1. Introduction
Porphyrins are macrocyclic compounds that are naturally found in diverse forms of life, where they perform distinct roles, including oxygen transport in cellular respiration processes, and mainly in catalytic processes, such as the detoxification of xenobiotic compounds and the oxidation of fatty acids [
1,
2].
Over the last 40 years, these compounds have been intensively studied due to their application as bio-inspired models of heme proteins that present catalytic activity, e.g., the cytochrome P450 enzyme family [
3]. The cytochrome P450 enzymes are defined by the presence of a heme (iron protoporphyrin IX) prosthetic group coordinated on the proximal side by a thiolate ion deriving from a protein residue [
4,
5,
6,
7].
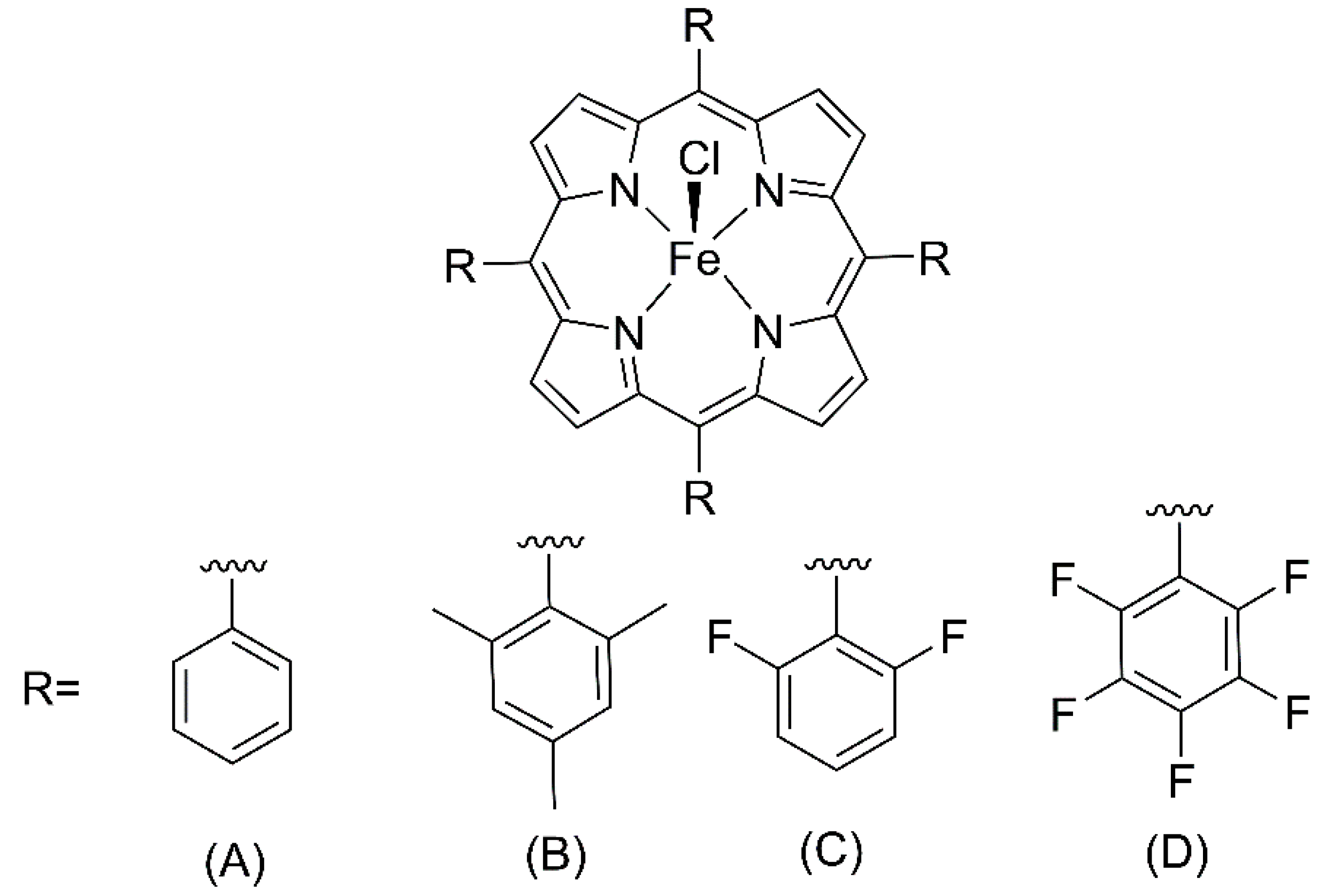
In 1979, Groves et al. demonstrated for the first time the catalytic activity of a cytochrome P450 biomimetic model based on a synthetic iron (III) porphyrin [Fe(TPP)]Cl (
Figure 1A), which was able to catalyze the hydroxylation and epoxidation of hydrocarbons with iodosylbenzene as an oxygen source under mild conditions [
8]. This transformation was indicated as occurring through the formation of a high valent metal–oxygen intermediate complex [
3,
9].
Although the biomimetic catalytic system developed by Groves et al. [
8] qualitatively reproduced the P450 reaction, the catalytic activity was rapidly diminished by extensive destruction of the metalloporphyrin. However, in 1981, the same group showed that the iron porphyrin ([Fe(TMP)]Cl (
Figure 1B) was able to resist oxidative destruction due to the steric hindrance imposed by the
meso substitutes present in the porphyrin structure [
10,
11].
