Abstract
The oxidation of alcohols to the corresponding carbonyl products is an important organic transformation and the products are used in a variety of applications. The development of catalytic methods for selective alcohol oxidation have garnered significant attention in an attempt to find a more sustainable method without any limitations. Copper, in combination with 2,2,6,6-tetramethyl-1-piperidine N-oxyl (TEMPO) and supported by organic ligands, have emerged as the most effective catalysts for selective alcohol oxidation and these catalyst systems are frequently compared to galactose oxidase (GOase). The efficiency of GOase has led to extensive research to mimic the active sites of these enzymes, leading to a variety of Cu/TEMPO· catalyst systems being reported over the years. The mechanistic pathway by which Cu/TEMPO· catalyst systems operate has been investigated by several research groups, which led to partially contradicting mechanistic description. Due to the disadvantages and limitations of employing TEMPO· as co-catalyst, alternative nitroxyl radicals or in situ formed radicals, as co-catalysts, have been successfully evaluated in alcohol oxidation. Herein we discuss the development and mechanistic elucidation of Cu/TEMPO· catalyst systems as biomimetic alcohol oxidation catalysts.
1. Introduction
Aldehydes and ketones are important intermediates in the pharmaceutical industry [1,2] as well as for the synthesis of fine chemicals such as fragrances and food additives [3]. One of the most widely used reactions to synthesise these carbonyl products is via the oxidation of alcohol substrates. However, several limitations are associated with these alcohol oxidation methods, such as the use of toxic or hazardous heavy metal salts or expensive catalysts containing transition metals such as Ru [4] and Pd [4,5] at high oxygen pressures. In addition, catalyst cost and efficiency, stability as well as the complexity of the reaction set-up are additional drawbacks of these systems. As a result, these catalysts pose environmental and safety concerns which led to the development of new environmentally benign Cu-based oxidation systems [4,6].
Cu-based catalyst systems, specifically in combination with 2,2,6,6-tetramethyl-1-piperidine N-oxyl (TEMPO·), at ambient temperatures and molecular O2 or air as the oxidant, are some of the most effective catalysts for the oxidation of alcohols to the corresponding aldehydes [2,3,7,8,9,10].
Initially it was believed that these Cu/TEMPO·-catalysed oxidation reactions proceeded via the TEMPO-based oxoammonium cation (TEMPO+) mechanistic pathway [7]. However, in ensuing years, the lack of reactivity between CuII and TEMPO· during the oxidation reaction was highlighted [11] and this led to the development of alternative mechanistic pathways, implicating TEMPO-H or TEMPO· as reactive intermediates.
A number of reviews on catalytic copper oxidation chemistry have appeared, with the focus specifically on the application of Cu-based catalyst systems in the aerobic functionalisation of C-H bonds [12]. Furthermore, some of the reviews also cover copper and other metal catalysts, in combination with a range of oxidants such as oxygen, as well as possible mechanistic pathways for selective reaction types [13,14,15,16].
This review specifically focuses on different Cu/TEMPO· catalyst systems for alcohol oxidation, up to 2018, the different partially contradicting mechanistic pathways proposed for this transformation, as well as the evaluation of alternative co-catalysts to replace TEMPO·. The importance of copper as metal, as well as in combination with TEMPO·, for alcohol oxidation reactions are described. Catalyst systems using water as the only solvent are mentioned, but not discussed in detail. The different proposed mechanistic pathways for alcohol oxidation, are discussed in detail, with particular emphasis on experimental investigations. Finally, recent advances in the development of alternative co-catalysts are described.
2. Copper in Biomimetic Oxidation Catalysis
Copper is a cheap and biocompatible metal, found in various metalloproteins, such as enzymes, which are associated with the binding of molecular O2 in mild and highly selective aerobic oxidative transformations [17]. One of these enzymes is galactose oxidase (GOase), a type-II mononuclear Cu-enzyme that mediates aerobic alcohol oxidation at a Cu-centre with a redox-active phenolate/phenoxyl radical ligand [11,18].
A tyrosine unit coordinates in the axial position of the square-bipyramidal coordination sphere of the CuII-species (Scheme 1, intermediate A) [19,20,21]. The equatorial ligand sites are occupied by two histidine imidazole units, a modified tyrosine unit with a cross-linked cysteine unit and either H2O or an acetate molecule. The alcohol substrate coordinates to the active Cu-centre, B, and is deprotonated by the phenolic Tyr-495. The phenoxyl radical is involved in the abstraction of a β-H-atom from the coordinated alcohol, C, to afford the aldehyde through a single electron transfer (SET) with simultaneous formation of a CuI-species, D. In the last step, CuI is re-oxidised to CuII, E, while the reduction of O2 to H2O2 results in the formation of the initial intermediate, A [22].
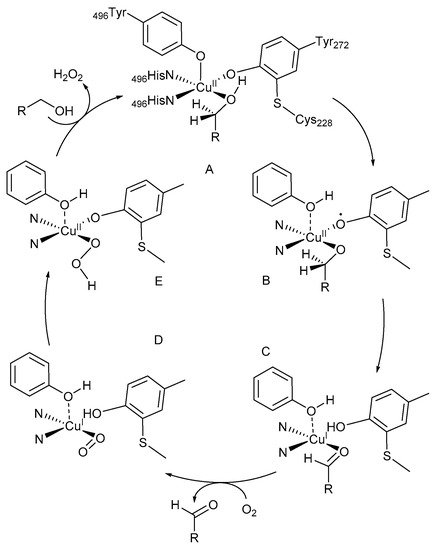
Scheme 1.
Proposed reaction mechanism for catalysis reactions by galactose oxidase (GOase). Reproduced with permission from Whittaker and co-workers [22]. Copyright Elsevier, 1993. For intermediates B–E, the ligands in the Cu coordination sphere are shown in their reduced form.
During the oxidation reaction, the CuII-ion coordinates with the tyrosyl radical to afford CuII-(·OR) intermediates [8,11,19,20,23,24]. These intermediates are used in the two-electron oxidation of primary alcohol substrates and the reduction of dioxygen to H2O2 (Scheme 1). The initial CuII-species is restored afterwards and available for a reaction with another tyrosine unit (Scheme 1, intermediate A).
The ability to successfully mimic particular geometries around the copper centre and the presence of a radical that can coordinate to the Cu-centre offers a platform for the development of different copper-based homogeneous catalyst systems. In addition, the low costs, ready availability of Cu and their interesting spectroscopic properties resulted in the development of numerous Cu-based catalysts for the oxidation of alcohols [19,25].
Consequently, it is surprising that only a few catalyst systems consisting of cheap and ‘green’ Cu-based catalysts and molecular oxygen [7,26,27,28] or ambient air [2,8,10] are known so far. However, the mechanistic pathway by which these Cu-based catalyst systems mediate the oxidation of alcohols is not yet fully understood [25].
3. Nitroxyl Radicals in Biomimetic Oxidation Catalysis
Nitroxyl radicals and their diamagnetic precursors are popular in the pharmaceutical and food industry in order to improve the quality of alcohols, fragrances and flavours [29]. These radicals contain N,N-disubstituted NO-groups possessing one unpaired electron, which is (usually) unreactive to air and moisture, allowing easy handling and storage [30,31]. In addition, these radicals act as antioxidants [32] and they are used as inhibitors in free radical processes. Their unique structural and electronic properties have been exploited as co-catalysts in synthetic GOase mimics.
Nitroxyl radicals may be classified, based on their properties and applications (Table 1) [33]. The first group consist of the stable radicals, including conjugated and non-conjugated radicals, while the second group consist of the so-called reactive radicals. Stable radicals scavenge free radicals and could therefore be used as inhibitors of free radical autoxidations. In contrast, reactive radicals such as N-hydroxyphthalimide (NHPI) would rather catalyse the autoxidation reactions through the formation of the phthalimide N-oxyl radical (PINO·) [33].

Table 1.
A summary of the two types of nitroxyl radicals, based on their properties and applications [33]. Reproduced with permission from Sheldon and co-workers. Copyright Elsevier, 2006.
An example of the conjugated radicals is the diphenyl nitroxyl radical where the unpaired electron is delocalised over the entire molecule and these radicals are not used for alcohol oxidation. Non-conjugated radicals include di-tert-alkyl nitroxyl radicals and α-substituted piperidin-1-oxyl radicals (TEMPO·), where the unpaired electron is only delocalised over the N-O bond. These radicals are only stable in the absence of α-hydrogens [34]. In the presence of α-hydrogens, the radical undergoes a disproportionation reaction to afford a hydroxylamine, 1, or a nitrone, 2, either or both of which may undergo further reaction (Scheme 2) [34].
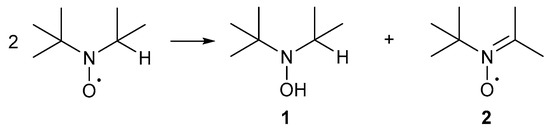
Scheme 2.
The disproportionation of di-tert-alkyl nitroxyl radicals affords hydroxylamine, 1, and nitrone, 2. Adapted from De Nooy and co-workers [34].
The most important sub-group of the stable non-conjugated radicals is TEMPO· and it was the first type to be synthesised. TEMPO· may oxidise a number of functionalities [3,29,35,36] and most of the studies on this stable radical, in combination with a metal such as copper, have been reported for the transformation of alcohols to the corresponding carbonyl compounds [2,8,10,35,36,37].
However, in the absence of a transition metal, TEMPO·-based systems are safer, cheaper and more environmentally friendly, but high TEMPO· loadings are required, which are not recoverable after the reaction [38] In terms of substrate selectivity, these TEMPO·-based catalyst systems are more selective for benzylic alcohols, while aliphatic alcohols either showed low oxidation activities or full conversion towards a mixture of products, which include aldehydes and esters (over-oxidation) [39].
4. Cu/TEMPO· Catalyst Systems
Several CuI/TEMPO·- and CuII/TEMPO·-based catalyst systems were reported in recent years and have proven to be highly efficient for the transformation of a broad range of primary and secondary alcohols to the corresponding aldehydes and ketones. The different Cu/TEMPO catalyst systems for aerobic alcohol oxidation of alcohols to aldehydes are summarised in Table 2 [2,7,8,10,28,40,41,42,43,44,45].
Table 2.
Different Cu/TEMPO· catalyst systems for the aerobic oxidation of alcohols. The different Cu-precursor, ligand, base and solvent system and also the ability to oxidise primary (benzylic, allylic and aliphatic) and secondary alcohols, are summarised.
Semmelhack and co-workers reported the first CuI/TEMPO· catalyst system in the absence of ligand in 1984 (Scheme 3) [7]. They used N,N-dimethylformamide (DMF) as the solvent under an O2 atmosphere.
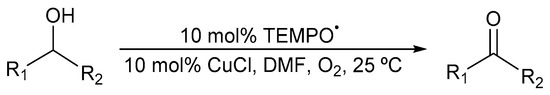
Scheme 3.
CuI/TEMPO· catalyst system in DMF for aerobic alcohol oxidation [7]. Reproduced with permission from Semmelhack and co-workers. Copyright American Chemical Society, 1984. Successful oxidation was possible for a variety of primary alcohols (85 to 100%). Selectivity: primary over secondary alcohols, even in the presence of an excess of secondary alcohols.
Complete conversion to the aldehyde was possible for primary benzylic and allylic alcohols, without any over-oxidation. However, aliphatic alcohols were largely unreactive [21] and stoichiometric quantities of Cu and TEMPO· were needed for optimal activity [7,47]. In addition, they also found that their catalyst system was more selective for the oxidation of primary alcohols than secondary alcohols, even in the presence of an excess of secondary alcohols [7].
In 2000, Knochel and co-workers reported a Cu/TEMPO· catalyst system using a mixture of chlorobenzene/perfluorooctane as a biphasic solvent system in combination with fluoroalkylsubstituted 2,2′-bipyridine (bpy) ligands [27,44]. Operating at 90 °C, a broad range of primary and secondary benzylic, allylic and aliphatic alcohols could be oxidised to the corresponding carbonyl products. The oxidation of benzylic alcohols was found to be faster than the oxidation of aliphatic alcohols, while for secondary alcohol substrates, the success of the oxidation was dependent on the steric environment of the alcohol functionality [44] Additionally, with the use of the biphasic solvent system, it was possible to reuse the catalyst up to eight times with only minor losses in catalytic activity between recycling runs [44].
In 2001, Minisci and co-workers, reported a highly efficient and cost-effective method for the oxidation of primary and secondary alcohols. They used TEMPO· in combination with Co/Mn or Cu/Mn nitrates and acetic acid as solvent, at ambient pressures and temperatures [37]. MnII nitrates was found to be more effective than CoII or CuII, however, the combination of MnII nitrates with either CoII or CuII nitrates resulted in an increase in the catalytic efficiency of the oxidation reaction. Furthermore, the acidic solution is used for the disproportionation of TEMPO· to afford the oxoammonium oxidant for alcohol oxidation, which does not occur in a non-acidic solution. After the reaction, TEMPO· is regenerated through the use of O2 and the metal salt. As a result, TEMPO· is responsible for the generation of the oxoammonium salt for the alcohol oxidation and it could also be used to inhibit further oxidation of the carbonyl products [37].
