Hydrodeoxygenation (HDO) of Aliphatic Oxygenates and Phenol over NiMo/MgAl2O4: Reactivity, Inhibition, and Catalyst Reactivation


 ,
,  and
and 
Abstract
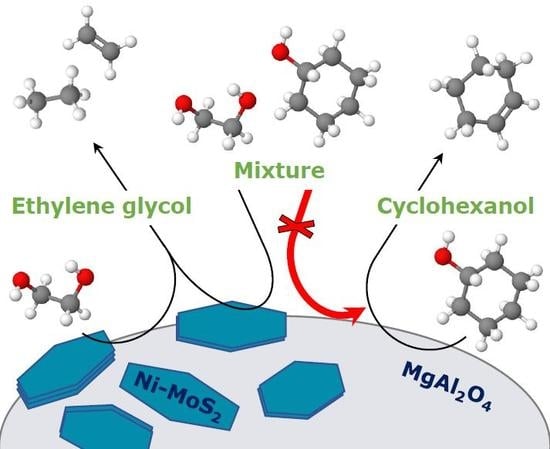
:
1. Introduction
2. Results
2.1. Reaction Thermodynamics
2.2. Reaction Schemes
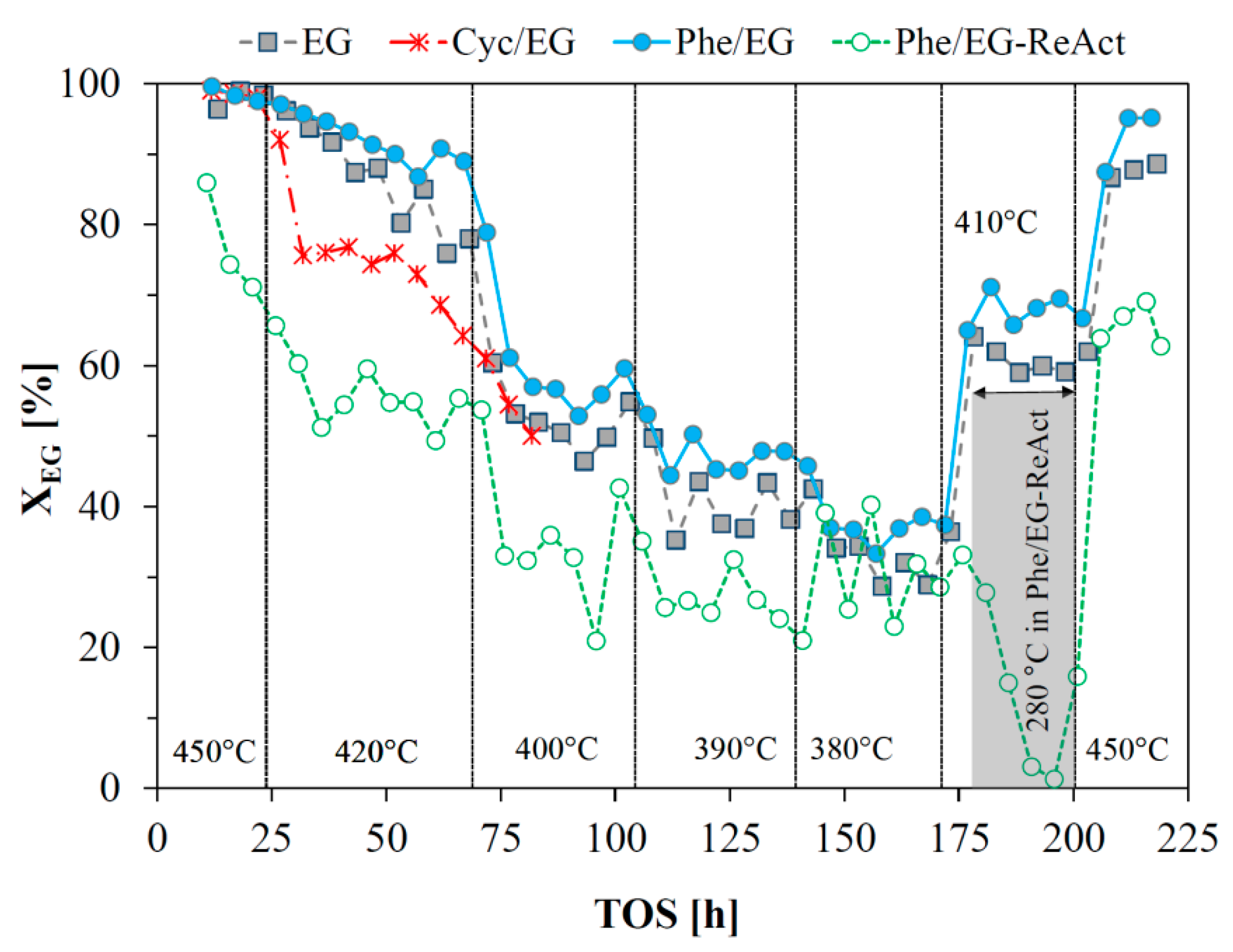
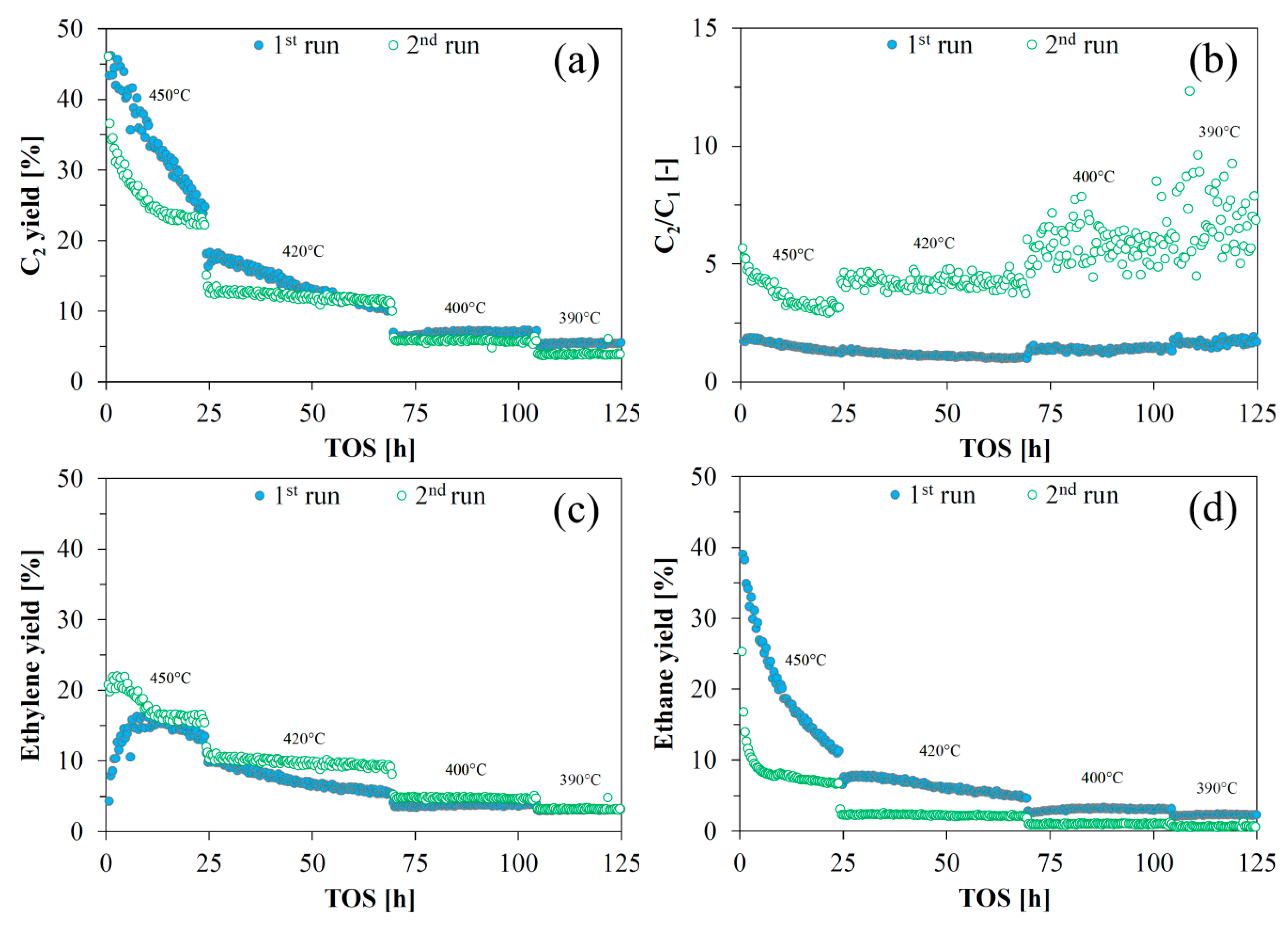
2.3. HDO of Ethylene Glycol
2.4. Catalyst Reactivation
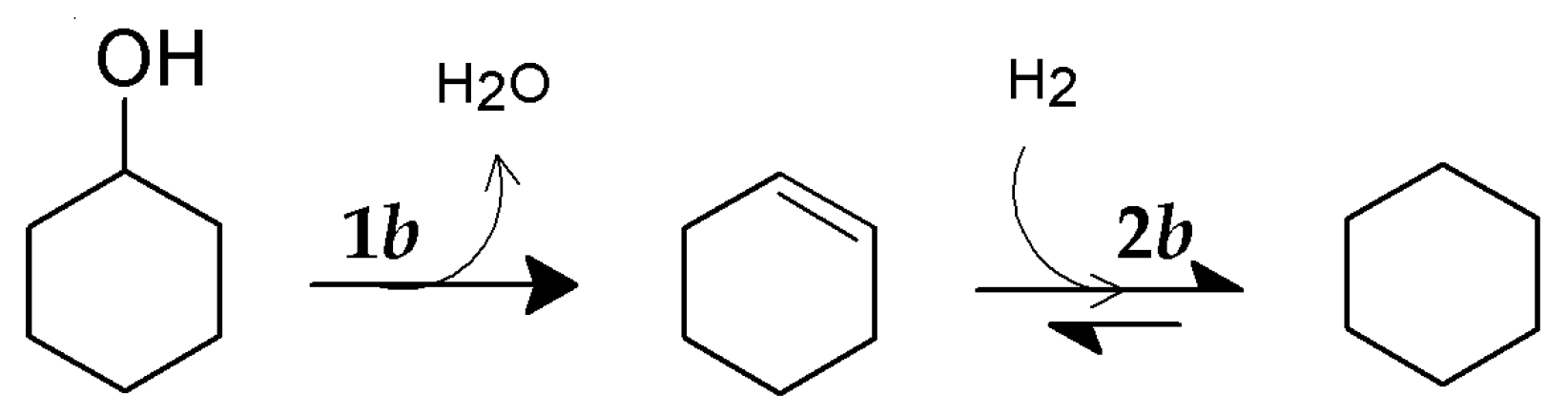
2.5. HDO of Cyclohexanol
2.6. HDO of Phenol
2.7. HDO of Acetic Acid
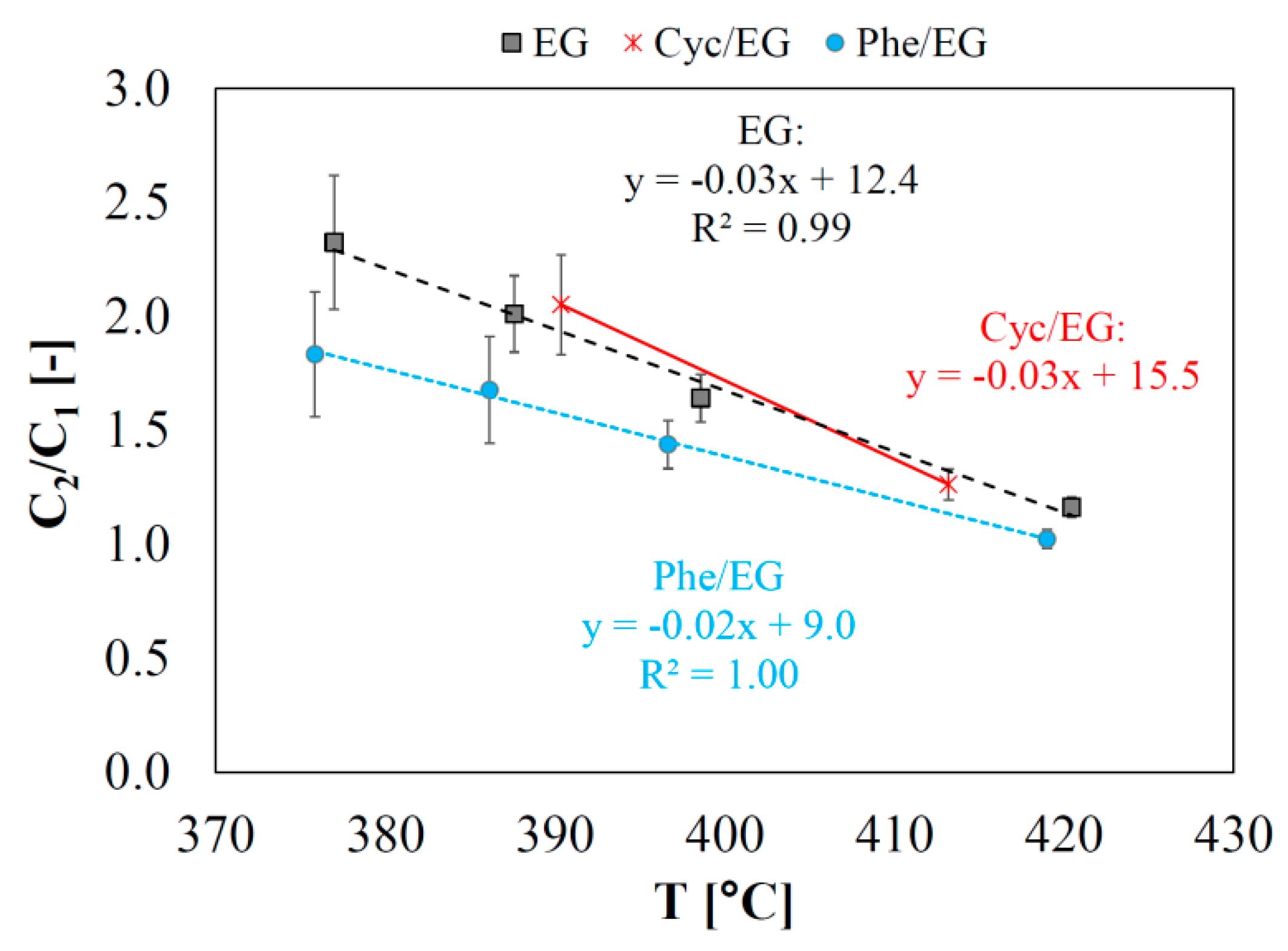
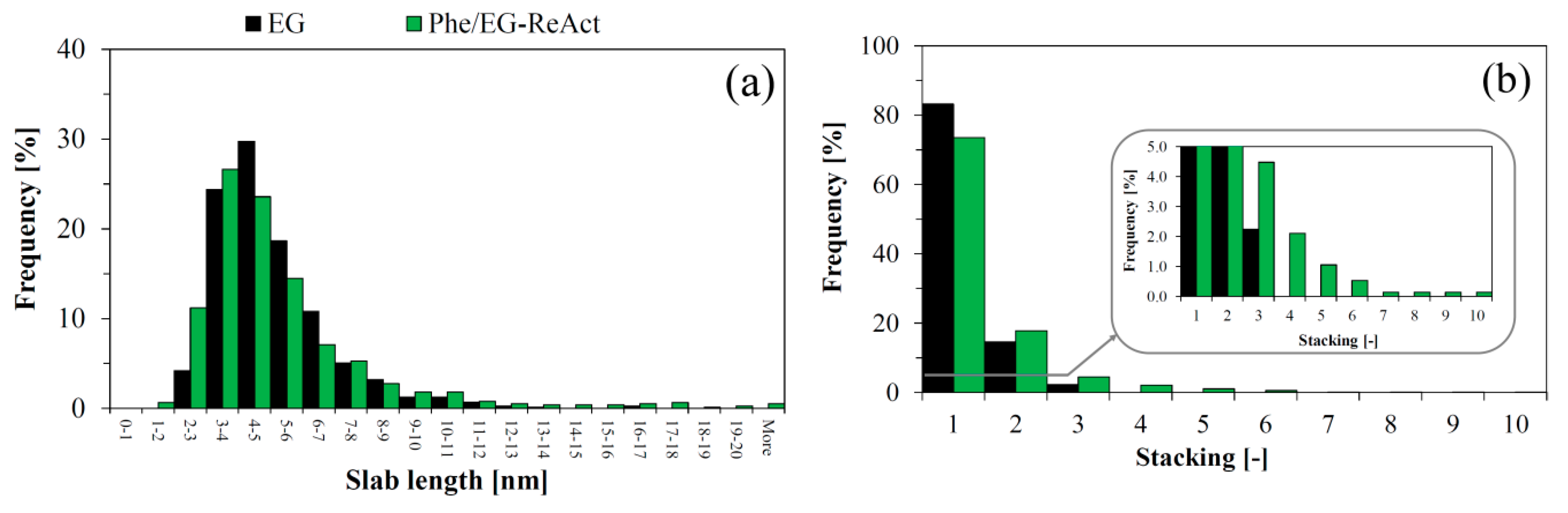
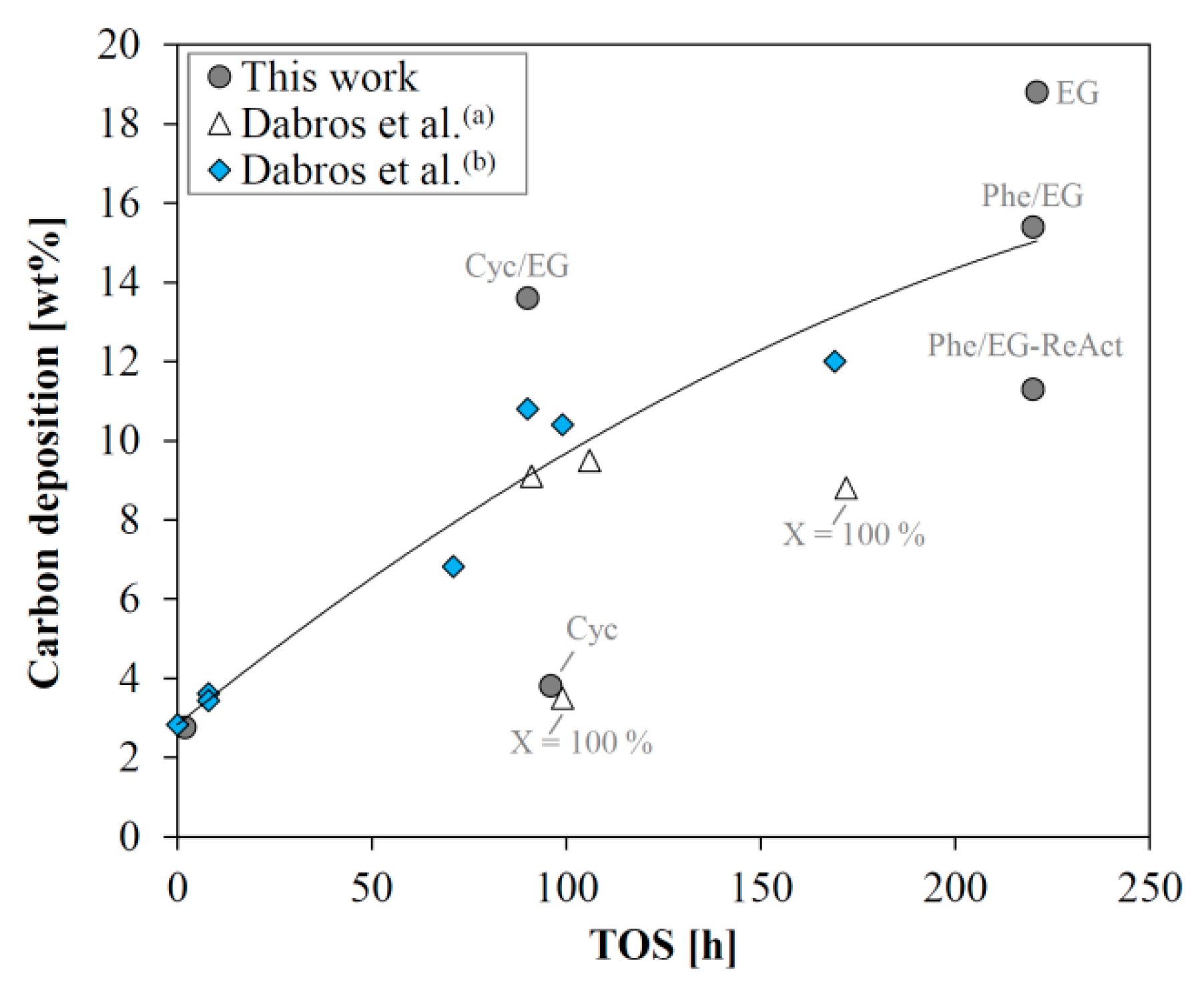
2.8. Investigation of Catalyst Deactivation
3. Discussion
4. Materials and Methods
4.1. Catalyst Preparation and Characterization
4.2. Activity Testing
4.3. Calculations
4.4. Reaction Conditions
4.5. Macro-Kinetic Model
- Reaction 1a:
- Reaction 2a:
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bridgwater, A.V. Review of fast pyrolysis of biomass and product upgrading. Biomass Bioenergy 2012, 38, 68–94. [Google Scholar] [CrossRef]
- Stummann, M.Z.; Høj, M.; Hansen, A.B.; Davidsen, B.; Wiwel, P.; Gabrielsen, J.; Jensen, P.A.; Jensen, A.D. New insights into the effect of pressure on catalytic hydropyrolysis of biomass. Fuel Process. Technol. 2019, 193, 392–403. [Google Scholar] [CrossRef]
- Venderbosch, R.H. A critical view on catalytic pyrolysis of biomass. Chem. Sus. Chem. 2015, 8, 1306–1316. [Google Scholar] [CrossRef]
- Resende, F.L.P. Recent advances on fast hydropyrolysis of biomass. Catal. Today. 2016, 269, 148–155. [Google Scholar] [CrossRef]
- Furimsky, E. Catalytic hydrodeoxygenation. Appl. Catal. A Gen. 2000, 199, 147–190. [Google Scholar] [CrossRef]
- Dabros, T.M.H.; Stummann, M.Z.; Høj, M.; Jensen, P.A.; Grunwaldt, J.-D.; Gabrielsen, J.; Mortensen, P.M.; Jensen, A.D. Transportation fuels from biomass fast pyrolysis, catalytic hydrodeoxygenation, and catalytic fast hydropyrolysis. Prog. Energy Combust. Sci. 2018, 68, 268–309. [Google Scholar] [CrossRef]
- Wang, H.; Male, J.; Wang, Y. Recent advances in hydrotreating of pyrolysis bio-oil and its oxygen-containing model compounds. ACS Catal. 2013, 3, 1047–1070. [Google Scholar] [CrossRef]
