Hierarchical H-MOR Zeolite Supported Vanadium Oxide for Dimethyl Ether Direct Oxidation

 ,
,
Abstract
:
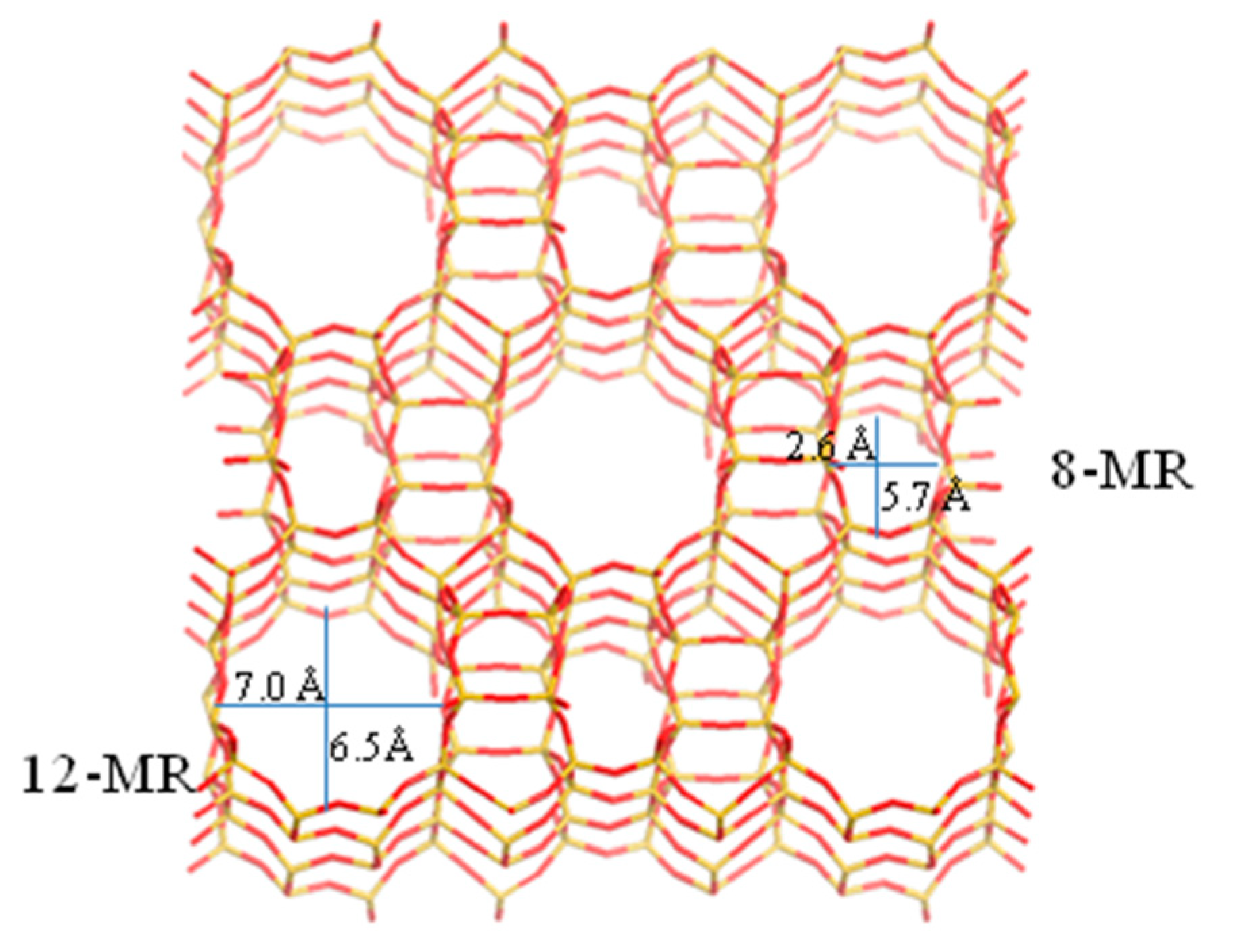
1. Introduction
2. Results and Discussion
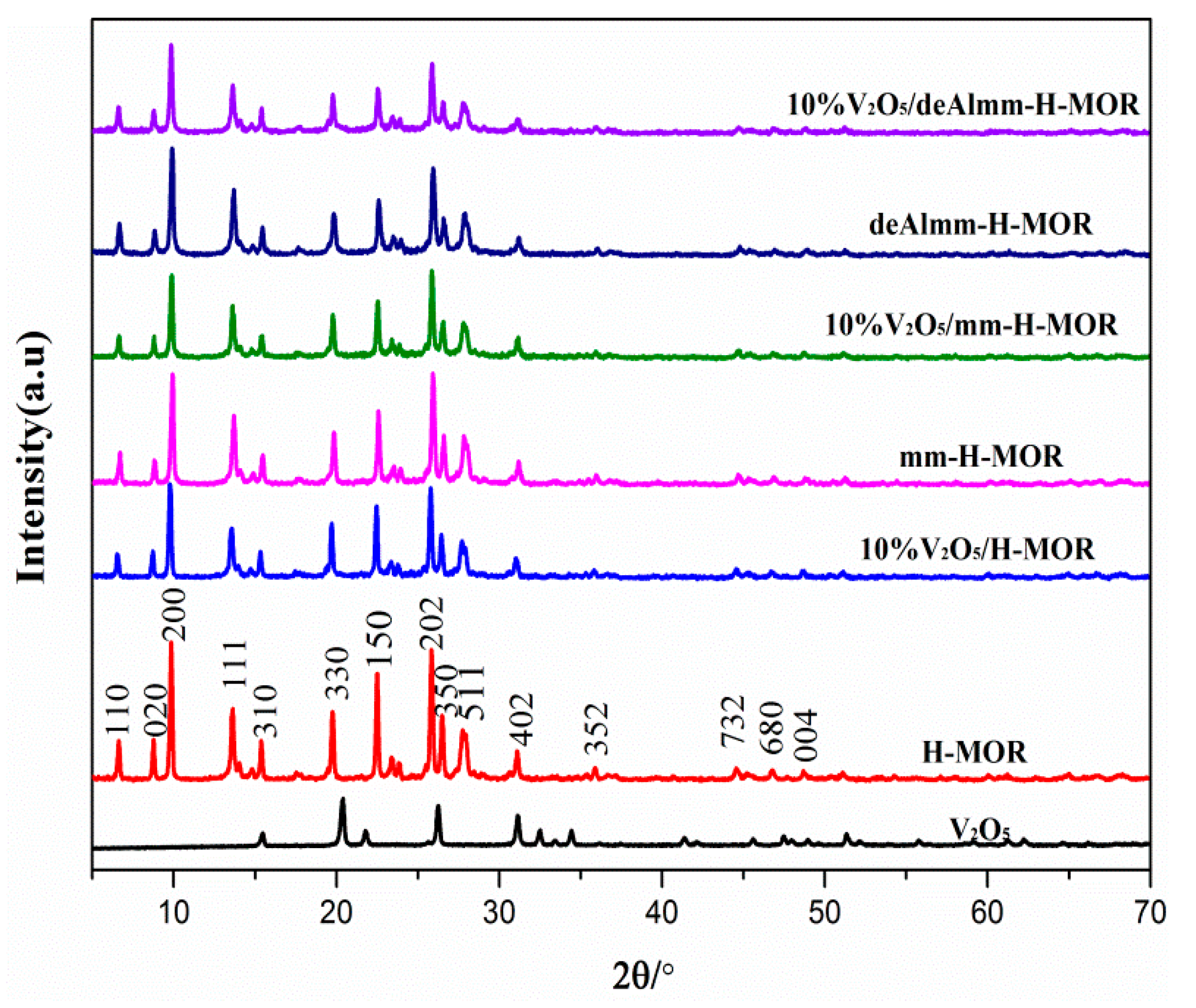
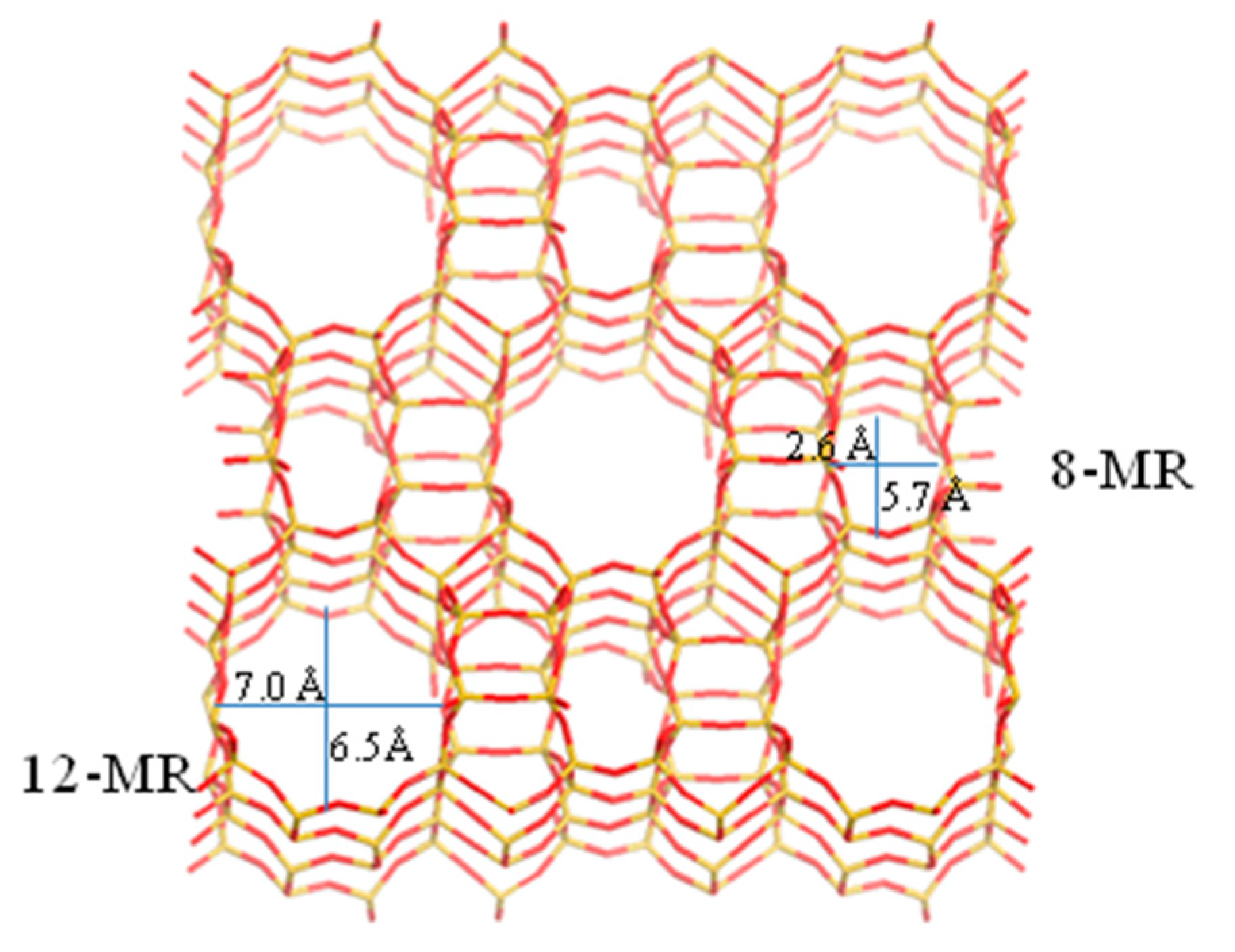
2.1. XRD Characterization
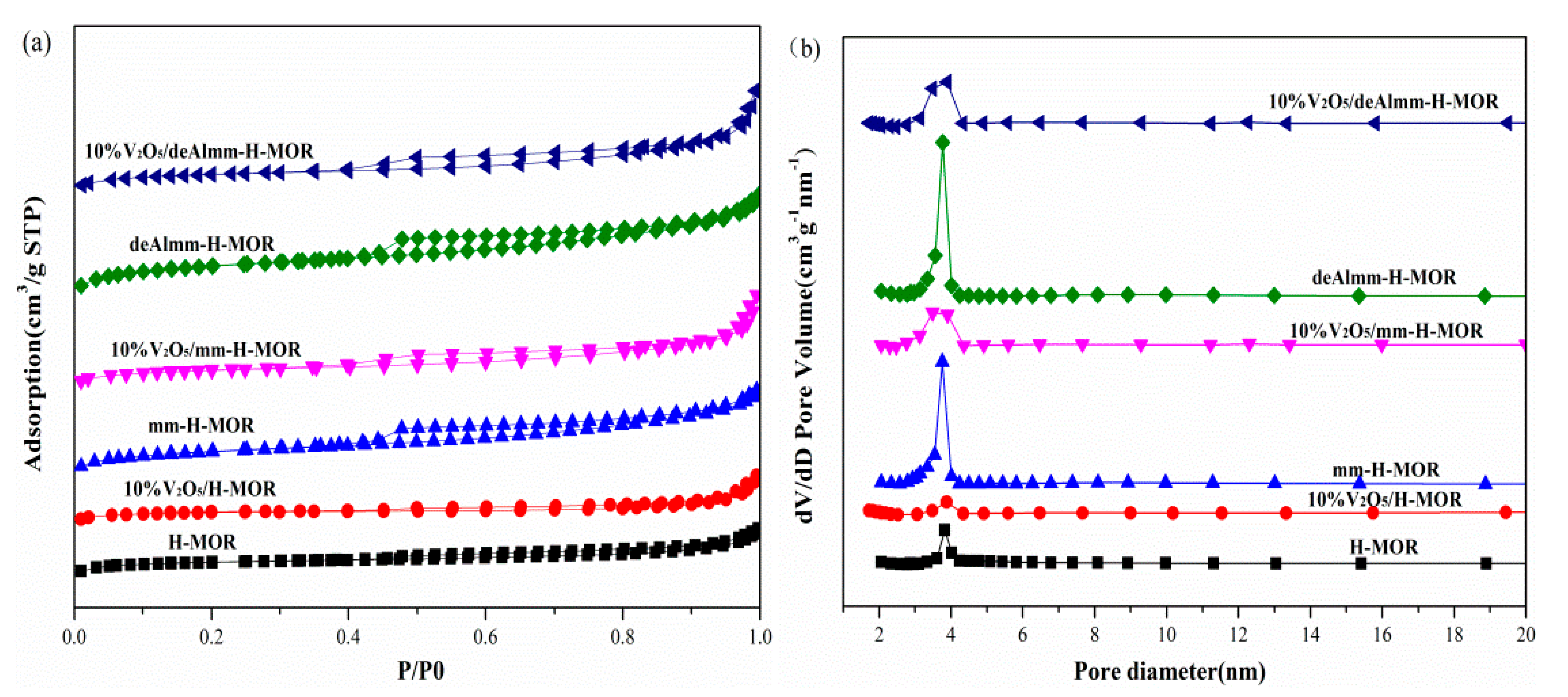
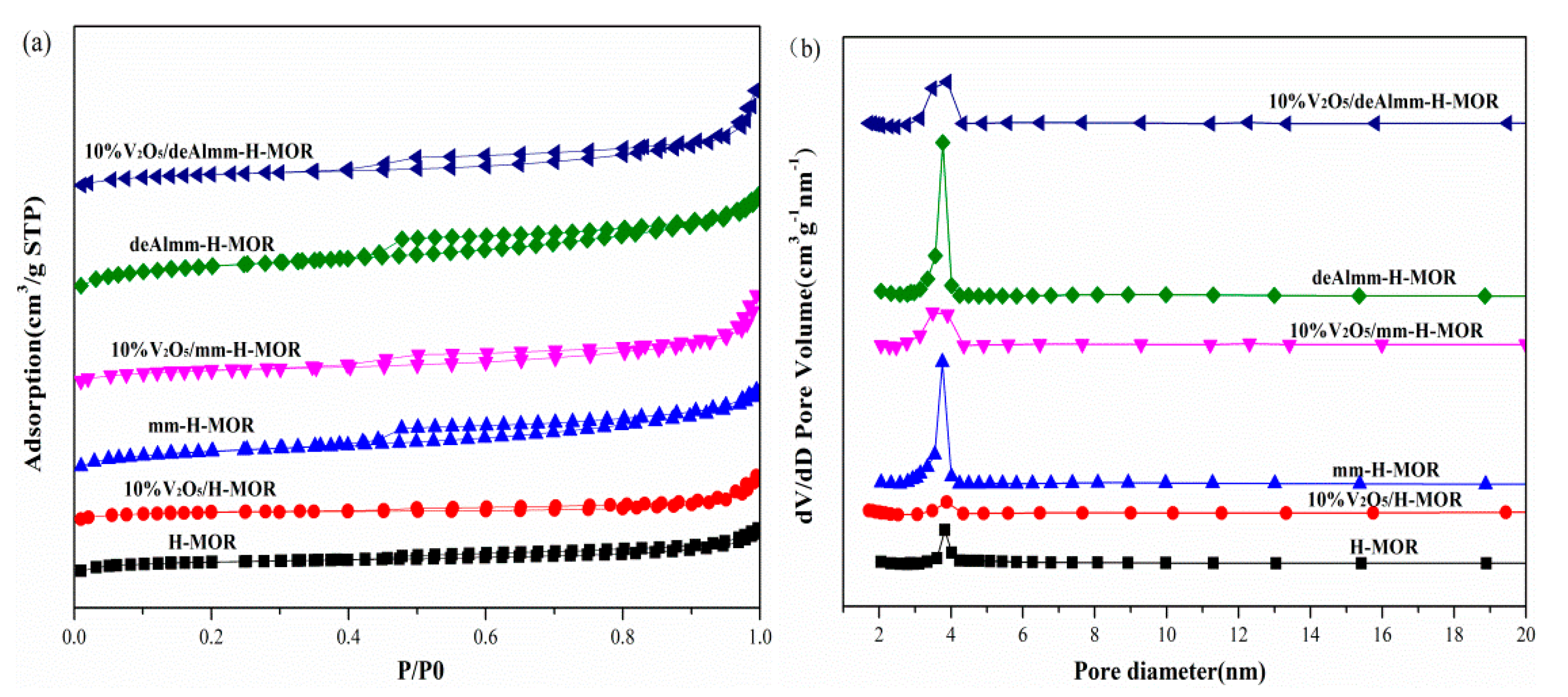
2.2. N2 Adsorption–Desorption Measurement
2.3. TEM
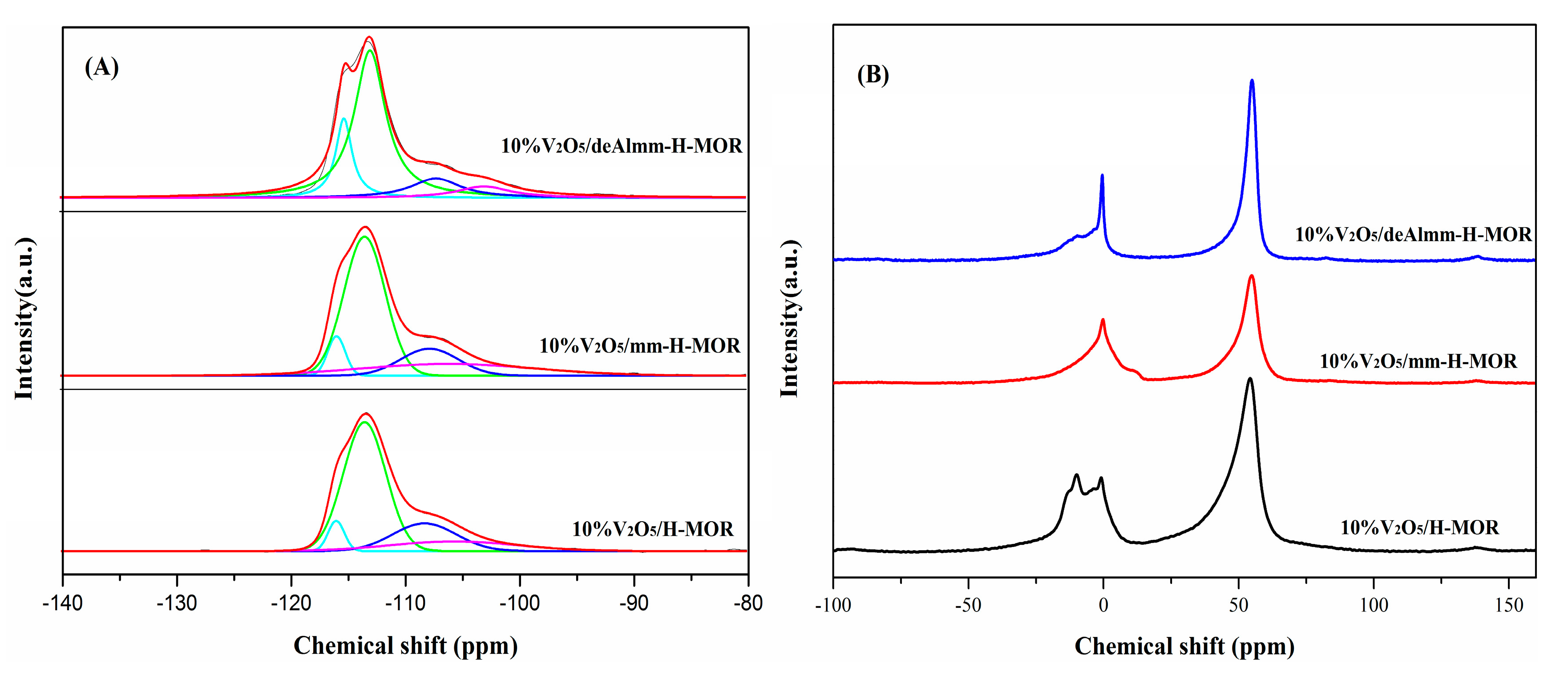
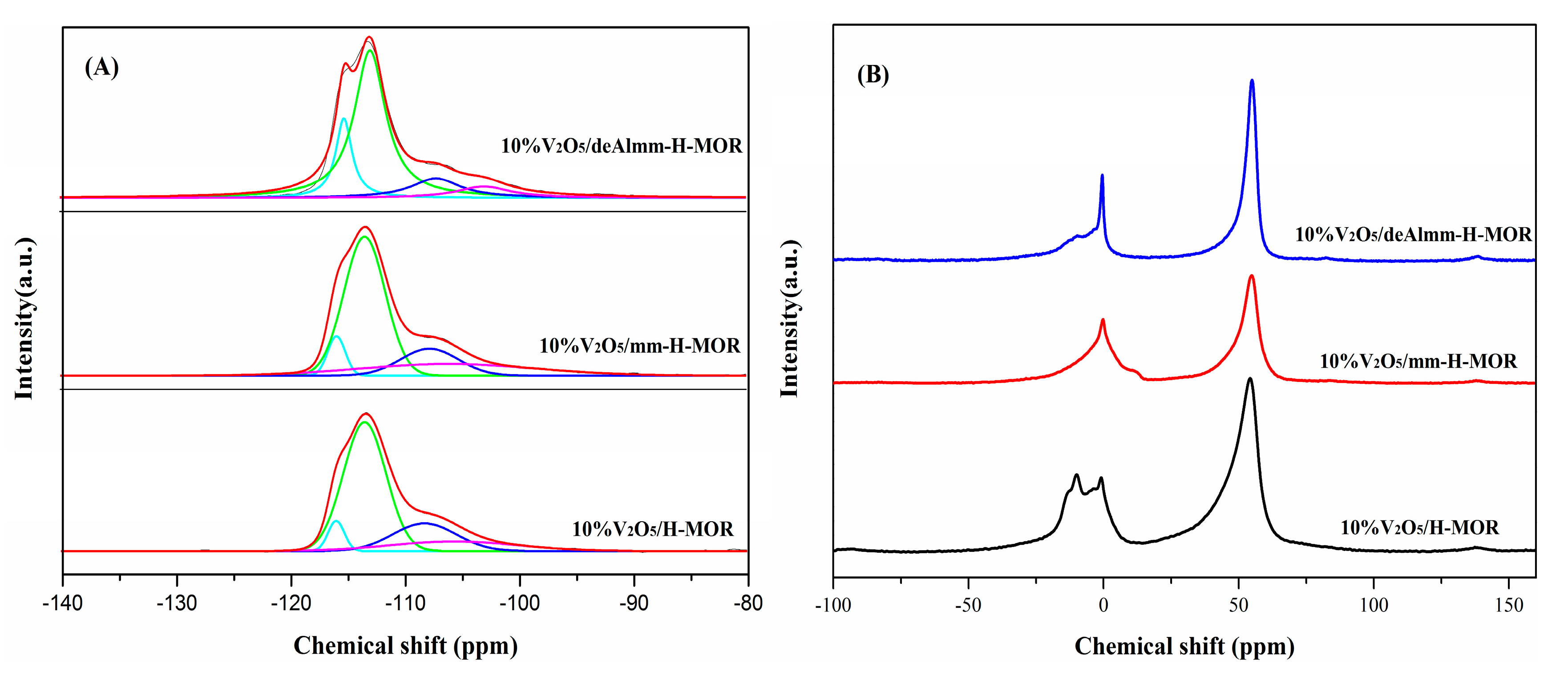
2.4. 29Si MAS NMR and 27Al MAS NMR
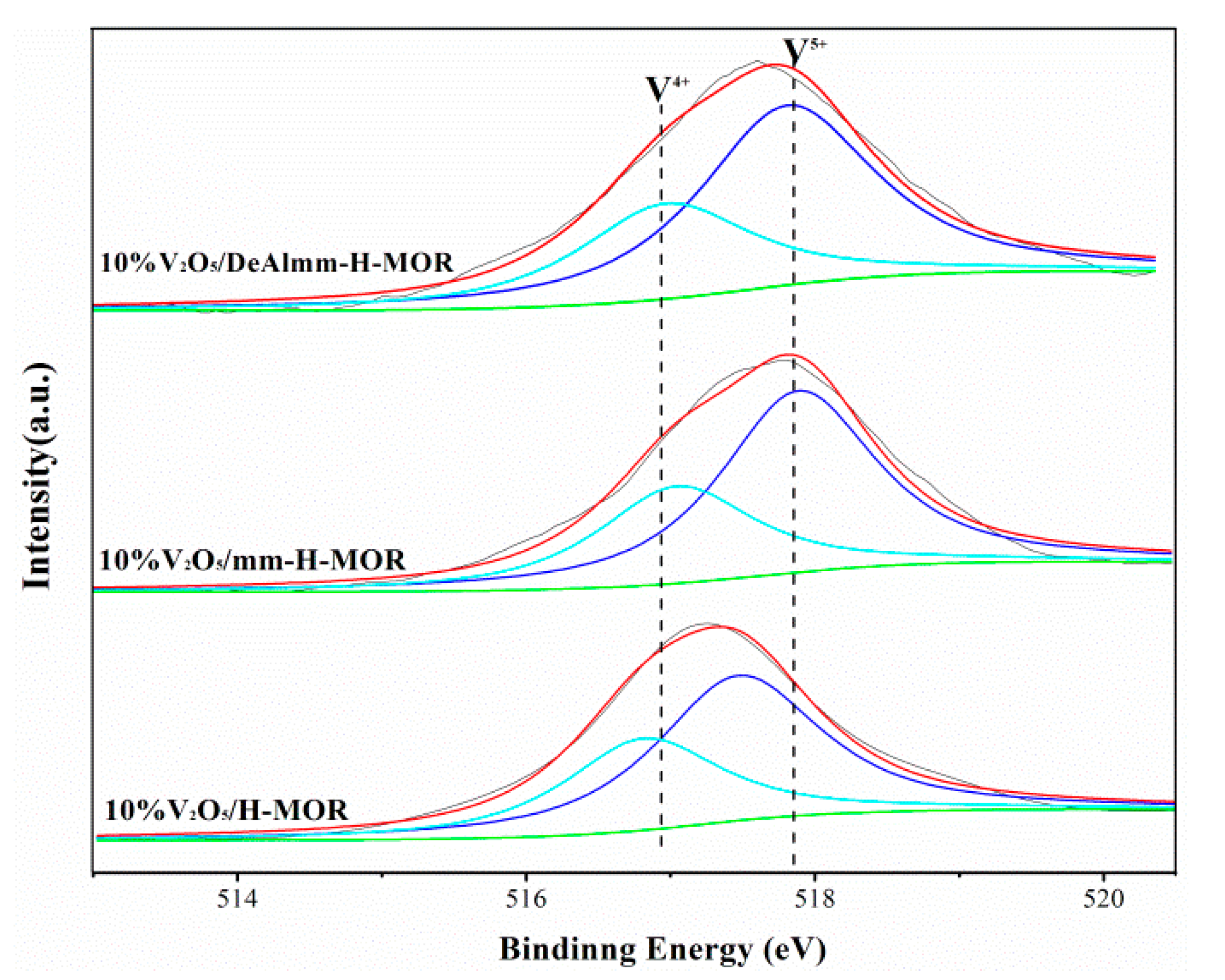
2.5. X-ray Photoelectron Spectra (XPS)
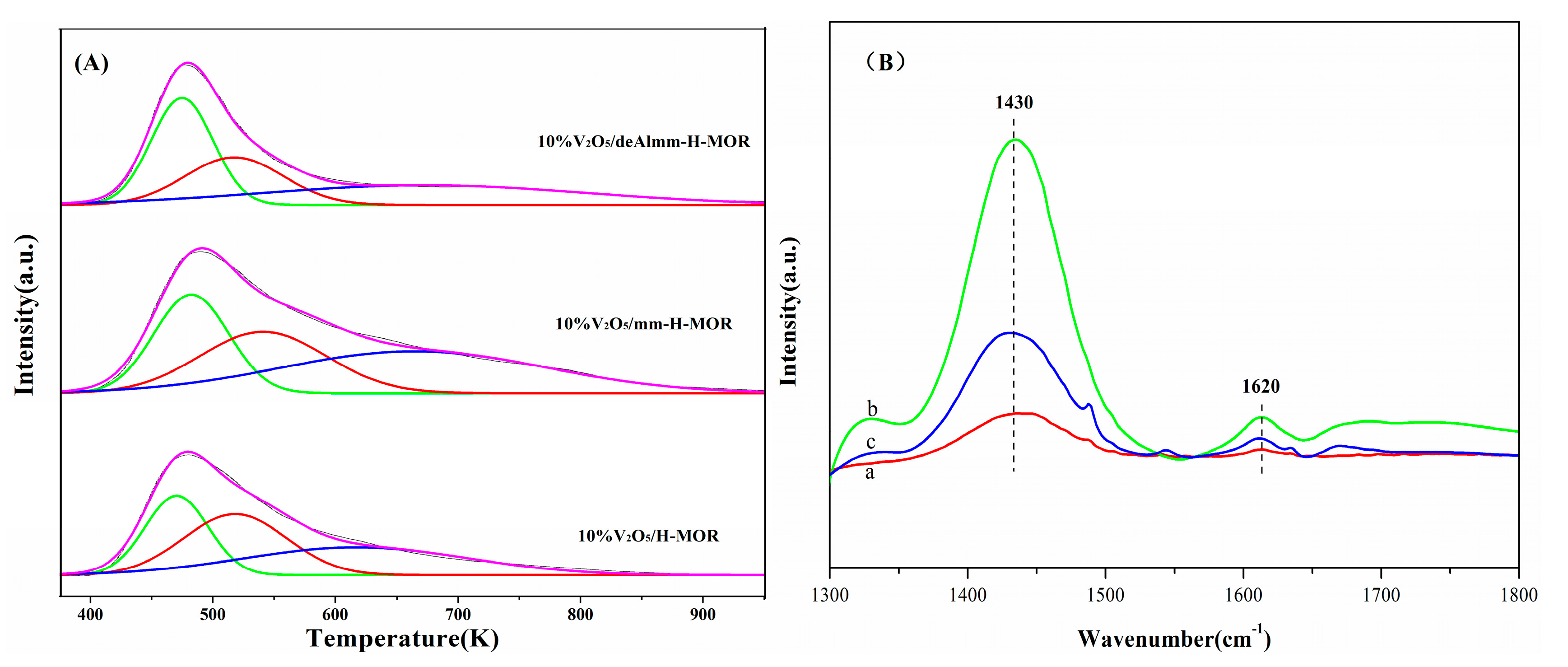
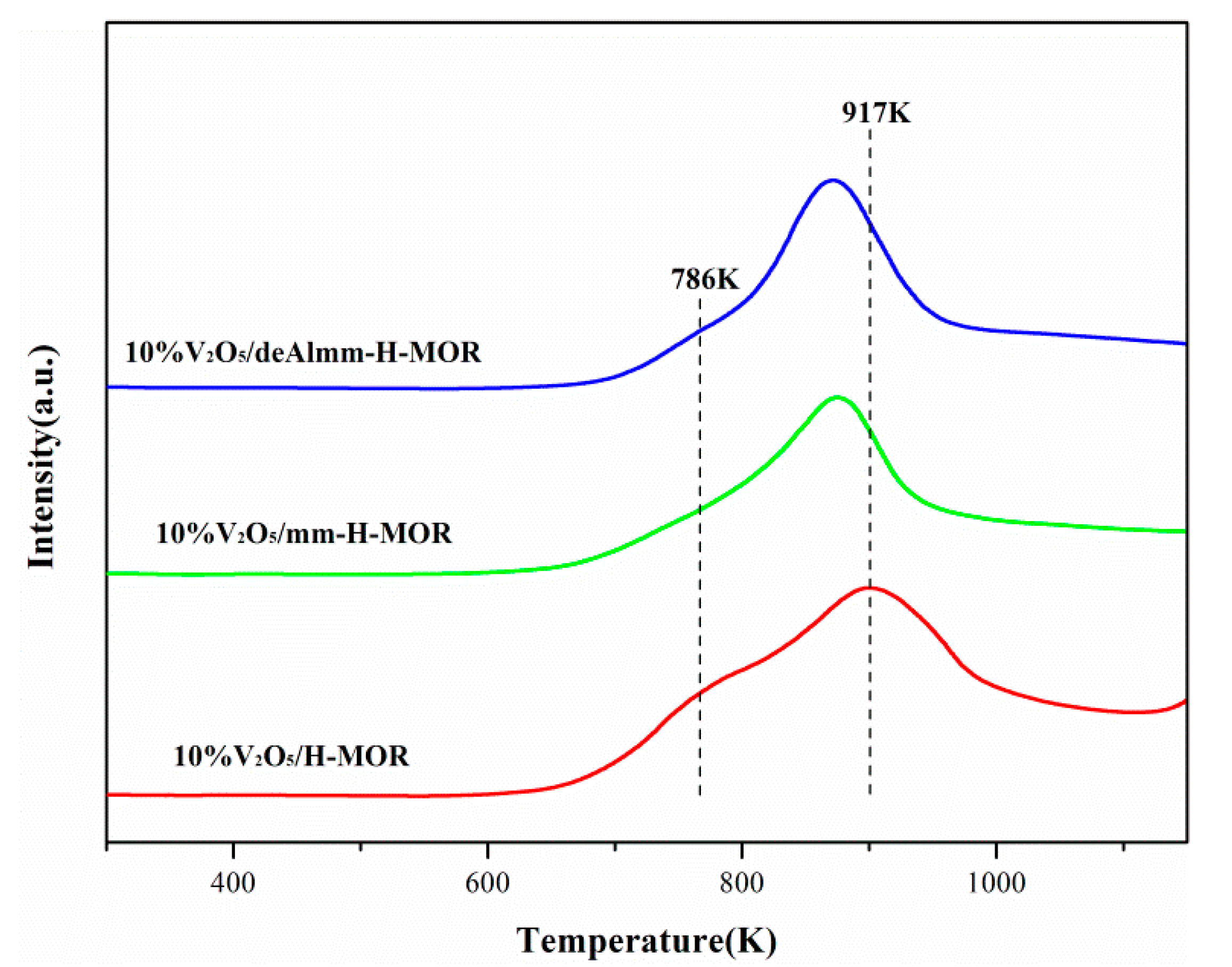
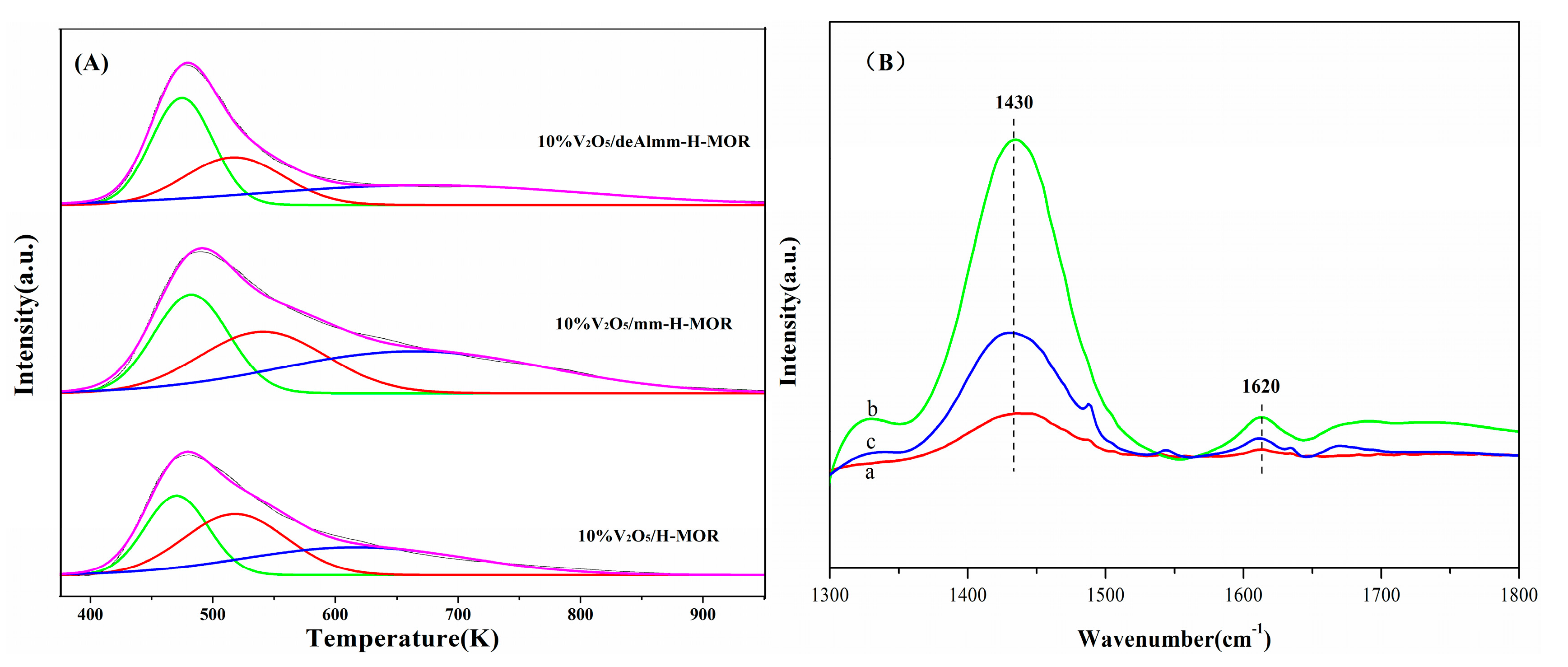
2.6. H2-TPR
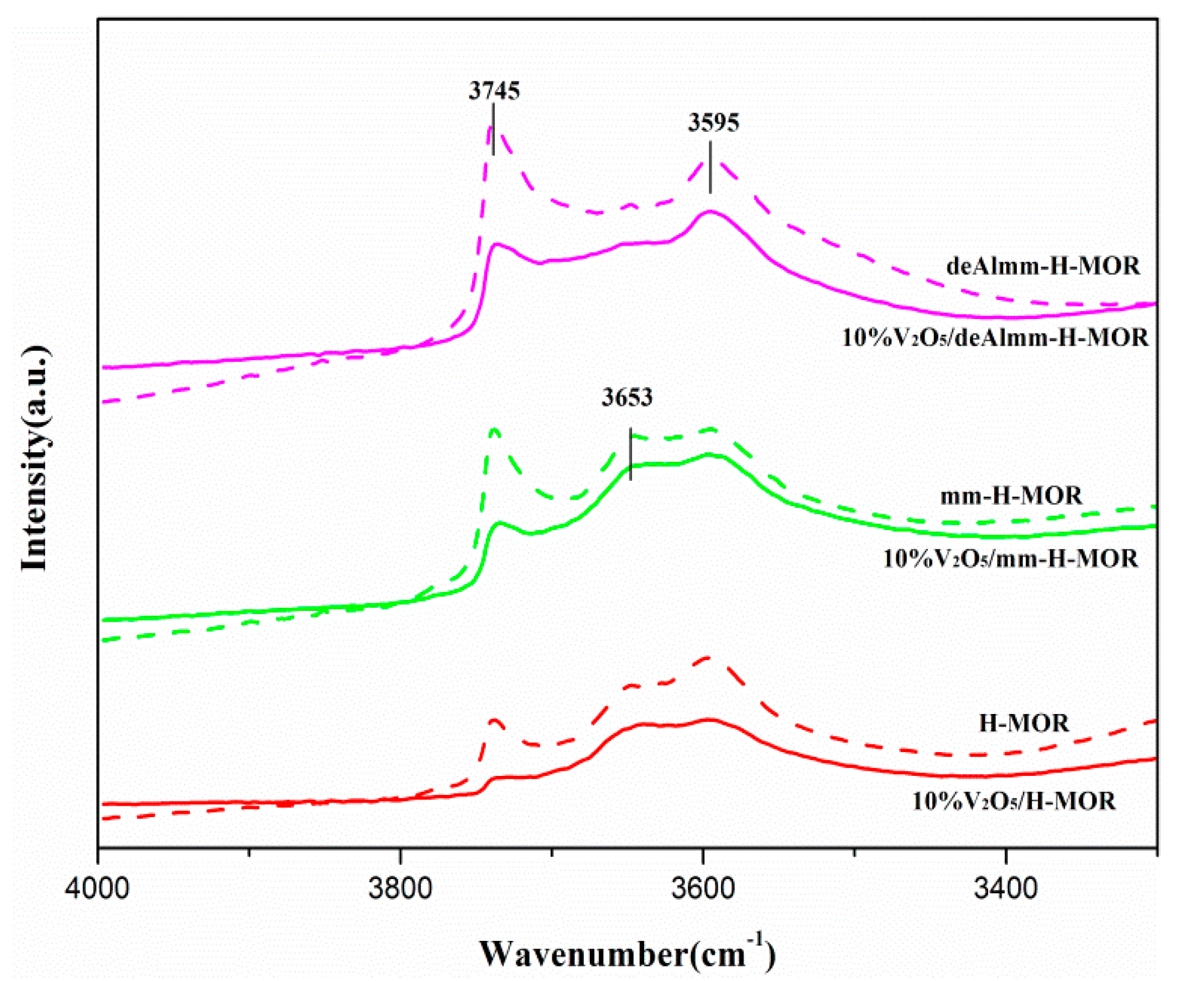
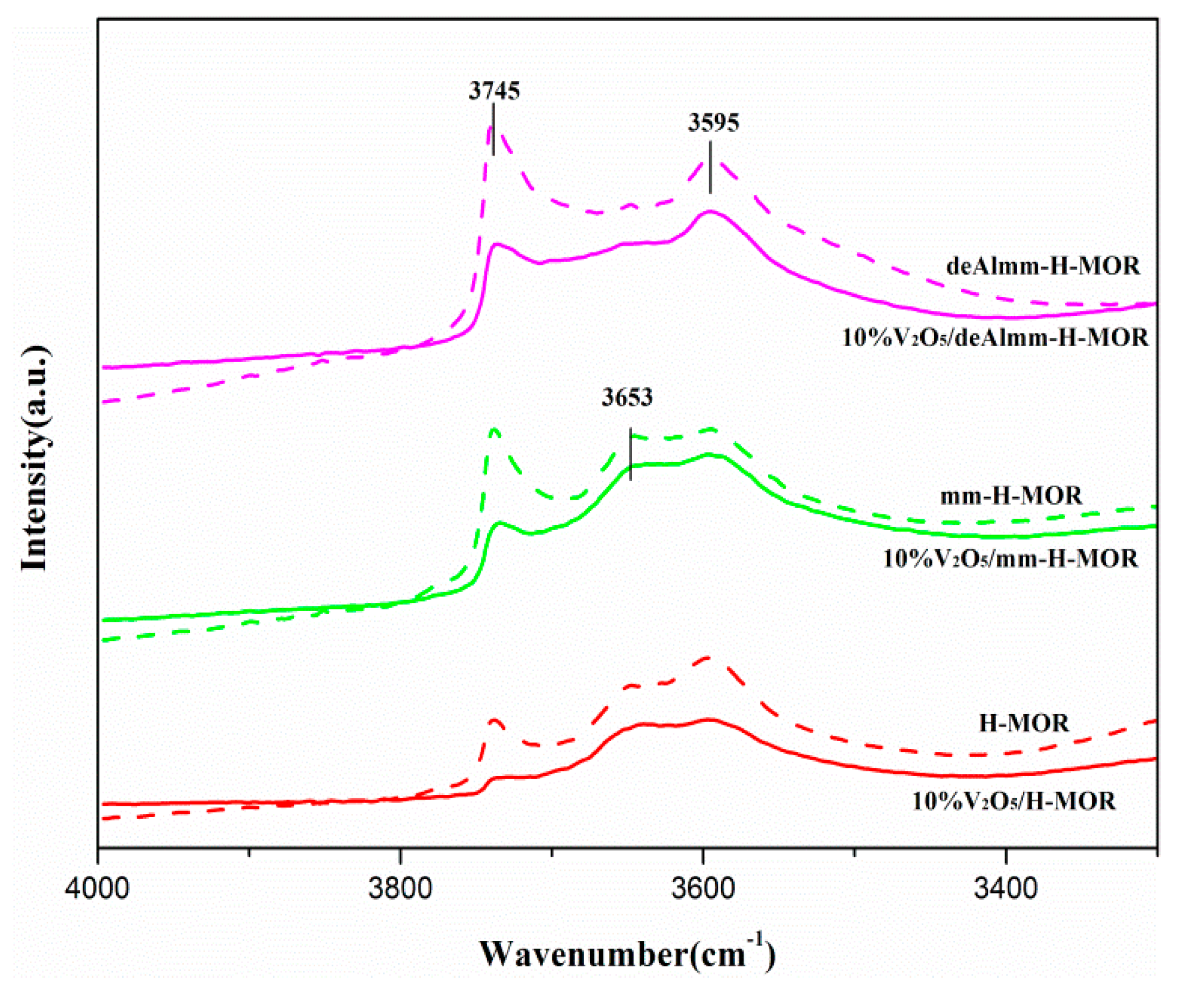
2.7. FT-IR
2.8. Acid Property
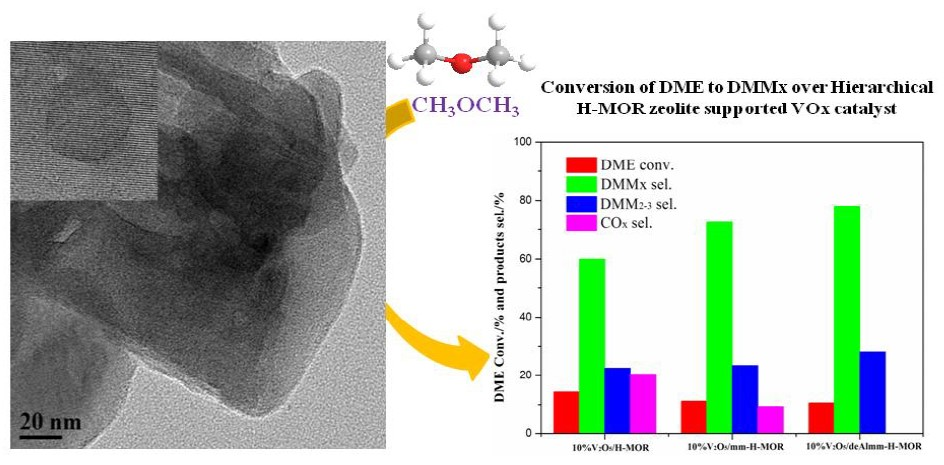
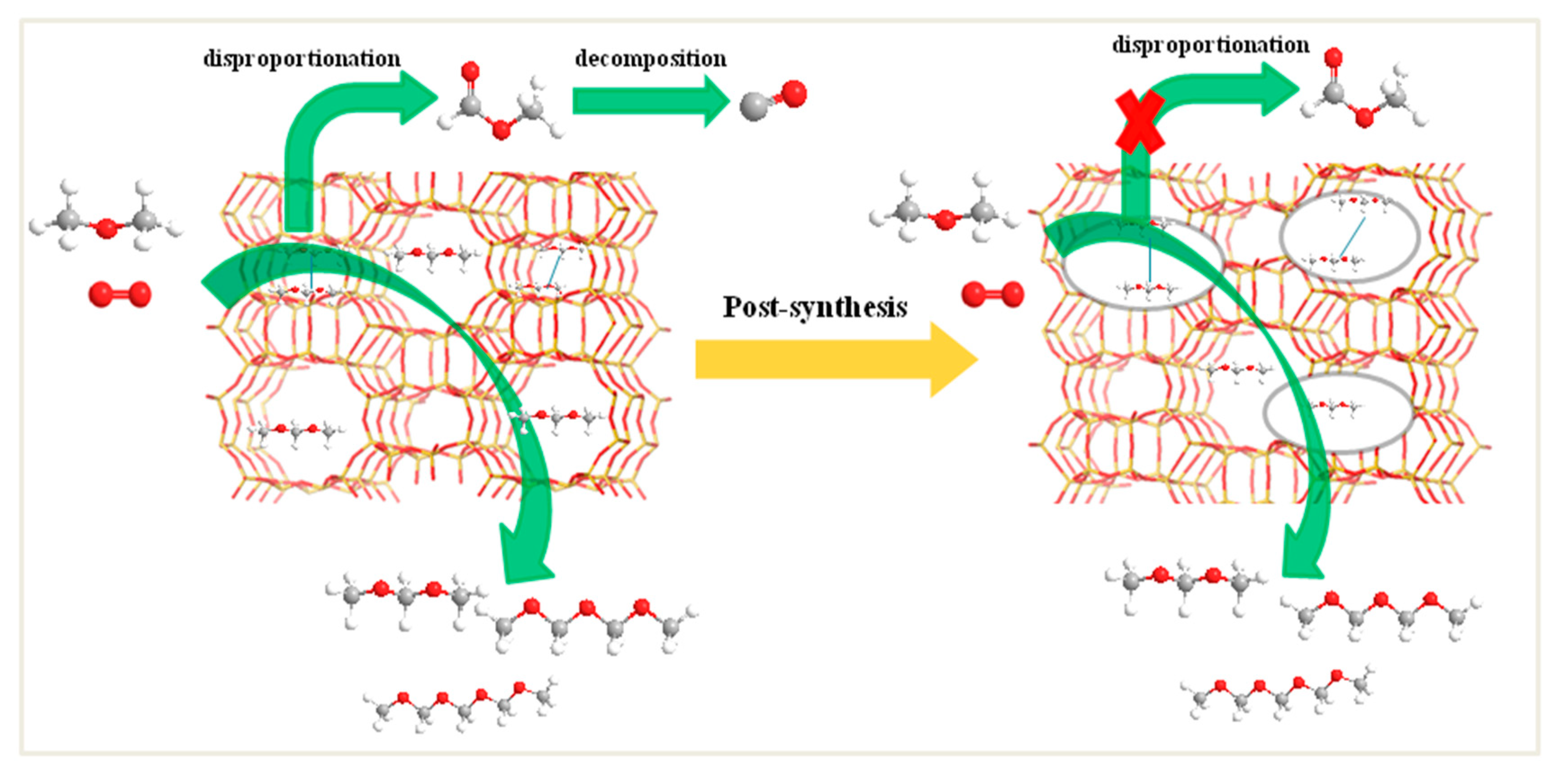
2.9. Catalytic Performance
3. Experimental
3.1. Catalysts Preparation
3.2. Catalyst Characterization
3.3. Catalytic Reaction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sánchez-Contador, M.; Ateka, A.; Aguayo, A.T.; Bilbao, J. Effects of diesel/PODE (polyoxymethylene dimethyl ethers) blends on combustion and emission characteristics in a heavy duty diesel engine. Fuel Process. Technol. 2018, 179, 258–268. [Google Scholar] [CrossRef]
- Song, F.; Tan, Y.; Xie, H.; Zhang, Q.; Han, Y. Direct synthesis of dimethyl ether from biomass-derived syngas over Cu-ZnO-Al2O3-ZrO2(x)/γ-Al2O3 bifunctional catalysts: Effect of Zr-loading. Fuel Process. Technol. 2014, 126, 88–94. [Google Scholar] [CrossRef]
