Reaction of Glycerol with Trimethyl Orthoformate: Towards the Synthesis of New Glycerol Derivatives





Abstract
:
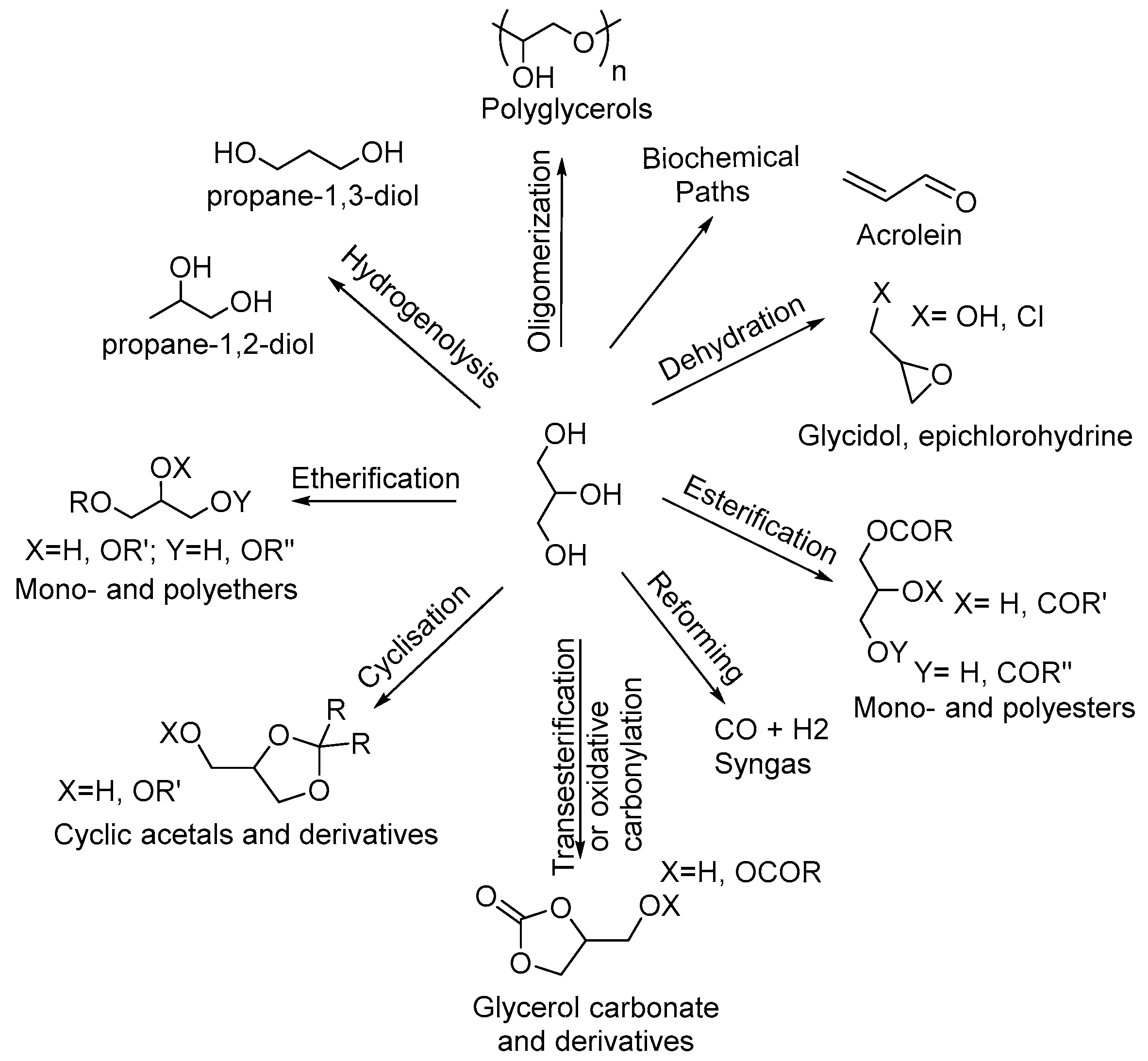
1. Introduction
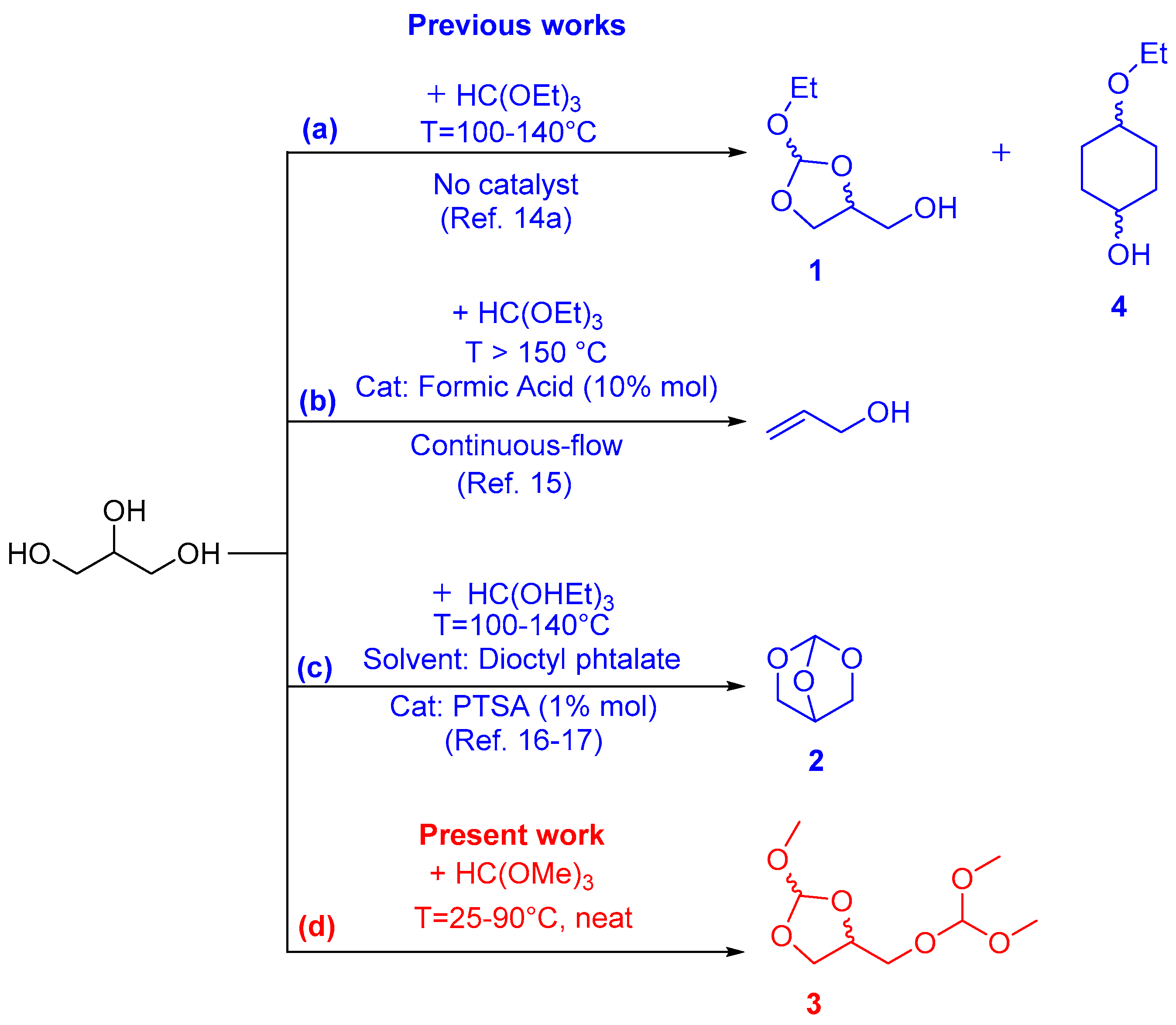
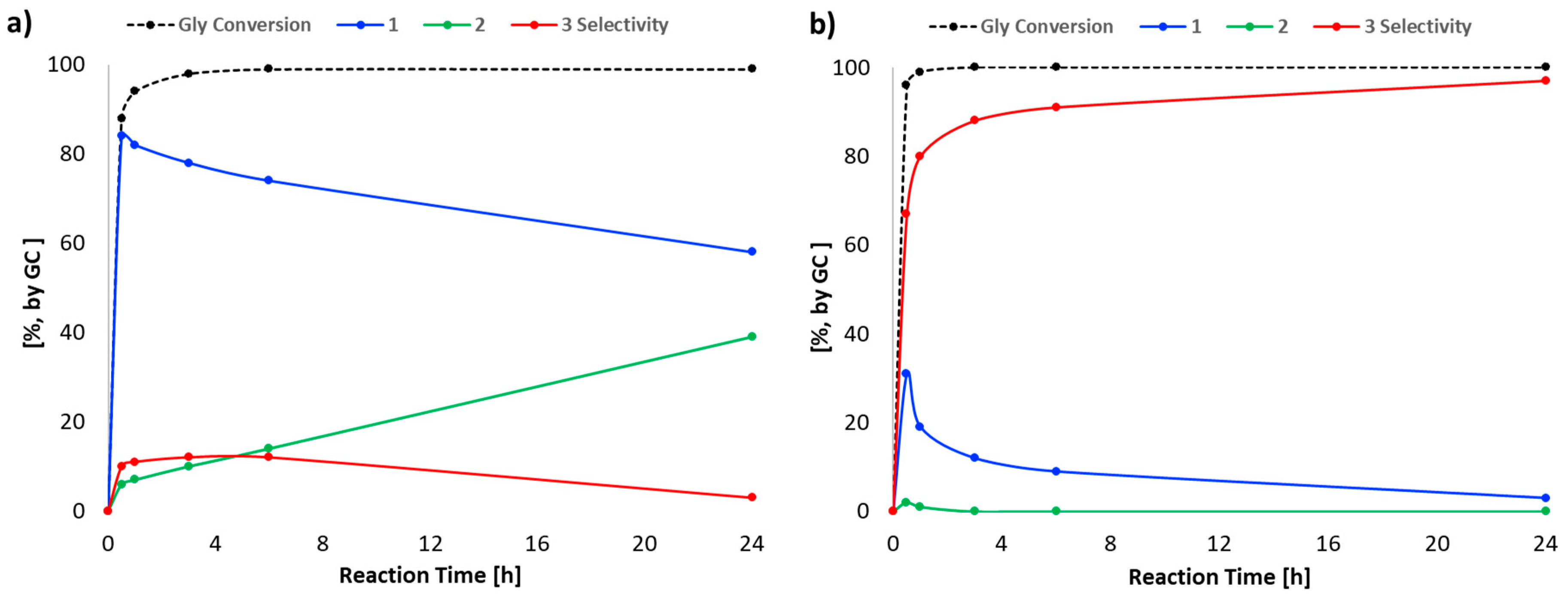
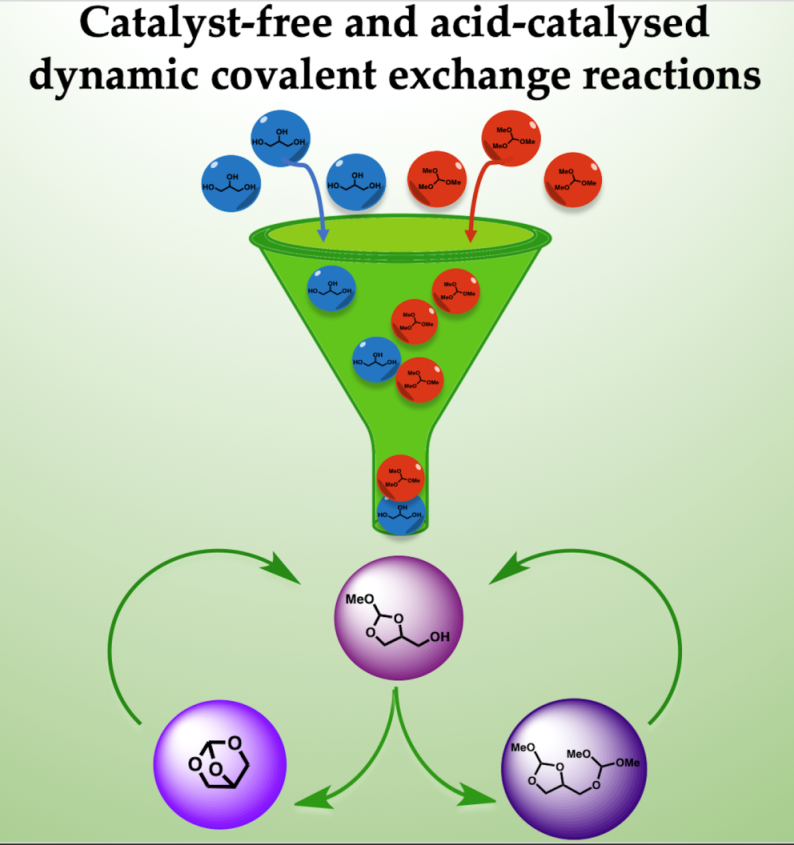
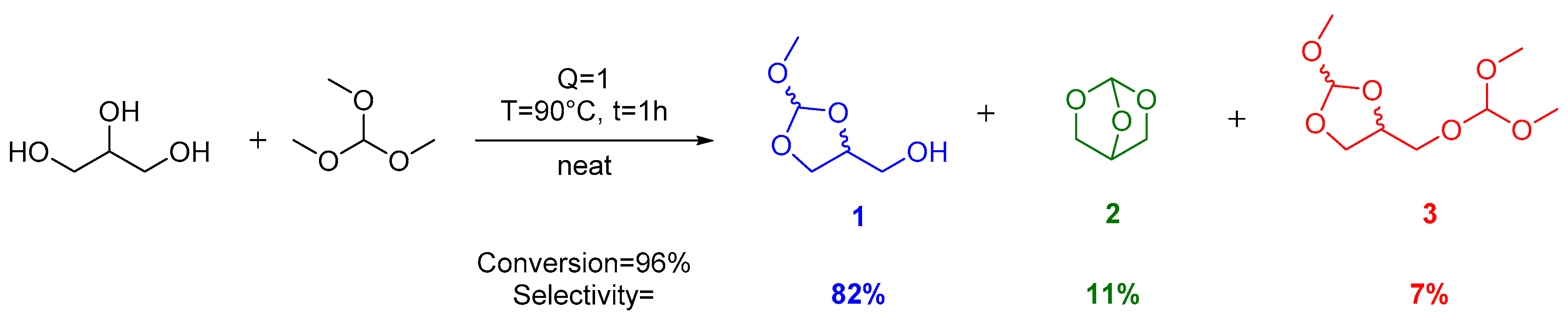
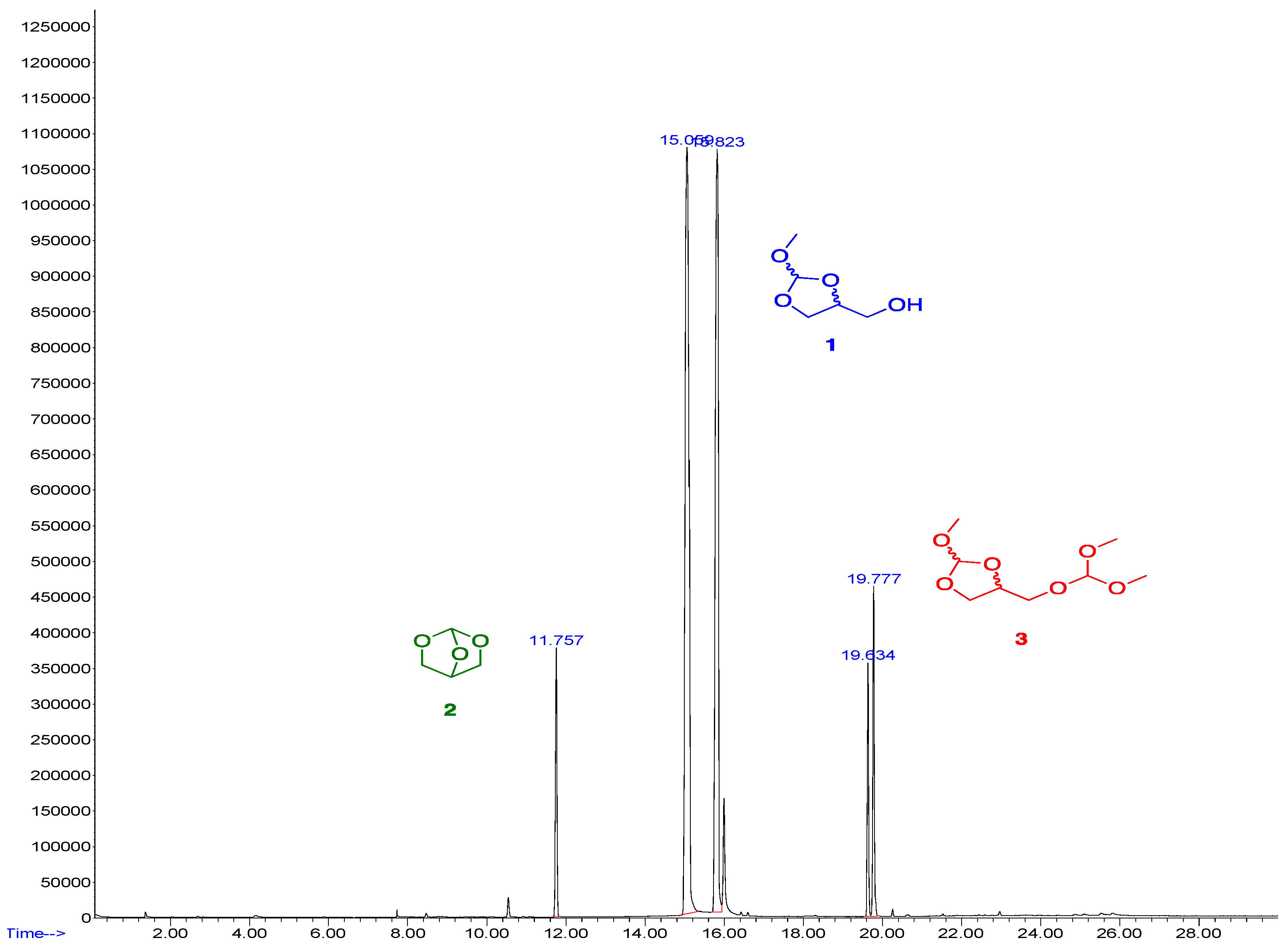
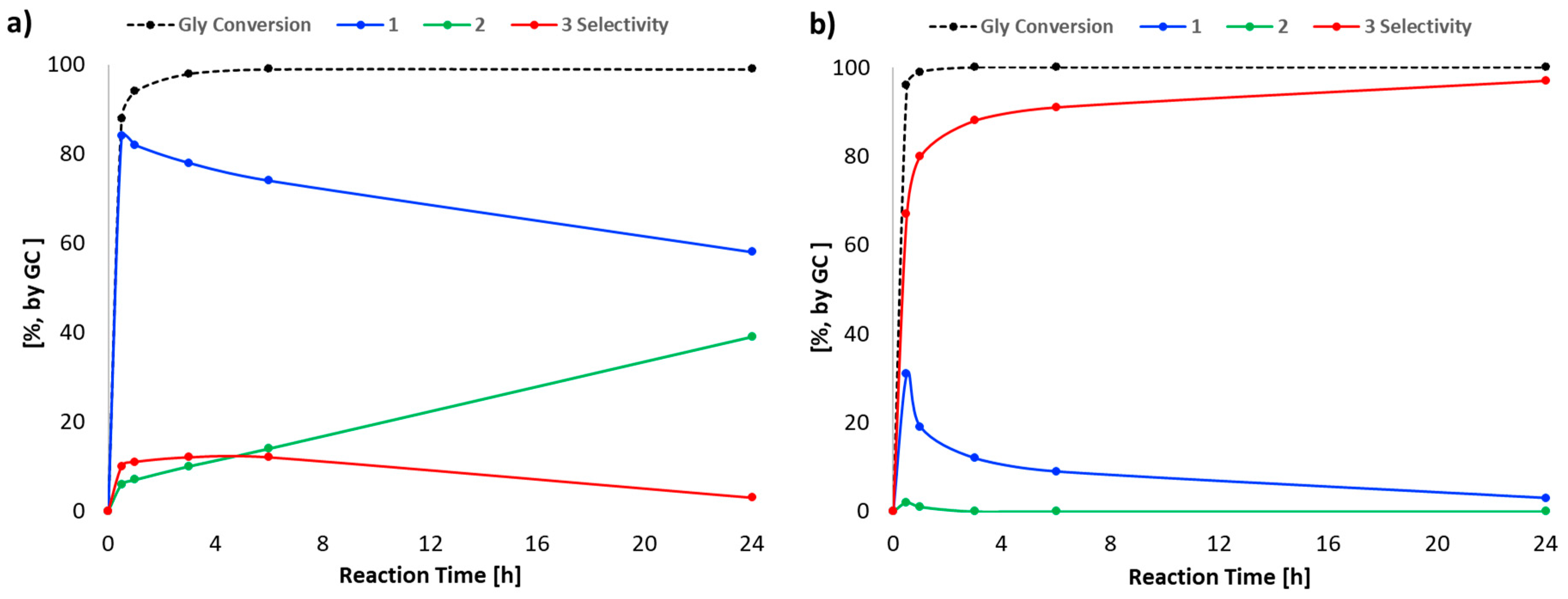
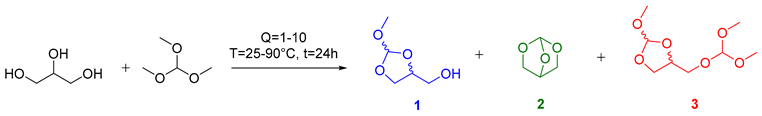
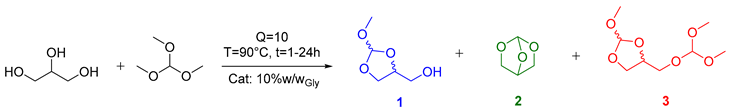
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthesis/Isolation of Reaction Products
3.2.1. Synthesis of (2-methoxy-1,3-dioxolan-4-yl) Methanol (1)
3.2.2. Synthesis of 2,6,7-Trioxabyciclo [2.2.1] Heptane (2)
3.2.3. Synthesis of 4-(Dimethoxymethoxy)Methyl)-2-Methoxy-1,3-Dioxolane (3)
3.3. Synthesis of the Acidic Catalysts
3.3.1. Synthesis of Pyridinium Para-Toluensulfonate (PPTS)
3.3.2. Synthesis of Diazobicycloundecenium Bromide (DBUHBr)
3.3.3. Synthesis of Butylsulfonylmethylimidazolium Hydrogensulfate (BSMImHSO4)
3.3.4. Synthesis of Butylsulfonylmethylimidazolium Bromide (BSMImBr)
3.4. General Procedures for the Gly-HC(OMe)3 Reactions
3.4.1. Catalyst-Free Conditions
3.4.2. Catalyzed Reaction
3.4.3. Catalytic Procedure for the Synthesis of 3
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Glyc | glycerol |
| OEs | orthoesters |
| Q | molar ratio HC(OMe)3:glycerol |
| PPTS | pyridinium paratoluensulfonate |
| DBUHBr | diazabicycloundeconium bromide |
| A-15 | Amberlyst 15 |
| A-36 | Amberlyst 36 |
| BSMIMHSO4 | butylsulfonylmethylimidazolium hydrogensulfate |
| BSMIMBr | butylsulfonylmethylimidazolium bromide |
| ([P1888]CH3OCO2−) | trioctylmethylphosphonium methylcarbonate |