- Azeez, A.M.; Meier, D.; Odermatt, J.; Willner, T. Fast pyrolysis of African and European lignocellulosic biomasses using Py-GC/MS and fluidized bed reactor. Energy Fuels 2010, 24, 2078–2085. [Google Scholar]
- Mante, O.D.; Babu, S.P.; Amidon, T.E. A comprehensive study on relating cell-wall components of lignocellulosic biomass to oxygenated species formed during pyrolysis. J. Anal. Appl. Pyrolysis 2014, 108, 56–67. [Google Scholar] [CrossRef]
- Oasmaa, A.; Kuoppala, E.; Solantausta, Y. Fast pyrolysis of forestry residue. 2. Physicochemical composition of product liquid. Energy Fuels 2003, 17, 433–443. [Google Scholar]
- Elliott, D.C. Historical developments in hydroprocessing bio-oils. Energy Fuels 2007, 21, 1792–1815. [Google Scholar]
- Yin, W.; Kloekhorst, A.; Venderbosch, R.H.; Bykova, M.V.; Khromova, S.A.; Yakovlev, V.A.; Heeres, H.J. Catalytic hydrotreatment of fast pyrolysis liquids in batch and continuous set-ups using a bimetallic Ni–Cu catalyst with a high metal content. Catal. Sci. Technol. 2016, 6, 5899–5915. [Google Scholar] [CrossRef]
- Dwiatmoko, A.A.; Lee, S.; Ham, H.C.; Choi, J.-W.; Suh, D.J.; Ha, J.-M. Effects of carbohydrates on the hydrodeoxygenation of lignin- derived phenolic compounds. ACS Catal. 2015, 5, 433–437. [Google Scholar] [CrossRef]
- Ryymin, E.-M.; Honkela, M.L.; Viljava, T.-R.; Krause, A.O.I. Competitive reactions and mechanisms in the simultaneous HDO of phenol and methyl heptanoate over sulphided NiMo/γ-Al2O3. Appl. Catal. A Gen. 2010, 389, 114–121. [Google Scholar] [CrossRef]
- Boscagli, C.; Raffelt, K.; Grunwaldt, J.-D. Reactivity of platform molecules in pyrolysis oil and in water during hydrotreatment over nickel and ruthenium catalysts. Biomass Bioenergy 2017, 106, 63–73. [Google Scholar] [CrossRef]
- Stummann, M.Z.; Høj, M.; Schandel, C.B.; Hansen, A.B.; Wiwel, P.; Gabrielsen, J.; Jensen, P.A.; Jensen, A.D. Hydrogen assisted catalytic biomass pyrolysis. Effect of temperature and pressure. Biomass Bioenergy 2018, 115, 97–107. [Google Scholar] [CrossRef]
- Trinh, T.N.; Jensen, P.A.; Dam-Johansen, K.; Knudsen, N.O.; Sørensen, H.R.; Hvilsted, S. Comparison of lignin, macroalgae, wood, and straw fast pyrolysis. Energy Fuels 2013, 27, 1399–1409. [Google Scholar]
