5–C8 Cycloalkanes
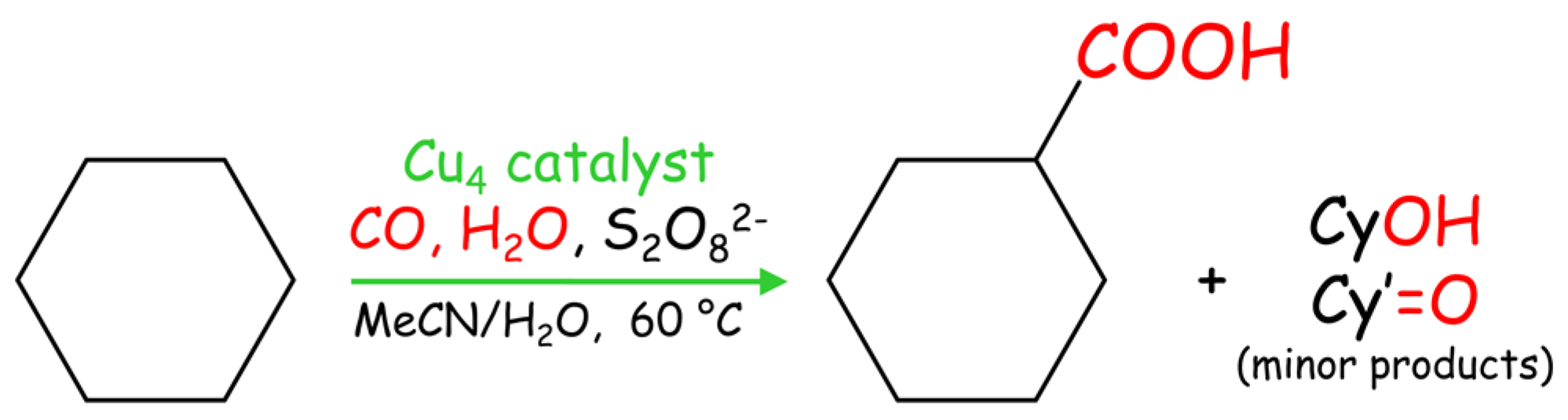
In addition to the oxidation reactions, catalysts 1–3 were tested in the single-pot carboxylation of cycloalkanes under mild conditions [8]. This reaction consists of the treatment of Cn cycloalkane with a carbonyl source (CO), a hydroxyl source (H2O), and an oxidant (potassium peroxodisulfate) to directly generate a Cn+1 cycloalkane carboxylic acid as the main product (Scheme 4); in addition, cyclic alcohols and ketones are formed as minor oxidation products.
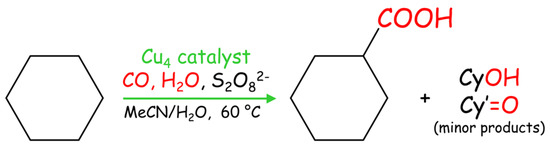
Scheme 4.
Single-pot Cu-catalyzed carboxylation of cycloalkanes (Cn) to give cycloalkane carboxylic acids (Cn+1); cyclohexane is shown as a model substrate.
Compounds 1–3 show very high activity (total product yields up to 46%) in the carboxylation of cycloalkanes (cyclooctane, cycloheptane, cyclohexane, and cyclopentane) to form cycloalkane carboxylic acid as a main product (Table 4). The catalytic activity of 1–3 is rather similar. In comparison to the oxidation reactions of cycloalkanes with H2O2, the Cu-catalyzed alkane carboxylation reactions proceed in the absence of an acid promoter.
Table 4.
Single-pot Cu-catalyzed carboxylation of cycloalkanes a.
C5 and C6 cycloalkanes represent the highest reactivity, resulting in cyclopentane carboxylic and cyclohexane carboxylic acids with yields of 31 and 43%, respectively (hereinafter, yields are relative to alkane substrate). Corresponding ketones and alcohols are also formed in low amounts (0.3–3.5%) as by-products of oxidation.
Carboxylation of cycloheptane and cyclooctane results in cyclooctane carboxylic (up to 27% yield) and cyclononane carboxylic (up to 14% yield) acids. Herein, the formation of alcohol and ketone by-products is more appreciable, especially in the case of C8H16 with the yields of cyclooctanol and cyclooctanone of up to 12% (Table 4).