References
- Bozell, J.J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates—The US Department of Energy’s “Top 10” revisited. Green Chem. 2010, 12, 539–554. [Google Scholar] [CrossRef]
- Mota, C.J.; Pinto, B.P.; de Lima, A.L. Glycerol, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 21–103. [Google Scholar]
- Hu, J.; Gu, Y.; Guan, Z.; Li, J.; Mo, W.; Li, T.; Li, G. An efficient palladium catalyst system for the oxidative carbonylation of glycerol to glycerol carbonate. ChemSusChem 2011, 4, 1767–1772. [Google Scholar] [CrossRef] [PubMed]
- Pearson, D.M.; Conley, N.R.; Waymouth, R.M. Palladium-catalyzed carbonylation of diols to cyclic carbonates. Adv. Synth. Catal. 2011, 353, 3007–3013. [Google Scholar] [CrossRef]
- Gabriele, B.; Mancuso, R.; Salerno, G. Oxidative carbonylation as a powerful tool for the direct synthesis of carbonylated heterocycles. Eur. J. Org. Chem. 2012, 2012, 6825–6839. [Google Scholar] [CrossRef]
- Casiello, M.; Monopoli, A.; Cotugno, P.; Milella, A.; Dell’Anna, M.M.; Ciminale, F.; Nacci, A. Copper (II) chloride-catalyzed oxidative carbonylation of glycerol to glycerol carbonate. J. Mol. Catal. A Chem. 2014, 381, 99–106. [Google Scholar] [CrossRef]
- Chavan, S.P.; Bhanage, B.M. Pd/C: An efficient and heterogeneous protocol for oxidative carbonylation of diols to cyclic carbonate. Tetrahedron Lett. 2014, 55, 1199–1202. [Google Scholar] [CrossRef]
- Guidi, S.; Calmanti, R.; Noè, M.; Perosa, A.; Selva, M. Thermal (catalyst-free) transesterification of diols and glycerol with dimethyl carbonate: A flexible reaction for batch and continuous-flow applications. ACS Sustain. Chem. Eng. 2016, 4, 6144–6151. [Google Scholar] [CrossRef]