- Dabros, T.M.H.; Kramer, H.; Høj, M.; Sprenger, P.; Gabrielsen, J.; Grunwaldt, J.-D.; Jensen, A.D. The influence of active phase loading on the hydrodeoxygenation (HDO) of ethylene glycol over promoted MoS2/MgAl2O4 catalysts. Top. Catal. 2019. [Google Scholar] [CrossRef]
- Dabros, T.M.H.; Gaur, A.; Pintos, D.G.; Sprenger, P.; Høj, M.; Hansen, T.W.; Studt, F.; Gabrielsen, J.; Grunwaldt, J.-D.; Jensen, A.D. Influence of H2O and H2S on the composition, activity, and stability of sulfided Mo, CoMo, and NiMo supported on MgAl2O4 for hydrodeoxygenation of ethylene glycol. Appl. Catal. A Gen. 2018, 551, 106–121. [Google Scholar] [CrossRef]
- Fogler, S.H. Elements of Chemical Reaction Engineering, 4th ed.; Pearson Education International, Prentice Hall: Westford, MA, USA, 2010. [Google Scholar]
- Popov, A.; Kondratieva, E.; Goupil, J.M.; Mariey, L.; Bazin, P.; Gilson, J.-P.; Travert, A.; Maugé, F. Bio-oils hydrodeoxygenation: Adsorption of phenolic molecules on oxidic catalyst supports. J. Phys. Chem. C 2010, 114, 15661–15670. [Google Scholar] [CrossRef]
- Popov, A.; Kondratieva, E.; Mariey, L.; Goupil, J.M.; El Fallah, J.; Gilson, J.-P.; Travert, A.; Maugé, F. Bio-oil hydrodeoxygenation: Adsorption of phenolic compounds on sulfided (Co)Mo catalysts. J. Catal. 2013, 297, 176–186. [Google Scholar] [CrossRef]
- Bruix, A.; Füchtbauer, H.G.; Tuxen, A.K.; Walton, A.S.; Andersen, M.; Porsgaard, S.; Besenbacher, F.; Hammer, B.; Lauritsen, J.V. In situ detection of active edge sites in single-layer MoS2 catalysts. ACS Nano. 2015, 9, 9322–9330. [Google Scholar] [CrossRef] [PubMed]
- Grønborg, S.S.; Šarić, M.; Moses, P.G.; Rossmeisl, J.; Lauritsen, J.V. Atomic scale analysis of sterical effects in the adsorption of 4,6-dimethyldibenzothiophene on a CoMoS hydrotreating catalyst. J. Catal. 2016, 344, 121–128. [Google Scholar] [CrossRef]
- Šarić, M.; Rossmeisl, J.; Moses, P.G. Modeling the adsorption of sulfur containing molecules and their hydrodesulfurization intermediates on the Co-promoted MoS2 catalyst by DFT. J. Catal. 2018, 358, 131–140. [Google Scholar] [CrossRef]