- Liu, F.; Wang, T.; Zheng, Y.; Wang, J. Synergistic effect of Brønsted and Lewis acid sites for the synthesis of polyoxymethylene dimethyl ethers over highly efficient SO42-/TiO2 catalysts. J. Catal. 2017, 355, 17–25. [Google Scholar] [CrossRef]
- Liu, J.; Wang, H.; Li, Y.; Zheng, Z.; Xue, Z.; Shang, H.; Yao, M. Effects of diesel/PODE (polyoxymethylene dimethyl ethers) blends on combustion and emission characteristics in a heavy duty diesel engine. Fuel 2016, 177, 206–216. [Google Scholar] [CrossRef]
- Qi, J.G.; Hu, Y.F.; Niu, J.G.; Ma, W.T.; Jiang, S.Q.; Wang, Y.C.; Zhang, X.M.; Zhang, Y.H. Evaluation of polyoxymethylene dimethyl ethers as a new type of diesel additives. Fuel 2018, 234, 135–141. [Google Scholar] [CrossRef]
- Baranowski, C.J.; Bahmanpour, A.M.; Kröcher, O. Catalytic synthesis of polyoxymethylene dimethyl ethers (OME): A review. Appl. Catal. B Environ. 2017, 217, 407–420. [Google Scholar] [CrossRef]
- Liu, H.C.; Iglesia, E. Selective one-step synthesis of dimethoxymethane via methanol or dimethyl ether oxidation on H3+nVnMo12-nPO40 keggin structures. J. Phys. Chem. B 2003, 107, 10840–10847. [Google Scholar] [CrossRef]
- Tsubaki, N.; Zhang, Q.; Wang, W.; Bai, Y.; Han, Y.; Tan, Y. Application of modified CNTs with Ti(SO4) 2 in selective oxidation of dimethyl ether. Catal. Sci. Technol. 2016, 6, 7193–7202. [Google Scholar]
- Zhang, Q.D.; Tan, Y.S.; Liu, G.B.; Zhang, J.F.; Han, Y.Z. Rhenium oxide-modified H3PW12O40/TiO2 catalysts for selective oxidation of dimethyl ether to dimethoxy dimethyl ether. Green Chem. 2014, 16, 4708–4715. [Google Scholar] [CrossRef]
- Gao, X.J.; Wang, W.F.; Gu, Y.Y.; Zhang, Z.Z.; Zhang, J.F.; Zhang, Q.D.; Tsubaki, N.; Han, Y.Z.; Tan, Y.S. Synthesis of polyoxymethylene dimethyl ethers from dimethyl ether direct oxidation over carbon-based catalysts. ChemCatChem 2018, 10, 273–279. [Google Scholar] [CrossRef]