Chang and Ebina, in 1981, demonstrated that fluorinated iron porphyrin [Fe(TPFPP)]Cl (
Figure 1D) had a superior catalytic activity toward cyclohexane hydroxylation in contrast with the [Fe(TPP)]Cl (
Figure 1A), achieving a 71% yield of cyclohexanol versus the 5% yield of the non-fluorinated compound. The electronic effects of fluorine atoms were attributed to causing the improvement of the catalytic activity in comparison to the results presented by the [Fe(TPP)]Cl used by Groves (8% cyclohexanol) [
8,
11,
12].
Both the iron porphyrins [Fe(TMP)]Cl and [Fe(TPFPP)]Cl (
Figure 1B,D) were more effective catalysts for hydrocarbon oxidation due to the structural modification of the porphyrin ring periphery. That modification increased the robustness of the complex during the catalytic reaction against the electrophilic attack that can occur in a solution (homogeneous catalysis), and it was able to form a reasonably stable high-valent iron porphyrin catalytic active species [
3,
11].
All of these distinct structural features of the tetrapyrrolic macrocycles have led to a better understanding and the development of more robust catalysts based on porphyrins, enabling their use as a sophisticated bulky, chiral, and surface-linked catalysts [
13].
In a multistep reaction, where multifunctional compounds are involved, sometimes it is necessary to protect a specific functional group to avoid undesired side reactions. Among the functional group protection reactions, the acetalization of aldehydes and ketones is an important step in fine chemistry and organic synthesis to protect carbonyl groups, leading to pharmaceutical compounds and intermediates of carbohydrates and steroids [
14,
15].
Wang et al. demonstrated the attainment of a mono and a disubstituted optically active α-hydroxy acid with high enantiomeric excesses, which are important intermediates for an asymmetric synthesis. By the transacetalization reaction using BF
3·Et
2O as a catalyst, the asymmetry of (1
S)-(+)-
N,N-diisopropyl-10-camphorsulfonamide was transferred to the α-hydroxy acid, leading to the formation of the desired product [
16].
Biasutto et al. synthesized acetal derivatives as prodrugs of resveratrol, a polyphenol compound that exhibits a range of activities of biomedical interest, such as reducing inflammation, (anti-inflammatory), neuroprotection, protection of the cardiovascular system, and the improvement of glucose processing in diabetes, among others. They observed that the use of acetal derivatives may be a convenient strategy to protect polyphenols and possibly other drugs from metabolism while still allowing a sustained absorption [
17].
The acetalization reaction is usually carried out by treating aldehydes or ketones with an excess of the corresponding alcohol or glycol in an acid medium, and continuously removing the water formed to shift the equilibrium [
18]. Inorganic acids, oxides, zeolites, porous materials, and inorganic complexes are some examples of acid catalysts that can be used for the protection of carbonyl compounds [
18,
19,
20,
21,
22,
23].
Metalloporphyrins are widely used as efficient and selective catalysts for oxidative processes, both in homogeneous and heterogeneous phases [
24]. However, there are few studies that have explored their use as catalysts in functional groups’ protection and reaction and their activity as Lewis acid catalysts.
Suda et al. reported the use of a high-valent manganese porphyrin [Mn(TPP)]Cl as a mild acid catalyst for the rearrangement of
N-phenyl-spirooxaziridines into lactams [
25]. Subsequently, the same group demonstrated the use of a series of metalloporphyrins that are able to effectively catalyze the isomerization reaction of epoxides into carbonyl compounds, where an iron (III) porphyrin [Fe(TPP)]ClO
4 was the most efficient catalyst [
26]. In 1997, Firouzabadi et al. utilized [Fe(TPP)]Cl as an acid catalyst for the silylation reaction of hydroxyl groups in the presence of hexamethyldisilazane (HMDS) as a silylation agent. They found that diphenylcarbinol, which is resistant to silylation with HMDS, could undergo this transformation very easily and with excellent yields in the presence of [Fe(TPP)]Cl at room temperature [
27].
Tangestaninejad et al. used [Fe(TPFPP)]Cl as a new and efficient acid catalyst for the conversion of epoxides into thiiranes, obtaining more than 90% conversion in 15 min of reaction [
28]. The reduction of cyclohexanone to cyclohexanol by the Meerwein–Ponndorf–Verley (MPV) reaction catalyzed by an aluminum(III) porphyrin, [Al(TPP)]Cl, was demonstrated by Inoue et al. The aluminum porphyrin was an efficient acid catalyst with good results [
29].
There are some examples in the literature where metalloporphyrins were employed as catalysts to open epoxides and generate protected diols through the reaction of acetolysis [
30,
31,
32]. However, there are no systematic studies on the acetalization reaction with metalloporphyrins acting as acid catalysts.
In this work, electron-deficient metalloporphyrins iron (III) porphyrins (
Figure 1A,C,D) were used as mild Lewis acid catalysts in the acetalization of benzaldehyde with different protective groups. To the best of our knowledge, this is the first systematic study of this type of reaction catalyzed by metalloporphyrins aiming to protect carbonyl groups.
2. Results and Discussion
2.1. Study of the Dependence on the Number of Fluorine Atoms in the Macrocycle Structure and the Catalytic Activity
The investigation of the catalytic activity of metalloporphyrins for benzaldehyde acetalization started with studies about the influence of peripheral fluorine atoms in the structure of three different iron (III) porphyrins, since these electronegative atoms can withdraw electron density from the macrocycle structure, modulating the charge density over the complex. For this purpose, a first generation porphyrin with no fluorine atoms was selected, [Fe(TPP)]Cl (named
Fe0F), along with two second-generation chromophores: [Fe(TDFPP)]Cl (
Fe2F) and [Fe(TPFPP)Cl] (
Fe5F) (
Figure 1A,C,D respectively) [
7,
11]. These two second-generation macrocycle porphyrins possess two and five fluorine atoms, respectively, for each of the four
meso-aryl rings of the macrocycle structure. For a clear comparison of catalytic tests, these three complexes have chloride as counter ions, avoiding misinterpretation of the role of axial ligands.