- Furimsky, E.; Massoth, F.E. Deactivation of hydroprocessing catalysts. Catal. Today 1999, 52, 381–495. [Google Scholar] [CrossRef]
- Dufresne, P. Hydroprocessing catalysts regeneration and recycling. Appl. Catal. A Gen. 2007, 322, 67–75. [Google Scholar] [CrossRef]
- Smolik, G.R.; Petti, D.A.; Schuetz, S.T. Oxidation, volatilization, and redistribution of molybdenum from TZM alloy in air. INEEL/EXT-99-01353; Idaho National Engineering and Environmental Laboratory, 2000. Available online: https://inldigitallibrary.inl.gov/sites/sti/sti/3318122.pdf (accessed on 9 June 2019).
- Schachtl, E.; Yoo, J.S.; Gutiérrez, O.Y.; Studt, F.; Lercher, J.A. Impact of Ni promotion on the hydrogenation pathways of phenanthrene on MoS2/γ-Al2O3. J. Catal. 2017, 352, 171–181. [Google Scholar] [CrossRef]
- Prins, R. Chapter 13.2: Hydrotreating. In Handbook of Heterogeneous Catalysis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; pp. 2695–2718. [Google Scholar]
- Bui, V.N.; Laurenti, D.; Afanasiev, P.; Geantet, C. Hydrodeoxygenation of guaiacol with CoMo catalysts. Part I: Promoting effect of cobalt on HDO selectivity and activity. Appl. Catal. B Environ. 2011, 101, 239–245. [Google Scholar] [CrossRef]
- Şenol, O.İ.; Ryymin, E.-M.; Viljava, T.-R.; Krause, A.O.I. Effect of hydrogen sulphide on the hydrodeoxygenation of aromatic and aliphatic oxygenates on sulphided catalysts. J. Mol. Catal. A Chem. 2007, 277, 107–112. [Google Scholar] [CrossRef]
- Bouvier, C.; Romero, Y.; Richard, F.; Brunet, S. Effect of H2S and CO on the transformation of 2-ethylphenol as a model compound of bio-crude over sulfided Mo-based catalysts: propositions of promoted active sites for deoxygenation pathways based on an experimental study. Green Chem. 2011, 13, 2441–2451. [Google Scholar] [CrossRef]
- Gonçalves, V.O.O.; Brunet, S.; Richard, F. Hydrodeoxygenation of cresols over Mo/Al2O3 and CoMo/Al2O3 sulfided catalysts. Catal. Letters 2016, 146, 1562–1573. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Gardini, D.; Damsgaard, C.D.; Grunwaldt, J.-D.; Jensen, P.A.; Wagner, J.B.; Jensen, A.D. Deactivation of Ni-MoS2 by bio-oil impurities during hydrodeoxygenation of phenol and octanol. Appl. Catal. A Gen. 2016, 523, 159–170. [Google Scholar] [CrossRef]