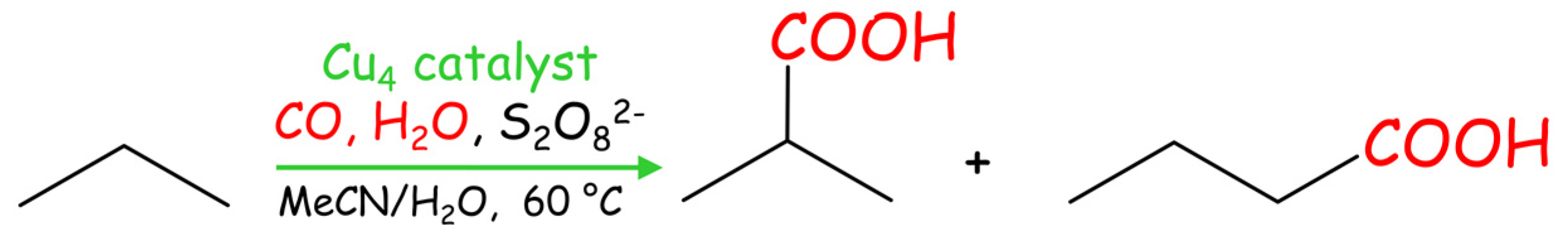
2.7. Mild Cu-Catalyzed Carboxylation of Propane
Compounds 1–3 were also applied as catalysts for the carboxylation of propane (Table 5, Scheme 5) to give iso-butyric acid (main product, 23–33% yield) and n-butyric acid (minor product, 5–7% yield), as expected for the superior reactivity of the C(2) atom in propane. Total yields of butyric acids attain 40.1% (catalyst 3), followed by 33.9 and 27.9% when using catalysts 1 and 2, respectively.
Table 5.
Single-pot Cu-catalyzed carboxylation of propane a.
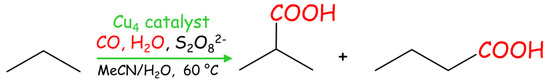
Scheme 5.
Single-pot Cu-catalyzed carboxylation of propane to butyric acids.
On the basis of experimental data and taking into consideration prior literature background for related reactions catalyzed by copper compounds [8,34,35,36], we can propose a simplified mechanism (free-radical type) which includes several steps as follows: (1) homolysis of K2S2O8 to give sulfate radical SO4•−; (2) interaction of SO4•− with alkane to abstract a hydrogen atom and generate R• (alkyl radical); (3) formation of RCO• (acyl radical) via carbonylation of R• with CO; (4) generation of RCO+ (acyl cation) by oxidation of acyl radical (with an involvement of CuII/CuI redox couple; CuII form is then regenerated through the oxidation with peroxodisulfate); (5) formation of a carboxylic acid product via hydrolysis of acyl cation by water.
3. Experimental
3.1. Materials and Methods
All reagents and solvents were acquired from commercial suppliers. FTIR spectra were recorded (4000–400 cm−1, KBr pellets) on a JASCO FT/IR-4100 Type A or Shimadzu IRAffinity-1S apparatus (abbreviations: vs—very strong, s—strong, m—medium, w—weak, br—broad, sh—shoulder). Elemental analyses (EA) were carried out on a Perkin Elmer PE 2400 Series II analyzer (Laboratory of Analyses, IST). ESI-MS spectra were obtained on an LCQ Fleet apparatus equipped with an electrospray (ESI) ion source (Thermo Scientific, San Jose, CA, USA). Gas chromatography (GC) analyses were carried out on an Agilent Technologies 7820A series gas chromatograph (Flame ionization detector; capillary column: BP20/SGE; carrier gas: He;).
3.2. Synthetic Procedure and Analytical Data for 1–3
Firstly, N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid (H3bes, 1 mmol, 213 mg) was added to an aqueous solution of Cu(NO3)2·3H2O (1 mmol, 0.1 M, 10 mL) with stirring under aerobic conditions at room temperature (reaction solution A). Then, benzene carboxylic acid (1 mmol; benzoic acid for 1 (Hba, 122 mg), 4-hydroxybenzoic acid for 2 (Hfhba, 138 mg), or 3-hydroxybenzoic acid for 3 (Hthba, 138 mg)) was dissolved in aqueous NH4OH (25% m/m, 4–12 mmol, up to 1.87 mL) to produce reaction solution B. This was slowly added to solution A and the obtained solution was further stirred for 25 minutes, followed by filtration. Slow evaporation of filtrate at room temperature resulted in the self-assembly of green crystals (these were formed within 1–2 weeks). Crystals were collected and dried in air to give complexes 1–3 (yield ~50% based on copper(II) nitrate).