- Calmanti, R.; Galvan, M.; Amadio, E.; Perosa, A.; Selva, M. High-temperature batch and continuous-flow transesterification of alkyl and enol esters with glycerol and its acetal derivatives. ACS Sustain. Chem. Eng. 2018, 6, 3964–3973. [Google Scholar] [CrossRef]
- Cattelan, L.; Perosa, A.; Riello, P.; Maschmeyer, T.; Selva, M. Continuous-flow o-alkylation of biobased derivatives with dialkyl carbonates in the presence of magnesium–aluminium hydrotalcites as catalyst precursors. ChemSusChem 2017, 10, 1571–1583. [Google Scholar] [CrossRef]
- Perosa, A.; Moraschini, A.; Selva, M.; Noè, M. Synthesis of the fatty esters of Solketal and glycerol-formal: Biobased specialty chemicals. Molecules 2016, 21, 170. [Google Scholar] [CrossRef]
- Guidi, S.; Noè, M.; Riello, P.; Perosa, A.; Selva, M. Towards a rational design of a continuous-flow method for the acetalization of crude glycerol: Scope and limitations of commercial amberlyst 36 and AlF3·3H2O as model catalysts. Molecules 2016, 21, 657. [Google Scholar] [CrossRef] [PubMed]
- Selva, M.; Guidi, S.; Noè, M. Upgrading of glycerol acetals by thermal catalyst-free transesterification of dialkyl carbonates under continuous-flow conditions. Green Chem. 2015, 17, 1008–1023. [Google Scholar] [CrossRef]
- Cordes, E.; Bull, H. Mechanism and catalysis for hydrolysis of acetals, ketals, and ortho esters. Chem. Rev. 1974, 74, 581–603. [Google Scholar] [CrossRef]
- Greene, T.W.; Wuts, P.G.M. Protective Groups in Organic Synthesis, 4th ed.; Wiley: New York, NY, USA, 2014. [Google Scholar]
- Kociensky, P.J. Protecting Groups, 3rd ed.; Thieme-Verlag: New York, NY, USA, 2005. [Google Scholar]