- Romero, Y.; Richard, F.; Brunet, S. Hydrodeoxygenation of 2-ethylphenol as a model compound of bio-crude over sulfided Mo-based catalysts: Promoting effect and reaction mechanism. Appl. Catal. B Environ. 2010, 98, 213–223. [Google Scholar] [CrossRef]
- Baldauf, W.; Balfanz, U.; Rupp, M. Upgrading of flash pyrolysis oil and utilization in refineries. Biomass Bioenergy 1994, 7, 237–244. [Google Scholar] [CrossRef]
- Elliott, D.C.; Hart, T.R.; Neuenschwander, G.G.; Rotness, L.J.; Zacher, A.H. Catalytic hydroprocessing of biomass fast pyrolysis bio-oil to produce hydrocarbon products. Environ. Prog. Sustain. Energy 2009, 28, 441–449. [Google Scholar] [CrossRef]
- Cordero-Lanzac, T.; Palos, R.; Arandes, J.M.; Castaño, P.; Rodríguez-Mirasol, J.; Cordero, T.; Bilbao, J. Stability of an acid activated carbon based bifunctional catalyst for the raw bio-oil hydrodeoxygenation. Appl. Catal. B Environ. 2017, 203, 389–399. [Google Scholar] [CrossRef]
- Lemonidou, A.A.; Kechagiopoulos, P.; Heracleous, E.; Voutetakis, S. Chapter 14: Steam reforming of bio-oils to hydrogen. In The Role of Catalysis for the Sustainable Production of Bio-Fuels and Bio-Chemicals; Triantafyllidis, K., Lappas, A., Stöcker, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 467–493. [Google Scholar]
- Van Doorn, J.; Moulijn, J.A.; Djéga-Mariadassou, G. High-resolution electron microscopy of spent Ni-Mo/Al2O3 hydrotreating catalysts. Appl. Catal. 1990, 63, 77–90. [Google Scholar] [CrossRef]
- Liu, G.; Robertson, A.W.; Li, M.M.-J.; Kuo, W.C.H.; Darby, M.T.; Muhieddine, M.H.; Lin, Y.-C.; Suenaga, K.; Stamatakis, M.; Warner, J.H.; et al. MoS2 monolayer catalyst doped with isolated Co atoms for the hydrodeoxygenation reaction. Nat. Chem. 2017, 9, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Nie, T.; Lai, W.; Yang, W.; Jiang, X. Tailoring the morphology of Co-doped MoS2 for enhanced hydrodeoxygenation performance of p-cresol. Cryst. Eng. Comm. 2018, 20, 4069–4074. [Google Scholar] [CrossRef]