[Cu4(µ-Hbes)3(µ-H2bes)(µ-ba)]·2H2O (1). Analysis calculated for 1 + NH4OH: Cu4C31H68N5O25S4 (molecular weight: MW = 1292): C, 28.79%; H, 5.26%; N, 5.42%; S, 9.91%; found: C, 28.99%; H, 5.13%; N, 5.52%; S, 9.73%. FTIR (KBr, cm−1): 3568 (m br) ν(OH/H2O), 2872 (w) ν(CH), 1598 (s) δ(OH/H2O), 1546 (vs) νas(COO), 1401 (s) νs(COO), 1241 (s) ν(C–C), 1151 (vs) ν(C–N), 1025 (vs) ν(C–S), 907 (m), 802 (m), 782 (w), 750 (m), 675 (m), 559 (w), 501 (w), 419 (w). ESI-MS(±) (H2O), selected fragments, MS(+): m/z: 1099 (20%) [{Cu4(µ-Hbes)4} + H]+, 1038 (60%) [{Cu3(µ-Hbes)2(µ-H2bes)2} + H]+, 977 (42%) [{Cu2(µ-H2bes)4} + H]+, 825 (20%) [{Cu3(µ-Hbes)3} + H]+, 764 (70%) [{Cu2(µ-Hbes)(µ-H2bes)2} + H]+, 701 (95%) [{Cu(µ-H2bes)2(µ-H3bes)} + H]+, 551 (18%) [{Cu2(µ-Hbes)2} + H]+, 488 (88%) [{Cu(µ-H2bes)2} + H]+, 427 (64%) [(H3bes)2 + H]+, 214 (100%) [H3bes + H]+. MS(−), m/z: 760 (5%) [Cu2(µ-Hbes)2(µ-H2bes)]–, 699 (8%) [Cu(µ-H2bes)3]–, 547 (5%) [Cu2(µ-Hbes)(µ-bes)]−, 425 (42%) [(H3bes)(H2bes)]−, 212 (100%) [H2bes]−, 121 (35%) [ba]−.
[Cu4(µ-Hbes)3(µ-H2bes)(µ-fhba)]·2H2O (2). Analysis calculated for 2 + NH4OH: (Cu4C31H70N6O27S4) (molecular weight: MW = 1325): C, 28.08%; H, 5.28%; N, 5.81%; S, 9.66%. found: C, 28.20%; H, 5.03%; N, 6.06%; S, 9.93%. FTIR (KBr, cm−1): 3541 (m br) ν(OH/H2O), 2880 (w) ν(CH), 1609 (s) δ (OH/H2O), 1540 (s) νas(COO), 1398 (vs) νs(COO), 1242 (vs) ν(C–C), 1150 (vs) ν(C–N), 1024 (vs) ν(C–S), 905 (m), 864 (w), 794 (w), 750 (w), 676 (w), 630 (w), 567 (w), 503 (w), 460 (w), 423 (w). ESI-MS(±) (H2O), selected fragments, MS(+), m/z: 1376 (5%) [{Cu4(µ-Hbes)3(µ-H2bes)}{µ-Hfhba}2 + H]+, 1099 (16%) [{Cu4(µ-Hbes)4} + H]+, 1099 (18%) [{Cu4(µ-Hbes)4} + H]+, 1038 (58%) [{Cu3(µ-Hbes)2(µ-H2bes)2} + H]+, 977 (50%) [{Cu2(µ-H2bes)4} + H]+, 825 (20%) [{Cu3(µ-Hbes)3} + H]+, 764 (68%) [{Cu2(µ-Hbes)(µ-H2bes)2} + H]+, 701 (100%) [{Cu(µ-H2bes)2(µ-H3bes)} + H]+, 551 (15%) [{Cu2(µ-Hbes)2} + H]+, 488 (66%) [{Cu(µ-H2bes)2} + H]+, 427 (52%) [(H3bes)2 + H]+, 275 (18%) [{Cu(µ-Hbes)} + H]+, 214 (54%) [{bes} + H]+. MS(−), m/z: 760 (5%) [Cu2(µ-Hbes)2(µ-H2bes)]–, 699 (8%) [Cu(µ-H2bes)3]–, 547 (5%) [Cu2(µ-Hbes)(µ-bes)]−, 425 (42%) [(H3bes)(H2bes)]−, 212 (100%) [H2bes]−, 137 (34%) [fhba]−.