Later, in 2002, Ansari and Gree reported a copper(I) chloride (CuCl)/TEMPO· catalyst system in an ionic liquid, 1-butyl-3-methylimidazolium hexafluorophosphate ([bmim]PF6), rather than a traditional organic solvent for aerobic alcohol oxidation. This catalyst system was used for the efficient oxidation of alcohol substrates to their corresponding carbonyl products, with no evidence of over-oxidation, in the case of primary alcohols, to carboxylic acids [43]. The conversion of benzylic and allylic alcohols was found to be faster and more efficient than for aliphatic alcohols, which is in agreement with reactions performed in classical organic solvents due to the instability of the nitrosonium ion in the presence of aliphatic alcohols and O2 [7]. Furthermore, they found good solubility of gases such as O2 in the ionic solvent, as well as the recyclability and the ability to reuse the ionic solvent in multiple reactions. They also demonstrated the recyclability of the ionic solvent in the oxidation of different types of alcohol substrates. Unfortunately, they could not recycle the rest of the catalytic system, due to the slow decomposition of the TEMPO· co-catalyst during the oxidation reaction. Independently, Jiang and Ragauskas reported a Cu-free hydrogen bromide (HBr)/TEMPO·/hydrogen peroxide (H2O2) catalyst system in [bmim]PF6 for the selective oxidation of electron-deficient and electron-neutral benzylic alcohols with excellent yields [48]. Their catalyst system was inspired by the TEMPO·-Br2/I2 catalyst system for alcohol oxidation [49], as well as the H2O2-HBr system for benzylic bromination [50]. An ionic solvent was used due to its immiscibility with water, but unfortunately TEMPO· could not be recycled after the reaction. Due to the costs associated with TEMPO· and its lack of recyclability, it was replaced with the commercially available acetamido-TEMPO·. Acetamido- TEMPO· co-catalyst could be recycled and reused in multiple oxidation reactions without negatively affecting the conversion, selectivity or yields of the corresponding aldehyde product [48].
Punniyamurthy and co-workers reported a catalyst system, in 2003, employing a salen-type CuII-complex and H2O2 as the oxidant [51]. While primary alcohols were over-oxidised to their corresponding carboxylic acid analogues, the oxidation of secondary alcohols to the corresponding ketones was fast. Sheldon and co-workers improved their previously reported catalyst system by developing an uncomplicated and easily handled copper(II)bromide (CuIIBr2)/TEMPO· catalyst system with 2,2′-bipyridine (bpy) as the ligand and potassium tert-butoxide (KOtBu) as the base in an acetonitrile (MeCN) and H2O (2:1 system) solvent mixture with ambient air as the oxidant (Scheme 4) [3,8,35]. They could successfully oxidise aliphatic alcohols at elevated temperatures and increased catalyst loading [47].
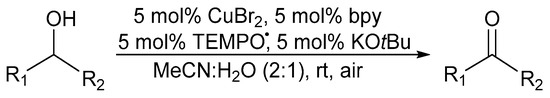
Scheme 4.
A (bpy)CuBr2/TEMPO·/KOtBu catalyst system in MeCN:H2O (2:1), as the solvent system, for the aerobic alcohol oxidation [3]. Reproduced with permission from Sheldon and co-workers. Copyright Royal Society of Chemistry, 2003. Successful oxidation was possible for a variety of primary alcohols (61 to 100%). Selectivity: primary over secondary alcohols (always >99% based on GC).
The role of the base is to deprotonate the alcohol substrate to afford an alkoxide species that can coordinate to the CuII-centre. Furthermore, TEMPO· coordinates in an η2 manner to a CuII-centre, 3, after which a β-H atom is transferred to TEMPO· to afford a CuII/TEMPO-H coordinated species, 4 (Scheme 5) [3]. The aldehyde, TEMPO-H and a CuI-species, 5, are formed in the last step through intramolecular one-electron transfer.
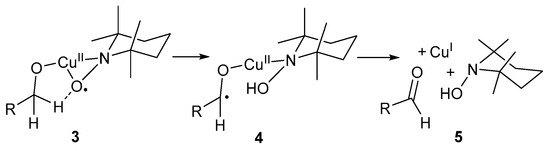
Scheme 5.
The proposed role of TEMPO· in the aerobic oxidation of aliphatic alcohols [3]. Reproduced with permission from Sheldon and co-workers. Copyright Royal Society of Chemistry, 2003.
This catalyst system was used for optimal oxidation of primary benzylic and allylic alcohol substrates [3]. In the case of primary alcohols, the second β-H atom can interact with the O-atom of TEMPO-H, stabilising the radical intermediate, 7 (Scheme 6).
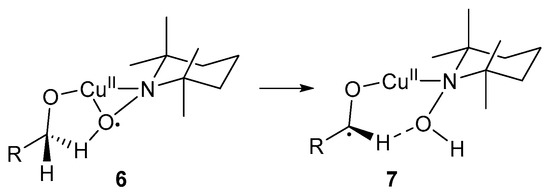
Scheme 6.
The interaction between the second β-H atom of primary alcohols and TEMPO-H [3]. Reproduced with permission from Sheldon and co-workers. Copyright Royal Society of Chemistry, 2003.
The oxidation of secondary alcohols is slower than that of primary alcohols, as observed in other TEMPO·-mediated systems [5,52]. This is possibly due to the lack of stabilisation of the radical species, as observed for primary alcohols, as well as the steric effects of the methyl group, which hinder the formation of the intermediate, 8 (Scheme 7) [3]. This intermediate is important in the C-H abstraction step from the alcohol substrate. Furthermore, the oxidation of activated alcohols is faster than those of aliphatic alcohols because the H-atom abstraction from the α-carbon by TEMPO· was the rate-determining step (Scheme 5, 4 and 5) [3].
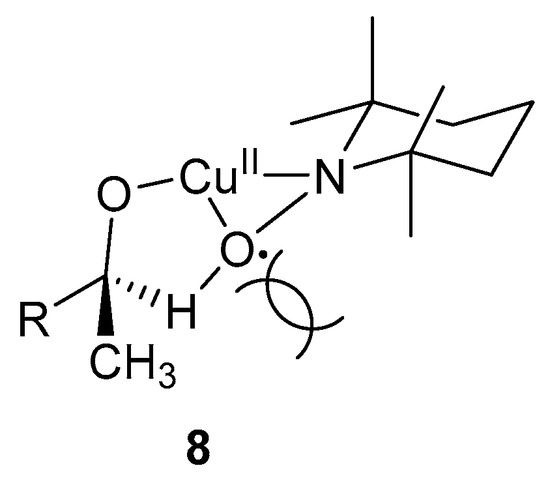
Scheme 7.
Possibilities for the lack of reactivity of secondary alcohol substrates due to (a) the stabilisation of radical species 7 (Scheme 6) by the second β–hydrogen of primary alcohols and (b) the methyl group of secondary alcohols cause steric hindrance, thereby preventing the formation of species 8 [3]. Reproduced with permission from Sheldon and co-workers. Copyright Royal Society of Chemistry, 2003.
Furthermore, they also found that the Cu-catalyst was still active after complete substrate consumption and with the addition of more alcohol substrate and TEMPO· to the reaction mixture, the oxidation reaction could continue. Therefore, their catalyst system is attractive from both economic and environmental viewpoints, but there are still some drawbacks concerning its industrial use, especially in terms of the high concentrations required, the costs of catalysts and co-catalysts and lastly the presence of an activator, KOtBu [42,53]. As a result, Geißlmeir and co-workers improved on these drawbacks, in 2005, by using elemental, fine powdered copper rather than a copper salt. In this way, the difference in reactivity observed for copper salts with different counter-ions is avoided [35]. They also used inductively coupled plasma (ICP) analysis to determine that only 0.22 mol % of the Cu-catalyst was active during the oxidation reaction, and by decreasing the concentration of the Cu-catalyst, they developed a more environmentally friendly Cu/TEMPO·-based catalyst system for alcohol oxidation [42]. The costly KOtBu base was also replaced by sodium hydroxide (NaOH), added continuously in small amounts, to maintain the optimum pH (13–13.5) for the oxidation reaction [42]. They also indicated the importance of water during the oxidation reaction, because the presence of water and OH-ions led to the formation of a mononuclear CuII-hydroxo-intermediate in combination with CuII-species and bpy, as previously reported by Wagner-Juaregg and co-workers [42,54]. Stahl and co-workers also proposed the formation of the hydroxo-intermediate, as a resting state during alcohol oxidation (vide infra) [11]. In the absence of water, the aldehyde conversion is equal to the initial amount of TEMPO· (5 mol %) added to the reaction. Finally, their catalyst system could be used for the fast and optimal oxidation of activated allylic and benzylic alcohols, with a selectivity for primary over secondary alcohols [42].
Mannam and co-workers reported a TEMPO·-mediated CuCl/1,4-diazabicyclo[2.2.2]octane (DABCO) catalyst system for the oxidation of alcohols in toluene at 100 °C, in 2007 [41]. Molecular oxygen served as the oxidant, and water was the only by-product of the reaction. They found that the efficiency of the reaction was nearly the same with or without base in a toluene solvent system, because the ligand employed served the dual function of stabilising the active species and acting as a base for the deprotonation of alcohol substrates. This catalyst system was used for the oxidation of primary benzylic and allylic alcohols, without over-oxidation; however, longer reaction times were required for secondary benzylic alcohols and the oxidation of aliphatic alcohols required the longest reaction time.
To further improve on the previous catalyst systems, the choice of solvent for alcohol oxidation was also considered. Water as non-organic solvent has recently been employed in these alcohol oxidation reactions [53,55]. Geißlmeir and co-workers highlighted the importance of water in the oxidation reaction [42]. Repo and co-workers found that alcohol oxidation was possible in a pure aqueous alkaline solution, using dioxygen as the oxidant [55]. They reported a CuII/diimine/TEMPO·/NaOH catalyst system which possesses high catalytic activity towards both primary and secondary benzylic alcohol substrates, due to the high stability of the formed CuII-complexes in water. The presence of TEMPO· improved the in situ formation of catalytically active CuII-diimine complexes. Unfortunately, their catalyst system still possessed several limitations such as the use of increased temperature and oxygen pressure.
The reaction mechanism for the oxidation reaction catalysed by the system reported by Repo and co-workers was investigated by Yang and co-workers computationally and it was found to consist of three steps, namely catalyst activation, substrate oxidation and catalyst regeneration [56]. The rate determining step for the oxidation reaction was calculated to be the proton transfer step. They found that increased amounts of water resulted in lower reaction rates. Their calculations supported the experimentally observed reactivity trends: oxidation of primary benzylic and allylic alcohols proceeded efficiently, however the oxidation of primary aliphatic alcohols was unsuccessful, due the formation of computationally identified dimeric CuII-hydroxo intermediates in the presence of water. This observation is consistent with the experimental findings of Stahl and co-workers [11,47].
In 2009, Koskinen further improved on the catalyst system of Sheldon and co-workers by replacing the MeCN:H2O solvent system with non-aqueous MeCN in order to avoid any solubility problems for highly hydrophobic alcohol substrates [28]. Evaluating various bases, moderate activities were obtained with N-methylimidazole (NMI), rather than KOtBu as base while the highest activity was observed when employing 1,8-diazabicycloundec-7-ene (DBU). As a result, they reported the (bpy)CuBr2/TEMPO·/NMI or DBU catalyst system under an oxygen atmosphere (Scheme 8).
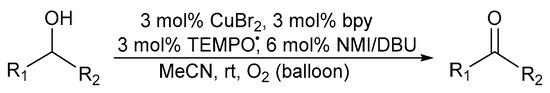
Scheme 8.
A (bpy)CuBr2/TEMPO/NMI and/or DBU catalyst system in a MeCN solvent system for the aerobic alcohol oxidation [28]. Reproduced with permission from Koskinen and co-workers. Copyright Wiley Materials, 2009. Successful oxidation was possible for primary allylic alcohols (87 to 100%) and more challenging primary aliphatic alcohols (84 to 100%) using CuBr2 and Cu(OTf)2, respectively.
While CuBr2 was used for allylic alcohol substrates, copper(II)-triflate (Cu(OTf)2) was suitable for more challenging alcohol substrates, particularly primary aliphatic alcohols [28]. A significant drawback was the need for pure O2 as oxidant.
Later, Stahl and co-workers found that the rate of the oxidation reaction could be increased when the CuII-catalyst is replaced with a CuI-catalyst [47,57]. In 2011, they reported a catalyst system that involves a bpy ligand that is coordinated to copper(I)triflate (CuOTf), in combination with TEMPO· as co-catalyst and NMI as base, under ambient air as oxidant (Scheme 9) [2,11].

Scheme 9.
A (bpy)CuOTf/TEMPO·/NMI catalyst system in MeCN under air for the aerobic alcohol oxidation [2]. Reproduced with permission from Stahl and co-workers. Copyright American Chemical Society, 2011. Successful oxidation was possible for a variety of primary alcohols (72 to >99%). Selectivity: primary over secondary alcohols (diols).
This catalyst system exhibits a high efficiency in the oxidation of benzylic, allylic and other activated alcohols, whereas aliphatic alcohols were found to oxidise more slowly [58,59]. The oxidation of aliphatic alcohols is hindered, thereby requiring longer reaction times, by the higher pKa of the hydroxyl group, in comparison to the lower pKa value of benzylic alcohols [11].
More recently, in 2017, our group reported the use of a bis(pyridyl)-N-alkylamine ligand (L3, Scheme 10) in our (L3)CuI/TEMPO·/NMI catalyst system. Even though the reaction rate for ligand L3 was slightly slower than the previously used bpy, quantitative conversion of 1-octanol was obtained in a shorter reaction time than was obtained by Stahl and co-workers [2,10]. Our catalyst system could be used for the oxidation of a variety of primary aliphatic, allylic, benzylic and heterocyclic alcohol substrates with excellent yields of the corresponding aldehydes, under synthetically relevant reaction conditions [10].
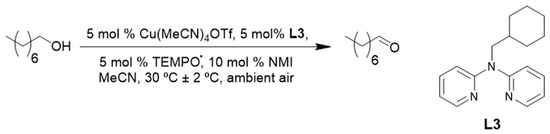
Scheme 10.