- Gaur, A.; Dabros, T.M.H.; Høj, M.; Boubnov, A.; Prüssmann, T.; Jelic, J.; Studt, F.; Jensen, A.D.; Grunwaldt, J.-D. Probing the active sites of MoS2 based hydrotreating catalysts using modulation excitation spectroscopy. ACS Catal. 2019, 9, 2568–2579. [Google Scholar] [CrossRef]
- Marker, T.L.; Felix, L.G.; Linck, M.B.; Roberts, M.J. Integrated hydropyrolysis and hydroconversion (IH2) for the directproduction of gasoline and diesel fuels or blending components from biomass, part 1: Proof of principle testing. Environ. Prog. Sustain. Energy 2012, 31, 191–199. [Google Scholar] [CrossRef]
- Marker, T.L.; Felix, L.G.; Linck, M.B.; Roberts, M.J.; Ortiz-toral, P.; Wangerow, J. Integrated hydropyrolysis and hydroconversion (IH2) for the directproduction of gasoline and diesel fuels or blending components from biomass, part 2: Continuous testing. Environ. Prog. Sustain. Energy 2014, 33, 762–768. [Google Scholar] [CrossRef]
- Stummann, M.Z.; Hansen, A.B.; Hansen, L.P.; Davidsen, B.; Rasmussen, S.B.; Wiwel, P.; Gabrielsen, J.; Jensen, P.A.; Jensen, A.D.; Høj, M. Catalytic hydropyrolysis of biomass using molybdenum sulfide based catalyst. Effect of promoters. Energy Fuels 2019, 33, 1302–1313. [Google Scholar]
- Stummann, M.Z.; Høj, M.; Davidsen, B.; Hansen, A.B.; Hansen, L.P.; Wiwel, P.; Schandel, C.B.; Gabrielsen, J.; Jensen, P.A.; Jensen, A.D. Effect of the catalyst in fluid bed catalytic hydropyrolysis. Catal. Today 2019, in press. [Google Scholar] [CrossRef]
- Dayton, D.C.; Hlebak, J.; Carpenter, J.R.; Wang, K.; Mante, O.D.; Peters, J.E. Biomass hydropyrolysis in a fluidized bed reactor. Energy Fuels 2016, 30, 4879–4887. [Google Scholar]
- Routray, K.; Barnett, K.J.; Huber, G.W. Hydrodeoxygenation of pyrolysis oils. Energy Technol. 2017, 5, 80–93. [Google Scholar] [CrossRef]
- Elliott, D.C.; Neuenschwander, G.G.; Hart, T.R. Hydroprocessing bio-oil and products separation for coke production. ACS Sustain. Chem. Eng. 2013, 1, 389–392. [Google Scholar] [CrossRef]
- Zacher, A.H.; Elliott, D.C.; Olarte, M.V.; Santosa, D.M.; Preto, F.; Iisa, K. Pyrolysis of woody residue feedstocks: Upgrading of bio-oils from mountain-pine-beetle-killed trees and hog fuel. Energy Fuels 2014, 28, 7510–7516. [Google Scholar]