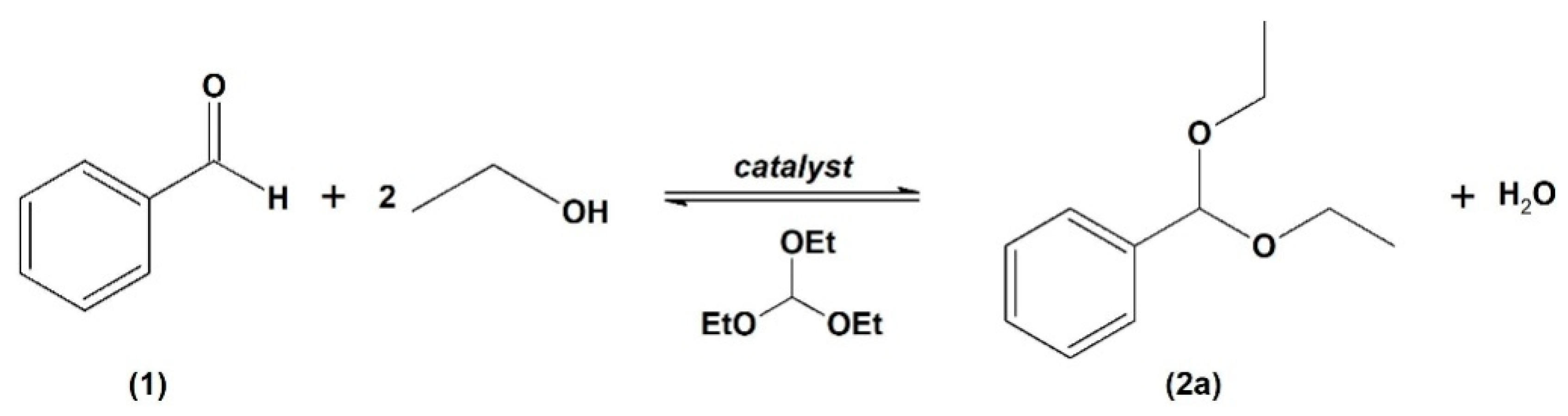
The acetalization of benzaldehyde (
1) was selected as the preliminary reaction using ethanol as a protective group in the presence of triethyl orthoformate (TEOF) as a drying agent, producing benzaldehyde diethyl acetal (
2a). Since water is a by-product of this reaction, a drying agent may be needed to avoid product hydrolysis (
Scheme 1) [
19].
Reactions using a 2% mol of catalyst
Fe0F,
Fe2F, or
Fe5F were carried out in homogeneous media at 70 °C and samples were analyzed after 1 h, 3 h, 6 h, and 24 h of reaction in these early studies (
Table 1). It was clear that the Lewis acid catalytic activity of the investigated iron (III) porphyrins is strictly dependent on the level of fluorine substitution of the aryl group at the
meso positions. No product was observed after 1 h of reaction with the use of
Fe0F and
Fe2F, while after the same time, 84% conversion was reached using
Fe5F, which is the most fluorinated catalyst. After 2 h, full conversion was observed for
Fe5F, and after 24 h, there was an increased conversion for the formation of
2a with rising catalyst fluorination levels. These catalytic results indicate that the inductive effect plays a key role in the regulation of the Lewis acidity of Fe(III), since this is considered the catalytic active species responsible for the activation of the carbonyl group for acetalization reactions, as previously observed by Tangestaninejad and Mirkhani in the conversion of epoxides to thiiranes, which is a Lewis acid-dependent reaction [
28].
Benzoic acid was not detected in the product composition, showing that the catalyst was not active for aldehyde oxidation in these conditions. Since Fe5F was the most active metalloporphyrin in this first screening, it was selected as the catalyst to be investigated in the following experiments.