- Celik, F.E.; Kim, T.J.; Bell, A.T. Effect of zeolite framework type and Si/Al ratio on dimethoxymethane carbonylation. J. Catal. 2010, 270, 185–195. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, W.; Zhang, Z.; Han, Y.; Tan, Y. Low-Temperature Oxidation of Dimethyl Ether to Polyoxymethylene Dimethyl Ethers over CNT-Supported Rhenium Catalyst. Catalysts 2016, 6, 43. [Google Scholar] [CrossRef]
- Zhang, Q.D.; Tan, Y.S.; Yang, C.H.; Han, Y.Z. MnCl2 modified H4SiW12O40/SiO2 catalysts for catalytic oxidation of dimethy ether to dimethoxymethane. J. Mol. Catal. A Chem. 2007, 263, 149–155. [Google Scholar] [CrossRef]
- Zhang, Q.D.; Tan, Y.S.; Yang, C.H.; Han, Y.Z. Research on catalytic oxidation of dimethyl ether to dimethoxymethane over MnCl2 modified heteropolyacid catalysts. Catal. Commun. 2008, 9, 1916–1919. [Google Scholar] [CrossRef]
- Zhang, Q.D.; Tan, Y.S.; Yang, C.H.; Liu, Y.Q.; Han, Y.Z. Catalytic oxidation of dimethyl ether to dimethoxymethane over MnCl2-H4SiW12O40/SiO2 catalyst. Chin. J. Catal. 2006, 27, 916–920. [Google Scholar] [CrossRef]
- Leng, K.; Wang, Y.; Hou, C.; Lancelot, C.; Lamonier, C.; Rives, A.; Sun, Y. Enhancement of catalytic performance in the benzylation of benzene with benzyl alcohol over hierarchical mordenite. J. Catal. 2013, 306, 100–108. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, L.; Wang, G.; Chen, F.; Zhu, J.; Wang, C.; Bian, C.; Pan, S.; Xiao, F.-S. Hierarchical Sn-Beta Zeolite Catalyst for the Conversion of Sugars to Alkyl Lactates. ACS Sustain. Chem. Eng. 2017, 5, 3123–3131. [Google Scholar] [CrossRef]