- Olarte, M.V.; Padmaperuma, A.B.; Ferrell, J.R., III; Christensen, E.D.; Hallen, R.T.; Lucke, R.B.; Burton, S.D.; Lemmon, T.L.; Swita, M.S.; Fioroni, G.; et al. Characterization of upgraded fast pyrolysis oak oil distillate fractions from sulfided and non-sulfided catalytic hydrotreating. Fuel 2017, 202, 620–630. [Google Scholar] [CrossRef]
- Schofield, K. The enigmatic mechanism of the flame ionization detector: Its overlooked implications for fossil fuel combustion modeling. Prog. Energy Combust. Sci. 2008, 34, 330–350. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | TOS | T | k′1a × 103 | k′2a × 103 | Ea,1a | Ea,2a | k′1a,mean × 103 | k′2a,mean × 103 |
|---|---|---|---|---|---|---|---|---|
| [h] | [°C] | [L/(min·g)] | [kJ/mol] | [L/(min·g)] | ||||
| EG | 90–104 | 399 | 16.4 | 271 | 93.3 | 8.15 | 15.0 | 269 |
| 134–139 | 388 | 12.4 | 264 | |||||
| 165–170 | 377 | 9.41 | 258 | |||||
| Phe/EG | 99–104 | 397 | 14.2 | 250 | 95.9 | 22.1 | 13.6 | 248 |
| 134–139 | 386 | 10.8 | 235 | |||||
| 165–170 | 376 | 8.15 | 220 | |||||
| Phe/EG-ReAct 1 | 148–153 | 388 | 12.6 | 78.4 | 141 | 32.3 | 16.4 | 83.3 |
| 173–178 | 377 | 8.19 | 71.1 | |||||
| Cyc/EG | 64–69 | 413 | 22.2 | 336 | 85.4 | 2.43 | 14.8 | 332 |
| 85–90 | 390 | 13.3 | 331 | |||||
| Experiment | TOS | Mo | Ni | Ni/Mo | S | C | S/Mo |
|---|---|---|---|---|---|---|---|
| [h] | [wt%] | [wt%] | [molar] | [wt%] | [wt%] | [molar] | |
| EG | 221 | 2.55 | 0.46 | 0.29 | 1.71 | 18.8 | 2.00 |
| Phe/EG-ReAct 1 | 220(2nd) | 1.45 | 0.44 | 0.49 | 2.40 | 11.3 | 4.96 |
| Experiment | TOS | T | k1b × 103 | k′2b × 103 | Ea,1b | Ea,2b | k1b,mean × 103 | k′2b,mean × 103 |
|---|---|---|---|---|---|---|---|---|
| [h] | [°C] | [L/(min·g)] | [kJ/mol] | [L/(min·g)] | ||||
| Cyc | 53–63 | 413 | ≥524 1 | 55.1 | - | 43.3 | - | 45.2 |
| 78–88 | 391 | ≥524 1 | 43.5 | |||||
| Cyc/EG | 54–64 | 413 | 21.4 | 74.4 | 121 | 82.9 | 12.1 | 50.3 |
| 74–84 | 390 | 10.3 | 44.9 | |||||
| Experiment | CH4 [%] | CO [%] | CO2 [%] | Ethylene [%] | Ethane [%] | Propylene [%] | Propane [%] | Total Carbon in Gas Phase [%] |
|---|---|---|---|---|---|---|---|---|
| EG | 8–9 | 12–13 | 4–5 | 8–11 | 28–29 | 4–7 | 3 | 67–74 |
| AcOH | 17–21 | 17–20 | 7–8 | 2 | 21–24 | 8–11 | 9–11 | 83–93 |