A (L3)CuI/TEMPO·/NMI catalyst system the aerobic oxidation of 1-octanol to 1-octanal under synthetically relevant reaction conditions [10]. Reproduced with permission from Swarts and co-workers. Copyright Royal Society of Chemistry, 2017. Successful oxidation was possible for a variety of primary alcohols (65 to 98%). The selectivity of the catalyst system was not evaluated.
Our catalyst system could also be used for the successful oxidation of several alcohol substrates which proved problematic in previous studies [2]. Stahl and co-workers observed low conversions during the oxidation of 2-pyridinemethanol, possibly due to the competition between the coordination of the alcohol substrate to copper and copper-catalysed substrate oxidation. Spectrometric (MS) analysis provided experimental evidence for the inhibitory effect for the oxidation of 2-pyridinemethanol, which results in a 2-pyridinemethanol-ligated CuII-hydroxo dimer (Scheme 11) [47,60,61,62,63,64].
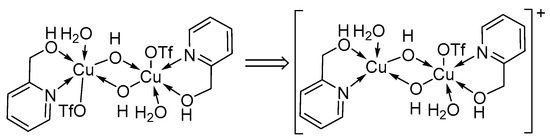
Scheme 11.
The Cu2-hydroxo dimer that is responsible for the inhibitory effect observed during the oxidation of 2-pyridinemethanol [10]. Reproduced with permission from Swarts and co-workers. Copyright Royal Society of Chemistry, 2017.
Unfortunately, our catalyst system could not be used for the oxidation of secondary alcohols, attributed to the bulky structure of TEMPO· and the steric pressure exerted during the TEMPO·-mediated C-H abstraction step [2,8,28,59].
Several reported homogeneous catalyst systems such as CuBr/bpy/TEMPO·/NMI form insoluble precipitates during the oxidation reaction and fail to reach completion [2], while others such as CuBr2/bpy/TEMPO·/KOH are deactivated once the reaction approach completion [28]. It was found that the nature of the external bases have an influence on the catalytic reactivity, selectivity and stability [28] which have led to the development of catalyst systems capable of operating in the absence of a base. In 2018, Han and co-workers reported a base-free catalyst system, using TEMPO·, [2,2′]-bipyridinyl-5,5′-dicarboxylic acid diethyl ester (BPYDCDE) as the organic ligand and Cu(OAc)2 as the metal precursor (Figure 1) [65].
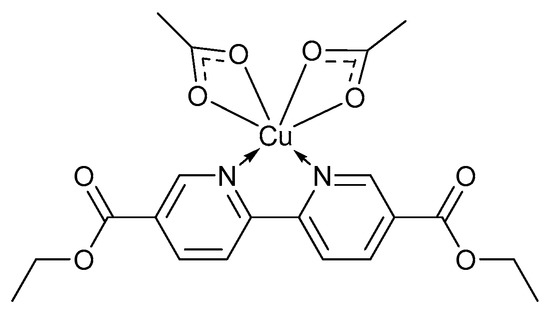
Figure 1.
The Cu(BPYDCDE)(OAc)2 complex for the base-free catalyst system for alcohol oxidation [65]. Reproduced with permission from Han and co-workers. Copyright American Chemical Society, 2018.
Their catalyst system, operating at ambient temperatures and pressures, could be used for the conversion of a variety of primary and secondary alcohols to the corresponding aldehydes and ketones in a short reaction time, without the addition of external base [65]. The coordination between the ligand and OAc- enriches the electron population on the Cu-centre, increasing the alkalinity of the OAc- counter ion to afford a better leaving group. The activity of the different complexes are governed by the metal, the alkalinity of the counter ion as well as the structure of the ligand. Furthermore, they also found that the CuII-complex could be recycled for up to five times without the loss of activity [65].
Independently, Liu and co-workers also reported a CuI/NMI/TEMPO· (ratio of 1:2:1) catalyst system, in the absence of an external base, for the oxidation of both primary and secondary (partially successful) alcohols to their corresponding carbonyl products, under ambient conditions and increased reaction times [66]. NMI was used as a ligand while they neglected its role as a base, because they suggested that the presence of a base may lead to a loss of copper species due to the formation of CuII-hydroxide species. Furthermore, they found that an increase in the electron density on the metal centre will lead to an increase in the catalysis and this can be achieved when a ligand with strong basicity is used. Therefore, they suggested that the activation of O2 may be one of the essential steps during the oxidation reaction.
In conclusion, a number of different Cu/TEMPO· catalyst systems have been reported for the oxidation of alcohols. The mechanistic pathway by which Cu/TEMPO· catalyst systems mediate the oxidation of alcohols, has been evaluated by several research groups [7,8,9,11,28]. The next section will detail the mechanistic understanding that has been established for Cu/TEMPO· catalysed alcohol oxidation.
5. Mechanistic Aspects of Cu/TEMPO·-Catalysed Alcohol Oxidation
Different, partially contradicting mechanistic pathways have been proposed, which involve the formation of TEMPO+, TEMPO-H or TEMPO· as the reactive intermediate (Table 3). These pathways are supported by experimental evidence for the postulated intermediates and reaction steps.
Table 3.
A summary of the different mechanistic pathways for the aerobic alcohol oxidation. The Cu-precursor as well as the reactive intermediate for each pathway is indicated.
5.1. Option 1: Oxoammonium Cation (TEMPO+) Mechanistic Pathway
Initially, it was believed the first reported Cu/TEMPO· catalyst system capable of mimicking GOase, proceeds via an oxoammonium (TEMPO+) pathway (Scheme 12) [7]. Semmelhack and co-workers proposed the TEMPO+-mediated mechanistic pathway in which CuII oxidises TEMPO-H to TEMPO+, which then serves as the reactive intermediate in the alcohol oxidation reactions [7].
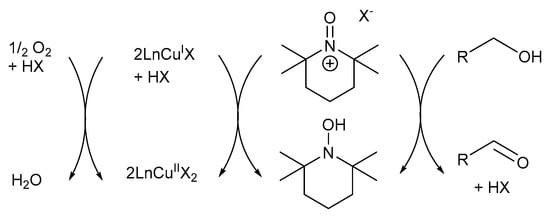
Scheme 12.
Semmelhack’s proposed oxoammonium cation (TEMPO+) mechanistic pathway where the CuII-precursor is used in the oxidation of TEMPO-H to TEMPO+ [7]. Reproduced with permission from Semmelhack and co-workers. Copyright American Chemical Society, 1984.
In addition, Sheldon and Arends suggested that TEMPO-H has a significant inhibitory effect on the aerobic alcohol oxidation reactions, but the corresponding TEMPO+ could be used as a relatively strong oxidant [33]. Initially, they generated TEMPO+ in situ through the addition of single oxidants, such as sodium hypochlorite [67] or m-chloroperbenzoic acid (m-CPBA) [68]. In addition, TEMPO+ could also be generated from TEMPO·, in a separate reaction, after which it was added to the oxidation reaction [69].
The mechanistic pathway can be divided into four reaction equations (Scheme 13) [7]: CuII oxidises TEMPO-H to TEMPO+ through a one-electron oxidation. TEMPO+ then serves as the mediator during the oxidation reaction (Equation (1)) [7,33], after which the alcohol substrate is oxidised to afford the corresponding aldehyde and TEMPO-H as products (Equation (2)) [7,67]. In addition, TEMPO+ is also used in the rapid oxidation of TEMPO-H to TEMPO· in the presence of O2, while the active CuII catalyst is regenerated (Equations (3) and (4)) [67,70]. Therefore, TEMPO· acts as a hydrogen transfer mediator during the deprotonation of the alcohol substrate. The net reaction involves the oxidation of an alcohol substrate in the presence of O2 to afford the corresponding aldehyde and water as products (Equation (5)) [7,67]. The addition of a base is not required as there is no net formation of an acid during the oxidation of alcohols [71].
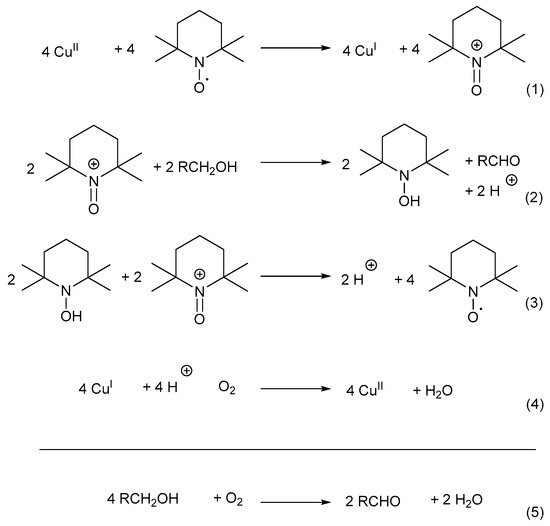
Scheme 13.
The oxoammonium cation mechanistic pathway which involves the oxidation of TEMPO-H to TEMPO+ (reactive intermediate) during alcohol oxidation. Reproduced with permission from Semmelhack and co-workers [7]. Copyright American Chemical Society, 1984.
Baerend and co-workers used density functional theory (DFT) calculations to investigate the mechanistic pathway for alcohol oxidation [72]. They only focused on the restricted singlet Cu-TEMPO· species as starting structures and proposed a reaction mechanism similar to that proposed by Semmelhack and co-workers [7,72]. Independently, Bailey and Bobbit also used DFT calculations to confirm the TEMPO+ mechanistic pathway [24]. They proposed the formation of pre-oxidation complexes during the coordination of alkoxide species and TEMPO+, via the attack of the alkoxide species on the nitrogen or oxygen atom of TEMPO+. The most stable pre-oxidation intermediate is found when the nitrogen atom of TEMPO+ is attacked at the equatorial site [7,24]. In addition, the equilibrium constants for the formation of the pre-oxidation complexes decreased with an increase in the steric bulk of the alkoxide species [24]. These large differences in the stability of the complex were proposed as a possible explanation for the successful oxidation of primary alcohols, rather than secondary alcohols.
However, Stahl and co-workers used electron paramagnetic resonance (EPR) measurements and cyclic voltammetry (CV) to indicate the lack of reactivity between the CuII-catalyst and TEMPO-H [11]. In addition, their kinetic studies indicated that TEMPO+ is not kinetically competent to act as the reactive intermediate during alcohol oxidations under their catalytic conditions [11]. Swarts and co-workers also confirmed the lower reaction rates in alcohol oxidation, when the oxoammonium cation, TEMPO+, was used as co-catalyst instead of TEMPO· [10]. In addition, Sheldon and co-workers used Hammett correlation studies to determine a ρ-value of -0.16 for Cu/TEMPO· oxidation reactions [8]; however, this is different to that observed in the stoichiometric oxidation with TEMPO+ (ρ ~ -0.3) [73]. These low ρ-values suggest that the mechanistic pathway involving the formation of TEMPO+, by the oxidation of the α-C-H bond, may be excluded.
Having established that TEMPO+ is not the reactive intermediate, alternative mechanistic pathways were evaluated where either TEMPO-H or TEMPO· is implicated as the reactive intermediate [8,9,10,11,28,74].
5.2. Option 2: Monomeric CuII-TEMPO-H Mechanistic Pathway
The catalyst system, as reported by Semmelhack and co-workers, could be used for the oxidation of benzylic and allylic alcohols [8]. However, the oxidation of aliphatic alcohols was unsuccessful, which is inconsistent with their TEMPO+-based mechanistic pathway, even though TEMPO+ is known for the efficient oxidation of a variety of aliphatic alcohols. As a result, Sheldon and co-workers investigated the possibilities for an alternative mechanistic pathway. They showed that the Ru-TEMPO· catalyst system for the aerobic oxidation of alcohols did not involve the TEMPO+ mechanistic pathway [21], but rather proceeded by Ru-centred oxidative dehydrogenation of the alcohol. This alternative metal-hydride mechanistic pathway is based on the results of stoichiometric oxidations with TEMPO·, kinetic isotope effect (KIE) and Hammett correlation studies [8], and involves the oxidative dehydrogenation of the alcohol substrate to afford Ru-hydride species and the re-oxidation of this species, facilitated by TEMPO· [8,21].
The rationale behind the Ru/TEMPO catalyst system led to the development of a Cu-centred mechanistic pathway for the catalyst system, as proposed by Sheldon and co-workers (Scheme 14) [8]. Their alternative mechanistic pathway involves two half-reactions: firstly, the CuI/TEMPO-H catalyst is oxidised by O2 and secondly, the oxidation of the alcohol substrate mediated by CuII and TEMPO-H [11].
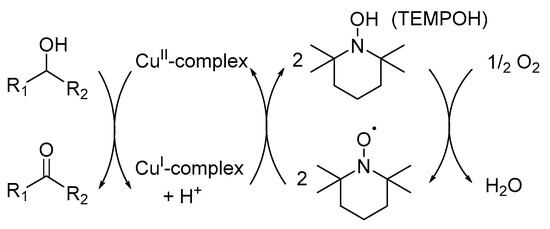
Scheme 14.
A summarised version of the Cu-centred mechanistic pathway for the Cu/TEMPO· catalysed aerobic oxidation of alcohols. Reproducedwith permission from Sheldon and co-workers [8]. Copyright Royal Society of Chemistry, 2003.
A detailed version of the mechanistic pathway (Scheme 15) involves the coordination of bpy to CuII to form complex I. The alcohol substrate is then deprotonated to afford the alkoxide species, the formation of which was confirmed by KIE and Hammett correlation studies [8]. The value of the primary KIE for the oxidation of α-deutero-p-methylbenzyl alcohol was determined to be 5.42 at 25 °C, for the C-H bond cleavage step [8]. This value is lower than the KIE value of aliphatic alcohols, where the C-H bond cleavage step is rate-determining, which influences the oxidation reaction. As a result, the KIE for α-deutero-p-methylbenzyl alcohol is consistent with the isotope effects observed with other metal-centred hydrogen abstractions, including GOase and the biomimetic Cu-complex as proposed by Stack [8,75].