- Wei, X.; Smirniotis, P.G. Development and characterization of mesoporosity in ZSM-12 by desilication. Microporous Mesoporous Mater. 2006, 97, 97–106. [Google Scholar] [CrossRef]
- Van Laak, A.N.; Gosselink, R.W.; Sagala, S.L.; Meeldijk, J.D.; De Jongh, P.E.; De Jong, K.P. Alkaline treatment on commercially available aluminum rich mordenite. Appl. Catal. A Gen. 2010, 382, 65–72. [Google Scholar] [CrossRef]
- Keller, T.C.; Arras, J.; Wershofen, S.; Perez-Ramirez, J. Design of hierarchical zeolite catalysts for the manufacture of polyurethane intermediates. ACS Catal. 2015, 5, 734–743. [Google Scholar] [CrossRef]
- Sazama, P.; Sobalík, Z.; Dědeček, J.; Jakubec, I.; Parvulescu, V.; Bastl, Z.; Rathousky, J.; Jirglová, H. Enhancement of Activity and Selectivity in Acid-Catalyzed Reactions by Dealuminated Hierarchical Zeolites. Angew. Chem. 2013, 125, 2092–2095. [Google Scholar] [CrossRef]
- Pastvova, J.; Kaucky, D.; Moravkova, J.; Rathousky, J.; Sklenak, S.; Vorokhta, M.; Brabec, L.; Pilar, R.; Jakubec, I.; Tabor, E.; et al. Effect of Enhanced Accessibility of Acid Sites in Micromesoporous Mordenite Zeolites on Hydroisomerization of n-Hexane. ACS Catal. 2017, 7, 5781–5795. [Google Scholar] [CrossRef]
- Zhang, J.; Rao, C.; Peng, H.; Peng, C.; Zhang, L.; Xu, X.; Liu, W.; Wang, Z.; Zhang, N.; Wang, X. Enhanced toluene combustion performance over Pt loaded hierarchical porous MOR zeolite. Chem. Eng. J. 2018, 334, 10–18. [Google Scholar] [CrossRef]
- Zhao, H.Y.; Bennici, S.; Shen, J.Y.; Auroux, A. Nature of surface sites of catalysts and reactivity in selective oxidation of methanol to dimethoxymethane. J. Catal. 2010, 272, 176–189. [Google Scholar] [CrossRef]