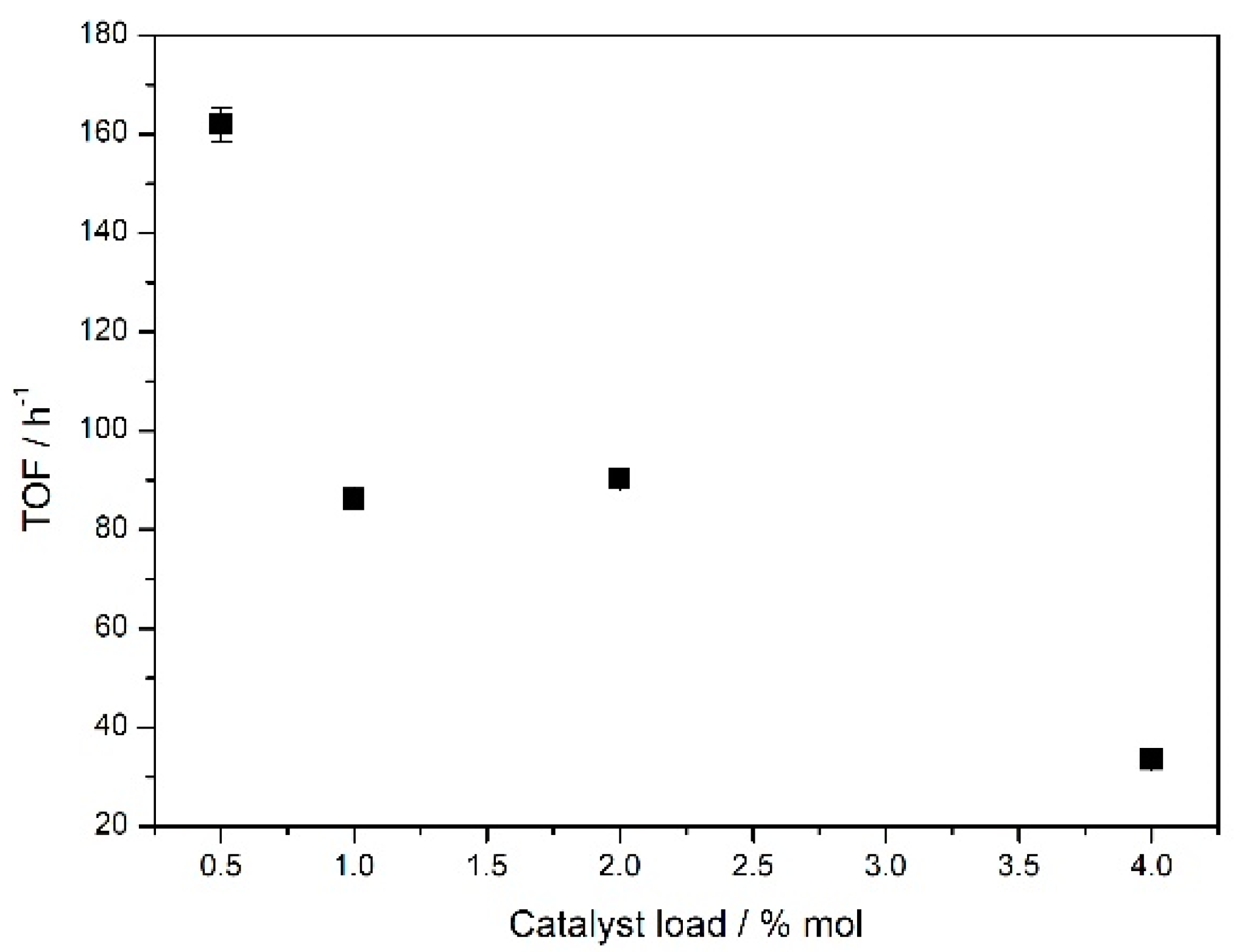
To optimize the catalyst load, reactions were conducted with variations of the
Fe5F/benzaldehyde ratio. Reactions with 0.5%, 1.0%, 2.0%, and 4.0% mol of
Fe5F in relation to
1 were carried out, and all of these compositions led to the formation of
2a (
Figure 2 and
Table S1; Supplementary Materials (SM)). In order to make better comparisons, turnover frequencies (TOF) were determined after 1 h of reaction at 70 °C [
33].
Figure 2 shows that the lowest amount of catalyst (0.5% mol) led to the highest TOF value, while the opposite was observed for the highest catalyst load, 4.0% mol of
Fe5F, yielded TOF equal to 33 h
−1. The more diluted the reaction media, the less aggregated the metalloporphyrins were, which can explain this pattern if one considers that the catalytic species is an isolated and soluble molecule of iron (III) porphyrin in the solution. The lowest TOF value for the use of 4.0% mol of
Fe5F can also be attributed to the total non-solubilization of this amount of catalyst in the reaction medium.
Intermediate values of TOF are appropriate to study the kinetic profiles and other features, since the reaction is not too fast or too slow to determine physicochemical parameters. Reactions conducted with 1.0% and 2.0% mol of catalyst yielded TOF values equal to 86 h−1 and 91 h−1, respectively, inducing the choice of 1.0% mol catalyst for further studies, since this amount resulted in the same TOF of 2.0% mol and used a lesser amount of iron (III) porphyrin.
2.2. Kinetic Studies for 2a Synthesis at Different Temperatures
To evaluate the influence of temperature and time on benzaldehyde acetalization, studies were conducted for the same reaction using 1.0% mol of
Fe5F as catalyst at 70 °C, 65 °C, 60 °C, 55 °C, and 50 °C, and the samples were analyzed at different times up to 6 h (
Table 2 and
Figure S1, SM). A control reaction, in the absence of catalyst, was also performed at 70 °C.
The catalyst Fe5F was active for 2a synthesis at all of the evaluated temperatures, with an increasing rate of acetal formation with rising temperature, and the best result was observed at 70 °C. In contrast, at the same temperature in the control reaction, only 1.8% benzaldehyde was converted to the desired product after 4 h, indicating that Fe5F is the active species for this reaction.
All of the data could be fitted in a first-order exponential equation. After data linearization from zero to 45 min, it was possible to estimate values for pseudo-first order rate constants (
kobs), because ethanol was in large excess in the reaction media (
Table 2). The ratio between the rate constants for the catalyzed and non-catalyzed reactions performed at 70 °C (
kcat/
knon-cat) gives an idea of how fast the catalyst promotes the reaction in comparison to the stoichiometric process. In this case, benzaldehyde acetalization in the presence of 1.0% mol of
Fe5F accelerated 576 times in the homogeneous phase compared to the non-catalyzed reaction.
The activation parameters for this reaction could be determined from kinetic data. From the linearized form of the Arrhenius equation (Equation (1)), activation energy (
Ea) was estimated to be 126 ± 17.2 kJ·mol
−1. Using the Eyring equation in its linearized form (Equation (2)) [
34,
35], it was possible to estimate the activation enthalpy and entropy, ∆
H‡ and Δ
S‡, respectively. In both equations,
A is the pre-exponential factor,
k is the rate constant,
R is the universal gas constant,
T is the temperature,
kB is the Boltzmann constant, and
h is the Planck constant.