[Cu4(µ-Hbes)3(µ-H2bes)(µ-thba)]·2H2O (3). Analysis calculated for 3 + 0.75NH4OH: (Cu4C31H66.75N4.75O25.75S4) (molecular weight: MW = 1299.25): C, 28.63%; H, 5.16%; N, 5.12%; S, 9.85%. found: C, 28.86%; H, 5.09%; N, 5.23%; S, 9.70%. FTIR (KBr, cm-1): 3568 (m br) ν(OH/H2O), 2870 (w) ν(CH), 1600 (s) δ(OH/H2O), 1546 (s) νas(COO), 1401 (s) νs(COO),1240 (m) ν(C–C), 1151 (vs) ν(C–N), 1025 (vs) ν(C–S), 907 (m), 802 (w), 782 (w), 750 (m), 675 (w), 560 (w), 501 (w), 419 (w). ESI-MS(±)(H2O), selected fragments, MS(+), m/z: 1376 (5%) [{Cu4(µ-Hbes)3(µ-H2bes)}{µ-Hthba}2 + H]+, 1099 (20%) [{Cu4(µ-Hbes)4} + H]+, 1038 (60%) [{Cu3(µ-Hbes)2(µ-H2bes)2} + H]+, 977 (55%) [{Cu2(µ-H2bes)4} + H]+, 825 (30%) [{Cu3(µ-Hbes)3} + H]+, 764 (90%) [{Cu2(µ-Hbes)(µ-H2bes)2} + H]+, 701 (100%) [{Cu(µ-H2bes)2(µ-H3bes)} + H]+, 551 (18%) [{Cu2(µ-Hbes)2} + H]+, 488 (86%) [{Cu(µ-H2bes)2} + H]+, 427 (52%) [(H3bes)2 + H]+, 275 (18%) [{Cu(µ-Hbes)} + H]+, 214 (47%) [H3bes + H]+. MS(−), m/z: 823 (5%) [Cu3(µ-H2bes)2(µ-Hbes)]–, 760 (5%) [Cu2(µ-H2bes)2(µ-Hbes)]–, 699 (8%) [Cu(µ-H2bes)3]–, 547 (10%) [Cu2(µ-Hbes)(µ-bes)]−, 425 (32%) [(H3bes)(H2bes)]−, 274 (5%) [Cu(µ-Hbes)]−, 212 (100%) [H2bes]−, 137 (22%) [thba]−.
3.3. X-ray Diffraction
Single crystals of 1–3 were mounted with Fomblin© in a cryoloop. Diffraction data were obtained on Bruker AXS-KAPPA APEX II or BRUKER D8 QUEST diffractometers (graphite-monochromated radiation, Mo Kα, λ = 0.7107 Å, 298 K). X-ray generator parameters were 50 kV and 30 mA. APEX2 [42] and APEX3 [43] programs were used to monitor the collection of X-ray data. SAINT [44] and SADABS [45] were applied for correction of all data for Lorentzian, polarization, and absorption effects. SHELXT was applied to solve the structures, while SHELXL-97 was used for refinement (full-matrix least-squares on F2) [46]. This software is a part of the WINGX-Version 2014.1 package [47]. All atoms (with the exception of H atoms) were refined anisotropically. For non-H atoms, full-matrix least-squares refinement was used with anisotropic thermal parameters. In 3, water O atoms were refined with 0.5 occupancy. A twin matrix was applied to 3. All the hydrogen atoms bonded to carbon atoms were inserted in idealized positions and allowed to refine at the parent carbon atom. H atoms on the aminoalcohol moieties were localized by the electron density map and their positions were fixed. Regarding the hydroxyl groups in the benzene carboxylate in 3 and 2, the hydrogen atoms were inserted in idealized positions and allowed to refine at the parent O atom. With the exception of 2, the water hydrogen atoms were not possible to locate. Despite several attempts to recrystallize the compounds and several data collections for each sample, crystals of 1 and 3 were of low quality and the refinement of these crystal structures resulted in high Rint and final R1 and wR2 values or completeness issues (for 3). The structure of 2 represents the best refinement parameters (Table S1, Supplementary Materials) and, thus, is discussed in detail as a representative example. The structural data for 1–3 were deposited as CIF files within the Cambridge Crystallographic Data Base (CCDC 1899840-1899842).
3.4. Catalytic Oxidation of Cycloalkanes
Catalytic tests were performed in glass reactors (equipped with a condenser) under aerobic conditions and under vigorous stirring at 50 °C, using MeCN as a solvent (up to 2.5 mL of total reaction volume). Total volume refers to all reagents and solvent (H2O2, alkane, GC standard, H2O, and MeCN). The typical procedure was as follows: catalyst 1–3 (2.5 μmol) was introduced into a MeCN solution, followed by the addition of an acid promoter (50 μmol) and GC internal standard (MeNO2, 250 μL). Then, an alkane substrate (1 mmol) and hydrogen peroxide (50% in H2O, 5 mmol) were added. The oxidation reactions were followed by withdrawing small aliquots of the reaction mixture at different time periods. Prior to GC (gas chromatography) analysis, the aliquots were treated with solid PPh3 for reducing alkyl hydroperoxides (primary products in cycloalkane reactions) and remaining H2O2; all the product yields were calculated based on GC data after the treatment with solid PPh3. Generation of alkyl hydroperoxides as primary products was corroborated by doing the GC analyses of selected samples twice, before and after the treatment with solid PPh3 (Shul’pin’s method) [5,15]. Peaks were attributed by comparing the obtained chromatograms with those of commercially available samples of products.