- Chen, S.; Wang, S.; Ma, X.; Gong, J. Selective oxidation of methanol to dimethoxymethane over bifunctional VOx/TS-1 catalysts. Chem. Commun. 2011, 47, 9345. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.M.; Li, Y.G.; Xu, Y.D.; Shen, W.J. Kinetics of selective oxidation of dimethyl ether to formaldehyde over Al2O3-supported VOx and MoOx catalysts. Catal. Lett. 2004, 97, 185–190. [Google Scholar] [CrossRef]
- Cheung, P.; Liu, H.; Iglesia, E. Effects of Al2O3 support modifications on MoOx and VOx catalysts for dimethyl ether oxidation to formaldehyde. Phys. Chem. Chem. Phys. 2003, 5, 3795. [Google Scholar]
- Zhang, W.; Meng, T.; Tang, J.; Zhuang, W.; Zhou, Y.; Wang, J. Direct Synthesis of 2, 5-Diformylfuran from Carbohydrates Using High-Silica MOR Zeolite-Supported Isolated Vanadium Species. ACS Sustain. Chem. Eng. 2017, 5, 10029–10037. [Google Scholar] [CrossRef]
- Kowalska-Kus, J.; Held, A.; Frankowski, M.; Nowinska, K. Solketal formation from glycerol and acetone over hierarchical zeolites of different structure as catalysts. J. Mol. Catal. A Chem. 2017, 426, 205–212. [Google Scholar] [CrossRef]
- Fu, W.H.; Liang, X.M.; He, M.Y.; Zhang, H.; Wang, Y.M. Shape selectivity extending to ordered supermicroporous aluminosilicates. Chem. Commun. 2015, 51, 1449–1452. [Google Scholar] [CrossRef]
- Góra-Marek, K.; Tarach, K.A.; Tekla, J.; Olejniczak, Z.; Kuśtrowski, P.; Cheng, L.; Martínez-Triguero, J.; Rey, F.; Liu, L. Hierarchical Mordenite Dedicated to the Fluid Catalytic Cracking Process: Catalytic Performance Regarding Textural and Acidic Properties. J. Phys. Chem. C 2014, 118, 28043–28054. [Google Scholar] [CrossRef]
- Harlin, M.; Niemi, V.; Krause, A. Alumina-Supported Vanadium Oxide in the Dehydrogenation of Butanes. J. Catal. 2000, 195, 67–78. [Google Scholar] [CrossRef]