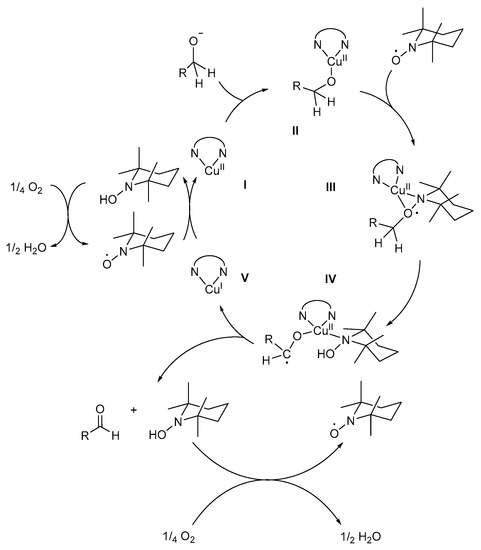
Scheme 15.
The proposed Cu-mediated mechanistic pathway for the (bpy)CuBr2/TEMPO·/KOtBu catalyst system for the aerobic alcohol oxidation. Reproduced with permission from Sheldon and co-workers [8]. Copyright Royal Society of Chemistry, 2003.
The alkoxide species coordinates to complex I to afford the (bpy)Cu/alkoxide-intermediate, II, after which TEMPO· also coordinates, in a η2 manner, to afford (bpy)Cu/TEMPO/alkoxide intermediate, III, confirmed by single crystal X-ray diffraction [8,35,76,77]. The role of TEMPO· is still unclear, but Sheldon and co-workers suggested that it might act as a hydrogen acceptor for the abstraction of the β-hydrogen of the alcohol to afford a ketyl radical intermediate, IV, which is the rate-determining step [3]. Intramolecular one-electron transfer from the intermediate, IV, to CuII affords CuI-species, V, the corresponding aldehyde and TEMPO-H [8,35].
In addition, spectroscopic studies showed that TEMPO· re-oxidises CuI to CuII, in the presence of molecular O2 [8,16,35,42]. During the protonation of the intermediate, IV, unstable TEMPO-H is formed [21], which is rapidly oxidised to TEMPO· in the presence of O2. However, this observation differs from the findings of Neumann and co-workers where they postulated that the oxidation of TEMPO-H to TEMPO· was the slower, rate-determining step for reactions catalysed by TEMPO· and catalytic amounts of polyoxometalate (H5PV2Mo10O40) as co-catalysts [78]. Sheldon and co-workers found that the difference in their observations could be rationalised on the basis of the pH-dependence of the oxidation reaction [8]. At basic pH, the rate of aerobic oxidation of TEMPO-H to TEMPO· is high, while at acidic pH, it is relatively low and might be rate-determining. In addition, the oxidation of TEMPO-H is expected to result in the formation of H2O2, which was not detected likely due to Cu-catalysed disproportionation [8].
Wu and co-workers used DFT calculations to re-examine the starting structures for alcohol oxidation, as proposed by Baerends and co-workers [72], and they found that open shell singlet and triplet spin states are also possible [6]. The open shell state species was found to be better starting structures due to lower energies, however, by employing the restricted singlet state species, they obtained results that support the mechanistic pathway as proposed by Baerend and co-workers [72]. In addition, they also proposed three different pathways for TEMPO· regeneration during the alcohol oxidation reaction [6]. They found that the concentration of TEMPO· stays constant during the oxidation reaction, but a decrease in the concentration is observed once all the alcohol substrate is consumed. This observation could not be explained experimentally by Sheldon and co-workers [3,35], but it is closely related to the TEMPO· regeneration, as proposed by Wu and co-workers [6]. As a result, even though their calculations supported the mechanistic pathway proposed by Baerends and co-workers, their findings also confirmed the presence of CuII-TEMPO· species rather than CuI-TEMPO+, providing support for the mechanistic pathway proposed by Sheldon and co-workers (Scheme 16) [6,8]. Furthermore, the catalyst system and mechanistic insights reported by Geißlmeir and co-workers also supports the Cu-centred mechanistic pathway as proposed by Sheldon and co-workers [42].
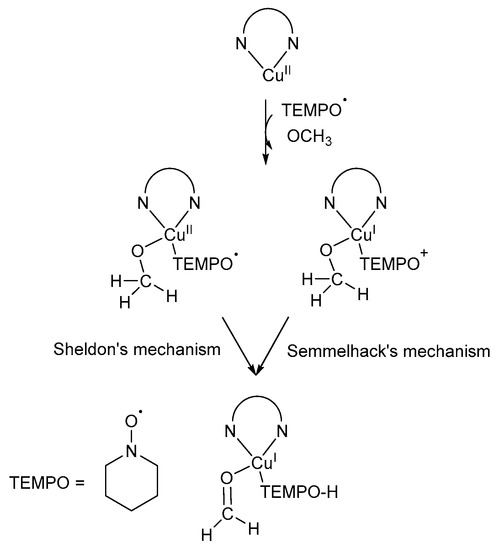
Scheme 16.
A summary of the mechanistic pathways as proposed by the groups of Semmelhack and Sheldon and their co-workers [7,8]. Reproduced with permission from Wu and co-workers [6]. Copyright American Chemical Society, 2010.
5.3. Option 3: A Dimeric CuII-Species-Based Mechanistic Pathway
The above-mentioned mechanistic pathways were further evaluated by the groups of Koskinen and Stahl [11,28]. Their mechanistic pathway involves the oxidation of TEMPO· to TEMPO-H and the formation of dimeric CuII species, which is not in agreement with GOase mimicry [28]. Instead, their mechanistic pathway mimics the mode of activity of tyrosinase enzymes.
Their proposed mechanistic pathway excluded the use of water as a co-solvent for MeCN, because they found that the presence of water is responsible for decreased oxidation rates [11,28]. Koskinen and co-workers used a CuII-salt, which is reduced to a CuI-complex, during the oxidation reaction and they found that by incorporating a non-coordinating counter-ion in the CuII-catalyst, they could oxidise a wide variety of alcohol substrates [28,35]. In addition, it became clear that the rate and the reactivity of the reaction increased when the CuII-catalyst was replaced by a CuI-catalyst in the alcohol oxidation [2,10,28,35]. CuI-precursors with a non-coordinating anion such as triflate displayed higher activity than the analogues containing coordinating anions such as CuBr and CuCl, attributed to faster dissociation rates of non-coordinating anions in the former [2,10,28,35].
The simplified version of their proposed TEMPO·-based mechanistic pathway (Scheme 17) consists of two key steps: Step 1: Catalyst oxidation – CuI enters the catalytic cycle and coordinates with bpy and NMI to afford a CuI-intermediate, (bpy)(NMI)CuI, which is oxidised to the CuII-hydroxide species, (bpy)(NMI)CuII-OH, in the presence of O2 [11]. This is accompanied by the oxidation of TEMPO-H to TEMPO·. Step 2: Substrate oxidation – CuII-hydroxide species and TEMPO· mediate the oxidation of the alcohol substrate to the corresponding aldehyde [11].
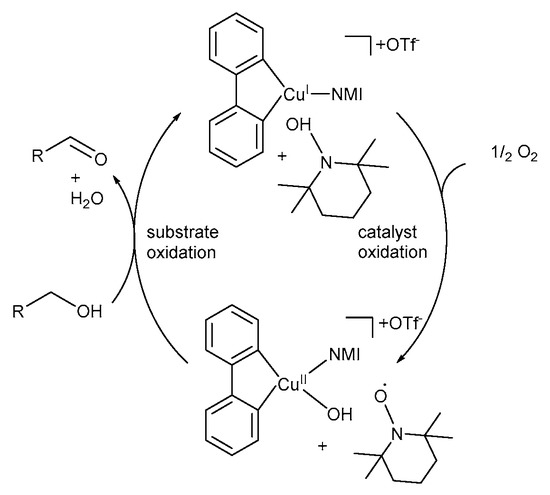
Scheme 17.
A simplified version of the mechanistic pathway for the (bpy)CuI/TEMPO·/NMI-catalysed aerobic alcohol oxidation. Reproduced with permission from Stahl and co-workers [11]. Copyright American Chemical Society, 2013.
Kinetic studies were employed to confirm the proposed mechanistic pathway where both aromatic and aliphatic alcohols exhibited similar kinetic dependencies on [Cu] and [O2]. The observed first-order dependence on [O2] and mixed second-order/first-order dependence on [Cu] was used to confirm the reaction between O2 and the biomimetic N-chelated CuI-intermediates [19,45,79]. These data are consistent with the oxidation of CuI to CuII under O2, followed by a reaction with a second CuI-centre to afford peroxo-bridged binuclear CuII-dimeric species [11].
UV-visible and EPR spectroscopic studies were used to monitor the concentration of CuI, CuII and TEMPO· species available in the system. The EPR measurements were conducted on quenched samples, and not in operando [11]. For aromatic alcohols, the copper species was predominantly CuI with only a minor decrease in [CuI] observed early in the reaction and a rapid decrease in CuI later in the reaction indicating complete conversion of the alcohol [11,47]. After complete conversion, EPR measurements showed that 70% of the Cu-catalyst was present as CuII, while UV-visible data showed that all the Cu existed as CuII [11,47]. A possible reason for this discrepancy was attributed to the formation of EPR-silent CuII species such as hydroxo-bridged binuclear CuII-dimeric species (Figure 2). The formation of the dimeric species was confirmed by single crystal X-ray diffraction analysis [47].
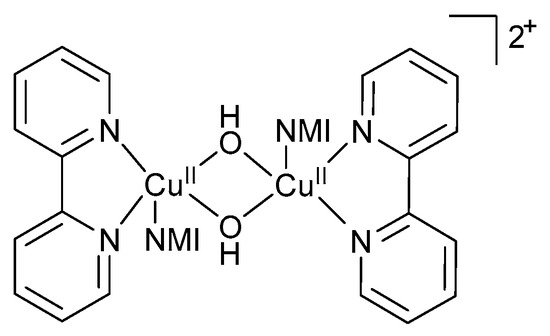
Figure 2.
Formation of hydroxo-bridged binuclear CuII-dimeric species during the aerobic alcohol oxidation reaction, as proposed by the groups of Koskinen and Stahl [11,28]. Copyright Wiley Materials, 2009 and American Chemical Society, 2013.
The concentration of TEMPO· remains low throughout the oxidation by O2, as observed during EPR measurements on quenched samples (17–32%) [11]. However, for the oxidation of aliphatic alcohols, CuI evolves gradually into CuII throughout the reaction, as confirmed by UV-visible and EPR spectroscopy. In addition, the concentration of TEMPO· is relatively high throughout the oxidation of aliphatic alcohols (70%). As a result, Stahl and co-workers suggested that the catalyst resting state changes, depending on the identity of the alcohol substrate [11]. For aromatic alcohols, copper is predominantly present as CuI and therefore, catalyst oxidation (Scheme 17, step 1) is the rate-limiting step. For aliphatic alcohols, both CuI and CuII is present in varying ratios during the oxidation reaction and therefore, both the catalyst and substrate oxidation steps (Scheme 17, steps 1 and 2) contribute to the rate-limiting step [11]. While both groups used NMI as base, Koskinen and co-workers also used DBU as a base in the oxidation reaction, which led to a small increase in the oxidation rate [28].
The more detailed version of the mechanistic pathway (Scheme 18) proposed by the groups of Koskinen and Stahl involves the coordination of bpy and NMI to CuI to stabilise the (bpy)(NMI)CuI intermediate, VI, which is then oxidised to (bpy)(NMI)CuIIO2·−, VII, under O2 [11,47,57]. The (bpy)(NMI)CuIIO2·− intermediate, VII, reacts with a second (bpy)(NMI)CuI intermediate to afford peroxo-bridged binuclear CuII-dimeric species, VIII [11,28]. However, Koskinen and co-workers proposed that the alkoxide species coordinates with the (bpy)(NMI)CuI intermediate, VI, after which it is oxidised by dioxygen to afford the CuII-dimeric species, VIII [13,28]. Analogous CuII-dimer species, in the absence of alcohol substrate, was also confirmed by Swarts and co-workers [10]. However, with the addition of the alcohol substrate, the CuII-dimer disappeared, and no further formation was observed during the oxidation reaction.
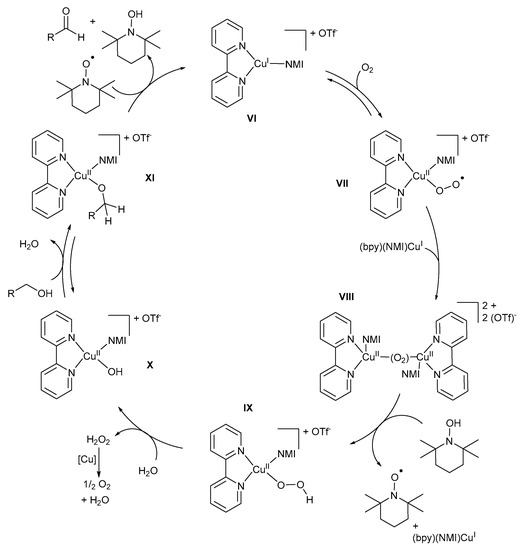
Scheme 18.
The proposed mechanistic pathway for the (bpy)CuI/TEMPO·/NMI catalyst system. Reproduced with permission from the groups of Koskinen and Stahl [11,28]. Copyright Wiley Materials, 2009 and American Chemical Society, 2013.