3.5. Catalytic Oxidation of Propane
The typical procedure was as follows: catalyst 1–3 (2.5 µmol) was introduced in an acetonitrile solution, and then an acid promoter (50 µmol), MeNO2 (GC internal standard, 250 μL), and hydrogen peroxide (50% in H2O, 5 mmol) were added. A stainless-steel autoclave (total volume of 20.0 mL) was closed, pressurized with 3 atm of propane substrate, and kept under stirring at 50 °C (oil bath and magnetic stirrer) for 4 h. The autoclave was then cooled down and degassed. Samples of the reaction mixture were treated with PPh3 and analyzed by GC for quantification of products (internal standard method).
3.6. Catalytic Carboxylation of Alkanes
The typical procedure was as follows: catalyst 1–3 (2.5 µmol), H2O (2.0 mL), MeCN (4.0 mL), cycloalkane (1.0 mmol), and K2S2O8 (1.50 mmol) were introduced into a stainless-steel autoclave (total volume of 20.0 mL). Then, the autoclave was closed and flushed three times with CO for air removal and pressurized with carbon monoxide (20 atm). In the case of propane carboxylation, the reactor was first flushed and pressurized with propane followed by the addition of CO. The reaction mixture was stirred at 60 °C for 4 h (oil bath and magnetic stirrer). After this period, the autoclave was cooled in an ice bath, degassed, and opened. The reaction mixture was transferred to a glass flask. Et2O (9.0 mL) and GC internal standard (cycloheptanone, 45 µL) were introduced; cyclohexanone was the GC standard in the carboxylation of cycloheptane. After stirring the obtained mixture for 10 min, the aliquots were taken from an organic layer. These were subjected to GC analysis for the quantification (internal standard method) of carboxylic acids as principal products (ketones and alcohols were also formed as by-products in minor amounts). Peaks were attributed by comparing the obtained chromatograms with those of commercially available samples of products.
4. Conclusions
In this study, we used a self-assembly synthesis to prepare three new tetracopper(II) complexes, derived from a trifunctional aminoalcohol sulfonic acid (main building block) and a benezene carboxylic acid (supporting ligand). The obtained products [Cu4(µ-Hbes)3(µ-H2bes)(µ-L)]·2H2O (L = ba− (1), fhba− (2), and thba− (3)) were fully characterized and reveal a similar type of tetracopper(II) core. These cores are soluble in aqueous medium and were used as catalysts for oxidative C–H functionalization of gaseous (propane) and liquid alkanes (C5–C8 cycloalkanes).
In fact, the obtained compounds 1–3 act as efficient and versatile homogeneous catalysts for the oxidation of cycloalkanes with hydrogen peroxide to produce cyclic alcohols and ketones, with total yields attaining 27% (yields are based on cycloalkane substrate). Propane oxidation into a mixture of the C3 oxidation products was also achieved. The influence of various parameters of the oxidation reaction was investigated in detail, in addition to mechanistic and selectivity features. Moreover, compounds 1–3 also catalyze the mild carboxylation of propane and C5–C8 cycloalkanes to give carboxylic acids in as high as 41% yield (based on alkane substrate). The obtained yields are remarkable considering the inertness of alkane substrates and mild reaction conditions applied [1,2,3,4,5,6,7,8].
Given the multicopper nature of active sites in some enzymes (i.e., multicopper oxidases, particulate methane monooxygenase) [20,21,22,48], the mild oxidative functionalization of alkanes using such bioinspired multicopper catalysts represents a particularly interesting research direction. Future research will also focus on the design of new structurally related coordination compounds and their application as catalysts in the mild functionalization of saturated hydrocarbons. The search for more efficient systems, optimization of reaction parameters, and widening of the substrate scope will be pursued.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4344/9/4/321/s1: Table S1: crystal data for 1–3; Figure S1: crystal structures of 1 and 3; Figure S2: IR spectra and their discussion; Figures S3–S7: ESI-MS data and their discussion; Figures S8–S10: additional catalysis data.
Author Contributions
Conceptualization, M.V.K. and A.M.K.; data curation, I.F.M.C, M.V.K., V.A., and T.A.F.; funding acquisition, M.V.K. and A.M.K.; investigation, I.F.M.C., M.V.K., V.A., and T.A.F.; methodology, M.V.K.; supervision, A.M.K.; visualization, A.M.K. and M.V.K.; writing—original draft, I.F.M.C., T.A.F., M.V.K., V.A., and A.M.K.; writing—review and editing, A.M.K.