| Experiment | TOS | Mo | Ni | Ni/Mo | S | C | S/Mo |
|---|---|---|---|---|---|---|---|
| [h] | [wt%] | [wt%] | [molar] | [wt%] | [wt%] | [molar] | |
| Phe/EG | 220(1st) | - | - | - | - | 15.4 | - |
| Cyc/EG | 90 | 2.63 | 0.42 | 0.26 | 1.69 | 13.6 | 1.92 |
| Cyc | 96 | 2.96 | 0.51 | 0.28 | 2.18 | 3.81 | 2.51 |
| AcOH | 2 | 3.09 | 0.52 | 0.27 | 2.03 | 2.76 | 1.96 |
| Experiment/Parameter | EG | Phe/EG | Phe/EG-ReAct | Cyc/EG | Cyc |
|---|---|---|---|---|---|
| Molar Flow Rates [mmol/min] | |||||
| Ethylene glycol | 2.5 | 2.2 | 2.2 | 2.2 | 0 |
| Phenol | 0 | 0.4 | 0.4 | 0 | 0 |
| Cyclohexanol | 0 | 0 | 0 | 0.4 | 1.5 |
| Oxygen | 4.9 | 4.9 | 4.9 | 4.8 | 1.5 |
| Carbon | 4.9 | 6.9 | 6.9 | 6.7 | 9.0 |
| Total flow rate [mL/min] | 0.14 | 0.16 | 0.16 | 0.16 | 0.16 |
| Total WHSV [h−1] | 19 | 21 | 21 | 21 | 18 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dabros, T.M.H.; Andersen, M.L.; Lindahl, S.B.; Hansen, T.W.; Høj, M.; Gabrielsen, J.; Grunwaldt, J.-D.; Jensen, A.D. Hydrodeoxygenation (HDO) of Aliphatic Oxygenates and Phenol over NiMo/MgAl2O4: Reactivity, Inhibition, and Catalyst Reactivation. Catalysts 2019, 9, 521. https://doi.org/10.3390/catal9060521
Dabros TMH, Andersen ML, Lindahl SB, Hansen TW, Høj M, Gabrielsen J, Grunwaldt J-D, Jensen AD. Hydrodeoxygenation (HDO) of Aliphatic Oxygenates and Phenol over NiMo/MgAl2O4: Reactivity, Inhibition, and Catalyst Reactivation. Catalysts. 2019; 9(6):521. https://doi.org/10.3390/catal9060521
Chicago/Turabian StyleDabros, Trine Marie Hartmann, Mads Lysgaard Andersen, Simon Brædder Lindahl, Thomas Willum Hansen, Martin Høj, Jostein Gabrielsen, Jan-Dierk Grunwaldt, and Anker Degn Jensen. 2019. "Hydrodeoxygenation (HDO) of Aliphatic Oxygenates and Phenol over NiMo/MgAl2O4: Reactivity, Inhibition, and Catalyst Reactivation" Catalysts 9, no. 6: 521. https://doi.org/10.3390/catal9060521
APA StyleDabros, T. M. H., Andersen, M. L., Lindahl, S. B., Hansen, T. W., Høj, M., Gabrielsen, J., Grunwaldt, J.-D., & Jensen, A. D. (2019). Hydrodeoxygenation (HDO) of Aliphatic Oxygenates and Phenol over NiMo/MgAl2O4: Reactivity, Inhibition, and Catalyst Reactivation. Catalysts, 9(6), 521. https://doi.org/10.3390/catal9060521