Activation enthalpy was estimated to be 119 ± 18.3 kJ·mol
−1 and activation entropy was equal to 80.4 ± 15.9 J·mol
−1·K
−1. Considering experimental errors,
Ea and ∆
H‡ values are close, indicating a convergence between the activation parameters. A positive value of ∆
S‡ for this reaction indicates that the formation of the transition state is a dissociative process with an increase of the system’s disorder [
36].
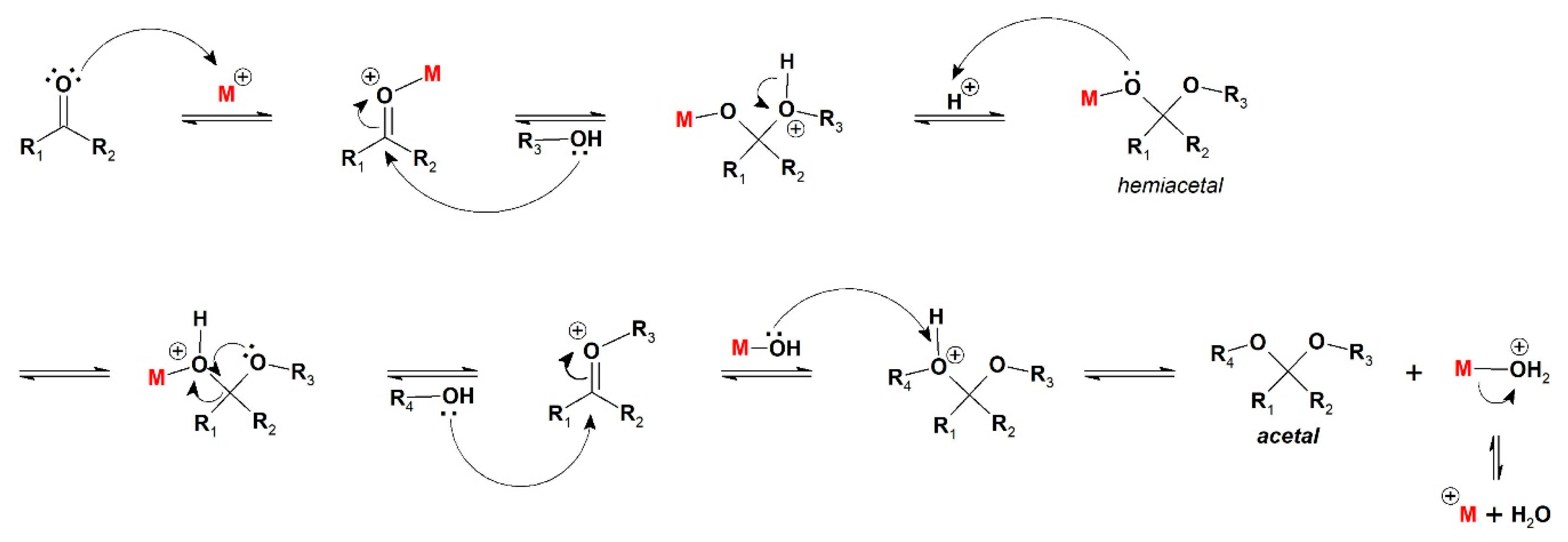
Based on the accepted mechanism of acetal formation from carbonyl compounds and alcohols catalyzed by acids [
37], the first steps are all associative, evolving carbonyl activation by the acid catalyst, nucleophilic attack, hemiacetal formation, and a consecutive nucleophilic attack (
Scheme 2).
Considering
Fe5F as the active catalytic species, and based on the catalytic results observed (
Table 2 and
Figure S1-SM), the coordination of the oxygen atom of carbonyl to Fe(III) center (M+) can enhance carbonyl’s electrophilicity. As ∆
S‡ has a positive value, the product release from catalyst can be considered the determining step in this case, as it is a dissociative step.
2.3. Studies of Solvent Composition and the Role of TEOF
To clarify the real role of TEOF in the acetalization reaction and its influence on the final product, some experiments were performed with different solvent composition media. Reactions were carried out at 60 °C, in the presence or absence of the catalyst
Fe5F, with and without TEOF and in different solvents: ethanol, methanol, acetonitrile (CH
3CN), carbon tetrachloride (CCl
4), and excess of TEOF. The reaction conditions are summarized in
Table 3.
Analysis of the three tests shows that the synthesis of 2a is dependent on ethanol in the reaction medium. The presence of TEOF with ethanol and Fe5F (Test I) yielded higher conversion than the same reaction without the drying agent (TOF = 68 h−1 for Test I and 54 h−1 for Test II), while the absence of alcohol and a large excess of TEOF (Test III, molar ratio TEOF/benzaldehyde = 60) didn’t yield results. A no-product reaction was observed when TEOF was mixed with 1 in the presence of catalyst and with acetonitrile (Test IV) and carbon tetrachloride (Test V) as solvents—polar and non-polar respectively—indicating the metalloporphyrin catalytic activity.
The reaction of
1 with methanol in the presence of
Fe5F and absence of TEOF (Test VI) rendered benzaldehyde dimethyl acetal (
2b) as the single product, with a TOF value equal to 23 h
−1, showing that alcohol is the protecting group. When triethyl orthoformate was added to the reaction medium (Test VII),
2b was produced at TOF = 21 h
−1 and benzaldehyde ethyl methyl acetal (
2c) was detected in lower concentration (TOF = 1.7 h
−1). The presence of
2c, a mixed acetal, in the product composition indicates that ethanol produced from TEOF hydrolysis can serve as a protective group. It is important to state that
2c started to be detected after 20 min of reaction. In previous reactions, TEOF was not able to act as an acetalizing group (see tests III to V in
Table 3), so we assumed that the ethyl group was derived from TEOF hydrolysis. The formation of
2c can be explained by the acid catalysis mechanism [
37], where the formation of hemiacetal is an intermediate step, corroborating the evidence that
Fe5F acts as a Lewis acid catalyst. Diethyl acetal
2a was not observed as a product from these reactions. Control reactions in the absence of
Fe5F (tests VIII to XI) didn’t result in any product, confirming the catalytic action of the compound
Fe5F as a Lewis acid catalyst.