- Gabriele, B.; Mancuso, R.; Salerno, G.; Veltri, L.; Costa, M.; Dibenedetto, A. A general and expedient synthesis of 5- and 6-membered cyclic carbonates by palladium-catalyzed oxidative carbonylation of 1, 2- and 1, 3- diols. ChemSusChem 2011, 4, 1778–1786. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Luo, S.; Tang, R.; Wang, R.; Wang, J. Diblock copolymers of polyethylene glycol and a polymethacrylamide with side-chains containing twin ortho ester rings: Synthesis, characterization, and evaluation as potential pH-responsive micelles. Macromol. Biosci. 2015, 15, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Heller, J.; Barr, J. Poly(ortho esters) from concept to reality. Biomacromolecules 2004, 5, 1625–1632. [Google Scholar] [CrossRef] [PubMed]
- Brachvogel, R.-C.; Hampel, F.; Von Delius, M. Self-assembly of dynamic orthoester cryptates. Nat. Commun. 2015, 6, 7129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löw, H.; Mena-Osteritz, E.; von Delius, M. Self-assembled orthoester cryptands: Orthoester scope, post-functionalization, kinetic locking and tunable degradation kinetics. Chem. Sci. 2018, 9, 4785–4793. [Google Scholar] [CrossRef] [PubMed]
- Crank, G.; Eastwod, F. Derivatives of orthoacids. I. Bicyclic orthoesters. Aust. J. Chem. 1964, 17, 1385–1391. [Google Scholar] [CrossRef]
- Crank, G.; Eastwood, F. Derivatives of orthoacids. II. The preparation of olefins from 1, 2-diols. Aust. J. Chem. 1964, 17, 1392–1398. [Google Scholar] [CrossRef]
- Tshibalonza, N.N.; Monbaliu, J.-C.M. Revisiting the deoxydehydration of glycerol towards allyl alcohol under continuous-flow conditions. Green Chem. 2017, 19, 3006–3013. [Google Scholar] [CrossRef]