- Meng, Y.L.; Wang, T.; Chen, S.; Zhao, Y.J.; Ma, X.B.; Gong, J.L. Selective oxidation of methanol to dimethoxymethane on V2O5-MoO3/gamma-Al2O3 catalysts. Appl. Catal. B 2014, 160, 161–172. [Google Scholar] [CrossRef]
- Lim, T.H.; Nam, K.; Song, I.K.; Lee, K.-Y.; Kim, D.H. Effect of Si/Al 2 ratios in Mo/H-MCM-22 on methane dehydroaromatization. Appl. Catal. A Gen. 2018, 552, 11–20. [Google Scholar] [CrossRef]
- Steinfeldt, N.; Muller, D.; Berndt, H. VOx species on alumina at high vanadia loadings and calcination temperature and their role in the ODP reaction. Appl. Catal. A Gen. 2004, 272, 201–213. [Google Scholar] [CrossRef]
- Arena, F.; Frusteri, F.; Martra, G.; Parmaliana, A.; Coluccia, S. Surface structures, reduction pattern and oxygen chemisorption of V2O5/SiO2catalysts. J. Chem. Soc. Faraday Trans. 1997, 93, 3849–3854. [Google Scholar] [CrossRef]
- Wang, W.; Gao, X.; Yang, Q.; Wang, X.; Song, F.; Zhang, Q.; Han, Y.; Tan, Y. Vanadium oxide modified H-beta zeolite for the synthesis of polyoxymethylene dimethyl ethers from dimethyl ether direct oxidation. Fuel 2019, 238, 289–297. [Google Scholar] [CrossRef]
- Ma, M.; Zhan, E.; Huang, X.; Ta, N.; Xiong, Z.; Bai, L.; Shen, W. Carbonylation of dimethyl ether over Co-HMOR. Catal. Sci. Technol. 2018, 8, 2124–2130. [Google Scholar] [CrossRef]
- Khandan, N.; Kazemeini, M.; Aghaziarati, M. Determining an optimum catalyst for liquid-phase dehydration of methanol to dimethyl ether. Appl. Catal. A Gen. 2008, 349, 6–12. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substances | Molecular Formula | Molecular Structure | Substances | Molecular Formula | Molecular Structure |
|---|---|---|---|---|---|
| DME | CH3OCH3 |  | CH3OH | CH3OH | 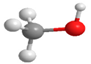 |