2.4. Study of the Scope of Acetalization Reactions Catalyzed by Fe5F
The investigation of the scope of carbonyl compounds acetalization catalyzed by
Fe5F took place using
1 and acetophenone (
3)—an aldehyde and a ketone, respectively—and as protective groups, we used ethanol, methanol, TEOF, ethylene glycol, propylene glycol, glycerol, and cyclooctene oxide. All of the reactions were carried out with 1% mol of
Fe5F related to aldehyde or ketone at 70 °C (except for methanol, where the temperature was 60 °C due to its boiling point) and the molar ratio between protective agent and carbonyl compound was of 60—the reactions were performed without solvent (
Table 4).
All of the reactions with 1 yielded the expected acetal products, with no by-products detected after 3 h. Control reactions were performed in the same conditions in the absence of catalyst, and no conversion was detected after the same time, reinforcing the concept that Fe5F is the true catalytic active species. Higher conversion values were observed when glycols were applied as protective agents; products 2-phenyl-1,3-dioxolane (2d) and 4-methyl-2-phenyl-1,3-dioxolane (2e), which were obtained from the reaction of 1 with ethylene and propylene glycol, respectively, were synthesized with >99% conversion in 1 h (TOF = 99 h−1). In contrast, acetals 2a and 2b, from ethanol and methanol, were produced with TOF values of 51 h−1 and 31 h−1, respectively.
Both isomers from reaction between
1 and glycerol, one from 1,3-addition (
2f) and the other from 1,2-addition (
2g), were detected but not separately quantified: the conversion for the mixture was 36% after 1 h of reaction. In addition, an epoxide, cyclooctene oxide, was a good protective agent for
1, giving acetal
2h with a TOF value of 18 h
−1. This product can be obtained from the hydrolysis of epoxide in a first step, generating 1,2-diol, which can then serve as the nucleophile for acetalization [
30,
31,
32].
No conversion was observed, even after 3 h of reaction, when ketone
3 was used as a substrate in the presence of ethanol, methanol, TEOF, and cyclooctene oxide. Cyclic acetals derived from acetophenone, 2-methyl-2-phenyl-1,3-dioxolane (
4d) and 2,4-dimethyl-2-phenyl-1,3-dioxolane (
4e) were obtained at TOF values of 18 h
−1 and 12 h
−1, respectively. Only a small amount of the mixture of isomers
4f and
4g was detected after 1 h of reaction (TOF = 2.1 h
−1). The higher activity of
Fe5F toward benzaldehyde acetalization in comparison to acetophenone can be related to the carbonyl electronic features. From the accepted mechanism for acetal formation [
37] and by the perspective of an iron (III) porphyrin acting as a Lewis acid (
Fe5F being represented by M
+ in
Scheme 2), carbonyl activation would come from the coordination of its oxygen atoms to the metalloporphyrin, making the carbon atoms more electrophilic. The presence of a methyl group adjacent to C=O in the ketone reduces the positive charge of carbon due to the inductive effect and hyperconjugation effect. This set of effects may lead to lower reactivity when compared to aldehyde [
37].
2.5. Study of the Selectivity of Benzaldehyde Acetalization in Competitive Reactions
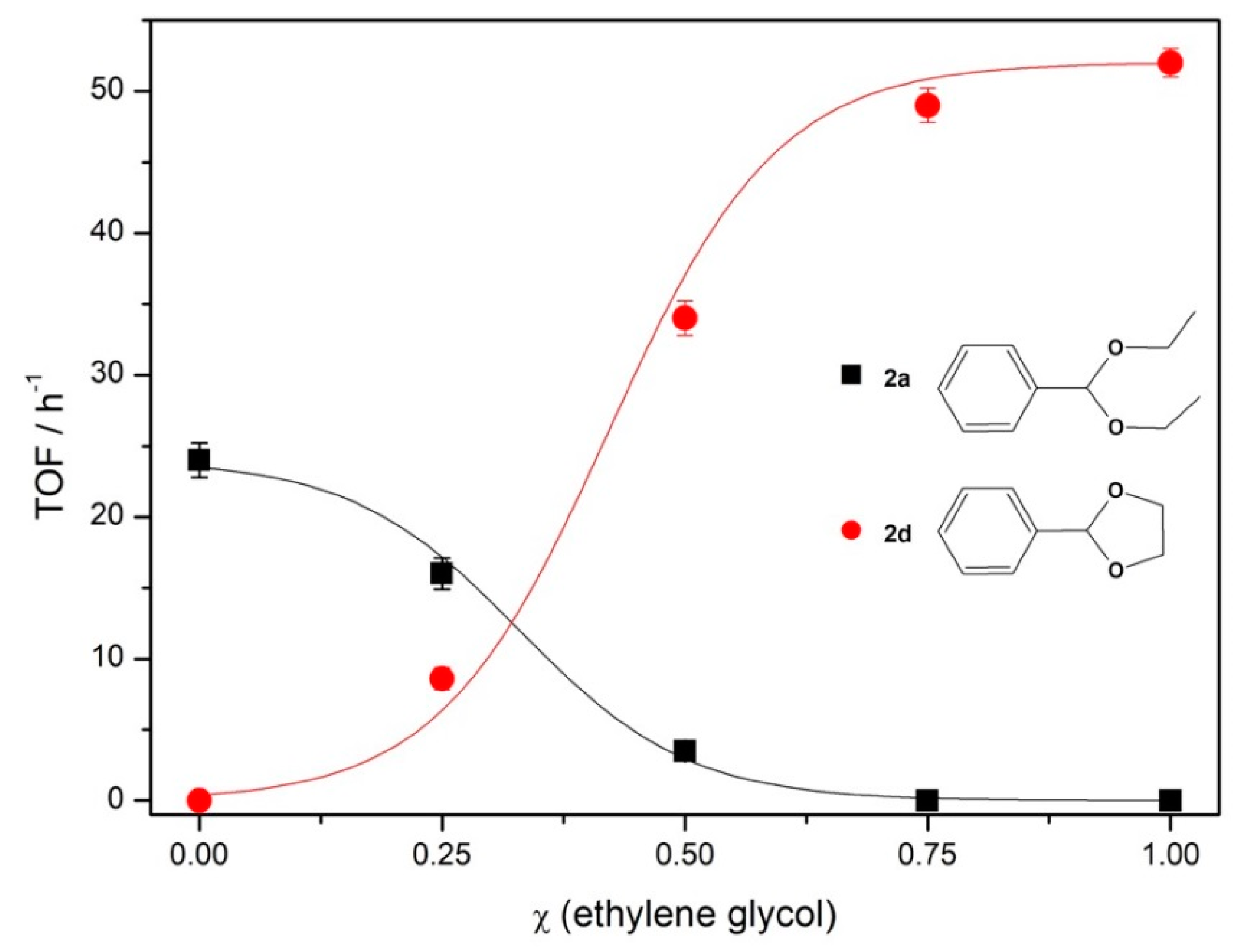
To evaluate the selectivity for benzaldehyde acetalization using glycols instead of alcohols, competitive reactions were performed where
1 was reacted simultaneously with ethanol and ethylene glycol in different molar fractions, using 1% mol of
Fe5F at 70 °C during 30 min (
Figure 3).
From the results of the TOF variation in the function of molar fraction of ethylene glycol in ethanol (
Figure 3), it was possible to verify that competitive reactions are selective to the formation of cyclic acetal
2d when compared to the aliphatic
2a. In the presence of equimolar amounts of alcohol and glycol (molar ratio of benzaldehyde/ethylene glycol/ethanol = 1:30:60, because two molecules of ethanol are necessary to produce one acetal while just one of glycol is needed), the TOF of 34 h