- Hall, H., Jr.; DeBlauwe, F.; Pyriadi, T. 2,6,7-Trioxabicyclo[2.2.1]heptane. J. Am. Chem. Soc. 1975, 97, 3854. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Padias, A.B.; De Blauwe, F.; Hall, H., Jr. Synthesis and polymerization of 2, 6, 7-trioxabicyclo[2.2.1]heptane and 1-Methyl-2, 6, 7-trioxabicyclo [2.2.1]heptane. Macromolecules 1980, 13, 252–261. [Google Scholar] [CrossRef]
- Heller, J.; Barr, J.; Ng, S.Y.; Abdellauoi, K.S.; Gurny, R. Poly (ortho esters): Synthesis, characterization, properties and uses. Adv. Drug Del. Rev. 2002, 54, 1015–1039. [Google Scholar] [CrossRef]
- Padias, A.B.; Szymanski, R.; Hall, H.K. Synthesis and polymerization of atom-bridged bicyclic acetals and ortho esters. In Ring-Opening Polymerization; American Chemical Society: Washington, DC, USA, 1985; Volume 286, pp. 313–333. [Google Scholar]
- Yokoyama, Y.; Hall, H. Ring-opening polymerization of atom-bridged and bond-bridged bicyclic ethers, acetals and orthoesters. In New Polymerization Reactions; Springer: Berlin/Heidelberg, Germany, 1982; pp. 107–138. [Google Scholar]
- Zhang, L.; Yu, M.; Wang, J.; Tang, R.; Yan, G.; Yao, W.; Wang, X. Low molecular weight PEI-based vectors via acid-labile ortho ester linkage for improved gene delivery. Macromol. Biosci. 2016, 16, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Amarasekara, A.S. Acidic ionic liquids. Chem. Rev. 2016, 116, 6133–6183. [Google Scholar] [CrossRef]
- Cox, J. A bond energy scheme—II: Strain and conjugation energies in cyclic compounds. Tetrahedron 1963, 19, 1175–1184. [Google Scholar] [CrossRef]
- Fletcher, S.E.; Mortimer, C.T.; Springall, H.D. 118. Heats of combustion and molecular structure. Part VII. 1:3-Dioxane and 1:3:5-trioxa-cycloalkenes. J. Chem. Soc. 1959, 580–584. [Google Scholar] [CrossRef]
