Abstract
Photopolymerization processes, and especially those carried out under visible light, are more and more widespread for their multiple advantages compared to thermal processes. In the present paper, new 1,8-naphthalimide derivatives are proposed as photoinitiators for free-radical polymerization upon visible light exposure using light-emitting diodes (LEDs) at 395, 405, and 470 nm. These photoinitiators are used in combination with both iodonium salts and phosphine. The synthesis of these compounds as well as their excellent polymerization initiation ability for methacrylate monomers are presented in this article. A full picture of the involved chemical mechanisms is also provided thanks to photolysis, radical characterization, and redox measurements.
1. Introduction
For polymer research, the development of more eco-friendly initiating systems has received enormous attention in recent times [1,2]. Polymers are unavoidable materials. Synthetic or natural, their role is essential in everyday life. Because of their numerous properties, such as flexibility and chemical resistance, polymers constitute the building blocks of numerous materials. However, polymers, and in particular polymers formed by a thermal process, show a strong negative impact on Earth related to their high energy consumption for production, emission of volatile organic compounds (VOCs), and problem of after-used wastes [3]. In this context, the photopolymerization process can represent an important progress for polymer synthesis in an eco-friendlier way. Usually, formation of the polymer is induced by a thermal process, but the use of light to initiate polymerization is becoming less and less marginal. Indeed, it offers significant benefits compared to other initiation pathways such as low energy consumption, no heating needed, no or almost no VOC emission, high spatial resolution, and temporal control of the reaction [4]. Up to now, most photopolymerization processes were performed under UV (ultraviolet) light exposure. However, these types of irradiation sources lead to major concerns, notably the dangerousness of the irradiation wavelength (risk of skin cancer, skin ageing, and eye damage when wavelengths are shorter than that of visible light) and ozone release [5]. Therefore, a huge challenge today is to perform photopolymerization under milder conditions of irradiation. In this way, new efficient photoinitiating systems under safe irradiation conditions are actively researched. Light-emitting diodes (LEDs) are really attractive new irradiation devices for photopolymerization due to their low cost and low energy consumption compared to traditional UV lamps, such as mercury bulbs, currently used in industry. Moreover, LEDs can deliver longer wavelengths of irradiation than the traditional irradiation setups. Therefore, the search for new photoinitiating systems with high performance under such types of irradiation is today a huge challenge. Different photoinitiating systems for LEDs were proposed recently; they are based on, for example, acylgermanes [6,7], acylstannanes [8,9], silyl glyoxylates [10], or different dye structures such as camphorquinone or anthraquinone derivatives reported in [11]. In the present paper, we propose 1,8-naphthalimide derivatives with original moieties as photoinitiators in a three-component system for the polymerization of methacrylates upon near-UV or visible LED irradiation. In the present work, the wavelengths of irradiation are 395, 405, and 470 nm. This range of irradiation can find applications in fields such as coatings, inks, dental composites, cheap 3D printing devices, and so on [12,13].
Some 1,8-naphthalimides derivatives have already been reported in the literature for their excellent photophysical properties. Herein, we propose 1,8-naphthalimide structures with original substituents, and their chemical structures are represented in the Scheme 1. 1,8-Naphthalimide derivatives are constituted of fused aromatic cycles with carbonyl groups. More particularly, these molecules are characterized by a delocalized π-electronic system, bearing an electron donating group and an electron withdrawing group. Thus, they have relatively high electron affinity and intramolecular charge transfer capacity, giving rise to a strong absorption band in the visible range. [14] They are also characterized by a quite good chemical stability, a large Stokes shift, and high fluorescent quantum yields [15]. Synthesized from the corresponding 1,8-naphthalic anhydride with a suitable amine, a large family of derivatives is possible. Additionally, substitution on the naphthalimide ring is possible at the 3–6 positions. Functionalization of the naphthalimide core and modulation of the photophysical properties are also highly possible, leading to a good versatile platform for photoinitiator design [14].
Scheme 1.
Chemical structure of 1,8-naphthalimide.
Because of these properties, 1,8-naphthalimide derivatives have been reported in the literature for various applications ranging from medicine [16] to fluorescence sensors and switchers [17]. As far as polymerization is concerned, dyes of similar architectures have also been reported by Grabchev for copolymerization with styrene and methacrylate resin [18]. The efficiency of other naphthalimides as photoinitiators for both radical or cationic polymerization has been reported in the literature. Polymerization is possible under various irradiation sources and, in particular, under blue LEDs with low power consumption. Different derivatives have been reported in the literature. Notably, in [19], a methacryloyl group was added on the molecule to address the migration issue of the naphthalimide derivatives in cured acrylate. Other structures are also reported where the R2 functional group (cf. Scheme 1) is modified such as in [20,21] or in [22]. This allows to finely tune the absorption properties of the compounds, and excellent photoinitiation abilities have been demonstrated when these latter were combined with an iodonium salt (Iod). To end, substituents at the 3- and 6-positions of the naphthalimide core were also changed in [23,24] with the aim of enhancing the photoinitiation abilities.