| DMM | CH3OCH2OCH3 | 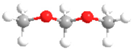 | MF | HCOOCH3 | 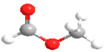 |
| DMM2 | CH3O(CH2O)2CH3 | 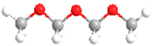 | CO | CO | 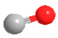 |
| DMM3 | CH3O(CH2O)3CH3 |  | CO2 | CO2 | 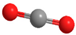 |
| HCHO | HCHO |  | CH4 | CH4 |  |
| Catalysts | Cryst./% a | Si/Al b | Si/Al c | SBET d (m2 g−1) | Vtotal e (cm3 g−1) | Vmicro f (cm3 g−1) | Vmeso g (cm3 g−1) |
|---|---|---|---|---|---|---|---|
| H-MOR | 100 | 9.6 | 8.0 | 542 | 0.276 | 0.189 | 0.087 |
| mm-H-MOR | 95 | 7.4 | 7.2 | 564 | 0.343 | 0.176 | 0.167 |
| deAlmm-H-MOR | 95 | 18.8 | 24.5 | 588 | 0.378 | 0.176 | 0.202 |
| 10%V2O5/H-MOR | 100 | 9.6 | 8.0 | 318 | 0.198 | 0.147 | 0.051 |
| 10%V2O5/mm-H-MOR | 95 | 7.4 | 7.2 | 335 | 0.247 | 0.137 | 0.110 |
| 10%V2O5/deAlmm-H-MOR | 94 | 18.8 | 24.5 | 318 | 0.251 | 0.128 | 0.123 |
| Catalysts | CS (ppm) | I (%) | CS (ppm) | I (%) | CS (ppm) | I (%) | CS (ppm) | I (%) |
|---|---|---|---|---|---|---|---|---|
| 10%V2O5/H-MOR | −115.9 | 5.5 | −113.4 | 61.3 | −108.2 | 19.6 | −105.6 | 13.7 |
| 10%V2O5/mm-H-MOR | −115.8 | 6.9 | −113.4 | 57.3 | −107.7 | 15.4 | −106.0 | 20.4 |
| 10%V2O5/deAlmm-H-MOR | −115.2 | 17.3 | −112.9 | 61.7 | −107.1 | 12.9 | −102.9 | 8.1 |
| Samples | V/(Si + Al) | V5+ | V4+ | ||
|---|---|---|---|---|---|
| BE/ev | Area/% | BE/ev | Area/% | ||
| 10%V2O5/H-MOR | 0.069 | 517.5 | 65.2 | 516.8 | 34.8 |
| 10%V2O5/mm-H-MOR | 0.055 | 517.9 | 64.9 | 517.1 | 35.1 |
| 10%V2O5/deAlmm-H-MOR | 0.032 | 517.8 | 65.6 | 517.0 | 34.4 |
| Catalysts | NH3-TPD | NH3-IR | ||||||
|---|---|---|---|---|---|---|---|---|
| WAS a | MAS b | SAS c | Total d | B e | L f | Total g | B/L h | |
| 10%V2O5/H-MOR | 0.092 (470) | 0.116 (518) | 0.113 (617) | 0.321 | 64.2 | 47.5 | 111.7 | 1.35 |