- Brachvogel, R.-C.; von Delius, M. Orthoester exchange: A tripodal tool for dynamic covalent and systems chemistry. Chem. Sci. 2015, 6, 1399–1403. [Google Scholar] [CrossRef]
- Kaupmees, K.; Trummal, A.; Leito, I. Basicities of strong bases in water: A computational study. Croat. Chem. Acta 2014, 87, 385–395. [Google Scholar] [CrossRef]
- Mezheritskii, V.V.; Olekhnovich, E.P.; Dorofeenko, G.N. The properties of orthoesters and their applications in organic synthesis. Russ. Chem. Rev. 1973, 42, 392. [Google Scholar] [CrossRef]
- Cordes, H. Ortho esters. In Carboxylic Acids and Esters; Patai, S., Ed.; Wiley: Hoboken, NJ, USA, 1969. [Google Scholar]
- Fabris, M.; Lucchini, V.; Noè, M.; Perosa, A.; Selva, M. Ionic liquids made with dimethyl carbonate: solvents as well as boosted basic catalysts for the Michael reaction. Chem. Eur. J. 2009, 15, 12273–12282. [Google Scholar] [CrossRef] [PubMed]
- Thomazeau, C.; Olivier-Bourbigou, H.; Magna, L.; Luts, S.; Gilbert, B. Determination of an acidic scale in room temperature ionic liquids. J. Chem. Soc. 2003, 125, 5264–5265. [Google Scholar] [CrossRef] [PubMed]
- Tao, D.-J.; Wu, J.; Wang, Z.-Z.; Lu, Z.-H.; Yang, Z.; Chen, X.-S. SO3H-functionalized Brønsted acidic ionic liquids as efficient catalysts for the synthesis of isoamyl salicylate. RSC Adv. 2014, 4, 1–7. [Google Scholar] [CrossRef]
- Olah, A.; Prakash, G.K.S.; Sommer, J. Superacids; Wiley: Chichester, UK, 1985. [Google Scholar]
- Bringué, R.; Iborra, M.; Tejero, J.; Izquierdo, J.F.; Cunill, F.; Fité, C.; Cruz, V.J. Thermally stable ion-exchange resins as catalysts for the liquid-phase dehydration of 1-pentanol to di-n-pentyl ether (DNPE). J. Catal. 2006, 244, 33–42. [Google Scholar] [CrossRef]