−1 was obtained from
2d synthesis against 3.5 h
−1 from
2a. In comparison to the aliphatic acetal, the cyclic one was produced in a ratio of 13:1. No diethyl acetal was produced when the mixture of protective agents was 75% ethylene glycol and 25% ethanol.
This behavior can be related to both kinetic and thermodynamic effects. From the acetalization mechanism, after the formation of hemiacetal intermediate, there is a second nucleophilic attack from the acetalizing agent (
Scheme 2) [
37]. In the case of ethanol, another molecule of alcohol is required to promote the addition, while the second attack for a glycol is an intramolecular process, since the free hydroxyl group acts as the nucleophile, making the process faster and yielding the kinetic product.
From the thermodynamic perspective, to obtain acetals 2a and 2d, it is necessary to break a C=O bond and form two new C–O bonds, so there will be no difference in enthalpy variation (∆H) for both systems. For 2a synthesis, two molecules of ethanol are needed to react with one of 1, while for 2d, only one molecule of glycol is necessary. In both reactions, the products are one acetal and one water molecule. Therefore, the variation of the number of molecules for 2a synthesis is negative and for 2d is nil, which gives a negative entropy variation (∆S) in the first case. From the Gibbs free energy equation, a negative value of ∆S increases the free energy (∆G), making the process less favorable than another at the same temperature and with the same ∆H values.
2.6. Kinetic Studies for the Synthesis of Cyclic Benzaldehyde Acetals
Since cyclic acetals from benzaldehyde—1,3-substituted dioxolane rings—were selectively synthesized with the use of
Fe5F as catalyst, kinetic studies of the reactions between
1 and ethylene glycol or propylene glycol were done at different temperatures (30 °C, 35 °C, and 40 °C). Reactions were carried out in pseudo-first order conditions, using a 100-fold excess of glycol in relation to
1 (
Table 5 and
Figure S2; SM). Glycols were applied as solvents as well.
The kinetic profiles for both substrates converged to first-order exponential growth and values of
kobs could be determined, such as in the case of
2a synthesis (
Table 2) due to the pseudo-first order experimental conditions. The formation of cyclic acetals was faster than the formation of
2a at lower temperatures. When the synthesis of
2d and
2e was performed at 45 °C, more than 90% conversion was reached in the first five minutes, and it was not possible to determine the kinetic parameters at higher temperatures. Control reactions in the absence of a catalyst were also carried out at all of the temperatures, but no product was detected even after 3 h of reaction, such as those observed in previous tests (
Table 4).
Ethylene glycol provided dioxolane moiety faster than propylene glycol did, with
kobs values for
2d synthesis 6.7 times higher than those for
2e. Due to the elevated reaction rates, TOF values were estimated at 15 min and were higher for
2d than for
2e. These differences in reaction rates for different protective groups at all of the investigated temperatures can be related to the medium’s viscosity. Ethylene glycol is less viscous (16.1 mPa s at 25 °C) than propylene glycol (40.4 mPa s at 25 °C) [
38], which leads to more efficient stirring and a better diffusion of aldehyde and metalloporphyrin molecules dissolved in the medium, causing more frequent collisions than in propylene glycol. It is also known that the rate constants are inversely proportional to medium viscosity [
39], and this was confirmed by the comparison between the two glycols when applied as protective agents. These factors can lead to a higher product conversion in a shorter time, enhancing
kobs and TOF values, as can be seen (
Table 5 and
Figure S2; SM).