The presence of NMI is important to coordinate with CuI during the oxidation as it could hinder the formation and/or facilitate the dissociation of the intermediate in its absence [47]. TEMPO-H is oxidised to TEMPO· through H-atom transfer by the CuII-dimeric species, VIII, to afford (bpy)(NMI)CuII-OOH intermediates, IX, as well as (bpy)(NMI)CuII-OOH intermediates, X [11]. The formation of CuII-hydroperoxo intermediate IX, which is responsible for H2O2 formation after the oxidation reaction, was also confirmed by Swarts and co-workers [10]. Analogous species have been observed in Fe-catalysed alkane hydroxylation reactions [80]. From EPR measurements, it was determined that the (bpy)(NMI)CuII-OH intermediate, X, was the catalyst resting state [11], but Iron and co-workers suggested that it is only a theoretical resting state and it would not be present during the oxidation reaction [74].
Stahl and co-workers found that TEMPO-H is not required during the oxidation of CuI to CuII; however, it appears to be required for kinetically competent reaction rates [11]. In addition, when no TEMPO-H is present initially, the reaction between the alcohol substrate and the CuII-dimeric species is proposed to afford CuII-OOH and CuII-OCH2R intermediates [11], but no sufficient evidence to support the formation of these intermediates exists [9]. A further reaction between the (bpy)(NMI)CuII-OOH intermediate, IX, and H2O or the alcohol substrate leads to the formation of the (bpy)(NMI)CuII-OH, X, or (bpy)(NMI)CuII-OCH2R, XI, intermediates and H2O2. Hydrogen peroxide is known to rapidly disproportionate to H2O and O2, which accounts for the observed 2:1 alcohol:O2 stoichiometry of the overall reaction [11]. Alcohol substrates are oxidised by (bpy)(NMI)CuII-OH intermediates, X, to afford (bpy)(NMI)CuII-OCH2R intermediates, XI, via a pre-equilibrium step [2,28]. The pre-equilibrium step is followed by H-atom abstraction by TEMPO· to afford the aldehyde and TEMPO-H [11,74]. In addition, the reduction of CuII to CuI occurs through a two-electron/one-proton reaction [11]. The result of kinetic studies is consistent with a bimolecular reaction between TEMPO· and the (bpy)(NMI)CuII-OCH2R, XI [11].
Deuterium kinetic isotope studies revealed large KIEs for the C-H cleavage step (KIE = 6.06 and 10.9 respectively) for both aromatic and aliphatic alcohols [11,58]. These results clearly indicated that the C-H cleavage step is not the turnover limiting step for aromatic alcohols, but it contributes to the turnover rate of aliphatic alcohols. This was rationalised on the basis of the weaker α-C-H bonds of the activated and aromatic alcohols and therefore much more rapid C-H cleavage is expected [11].
Therefore, the mechanistic pathway proposed by the groups of Koskinen and Stahl, provides a reason for the historical challenges associated with the oxidation of aliphatic alcohols with Cu/TEMPO· and related catalyst systems. A pKa value of ~2 units higher for the O-H bond of aliphatic alcohols, than for aromatic alcohols [81,82], hinders the formation of (bpy)(NMI)CuII-OCH2R intermediates, 9, (Scheme 19), which results in lower oxidation rates [11].
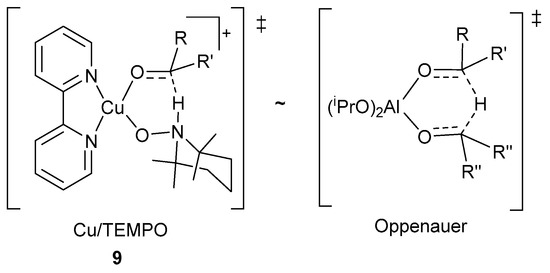
Scheme 19.
The structural similarities between the transition states for the H-atom transfer in the alcohol oxidation in Cu/TEMPO·, 9 and Oppenauer oxidation methods. Reproduced with permission from Stahl and co-workers [59]. Copyright American Chemical Society, 2014.
However, in 2014, Stahl and co-workers used DFT calculations to indicate the resemblance between the six-membered transition state for the H-atom transfer step, 9, and the transition state for Oppenauer oxidation of alcohols mediated by aluminium isopropoxide (Al(iOPr)3, Scheme 19) [59].
They specifically focused their studies on the (bpy)(NMI)CuII-OH intermediate, X, as the catalyst resting state and not the CuII-dimeric species, VIII, which is formed during the oxidation reaction (Scheme 18) [59]. Even though the transition states are structurally similar, the Cu/TEMPO·-mediated alcohol oxidation features a thermodynamic driving force associated with the reduction of CuII and TEMPO· to CuI and TEMPO-H [59].
More recently, in 2017, Stahl and co-workers synthesised different Cu/nitroxyl complexes and evaluated their electronic properties, using both spectroscopic and DFT computational studies [83]. They also used DFT computational studies to evaluate the electronic properties of the key proposed intermediate, 9, for the (bpy)Cu/TEMPO· catalyst system during aerobic alcohol oxidation (Scheme 19). Even though both the complexes and the proposed intermediate, 9, are in a CuII oxidation state, their investigation found that they exhibit CuI/TEMPO+ character. This was attributed to the ability of CuII and TEMPO· to achieve two-electron oxoammonium reactivity as two one-electron oxidants, resulting in an adduct with CuI/oxoammonium character and oxoammonium-like reactivity [83]. However, CuII is still not kinetically competent to promote one-electron oxidation of TEMPO· to afford TEMPO+, as the reactive intermediate, during aerobic alcohol oxidation. As such, even though they observed an oxoammonium-like reactivity during alcohol oxidation, their findings still does not support the oxoammonium cation (TEMPO+) mechanistic pathway as proposed by Semmelhack and co-workers [7,83].
Independently, Iron and co-workers also investigated the possibility for H-atom transfer to proceed via a six-membered ring (10), five-membered ring (11) or an intermolecular H-atom transfer step (12, Scheme 20) [74]. In further DFT computational studies, they focused on the six-membered transition state to investigate the mechanism of aerobic alcohol oxidation, which was found to be consistent with the one proposed by Stahl and co-workers [11].
Scheme 20.
The different transition states for the H-atom abstraction during the oxidation step. (10) Six-membered ring, (11) five-membered ring, (12) intermolecular hydrogen transfer. Reproduced with permission from Iron and co-workers [74]. Copyright John Wiley and Sons, 2016.
They suggested that the overall mechanism involves two possible pathways for the H-atom abstraction from the alcohol substrate, using TEMPO·. The first path involves the coordination of the H-atom to the O-atom of TEMPO· to afford TEMPO-H, while the second step involves the coordination of the H-atom to the N-atom of TEMPO· to afford TEMPO-NH-O [74]. Iron and co-workers also proposed that both TEMPO-H and TEMPO-NH-O would remain coordinated to the Cu-centre throughout the oxidation reaction [74]. However, Brückner and co-workers used UV-visible spectroscopy, where they found fluctuations in the intensity of the TEMPO· signal and therefore confirmed the continuous coordination and dissociation of TEMPO· to the Cu-centre [9].
5.4. Option 4: Mononuclear (bpy)(NMI)CuII-O2-TEMPO-Based Mechanistic Pathway
Stahl and co-workers developed the most efficient catalyst system [11,47,57,59] with the use of thorough mechanistic and kinetic studies, including in situ attenuated total reflection (ATR)-IR, UV-visible data and EPR measurements of quenched frozen samples [11]. More recently, Brückner and co-workers further improved our understanding of the TEMPO·-mediated mechanistic pathway [9]. They suggested alternative roles for TEMPO· and NMI, compared to those suggested by Stahl and co-workers. Previously, it was believed that TEMPO· was used for a one-electron oxidation of CuI to afford CuII-species [8,11,28]. However, the groups of Brückner and Wang used in operando EPR measurements and UV-visible spectroscopy, respectively, to confirm that there is no direct redox reaction between TEMPO· and the CuI/CuII-catalyst [9,46].
Based on their experimental data, the role of TEMPO· was identified to be for the fixation and activation of O2 via the formation of weakly coordinating TEMPO-O2 intermediates [9]. In addition, TEMPO· is also used in the stabilisation of the (bpy)(NMI)CuII-O2.- intermediate, formed by SET from the CuI-catalyst to molecular O2. Furthermore, Stahl and co-workers proposed the formation of Cu2O2-dimeric species [11], which is inconsistent with the results of Brückner and co-workers’ coupled operando studies [9].This observation is explained on the basis that the expected CuII-dimeric species would be EPR-silent. Instead, Brückner and co-workers found an increase in the CuII EPR signal during the reaction, which is a result of the possible formation of off-cycle CuII-dimeric species (such as hydroxo-bridged CuII-dimers) [9]. Stahl and co-workers also proposed the reaction between the alcohol substrate and CuII-dimeric species, in the absence of TEMPO-H, but no sufficient evidence to support the formation of the resulting CuII-OOH and CuII-OCH2R intermediates, exists [9]. The oxidation of both aromatic and aliphatic alcohols showed similar spectral features, although the rate of aldehyde formation and the rates of CuI/CuII conversion were much slower for aliphatic alcohols [9]. As a result, Brückner and co-workers proposed a mechanistic pathway which differs slightly from the one proposed by Stahl and co-workers (Scheme 21). This alternative mechanistic pathway is also supported by the experimental findings reported by the groups of Wang and Swarts [10,46]. UV-visible spectroscopic studies showed that the initial step involves coordination of bpy and the dissociation of acetonitrile solvent [9,46]. Upon introducing O2 into the solution, the intensity of the bands decreased and shifted slightly.
Scheme 21.
The proposed mechanistic pathway for the (bpy)CuI/TEMPO·/NMI catalyst system. Reproduced with permission from Brückner and co-workers [9]. Copyright John Wiley and Sons, 2015.
This was rationalised as a dipolar interaction between the (bpy)CuI intermediate and O2 rather than the oxidation of (bpy)CuI to tentative EPR-silent (bpy)CuII-O2· monomeric or (bpy)CuII-O2-CuII(bpy) dimeric species as postulated in previous reports [84,85]. UV-visible spectra supported the oxidation of CuI to CuII with the addition of NMI. NMI has a bifunctional role in the aerobic alcohol oxidation. Firstly, NMI is used to coordinate and stabilise the (bpy)CuI-intermediate to afford the (bpy)(NMI)CuI intermediate, XII, which is oxidised to (bpy)(NMI)CuIIO2·ˉ, XIII, through a SET to O2 [9,46,86]. The magnetic interaction between the paramagnetic CuII and O2·ˉ has an influence on the EPR studies, and therefore the (bpy)(NMI)CuIIO2·ˉ intermediate might not be seen [9]. Further reactions such as H-atom abstraction might take place to stabilise O2·ˉ, in the absence of TEMPO·, to afford a tentative EPR-active (bpy)(NMI)CuII-OOH intermediate and a possible increase in the EPR signal [9]. However, Brückner and co-workers proposed the stabilisation of (bpy)(NMI)CuIIO2·ˉ, XIII, in the presence of TEMPO· to afford the (bpy)(NMI)CuII-O2-TEMPO intermediate, XIV, which is confirmed by line broadening and a loss in the intensity of the EPR signal in the presence of O2 [9]. The formation of the (bpy)(NMI)CuII-O2-TEMPO intermediate, XIV, differs from the formation of the peroxo-CuII-dimeric species (Scheme 18, VIII), as proposed by the groups of Koskinen and Stahl [11,28]. NMI functions as base for the deprotonation of the alcohol substrate to afford alkoxide species that coordinates to XIV to afford the CuII-intermediate, XV [9,11,59]. H-atom abstraction from the alkoxide species affords the corresponding aldehyde and TEMPO·. This step is facilitated by the presence of O2·ˉ. Stahl and co-workers suggested that TEMPO· is used for the deprotonation step [11], which is in contrast with the experimental results of Brückner and co-workers highlighting the roles of NMI and TEMPO· during the oxidation reaction [9]. The consumption of alcohol substrate coincides with an accumulation of the (bpy)(NMI)CuII intermediate, but when more alcohol substrate is added, aldehyde formation continues, as indicated by an increase in the νC=O absorption band [9]. Swarts and co-workers employed spectrometric (ESI-MS) studies to confirm an increase in the intensity of the mass fragments for the CuI-intermediates after complete conversion of the second addition of alcohol substrate, and therefore no catalyst deactivation [10]. These results are in contrast with previous mechanistic pathways, where they suggest the reaction of TEMPO· with the (bpy)(NMI)CuII-alkoxide intermediate to afford TEMPO-H, the corresponding aldehyde and (bpy)(NMI)CuI [11,28,47]. If this is true, the EPR measurements for TEMPO· and CuII should decrease after the addition of more alcohol substrate, because CuI and TEMPO-H are EPR-silent species, however, the opposite was observed by Brückner and co-workers [9].
Lastly, CuII is reduced to CuI to afford the (bpy)(NMI)CuI-OOH intermediate, XVI, and H2O2 during the reaction with H+-NMI [9,11]. The resulting hydrogen peroxide rapidly disproportionates to H2O and O2. Swarts and co-workers used ESI-MS studies to provide experimental evidence for the formation of the CuII-hydroperoxo intermediate [10]. Furthermore, this is also supported by EPR measurements where the TEMPO· signal narrowed and the intensity increased after the addition of the alcohol substrate, thereby indicating the dissociation of the TEMPO-O2 adduct [9]. In addition, fluctuations in the TEMPO· EPR signal intensity confirmed the formation and decomposition of these TEMPO-O2 intermediates. Previous studies performed EPR measurements on quenched samples during the oxidation reaction and, as a result, the fluctuations in the TEMPO· signal were not observed [11].