- Harmer, M.A.; Sun, Q. Solid acid catalysis using ion-exchange resins. Appl. Catal. Gen. 2001, 221, 45–62. [Google Scholar] [CrossRef]
- Ni, L.; Xin, J.; Jiang, K.; Chen, L.; Yan, D.; Lu, X.; Zhang, S. One-step conversion of biomass-derived furanics into aromatics by Brønsted acid ionic liquids at room temperature. ACS Sustain. Chem. Eng. 2018, 6, 2541–2551. [Google Scholar] [CrossRef]
- Yao, L.; Liu, S.; Li, L.; Yu, S.; Liu, F.; Song, Z. Synthesis of hydroxymethylfurfural from sucrose using brönsted-lewis acidic ionic liquid. Bull. Chem. Soc. Ethiop. 2016, 30, 283–286. [Google Scholar] [CrossRef]
- Miyashita, M.; Yoshikoshi, A.; Grieco, P.A. Pyridinium p-toluenesulfonate. A mild and efficient catalyst for the tetrahydropyranylation of alcohols. J. Org. Chem. 1977, 42, 3772–3774. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Q | T (°C) | Conversion (%) a | Selectivity (%) a | ||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | ||||
| 1 | 1 | 90 | 96 | 58 | 39 | 3 |
| 2 | 1.5 | 90 | 96 | 56 | 9 | 35 |
| 3 | 2 | 90 | 96 | 51 | 3 | 46 |
| 4 | 5 | 90 | 99 | 24 | 2 | 74 |
| 5 | 10 | 90 | 100 | 3 | 0 | 97 |
| 6 | 1 | 25 | 63 | 79 | 8 | 13 |
| 7 | 10 | 25 | 95 | 74 | 5 | 21 |
| Entry | Catalyst | Time (h) | Conversion (%) a | Selectivity (%) a | H0 b | |||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | Others | |||||
| 1 | --- | 1 | 96 | 19 | 1 | 80 | - | |
| 2 | --- | 24 | >99 | 3 | 0 | 97 | - | |
| 3 | PPTS | 1 | >99 | 0 | 3 | 97 | - | |
| 4 | PPTS | 24 | >99 | 0 | 3 | 97 | - | |
| 5 | DBUHBr | 1 | 96 | 25 | 0 | 75 | - | |
| 6 | DBUHBr | 24 | >99 | 7 | 0 | 93 | - | |
| 7 | H2SO4 | 1 | >99 | 9 | 2 | 84 | 5 | −12.3 [41] |
| 8 | H2SO4 | 24 | >99 | 42 | 1 | 16 | 41 | |
| 9 | A-15 | 1 | >99 | 12 | 3 | 79 | 6 | −2.2 [42,43] |