Funding
This work was supported by the Foundation for Science and Technology (FCT) and Portugal 2020 (projects IF/01395/2013/CP1163/CT005, CEECIND/03708/2017, UID/QUI/00100/2013, LISBOA-01-0145-FEDER-029697, and REM2013). The publication was also prepared with the support of the RUDN University Program 5-100.
Acknowledgments
A.M.K. acknowledges the COST Action CA15106 (CHAOS). T.A.F. acknowledges the FCT for BPD grant SFRH/BPD/119980/2016. We thank S. Dias for experimental assistance, and M. C. Oliveira and A. Dias for ESI-MS(±) measurements.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Shilov, A.E.; Shul’pin, G.B. Activation and Catalytic Reactions of Saturated Hydrocarbons in the Presence of Metal Complexes; Kluwer Acad. Publ.: Dordrecht, The Netherlands, 2000. [Google Scholar]
- Olah, G.A.; Molnár, Á. Hydrocarbon Chemistry; Wiley: Hoboken, NJ, USA, 2003. [Google Scholar]
- Alkane, C.-H. Activation by Single-Site Metal Catalysis; Pérez, P.J., Ed.; Springer: New York, NY, USA, 2012. [Google Scholar]
- Nesterov, D.S.; Nesterova, O.V.; Pombeiro, A.J.L. Homo- and heterometallic polynuclear transition metal catalysts for alkane C-H bonds oxidative functionalization: Recent advances. Coord. Chem. Rev. 2018, 355, 199–222. [Google Scholar] [CrossRef]
- Shul’pin, G.B. Hydrocarbon Oxygenations with Peroxides Catalyzed by Metal Compounds. Mini-Rev. Org. Chem. 2009, 6, 95–104. [Google Scholar] [CrossRef]
- Fokin, A.A.; Schreiner, P.R. Selective Alkane Transformations via Radicals and Radical Cations: Insights into the Activation Step from Experiment and Theory. Chem. Rev. 2002, 102, 1551–1594. [Google Scholar] [CrossRef] [PubMed]
- Labinger, J.A.; Bercaw, J.E. Understanding and exploiting C–H bond activation. Nature 2002, 417, 507–514. [Google Scholar] [CrossRef]
- Kirillov, A.M.; Kirillova, M.V.; Pombeiro, A.J.L. Multicopper complexes and coordination polymers for mild oxidative functionalization of alkanes. Coord. Chem. Rev. 2012, 256, 2741–2759. [Google Scholar] [CrossRef]
- Nesterova, O.V.; Kopylovich, M.N.; Nesterov, D.S. Stereoselective oxidation of alkanes with m-CPBA as an oxidant and cobalt complex with isoindole-based ligands as catalysts. RSC Adv. 2016, 6, 93756–93767. [Google Scholar] [CrossRef]
- Antonangelo, A.R.; Grazia Bezzu, C.; McKeown, N.B.; Nakagaki, S. Highly active manganese porphyrin-based microporous network polymers for selective oxidation reactions. J. Catal. 2019, 369, 133–142. [Google Scholar] [CrossRef]
- Talsi, E.P.; Bryliakov, K.P. Chemo- and stereoselective CH oxidations and epoxidations/cis-dihydroxylations with H2O2, catalyzed by non-heme iron and manganese complexes. Coord. Chem. Rev. 2012, 256, 1418–1434. [Google Scholar] [CrossRef]
- Ottenbacher, R.V.; Talsi, E.P.; Bryliakov, K.P. Direct Selective Oxidative Functionalization of C–H Bonds with H2O2: Mn-Aminopyridine Complexes Challenge the Dominance of Non-Heme Fe Catalysts. Molecules 2016, 21, 1454. [Google Scholar] [CrossRef]
- Shilov, A.E.; Shul’pin, G.B. Activation of C−H Bonds by Metal Complexes. Chem. Rev. 1997, 97, 2879–2932. [Google Scholar] [CrossRef]
- Shul’pin, G.B. New Trends in Oxidative Functionalization of Carbon–Hydrogen Bonds: A Review. Catalysts 2016, 6, 50. [Google Scholar] [CrossRef]
- Shul’pin, G.B. Metal-catalyzed hydrocarbon oxygenations in solutions: The dramatic role of additives: A review. J. Mol. Catal. A Chem. 2002, 189, 39–66. [Google Scholar] [CrossRef]