In conclusion, various partially contradicting mechanistic pathways have been recently proposed for Cu/TEMPO· catalyst systems. These mechanistic pathways involve either TEMPO+, TEMPO· or TEMPO-H as the reactive intermediate during alcohol oxidation. However, the high costs of TEMPO· as co-catalyst, lack of recyclability and its steric influence on reactivity has resulted in attempts to replace TEMPO· with a sterically smaller and less expensive nitroxyl co-catalyst. In addition, the improvement of the system’s capability to oxidise both primary and secondary alcohols is also of importance. The next section will focus on alternative co-catalysts in combination with a Cu-catalyst for alcohol oxidation.
6. Alternative Co-Catalysts in Cu/Nitroxyl-Catalysed Alcohol Oxidation
Several alternative co-catalysts for the Cu-based catalyst systems, such as nitroxyl radicals and in situ radical formation from N-oxide co-catalysts, were reported in recent years. Employing alternative co-catalysts generate catalyst systems displaying higher catalytic activity devoid of the limitations described above.
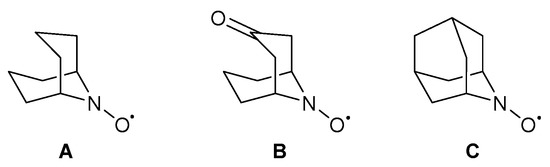
In 2013, Stahl and co-workers replaced TEMPO· with different nitroxyl co-catalysts varying in their electronic and steric properties [58]. Several nitroxyl derivatives in combination with a Cu-catalyst, have been reported previously [87,88,89,90,91,92]. They found that the reaction rates for the less sterically encumbered co-catalysts, such as 9-azabicyclo[3.3.1]nonane N-oxyl (ABNO·, A), ketoABNO· (B) and 2-azaadamantane-N-oxyl (AZADO·, C), were higher than the TEMPO· derivatives (Scheme 22). The increase in reactivity for sterically smaller co-catalysts was also observed electrochemically [93]. Furthermore, due to the lack of dependence on the rate of the nitroxyl redox potential, they proposed that the nitroxyl co-catalyst is not involved in the turnover-limiting step. These results support the findings of their Cu/TEMPO· catalyst system for activated alcohols but differ from the results for aliphatic alcohols [2] and therefore they suggest that less sterically hindered nitroxyl co-catalysts enhance the rate of the C-H cleavage step which leads to a new turnover-limiting step [58].
Scheme 22.
Different nitroxyl derivatives that can be used to replace TEMPO·. (A) ABNO·, (B) keto-ABNO· and (C) AZADO· [58]. Reproduced with permission from Stahl and co-workers. Copyright American Chemical Society, 2013.
Furthermore, they improved on the substrate scope of their Cu/TEMPO· catalyst system by developing a highly effective (bpy)CuI/ABNO·/NMI catalyst system which could be used for the successful oxidation of a variety of alcohol substrates, including aliphatic and secondary alcohols, within shorter reaction times and at room temperature. However, even though ABNO· is sterically less bulky than the traditional TEMPO· co-catalyst, it is more expensive, and therefore not the optimal alternative.
Iwabuchi and co-workers also focused on less sterically hindered nitroxyl radical co-catalysts, such as AZADO·, that exhibits a higher catalytic efficiency during alcohol oxidation [92,94]. Even though the sterically smaller ABNO· co-catalyst is kinetically more efficient and readily available, more bulky nitroxyl radical co-catalysts such as 1-Me-AZADO· resulted in higher activity, which is in contrast with previous studies [58,94,95]. They reported a (bpy)Cu/AZADO·/p-(N,N-dimethylamino)pyridine (DMAP) catalyst system for selective oxidation of amino alcohols to their corresponding amino carbonyl products [92]. This selectivity control was attributed to the preferential formation of a Cu-alkoxide intermediate rather than a Cu-amide intermediate. A lower loading for AZADO· as co-catalyst, relative to TEMPO· was used and they also found an increase in the reactivity when halide salts of CuI was used instead of CuOTf.
In 2015, Arndtsen and co-workers reported a catalyst system operating without the use of an N-oxide co-catalyst [96]. Tetrakisacetonitrilecopper(I) hexafluorophosphate ([Cu(MeCN)4]PF6) in combination with N,N′-di-tert-butyl-ethylenediamine (DBED) was employed as the catalyst system. DMAP was used as a base for non-activated aliphatic alcohols, while NMI was used for activated alcohols. Interestingly, the Cu/DBED catalyst system is selective for the oxidation of activated secondary over primary alcohols, which is in contrast with the Cu/TEMPO· catalyst system [2,96]. Initially they believed that their catalyst system acts as a type II Cu-based tyrosinase mimic, instead of GOase [96], but the groups of Stahl and Arndsten and co-workers used mechanistic studies to confirm the presence of a nitroxyl radical during the oxidation reaction [96,97]. They proposed that Cu/DBED-catalysed reactions involve both oxygenase- and oxidase-type reactivity. A Cu/O2 adduct is responsible for the in situ oxygenation of DBED, as previously observed in amine oxygenation with related Cu-oxygenase mimics [98,99]. The oxygenated ligand could then serve as a nitroxyl radical co-catalyst in the oxidase-type reaction that acts as a GOase, similar to those observed in Cu/nitroxyl reactions [8,11,59,100]. The approach to synthesise the nitroxyl radicals through an in situ process has significant implications for future catalyst development, because some of these radicals decompose readily when prepared and isolated.
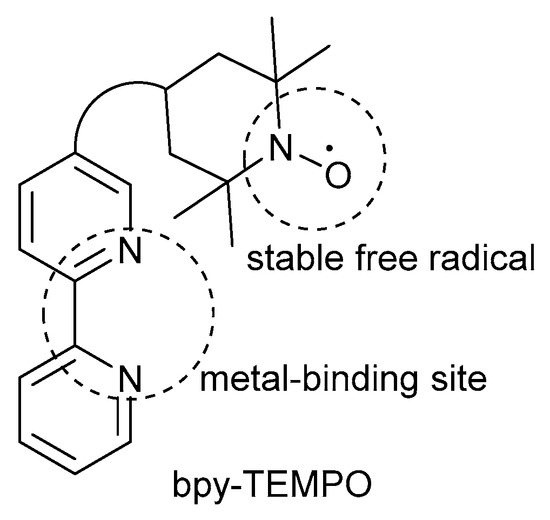
More recently, in 2016, Wang and co-workers rather decided to incorporate steric bulk into catalyst design by introducing a bifunctional ligand, which consists of bpy as metal-binding site covalently linked to a TEMPO· radical (Scheme 23) [46]. Previous groups also attempted to use a bifunctional ligand in their catalyst systems, but they encountered several limitations such as limited substrate scope, the need for high catalyst loadings as well as laborious preparation of the bifunctional ligand [101,102,103,104]. As a result, Wang and co-workers circumvented these limitations with the development of their CuI/bpy-TEMPO·/NMI catalyst system that could be used for the oxidation of a variety of primary (72 to >99%) and secondary (78 to 85%) alcohols, bearing selectivity toward primary alcohols over secondary alcohols [46]. Previous studies also showed selectivity for primary alcohols over secondary alcohols [2,7,8,28,42]. Unfortunately, as with many analogous systems, they failed to efficiently oxidise a broad range of secondary alcohol substrates.
Scheme 23.
Schematic presentation of the bpy-TEMPO·-based bifunctional ligand. Reproduced with permission from Wang and co-workers [46]. Copyright Royal Society of Chemistry, 2016.
In conclusion, several alternative nitroxyl radicals and other co-catalysts were used in recently reported catalyst systems. These alternative catalyst systems could improve on several limitations encountered with the Cu/TEMPO· catalyst systems, but some of the limitations still exists. As a result, even though sterically smaller co-catalysts result in higher catalytic activity, it is still expensive and therefore not the optimal replacement for TEMPO· during alcohol oxidation.
7. Summary and Outlook
This review sought to highlight the importance of copper as a metal, as well as in combination with TEMPO·, for alcohol oxidation reactions. The advantages, disadvantages and differences associated with the reported Cu/TEMPO· catalyst systems have been summarised, while the potential for new developments, such as a catalyst system for optimal oxidation of both primary and secondary alcohols, are also outlined. We have provided a detailed discussion of the previously proposed mechanistic pathways, which involve either TEMPO+, TEMPO· or TEMPO-H as the reactive intermediate. In particular, we sought to highlight contradiction which exist between mechanistic pathways. Lastly, recent efforts directed to the development of catalyst systems involving a copper precursor, in combination with several alternative nitroxyl radicals and in situ formed radicals, have been described. These reports encompass future catalyst design strategies for Cu-based catalyst systems, operating in the absence of a radical co-catalyst, for alcohol oxidation. Specifically, these catalyst systems should act as type II Cu-based tyrosinase mimics, instead of GOase. Similar co-catalyst free catalyst systems, for the conversion of phenolic substrates to afford quinones, have recently been reported [105,106,107]. Different ligands were recently evaluated, and it was found that DBED was the most efficient ligand for these oxygen atom transfer reactions [105,107]. In conclusion, different Cu/TEMPO· catalyst systems as well as partially contradicting mechanistic pathways were reported in recent years. Furthermore, the possibility to replace TEMPO· as co-catalyst with another nitroxyl radical or an in situ formed radical also exist, but the optimal replacement for TEMPO· could not yet be determined. Therefore, there is still a growing demand to develop an optimal Cu-based catalyst system for the oxidation of both primary and secondary alcohols. Keeping in mind the pace of advances in the development of Cu/TEMPO· catalyst systems and the understanding of the mechanistic pathway for alcohol oxidation in recent years, it is reasonable to expect that these challenges will soon be addressed, while new opportunities in Cu-based alcohol oxidation will be developed.
Funding
This work is based on the research supported by the National Research Foundation of South Africa (NRF) for the grant, Unique Grant No. 112545, 99305 and 117869, as well as the North-West University (NWU).
Acknowledgments
The support provided by the Focus Area for Chemical Resource Beneficiation (CRB) and in particular the Catalysis and Synthesis group at the NWU is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflicts of interest. Any opinion, finding and conclusion or recommendation expressed in this material is that of the author(s) and the NRF does not accept liability in this regard.