When TOF values for
2a synthesis in ethanol and triethyl orthoformate medium (
Table 2) are compared to TOF values for
2d or
2e (
Table 5), it is possible to note that even in more viscous systems (for comparison, ethanol viscosity is 1.07 mPa s at 25 °C [
38]) and at lower temperatures, Fe5F showed greater catalytic activity toward benzaldehyde acetalization.
In addition, for the synthesis of cyclic acetals, it was not necessary to use a drying agent such as TEOF, indicating that with a large excess of glycol, the medium stabilizes acetal without requiring the use of an apparatus for water removal, such as a Dean–Stark system [
18].
3. Materials and Methods
All of the solvents and reagents were of analytical grade, purchased from Aldrich (São Paulo, Brazil), Merck (São Paulo, Brazil), or Fluka (Mexico City, Mexico), and used without purification, unless otherwise stated. Benzaldehyde was treated with an aqueous NaHCO
3 solution to remove benzoic acid, distilled under reduced pressure, and kept under argon atmosphere [
40].
Free-base porphyrins 5,10,15,20-tetrakis(phenylporphyrin) [H
2(TPP)] and 5,10,15,20-tetrakis (2,6-difuorophenylporphyrin) [H
2(TDFPP)] were previously synthesized by our research group using Lindsey’s method [
41] and 5,10,15,20-tetrakis(pentafluorophenylporphyrin [H
2(TPFPP)] was prepared by the nitrobenzene method [
42]. The free-base ligands were purified by a column chromatography and characterized by UV-Vis, FTIR, and
1H NMR spectroscopies, as previously described [
43,
44,
45]. Iron (III) complexes were obtained from the Kobayashi procedure [
46], purified and characterized by UV-Vis, FTIR, and EPR spectroscopies as previously described for 5,10,15,20-tetrakis(phenylporphyrin) chloride, [Fe(TPP)]Cl (
Fe0F) [
43], 5,10,15,20-tetrakis (2,6-difluorophenylporphyrin) chloride, [Fe(TDFPP)Cl] (
Fe2F) [
44], and 5,10,15,20-tetrakis (pentafluorophenylporphyrin) chloride, [Fe(TPFPP)Cl] (
Fe5F) [
45].
For acetalization reactions of carbonyl compounds, standard experiments were carried out as described: a defined amount of iron porphyrin (the catalyst Fe0F, Fe2F, or Fe5F) was placed in a 1.5-mL screw top vial, followed by addition, in the next sequence, of the solvent (when necessary), the protective group (ethanol, methanol, triethyl orthoformate, ethylene glycol, propylene glycol, glycerol, or cyclooctene oxide) and the carbonyl compound (benzaldehyde or acetophenone). The system was placed in a thermostatic bath and kept under magnetic stirring and open atmosphere. Samples were extracted at different times, diluted in ethyl acetate, and analyzed by gas chromatography (GC), while the products were identified in a mass spectrometer coupled to a gas chromatograph (GC-MS) and quantified by GC by the area normalization method. Details about the load of catalyst, solvent, protective groups, and carbonyl compounds, such as temperature, time reactions, and other experimental parameters, were previously described in the Results and Discussion section. To quantify the products from the catalytic reactions, an Agilent 6850 gas chromatograph (flame ionization detector, or FID) equipped with a DB-WAX capillary column (30 m × 0.25 μm × 0.25 μm; J&W Scientific) was used. During analysis, the oven temperature was set at 80 °C for 2 min, and then raised to 220 °C at a rate of 20 °C min−1, which was kept isothermally for 5 min.
4. Conclusions
A systematic study regarding the catalytic activity of iron (III) porphyrins with different levels of fluorine substitution in the meso-aryl porphyrin ring positions for protection of benzaldehyde with ethanol and triethyl orthoformate (TEOF) was successfully performed, showing that the most substituted catalyst produced the highest conversion rates. This may be due to the increasing Lewis acidity of the Fe(III) center caused by the inductive effect of the peripheral electrons withdrawing atoms, if carbonyl activation occurs through coordination to the ferric center. The formation of benzaldehyde diethyl acetal (2a) converged to pseudo-first order kinetic behavior, and it was shown to be temperature-dependent. The presence of 1% mol of Fe5F enhanced the stoichiometric reaction rate 576 times, proving its catalytic activity. The presence of TEOF in the reaction media enhanced the reaction rate, which may have been due to the drying effect of TEOF, which can be hydrolyzed in the presence of water, which is the undesired by-product in the acetalization reaction.
It was not necessary to add a drying agent to obtain acetals with different protective groups. Benzaldehyde and acetophenone were converted into acetals in the presence of alcohols, glycols, glycerol, and an epoxide, but selectivity to cyclic acetals from ethylene and propylene glycol was observed, even in competitive reactions against ethanol. The kinetic and thermodynamic features may explain this behavior, even in systems that are more viscous and at lower temperatures.
To the best of our knowledge, this is the first report of a systematic investigation of the catalytic activity of metalloporphyrins for acetalization reactions. This work can be a proof-of-concept for further investigations, and the application of Fe(III) and other metalloporphyrins in sequential processes in which one of the steps depends on a Lewis acid or on the protection of carbonyl groups.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

