- Nesterov, D.S.; Nesterova, O.V.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Catalytic behaviour of a novel Fe(III) Schiff base complex in the mild oxidation of cyclohexane. Catal. Sci. Technol. 2015, 5, 1801–1812. [Google Scholar] [CrossRef]
- Nesterov, D.S.; Nesterova, O.V.; Kopylovich, M.N.; Pombeiro, A.J.L. Pronounced retention of stereoconfiguration upon sp3 C-H bonds hydroxylation of dimethylcyclohexanes and decahydronaphthalenes with m-CPBA oxidant and a Co-phthalocyanine catalyst. Mol. Catal. 2018, 459, 8–15. [Google Scholar] [CrossRef]
- Gupta, S.; Kirillova, M.V.; da Silva, M.F.C.G.; Pombeiro, A.J.L.; Kirillov, A.M. Alkali metal directed assembly of heterometallic Vv/M (M = Na, K, Cs) coordination polymers: Structures, topological analysis, and oxidation catalytic properties. Inorg. Chem. 2013, 52, 8601–8611. [Google Scholar] [CrossRef]
- Bäckvall, J.-E. (Ed.) Modern Oxidation Methods, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Karlin, K.D.; Tyeklar, Z. (Eds.) Bioinorganic Chemistry of Copper; Springer: Berlin, Germany, 2012. [Google Scholar]
- Itoh, S.; Rokita, S. (Eds.) Copper-Oxygen Chemistry; Wiley: New York, NY, USA, 2011. [Google Scholar]
- Brissos, R.S.; Garcia, S.; Presa, A.; Gamez, P. Bio-related copper-mediated oxidative processes. Comments Inorg. Chem. 2011, 32, 219–245. [Google Scholar] [CrossRef]
- Fernandes, T.A.; Santos, C.I.M.; André, V.; Kłak, J.; Kirillova, M.V.; Kirillov, A.M. Aqua-Soluble Copper(II) Coordination Polymers Self-assembled from Aminoalcohols and Pyromellitic Acid: Highly Active Pre-catalysts for the Mild Water-promoted Oxidation of Alkanes. Inorg. Chem. 2016, 55, 125–135. [Google Scholar] [CrossRef]
- Fernandes, T.A.; Santos, C.I.M.; André, V.; Dias, S.S.P.; Kirillova, M.V.; Kirillov, A.M. New aqua-soluble dicopper(II) aminoalcoholate cores for mild and water-assisted catalytic oxidation of alkanes. Catal. Sci. Technol. 2016, 6, 4584–4593. [Google Scholar] [CrossRef]
- Dias, S.S.P.; Kirillova, M.V.; André, V.; Kłak, J.; Kirillov, A.M. New tricopper(II) cores self-assembled from aminoalcohol biobuffers and homophthalic acid: Synthesis, structural and topological features, magnetic properties and mild catalytic oxidation of cyclic and linear C5–C8 alkanes. Inorg. Chem. Front. 2015, 2, 525–537. [Google Scholar] [CrossRef]
- Dias, S.S.P.; Kirillova, M.V.; André, V.; Kłak, J.; Kirillov, A.M. New Tetracopper (II) Cubane Cores Driven by a Diamino Alcohol: Self-assembly Synthesis, Structural and Topological Features, and Magnetic and Catalytic Oxidation Properties. Inorg. Chem. 2015, 54, 5204–5212. [Google Scholar] [CrossRef]
- Kulakova, A.N.; Bilyachenko, A.N.; Korlyukov, A.A.; Shul’pina, L.S.; Bantreil, X.; Lamaty, F.; Shubina, E.S.; Levitsky, M.M.; Ikonnikova, N.S.; Shul’pin, G.B. A new “bicycle helmet”-like copper(II) sodiumphenylsilsesquioxane. Synthesis, structure and catalytic activity. Dalton Trans. 2018, 47, 15666–15669. [Google Scholar] [CrossRef] [PubMed]
- Levitsky, M.M.; Yalymov, A.I.; Kulakova, A.N.; Petrov, A.A.; Bilyachenko, A.N. Cage-like metallsilsesquioxanes in catalysis: A review. J. Mol. Cat. A Chem. 2017, 426, 297–304. [Google Scholar] [CrossRef]
- Good, N.E.; Winget, G.D.; Winter, W.; Connolly, T.N.; Izawa, S.; Sing, R.M.M. Hydrogen ion buffers for biological research. Biochemistry 1966, 5, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, W.J.; Braunschweiger, K.I.; Braunschweiger, W.R.; Smith, J.R.; McCormick, J.J.; Wasmann, C.C.; Jarvis, N.P.; Bell, D.H.; Good, N.E. Hydrogen ion buffers for biological research. Anal. Biochem. 1980, 104, 300–310. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef]