References
- Cao, Q.; Dornan, L.M.; Rogan, L.; Hughes, N.L.; Muldoon, M.J. Aerobic oxidation catalysis with stable radicals. Chem. Commun. 2014, 50, 4524–4543. [Google Scholar]
- Hoover, J.M.; Stahl, S.S. Highly Practical Copper(I)/TEMPO Catalyst System for Chemoselective Aerobic Oxidation of Primary Alcohols. J. Am. Chem. Soc. 2011, 133, 16901–16910. [Google Scholar] [CrossRef] [PubMed]
- Gamez, P.; Arends, I.W.C.E.; Reedijk, J.; Sheldon, R.A. Copper(ii)-catalysed aerobic oxidation of primary alcohols to aldehydes. Chem. Commun. 2003, 2414–2415. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Arends, I.W.C.E.; Ten Brink, G.-J.; Dijksman, A. Green, Catalytic Oxidations of Alcohols. Acc. Chem. Res. 2002, 35, 774–781. [Google Scholar] [CrossRef]
- Stahl, S.S. Palladium Oxidase Catalysis: Selective Oxidation of Organic Chemicals by Direct Dioxygen-Coupled Turnover. Angew. Chem. Int. Ed. 2004, 43, 3400–3420. [Google Scholar] [CrossRef]
- Cheng, L.; Wang, J.; Wang, M.; Wu, Z. Mechanistic Insight into the Alcohol Oxidation Mediated by an Efficient Green [CuBr2 (2, 2′-bipy)]-TEMPO Catalyst by Density Functional Method. Inorg. Chem. 2010, 49, 9392–9399. [Google Scholar] [CrossRef]
- Semmelhack, M.F.; Schmid, C.R.; Cortes, D.A.; Chou, C.S. Oxidation of alcohols to aldehydes with oxygen and cupric ion, mediated by nitrosonium ion. J. Am. Chem. Soc. 1984, 106, 3374–3376. [Google Scholar] [CrossRef]
- Dijksman, A.; Arends, I.W.C.E.; Sheldon, R.A. Cu(ii)-nitroxyl radicals as catalytic galactose oxidase mimics. Org. Biomol. Chem. 2003, 1, 3232–3237. [Google Scholar] [CrossRef]
- Rabeah, J.; Bentrup, U.; Stößer, R.; Brückner, A. Selective Alcohol Oxidation by a Copper TEMPO Catalyst: Mechanistic Insights by Simultaneously Coupled Operando EPR/UV-Vis/ATR-IR Spectroscopy. Angew. Chem. Int. Ed. 2015, 127, 11957–11960. [Google Scholar] [CrossRef]
- Marais, L.; Bures, J.; Jordaan, J.H.L.; Mapolie, S.; Swarts, A.J. A bis(pyridyl)-N-alkylamine/Cu(i) catalyst system for aerobic alcohol oxidation. Org. Biomol. Chem. 2017, 15, 6926–6933. [Google Scholar] [CrossRef]
- Hoover, J.M.; Ryland, B.L.; Stahl, S.S. Mechanism of Copper(I)/TEMPO-Catalyzed Aerobic Alcohol Oxidation. J. Am. Chem. Soc. 2013, 135, 2357–2367. [Google Scholar] [CrossRef] [PubMed]
- Wendlandt, A.E.; Suess, A.M.; Stahl, S.S. Copper-Catalyzed Aerobic Oxidative C—H Functionalizations: Trends and Mechanistic Insights. Angew. Chem. Int. Ed. 2011, 50, 11062–11087. [Google Scholar]
- Allen, S.E.; Walvoord, R.R.; Padilla-Salinas, R.; Kozlowski, M.C. Aerobic copper-catalyzed organic reactions. Chem. Rev. 2013, 113, 6234–6458. [Google Scholar] [CrossRef]
- Punniyamurthy, T.; Velusamy, S.; Iqbal, J. Recent advances in transition metal catalyzed oxidation of organic substrates with molecular oxygen. Chem. Rev. 2005, 105, 2329–2364. [Google Scholar] [CrossRef]
- Punniyamurthy, T.; Rout, L. Recent advances in copper-catalyzed oxidation of organic compounds. Coord. Chem. Rev. 2008, 252, 134–154. [Google Scholar] [CrossRef]
- Schultz, M.J.; Sigman, M.S. Recent advances in homogeneous transition metal-catalyzed aerobic alcohol oxidations. Tetrahedron 2006, 62, 8227–8241. [Google Scholar] [CrossRef]
- Gamez, P.; Aubel, P.G.; Driessen, W.L.; Reedijk, J. Homogeneous bio-inspired copper-catalyzed oxidation reactions. Chem. Soc. Rev. 2001, 30, 376–385. [Google Scholar] [CrossRef]
- Whittaker, J.W. Free radical catalysis by galactose oxidase. Chem. Rev. 2003, 103, 2347–2364. [Google Scholar] [CrossRef]
- Hatcher, L.Q.; Karlin, K.D. Ligand influences in copper-dioxygen complex-formation and substrate oxidations. Adv. Inorg. Chem. 2006, 58, 131–184. [Google Scholar]
- Michel, F.; Thomas, F.; Hamman, S.; Philouze, C.; Saint-Aman, E.; Pierre, J.-L. Galactose Oxidase Models: Creation and Modification of Proton Transfer Coupled to Copper(II) Coordination Processes in Pro-Phenoxyl Ligands. Eur. J. Inorg. Chem. 2006, 2006, 3684–3696. [Google Scholar] [CrossRef]
- Dijksman, A.; Marino-Gonzalez, A.; Mairata i Payeras, A.; Arends, I.W.; Sheldon, R.A. Efficient and selective aerobic oxidation of alcohols into aldehydes and ketones using ruthenium/TEMPO as the catalytic system. J. Am. Chem. Soc. 2001, 123, 6826–6833. [Google Scholar] [CrossRef]
- Whittaker, M.M.; Whittaker, J.W. Ligand interactions with galactose oxidase: Mechanistic insights. Biophys. J. 1993, 64, 762–772. [Google Scholar] [CrossRef]
- Whittaker, M.M.; Ekberg, C.A.; Peterson, J.; Sendova, M.S.; Day, E.P.; Whittaker, J.W. Spectroscopic and magnetochemical studies on the active site copper complex in galactose oxidase. J. Mol. Catal. B Enzym. 2000, 8, 3–15. [Google Scholar] [CrossRef]
- Bailey, W.F.; Bobbit, J.M.; Wiberg, K.B. Mechanism of the Oxidation of Alcohols by Oxoammonium Cations. J. Org. Chem. 2007, 72, 4504–4509. [Google Scholar] [CrossRef]
- Sambiagio, C.; Marsden, S.P.; Blacker, A.J.; McGowan, P.C. Copper catalysed Ullmann type chemistry: From mechanistic aspects to modern development. Chem. Soc. Rev. 2014, 43, 3525–3550. [Google Scholar]
- Skibida, I.P.; Sakharov, A.M. Molecular oxygen as environmental acceptable, selective and the most strong oxidant in liquid-phase oxidation. Catal. Today 1996, 27, 187–193. [Google Scholar] [CrossRef]
- Ragagnin, G.; Betzemeier, B.; Quici, S.; Knochel, P. Copper-catalysed aerobic oxidation of alcohols using fluorous biphasic catalysis. Tetrahedron 2002, 58, 3985–3991. [Google Scholar] [CrossRef]
- Kumpulainen, E.T.; Koskinen, A.M. Catalytic activity dependency on catalyst components in aerobic copper-TEMPO oxidation. Chem. Eur. J. 2009, 15, 10901–10911. [Google Scholar] [CrossRef]
- Ciriminna, R.; Pagliaro, M. Industrial oxidations with organocatalyst TEMPO and its derivatives. Org. Process Res. Dev. 2009, 14, 245–251. [Google Scholar] [CrossRef]
- Rozantsev, E.G.; Sholle, V.D. Synthesis and Reactions of Stable Nitroxyl Radicals I. Synthesis. Synthesis 1971, 190–202. [Google Scholar] [CrossRef]
- Griller, D.; Ingold, K.U. Persistent carbon-centered radicals. Acc. Chem. Res. 1976, 9, 13–19. [Google Scholar] [CrossRef]
- Safaei, E.; Hajikhanmirzaei, L.; Karimi, B.; Wojtczak, A.; Cotič, P.; Lee, Y.-I. TEMPO-mediated aerobic oxidation of alcohols using copper (II) complex of bis (phenol) di-amine ligand as biomimetic model for Galactose oxidase enzyme. Polyhedron 2016, 106, 153–162. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Arends, I.W.C.E. Catalytic oxidation mediated by metal ions and nitroxyl radicals. J. Mol. Catal. A Chem. 2006, 251, 200–214. [Google Scholar] [CrossRef]
- De Nooy, A.E.; Besemer, A.C.; van Bekkum, H. On the use of stable organic nitroxyl radicals for the oxidation of primary and secondary alcohols. Synthesis 1996, 1996, 1153–1176. [Google Scholar] [CrossRef]
- Gamez, P.; Arends, I.W.C.E.; Sheldon, R.A.; Reedijk, J. Room Temperature Aerobic Copper-Catalysed Selective Oxidation of Primary Alcohols to Aldehydes. Adv. Synth. Catal. 2004, 346, 805–811. [Google Scholar] [CrossRef]
- Karimi, B.; Badreh, E. SBA-15-functionalized TEMPO confined ionic liquid: an efficient catalyst system for transition-metal-free aerobic oxidation of alcohols with improved selectivity. Org. Biomol. Chem. 2011, 9, 4194–4198. [Google Scholar] [CrossRef]
- Cecchetto, A.; Fontana, F.; Minisci, F.; Recupero, F. Efficient Mn–Cu and Mn–Co–TEMPO-catalysed oxidation of alcohols into aldehydes and ketones by oxygen under mild conditions. Tetrahedron Lett. 2001, 42, 6651–6653. [Google Scholar] [CrossRef]
- Karimi, B.; Biglari, A.; Clark, J.H.; Budarin, V. Green, Transition-Metal-Free Aerobic Oxidation of Alcohols Using a Highly Durable Supported Organocatalyst. Angew. Chem. Int. Ed. 2007, 46, 7210–7213. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Liang, X.; Dong, C.; Hu, X. Transition-metal-free: A highly efficient catalytic aerobic alcohol oxidation process. J. Am. Chem. Soc. 2004, 126, 4112–4113. [Google Scholar] [CrossRef] [PubMed]
- Brackman, W.; Gaasbeek, C. Studies in homogeneous catalysis.: Radicals of the R2NO-radical type as catalysts for the oxidation of methanol by cupric complexes and as promoters for various oxidations catalysed by copper. Recl. Trav. Chim. Pays-Bas 1966, 85, 221–241. [Google Scholar] [CrossRef]
- Mannam, S.; Alamsetti, S.K.; Sekar, G. Aerobic, Chemoselective Oxidation of Alcohols to Carbonyl Compounds Catalyzed by a DABCO-Copper Complex under Mild Conditions. Adv. Synth. Catal. 2007, 349, 2253–2258. [Google Scholar] [CrossRef]
- Geißlmeir, D.; Jary, W.G.; Falk, H. The TEMPO/copper catalyzed oxidation of primary alcohols to aldehydes using oxygen as stoichiometric oxidant. Monatsh. Chem. 2005, 136, 1591–1599. [Google Scholar] [CrossRef]
- Ansari Imtiaz, A.; Gree, R. TEMPO-Catalyzed Aerobic Oxidation of Alcohols to Aldehydes and Ketones in Ionic Liquid [bmim][PF6]. Org. Lett. 2002, 4, 1507–1509. [Google Scholar] [CrossRef]
- Betzemeier, B.; Cavazzini, M.; Quici, S.; Knochel, P. Copper-catalyzed aerobic oxidation of alcohols under fluorous biphasic conditions. Tetrahedron Lett. 2000, 41, 4343–4346. [Google Scholar] [CrossRef]
- Mirica, L.M.; Ottenwaelder, X.; Stack, T.D.P. Structure and spectroscopy of copper−dioxygen complexes. Chem. Rev. 2004, 104, 1013–1046. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Bie, Z.; Shang, S.; Lv, Y.; Li, G.; Niu, J.; Gao, S. Bioinspired aerobic oxidation of alcohols with a bifunctional ligand based on bipyridine and TEMPO. RSC Adv. 2016, 6, 35008–35013. [Google Scholar] [CrossRef]
- Hoover, J.M.; Ryland, B.L.; Stahl, S.S. Copper/TEMPO-Catalyzed Aerobic Alcohol Oxidation: Mechanistic Assessment of Different Catalyst Systems. ACS Catal. 2013, 3, 2599–2605. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Ragauskas, A.J. TEMPO-catalyzed oxidation of benzylic alcohols to aldehydes with the H2O 2/HBr/ionic liquid [bmim] PF 6 system. Tetrahedron Lett. 2005, 46, 3323–3326. [Google Scholar] [CrossRef]
- Miller, R.A.; Hoerrner, R.S. Iodine as a chemoselective reoxidant of TEMPO: Application to the oxidation of alcohols to aldehydes and ketones. Org. Lett. 2003, 5, 285–287. [Google Scholar] [CrossRef]
- Mestres, R.; Palenzuela, J. High atomic yield bromine-less benzylic bromination. Green Chem. 2002, 4, 314–316. [Google Scholar] [CrossRef]
- Velusamy, S.; Punniyamurthy, T. Copper (II)-Catalyzed Oxidation of Alcohols to Carbonyl Compounds with Hydrogen Peroxide. Eur. J. Org. Chem. 2003, 2003, 3913–3915. [Google Scholar] [CrossRef]
- Velusamy, S.; Srinivasan, A.; Punniyamurthy, T. Copper(II) catalyzed selective oxidation of primary alcohols to aldehydes with atmospheric oxygen. Tetrahedron Lett. 2006, 47, 923–926. [Google Scholar] [CrossRef]
- Figiel, P.J.; Sibaouih, A.; Ahmad, J.U.; Nieger, M.; Räisänen, M.T.; Leskelä, M.; Repo, T. Aerobic Oxidation of Benzylic Alcohols in Water by 2, 2, 6, 6-Tetramethylpiperidine-1-oxyl (TEMPO)/Copper (II) 2-N-Arylpyrrolecarbaldimino Complexes. Adv. Synth. Catal. 2009, 351, 2625–2632. [Google Scholar] [CrossRef]
- Wagner-Jauregg, T.; Hackley, B.E., Jr.; Lies, T.; Owens, O.; Proper, R. Model Reactions of Phosphorus-containing Enzyme Inactivators. IV. 1a The Catalytic Activity of Certain Metal Salts and Chelates in the Hydrolysis of Diisopropyl Fluorophosphate1b. J. Am. Chem. Soc. 1955, 77, 922–929. [Google Scholar] [CrossRef]
- Figiel, P.J.; Leskelä, M.; Repo, T. TEMPO-Copper(II) Diimine-Catalysed Oxidation of Benzylic Alcohols in Aqueous Media. Adv. Synth. Catal. 2007, 349, 1173–1179. [Google Scholar] [CrossRef]