| 10 | A-15 | 24 | >99 | 17 | 1 | 70 | 12 | |
| 11 | A-36 | 1 | >99 | 7 | 1 | 87 | 5 | −2.65 [42,43] |
| 12 | A-36 | 24 | >99 | 13 | 1 | 75 | 11 | |
| 13 | FeCl3 | 1 | >99 | 2 | 0 | 91 | 7 | |
| 14 | FeCl3 | 24 | >99 | 10 | 1 | 78 | 11 | |
| 15 | AlF3 | 1 | >99 | 9 | 1 | 87 | 3 | |
| 16 | AlF3 | 24 | >99 | 3 | 1 | 92 | 4 | |
| 17 | BSMImHSO4 | 1 | >99 | 0 | 2 | 98 | - | 1.02 [44] |
| 18 | BSMImHSO4 | 24 | >99 | 0 | 1 | 99 | - | |
| 19 | BSMImBr | 1 | >99 | 0 | 2 | 98 | - | 2.91 c [45] |
| 20 | BSMImBr | 24 | >99 | 0 | 1 | 99 | - | |
| 21 | BSMImHSO4 d | 1 | >99 | 17 | 2 | 81 | - | |
| 22 | BSMImBr d | 1 | >99 | 11 | 2 | 87 | - | |
| 23 | K2CO3 | 1 | 0 | 0 | 0 | 0 | - | |
| 24 | [P1888]CH3OCO2− | 1 | 0 | 0 | 0 | 0 | - | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calmanti, R.; Amadio, E.; Perosa, A.; Selva, M. Reaction of Glycerol with Trimethyl Orthoformate: Towards the Synthesis of New Glycerol Derivatives. Catalysts 2019, 9, 534. https://doi.org/10.3390/catal9060534
Calmanti R, Amadio E, Perosa A, Selva M. Reaction of Glycerol with Trimethyl Orthoformate: Towards the Synthesis of New Glycerol Derivatives. Catalysts. 2019; 9(6):534. https://doi.org/10.3390/catal9060534
Chicago/Turabian StyleCalmanti, Roberto, Emanuele Amadio, Alvise Perosa, and Maurizio Selva. 2019. "Reaction of Glycerol with Trimethyl Orthoformate: Towards the Synthesis of New Glycerol Derivatives" Catalysts 9, no. 6: 534. https://doi.org/10.3390/catal9060534
APA StyleCalmanti, R., Amadio, E., Perosa, A., & Selva, M. (2019). Reaction of Glycerol with Trimethyl Orthoformate: Towards the Synthesis of New Glycerol Derivatives. Catalysts, 9(6), 534. https://doi.org/10.3390/catal9060534