- Kirillov, A.M.; Coelho, J.A.S.; Kirillova, M.V.; Guedes da Silva, M.F.C.; Nesterov, D.S.; Gruenwald, K.R.; Haukka, M.; Pombeiro, A.J.L. Bringing an “Old” Biological Buffer to Coordination Chemistry: New 1D and 3D Coordination Polymers with [Cu4(Hbes)4] Cores for Mild Hydrocarboxylation of Alkanes. Inorg. Chem. 2010, 49, 6390–6392. [Google Scholar] [CrossRef]
- Kirillova, M.V.; Kirillov, A.M.; Martins, A.N.C.; Graiff, C.; Tiripicchio, A.; Pombeiro, A.J.L. Topologically Unique Heterometallic CuII/Li Coordination Polymers Self-Assembled from N,N-bis(2-Hydroxyethyl)-2-aminoethanesulfonic Acid Biobuffer: Versatile Catalyst Precursors for Mild Hydrocarboxylation of Alkanes to Carboxylic Acids. Inorg. Chem. 2012, 51, 5224–5234. [Google Scholar] [CrossRef]
- Kirillova, M.V.; Kirillov, A.M.; Pombeiro, A.J.L. Mild, single-pot hydrocarboxylation of gaseous alkanes to carboxylic acids in metal-free and copper-promoted aqueous systems. Chem. Eur. J. 2010, 16, 9485–9493. [Google Scholar] [CrossRef] [PubMed]
- Kirillova, M.V.; Kirillov, A.M.; Kuznetsov, M.L.; Silva, J.A.L.; Fraústo da Silva, J.J.R.; Pombeiro, A.J.L. Alkanes to carboxylic acids in aqueous medium: Metal-free and metal-promoted highly efficient and mild conversions. Chem. Commun. 2009, 2353–2355. [Google Scholar] [CrossRef]
- Kirillova, M.V.; Kirillov, A.M.; Pombeiro, A.J.L. Metal-free and copper-promoted single-pot hydrocarboxylation of cycloalkanes to carboxylic acids in aqueous medium. Adv. Synth. Catal. 2009, 351, 2936–2948. [Google Scholar] [CrossRef]
- Kirillov, A.M.; Haukka, M.; da Silva, M.F.C.G.; Pombeiro, A.J.L. Preparation and crystal structures of benzoylhydrazido- and -diazenidorhenium complexes with N,O-ligands and their catalytic activity towards peroxidative oxidation of cycloalkanes. Eur. J. Inorg. Chem. 2005, 2071–2080. [Google Scholar] [CrossRef]
- Schuchardt, U.; Cardoso, D.; Sercheli, R.; Pereira, R.; da Cruz, R.S.; Guerreiro, M.C.; Mandelli, D.; Spinace, E.V.; Pires, E.L. Cyclohexane Oxidation Continues to be a Challenge. Appl. Catal. A Gen. 2001, 211, 1–17. [Google Scholar] [CrossRef]
- Wittcoff, H.; Reuben, B.G.; Plotkin, J.S. Industrial Organic Chemicals, 2nd ed.; Wiley: New York, NY, USA, 2004. [Google Scholar]
- Kirillov, A.M.; Shul’pin, G.B. Pyrazinecarboxylic acid and analogs: Highly efficient co-catalysts in the metal-complex-catalyzed oxidation of organic compounds. Coord. Chem. Rev. 2013, 257, 732–754. [Google Scholar] [CrossRef]
- Armakola, E.; Colodrero, R.M.P.; Bazaga-García, M.; Salcedo, I.R.; Choquesillo-Lazarte, D.; Cabeza, A.; Kirillova, M.V.; Kirillov, A.M.; Demadis, K.D. Three-Component Copper-Phosphonate-Auxiliary Ligand Systems: Proton Conductors and Efficient Catalysts in Mild Oxidative Functionalization of Cycloalkanes. Inorg. Chem. 2018, 57, 10656–10666. [Google Scholar] [CrossRef] [PubMed]
- APEX2. Ver. 2014.11-0; Bruker-AXS: Billerica, MA, USA, 2014. [Google Scholar]
- APEX3. Ver. 2017.3-0; Bruker-AXS: Billerica, MA, USA, 2017. [Google Scholar]
- SAINT; Bruker-AXS: Billerica, MA, USA, 2014/2017.
- SADABS; Bruker-AXS: Billerica, MA, USA, 2014/2017.
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX—Version 1.80.05. J. Appl. Cryst. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Ayala, M.; Torres, E. Enzymatic activation of alkanes: Constraints and prospective. Appl. Catal. A 2004, 272, 1–13. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).