- Duan, R.; Cheng, L.; Zhang, Q.; Ma, L.; Ma, H.; Yang, J. Mechanistic insight into the aerobic oxidation of benzyl alcohol catalyzed by the Cu II–TEMPO catalyst in alkaline water solution. RSC Adv. 2015, 5, 83976–83984. [Google Scholar] [CrossRef]
- Ryland, B.L.; Stahl, S.S. Practical Aerobic Oxidations of Alcohols and Amines with Homogeneous Copper/TEMPO and Related Catalyst Systems. Angew. Chem. Int. Ed. 2014, 53, 8824–8838. [Google Scholar] [CrossRef]
- Steves, J.E.; Stahl, S.S. Copper(I)/ABNO-Catalyzed Aerobic Alcohol Oxidation: Alleviating Steric and Electronic Constraints of Cu/TEMPO Catalyst Systems. J. Am. Chem. Soc. 2013, 135, 15742–15745. [Google Scholar] [CrossRef]
- Ryland, B.L.; McCann, S.D.; Brunold, T.C.; Stahl, S.S. Mechanism of Alcohol Oxidation Mediated by Copper(II) and Nitroxyl Radicals. J. Am. Chem. Soc. 2014, 136, 12166–12173. [Google Scholar] [CrossRef]
- van Albada, G.A.; Mutikainen, I.; Roubeau, O.; Turpeinen, U.; Reedijk, J. Ferromagnetic trinuclear carbonato-bridged and tetranuclear hydroxo-bridged Cu (II) compounds with 4, 4′-dimethyl-2, 2′-bipyridine as ligand. X-ray structure, spectroscopy and magnetism. Inorg. Chim. Acta 2002, 331, 208–215. [Google Scholar] [CrossRef]
- Zheng, Y.-Q.; Lin, J.-L. Crystal structures of [Cu2(bpy)2(H2O)(OH)2(SO4)]·4H2O and [Cu(bpy)(H2O)2]SO4 with bpy = 2,2′-bipyridine. Z. Anorg. Allg. Chem. 2003, 629, 1622–1626. [Google Scholar] [CrossRef]
- Faucherre, J.; Petitfaux, C. Copper(II) chelates of pyridine-2-aldoxime. Bull. Soc. Chim. Fr. 1965, 347–359. [Google Scholar]
- Perlepes, S.P.; Huffman, J.C.; Christou, G. Preparation and characterization of dinuclear copper(II) complexes containing the [Cu2(μ-OAc)2]2+ core. Polyhedron 1992, 11, 1471–1479. [Google Scholar] [CrossRef]
- van Albada, G.A.; Mutikainen, I.; Turpeinen, U.; Reedijk, J. Structure, spectroscopy and magnetism of a strong ferromagnetically coupled dinuclear hydroxo-bridged Cu (II) compound with 4, 4′-dimethyl-2, 2′-bipyridine as a ligand. The first X-ray structure of a dinuclear Cu (II) compound with dmbipy. Inorg. Chim. Acta 2001, 324, 273–277. [Google Scholar] [CrossRef]
- Mei, Q.; Liu, H.; Yang, Y.; Liu, H.; Li, S.; Zhang, P.; Han, B. Base-Free Aerobic Oxidation of Alcohols over Copper-Based Complex under Ambient Condition. ACS Sustain. Chem. Eng. 2018, 6, 2362–2369. [Google Scholar]
- Liu, Z.; Shen, Z.; Zhang, N.; Zhong, W.; Liu, X. Aerobic Oxidation of Alcohols Catalysed by Cu (I)/NMI/TEMPO System and Its Mechanistic Insights. Catal. Lett. 2018, 148, 2709–2718. [Google Scholar] [CrossRef]
- Anelli, P.L.; Biffi, C.; Montanari, F.; Quici, S. Fast and selective oxidation of primary alcohols to aldehydes or to carboxylic acids and of secondary alcohols to ketones mediated by oxoammonium salts under two-phase conditions. J. Org. Chem. 1987, 52, 2559–2562. [Google Scholar] [CrossRef]
- Rychnovsky, S.D.; Vaidyanathan, R. TEMPO-catalyzed oxidations of alcohols using m-CPBA: The role of halide ions. J. Org. Chem. 1999, 64, 310–312. [Google Scholar] [CrossRef]
- Shibuya, M.; Tomizawa, M.; Iwabuchi, Y. Oxidative rearrangement of tertiary allylic alcohols employing oxoammonium salts. J. Org. Chem. 2008, 73, 4750–4752. [Google Scholar] [CrossRef]
- Bobbit, J.M. Oxoammonium Salts. 6.6 4-Acetylamino-2,2,6,6-tetramethylpiperidine-1-oxoammonium Perchlorate: A Stable and Convenient Reagent for the Oxidation of Alcohols. Silica Gel Catalysis. J. Org. Chem. 1998, 63, 9367–9374. [Google Scholar] [CrossRef]
- Semmelhack, M.; Chou, C.S.; Cortes, D.A. Nitroxyl-mediated electrooxidation of alcohols to aldehydes and ketones. J. Am. Chem. Soc. 1983, 105, 4492–4494. [Google Scholar] [CrossRef]
- Michel, C.; Belanzoni, P.; Gamez, P.; Reedijk, J.; Baerends, E.J. Activation of the C− H bond by electrophilic attack: theoretical study of the reaction mechanism of the aerobic oxidation of alcohols to aldehydes by the Cu (bipy) 2+/2, 2, 6, 6-tetramethylpiperidinyl-1-oxy cocatalyst system. Inorg. Chem. 2009, 48, 11909–11920. [Google Scholar] [CrossRef]
- Semmelhack, M.; Schmid, C.R.; Cortés, D.A. Mechanism of the oxidation of alcohols by 2, 2, 6, 6-tetramethylpiperidine nitrosonium cation. Tetrahedron Lett. 1986, 27, 1119–1122. [Google Scholar] [CrossRef]
- Iron, M.A.; Szpilman, A.M. Mechanism of the Copper/TEMPO-Catalyzed Aerobic Oxidation of Alcohols. Chem. Eur. J. 2017, 23, 1368–1378. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Stack, T. Galactose oxidase model complexes: Catalytic reactivities. J. Am. Chem. Soc. 1996, 118, 13097–13098. [Google Scholar] [CrossRef]
- Caneschi, A.; Grand, A.; Laugier, J.; Rey, P.; Subra, R. Three-center binding of a nitroxyl free radical to copper (II) bromide. J. Am. Chem. Soc. 1988, 110, 2307–2309. [Google Scholar] [CrossRef]
- Laugier, J.; Latour, J.M.; Caneschi, A.; Rey, P. Structural and redox properties of the Tempo adducts of copper (II) halides. Inorg. Chem. 1991, 30, 4474–4477. [Google Scholar] [CrossRef]
- Ben-Daniel, R.; Alsters, P.; Neumann, R. Selective Aerobic Oxidation of Alcohols with a Combination of a Polyoxometalate and Nitroxyl Radical as Catalysts. J. Org. Chem. 2001, 66, 8650–8653. [Google Scholar] [CrossRef]
- Lewis, E.A.; Tolman, W.B. Reactivity of dioxygen-copper systems. Chem. Rev. 2004, 104, 1047–1076. [Google Scholar] [CrossRef]
- Kim, C.; Chen, K.; Kim, J.; Que, L. Stereospecific Alkane Hydroxylation with H2O2 Catalyzed by an Iron (II)− Tris (2-pyridylmethyl) amine Complex. J. Am. Chem. Soc. 1997, 119, 5964–5965. [Google Scholar] [CrossRef]
- Bordwell, F.G.; Liu, W.-Z. Equilibrium Acidities and Homolytic Bond Dissociation Energies of N− H and/or O− H Bonds in N-Phenylhydroxylamine and Its Derivatives. J. Am. Chem. Soc. 1996, 118, 8777–8781. [Google Scholar] [CrossRef]
- Olmstead, W.N.; Margolin, Z.; Bordwell, F.G. Acidities of water and simple alcohols in dimethyl sulfoxide solution. J. Org. Chem. 1980, 45, 3295–3299. [Google Scholar] [CrossRef]
- Walroth, R.C.; Miles, K.C.; Lukens, J.T.; MacMillan, S.N.; Stahl, S.S.; Lancaster, K.M. Electronic Structural Analysis of Copper (II)–TEMPO/ABNO Complexes Provides Evidence for Copper (I)–Oxoammonium Character. J. Am. Chem. Soc. 2017, 139, 13507–13517. [Google Scholar] [CrossRef]
- Holland, P.L.; Rodgers, K.R.; Tolman, W.B. Is the Bis(µ-oxo) dicopper Core Capable of Hydroxylating an Arene? Angew. Chem. Int. Ed. 1999, 28, 1139–1142. [Google Scholar] [CrossRef]
- Kitajima, N.; Moro-oka, Y. Copper-dioxygen complexes. Inorganic and bioinorganic perspectives. Chem. Rev. 1994, 94, 737–757. [Google Scholar] [CrossRef]
- Adomeit, S.; Rabeah, J.; Surkus, A.E.; Bentrup, U.; Brückner, A. Effects of Imidazole-Type Ligands in CuI/TEMPO-Mediated Aerobic Alcohol Oxidation. Inorg. Chem. 2017, 56, 684–691. [Google Scholar] [CrossRef]
- Tebben, L.; Studer, A. Nitroxides: applications in synthesis and in polymer chemistry. Angew. Chem. Int. Ed. 2011, 50, 5034–5068. [Google Scholar]
- Bowman, D.; Gillan, T.; Ingold, K. Kinetic applications of electron paramagnetic resonance spectroscopy. III. Self-reactions of dialkyl nitroxide radicals. J. Am. Chem. Soc. 1971, 93, 6555–6561. [Google Scholar] [CrossRef]
- Malatesta, V.; Ingold, K. Protonated nitroxide radicals. J. Am. Chem. Soc. 1973, 95, 6404–6407. [Google Scholar] [CrossRef]
- Keana, J.F. Newer aspects of the synthesis and chemistry of nitroxide spin labels. Chem. Rev. 1978, 78, 37–64. [Google Scholar] [CrossRef]
- Nelsen, S.F.; Kessel, C.R.; Brien, D.J. Bredt’s rule kinetically stabilized nitrogen-centered radical cations and radicals in the 9-azabicyclo [3.3.1] nonyl system. J. Am. Chem. Soc. 1980, 102, 702–711. [Google Scholar] [CrossRef]
- Sasano, Y.; Nagasawa, S.; Yamazaki, M.; Shibuya, M.; Park, J.; Iwabuchi, Y. Highly chemoselective aerobic oxidation of amino alcohols into amino carbonyl compounds. Angew. Chem. Int. Ed. 2014, 53, 3236–3240. [Google Scholar] [CrossRef] [PubMed]
- Rafiee, M.; Konz, Z.M.; Graaf, M.D.; Koolman, H.F.; Stahl, S.S. Electrochemical Oxidation of Alcohols and Aldehydes to Carboxylic Acids Catalyzed by 4-Acetamido-TEMPO: An Alternative to “Anelli” and “Pinnick” Oxidations. ACS Catal. 2018, 8, 6738–6744. [Google Scholar] [CrossRef]
- Shibuya, M.; Tomizawa, M.; Sasano, Y.; Iwabuchi, Y. An expeditious entry to 9-Azabicyclo [3.3.1] nonane N-Oxyl (ABNO): Another highly active organocatalyst for oxidation of alcohols. J. Org. Chem. 2009, 74, 4619–4622. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Sasano, Y.; Nagasawa, S.; Shibuya, M.; Iwabuchi, Y. 9-Azanoradamantane N-oxyl (Nor-AZADO): A highly active organocatalyst for alcohol oxidation. Chem. Pharm. Bull. 2011, 59, 1570–1573. [Google Scholar] [CrossRef]
- Xu, B.; Lumb, J.P.; Arndtsen, B.A. A TEMPO-Free Copper-Catalyzed Aerobic Oxidation of Alcohols. Angew. Chem. Int. Ed. 2015, 54, 4208–4211. [Google Scholar] [CrossRef]
- McCann, S.D.; Lumb, J.-P.; Arndtsen, B.A.; Stahl, S.S. Second-Order Biomimicry: In Situ Oxidative Self-Processing Converts Copper(I)/Diamine Precursor into a Highly Active Aerobic Oxidation Catalyst. ACS Cent. Sci. 2017, 3, 314–321. [Google Scholar] [CrossRef]
- Chaloner, L.; Khomutovskaya, A.; Thomas, F.; Ottenwaelder, X. Supramolecular control of monooxygenase reactivity in a copper (II) cryptate. Dalton Trans. 2016, 45, 11109–11119. [Google Scholar] [CrossRef]
- Zhang, C.X.; Liang, H.-C.; Kim, E.-I.; Gan, Q.-F.; Tyeklár, Z.; Lam, K.-C.; Rheingold, A.L.; Kaderli, S.; Zuberbühler, A.D.; Karlin, K.D. Dioxygen mediated oxo-transfer to an amine and oxidative N-dealkylation chemistry with a dinuclear copper complex. Chem. Commun. 2001, 631–632. [Google Scholar] [CrossRef]
- Belanzoni, P.; Michel, C.; Baerends, E.J. Cu (bipy) 2+/TEMPO-catalyzed oxidation of alcohols: Radical or nonradical mechanism? Inorg. Chem. 2011, 50, 11896–11904. [Google Scholar] [CrossRef]
- Lu, Z.; Costa, J.S.; Roubeau, O.; Mutikainen, I.; Turpeinen, U.; Teat, S.J.; Gamez, P.; Reedijk, J. A copper complex bearing a TEMPO moiety as catalyst for the aerobic oxidation of primary alcohols. Dalton Trans. 2008, 3567–3573. [Google Scholar] [CrossRef]
- Lu, Z.; Ladrak, T.; Roubeau, O.; Van Der Toorn, J.; Teat, S.J.; Massera, C.; Gamez, P.; Reedijk, J. Selective, catalytic aerobic oxidation of alcohols using CuBr2 and bifunctional triazine-based ligands containing both a bipyridine and a TEMPO group. Dalton Trans. 2009, 3559–3570. [Google Scholar] [CrossRef]
- Liu, X.; Xia, Q.; Zhang, Y.; Chen, C.; Chen, W. Cu-NHC-TEMPO catalyzed aerobic oxidation of primary alcohols to aldehydes. J. Org. Chem. 2013, 78, 8531–8536. [Google Scholar] [CrossRef]
- Prathap, K.J.; Maayan, G. Metallopeptoids as efficient biomimetic catalysts. Chem. Commun. 2015, 51, 11096–11099. [Google Scholar] [CrossRef]
- Kwon, O.; Esguerra, K.V.N.; Glazerman, M.; Petitjean, L.; Xu, Y.; Ottenwaelder, X.; Lumb, J.-P. Development of 3, 5-Di-tert-butylphenol as a Model Substrate for Biomimetic Aerobic Copper Catalysis. Synlett 2017, 28, 1548–1553. [Google Scholar]
- Hamann, J.N.; Tuczek, F. New catalytic model systems of tyrosinase: fine tuning of the reactivity with pyrazole-based N-donor ligands. Chem. Commun. 2014, 50, 2298–2300. [Google Scholar] [CrossRef]
- Esguerra, K.V.N.; Fall, Y.; Petitjean, L.; Lumb, J.-P. Controlling the Catalytic Aerobic Oxidation of Phenols. J. Am. Chem. Soc. 2014, 136, 7662–7668. [Google Scholar] [CrossRef]




























© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).