| 10%V2O5/mm-H-MOR | 0.127 (481) | 0.133 (540) | 0.192 (663) | 0.452 | 359.2 | 239.4 | 598.6 | 1.50 |
| 10%V2O5/deAlmm-H-MOR | 0.106 (473) | 0.075 (516) | 0.103 (671) | 0.285 | 173.1 | 168.9 | 342.0 | 1.02 |
| Catalysts | DME Conv. (%) | Selectivity (C-mol%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| DMM1-3 | DMM | DMM2 | DMM3 | HCHO | MF | CH3OH | CO | CO2 | CH4 | ||
| 10%V2O5/H-MOR | 14.4 | 60.0 | 37.4 | 20.4 | 2.2 | 3.2 | 4.2 | 12.1 | 20.4 | 0 | 0 |
| 10%V2O5/mm-H-MOR | 11.2 | 72.5 | 49.0 | 22.0 | 1.5 | 0.8 | 6.1 | 11.3 | 9.4 | 0 | 0 |
| 10%V2O5/deAlmm-H-MOR | 10.6 | 78.2 | 50.1 | 26.8 | 1.3 | 0.4 | 6.8 | 14.7 | 0 | 0 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Gao, X.; Feng, R.; Yang, Q.; Zhang, T.; Zhang, J.; Zhang, Q.; Han, Y.; Tan, Y. Hierarchical H-MOR Zeolite Supported Vanadium Oxide for Dimethyl Ether Direct Oxidation. Catalysts 2019, 9, 628. https://doi.org/10.3390/catal9070628
Wang W, Gao X, Feng R, Yang Q, Zhang T, Zhang J, Zhang Q, Han Y, Tan Y. Hierarchical H-MOR Zeolite Supported Vanadium Oxide for Dimethyl Ether Direct Oxidation. Catalysts. 2019; 9(7):628. https://doi.org/10.3390/catal9070628
Chicago/Turabian StyleWang, Wenfeng, Xiujuan Gao, Ru Feng, Qi Yang, Tao Zhang, Junfeng Zhang, Qingde Zhang, Yizhuo Han, and Yisheng Tan. 2019. "Hierarchical H-MOR Zeolite Supported Vanadium Oxide for Dimethyl Ether Direct Oxidation" Catalysts 9, no. 7: 628. https://doi.org/10.3390/catal9070628
APA StyleWang, W., Gao, X., Feng, R., Yang, Q., Zhang, T., Zhang, J., Zhang, Q., Han, Y., & Tan, Y. (2019). Hierarchical H-MOR Zeolite Supported Vanadium Oxide for Dimethyl Ether Direct Oxidation. Catalysts, 9(7), 628. https://doi.org/10.3390/catal9070628