Herein, our goal is to extend the scope of 1,8-naphthalimide derivatives by introducing original electron donating or withdrawing moieties on the structure and then to show their excellent abilities to initiate free-radical polymerization. Synthesis of these new naphthalimide derivatives (Scheme 2) will be presented, and their uses in a three-component system in combination with an iodonium salt and a phosphine are reported. More particularly, we will see that the naphthalimide derivatives presented in this work are able to react with the other compounds to form a charge transfer complex, as reported for other systems in [25]. Polymerization of methacrylate with only a small amount of photoinitiator will be presented here. Mechanisms will be fully described by combining several techniques such as photolysis or ESR (electron spin resonance) measurements.
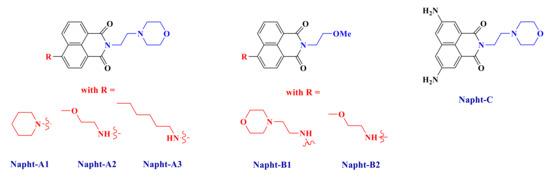
Scheme 2.
Chemical structures of 1,8-naphthalimide derivatives investigated in this study.
2. Results and Discussion
2.1. Light Absorption Properties of the Naphthalimide Derivatives.
Absorption spectra of the different 1,8-naphthalimides investigated in this work are reported in Figure 1. Extinction coefficients at the emission wavelengths of different LEDs used are reported in Table 1 and are compared to the extinction coefficients of camphorquinone (CQ). We observed that, at the wavelength of irradiations of the different light sources used, our derivatives presented absorptions with higher extinction coefficients than CQ.
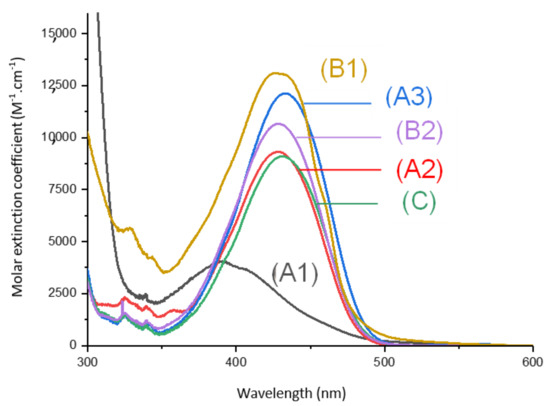
Figure 1.
UV-visible absorption spectra of (1) Napht-A1, (2) Napht-A2, (3) Napht-A3, (4) Napht-B2, (5) Napht-C, (6) Napht-B1; in acetonitrile.

Table 1.
Maximum absorption wavelengths (in the near visible range) and associated extinction coefficients and molar extinction coefficients for the investigated compounds at the wavelengths of the different light-emitting diodes (LEDs) used (in acetonitrile).
The different bands detected in the UV-vis absorption spectra between 350 and 500 nm for the different photoinitiators are characteristic of a π→π * transition. This assignment is supported by molecular modeling carried out on Napht-B1. As evidenced in Figure 2 with the contour plots of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), both orbitals were located on the naphthalimide moiety showing the π→π * transition. For all photoinitiators, the shape of the absorption spectra was rather similar, except for Napht-A1 where a hypsochromic shift of the absorption maximum was observed compared to the others. This difference could be predicted by comparing the different donor and acceptor moieties grafted on the naphthalimide core. The strength of the moiety influenced the HOMO/LUMO energy gap, which directly related to the absorption behavior of the compound. The moiety grafted on the naphthalimide core for Napht-A1 comprised a ring and an azote, in favor of an electron delocalization.
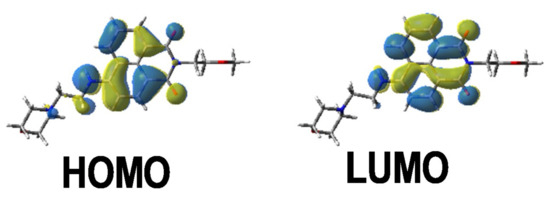
Figure 2.
Contour plots of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels of Napht-B1 optimized at the B3LYP/6-31G * level of theory.
2.2. Initiation Efficiency of the Free-Radical Polymerization of Methacrylates
2.2.1. Photopolymerization Using the New 1,8-Naphthalimide Derivatives
Because of their good light absorption properties, these derivatives were tested as photoinitiators for the free-radical polymerization of a benchmarked methacrylate blend and were compared to the reference initiating system. Thus, photoinitiators (0.1 wt%) were mixed into the Mix-MA monomer in combination with Ar2I+PF6− (Iod-3 wt%) and 4-dppba (2 wt%) to form a three-component photoinitiating system (See Scheme 3 and Scheme 4).
Scheme 3.
Composition of the reference polymerizable system (Mix-MA).
Scheme 4.
Chemical structures of the two additives.
Conversion versus time measurements were recorded upon irradiation by LED@395 nm, LED@405 nm, and LED@470 nm. Photopolymerization profiles are depicted in Figure 3. The new proposed systems were compared to the camphorquinone (CQ)/ethyl 4-(dimethylamino) benzoate (EDB) reference (Scheme 5) widely used in industry. In particular, this is the reference system for dental composites [26]. Excellent photopolymerization efficiencies were found for the new proposed systems with final C=C function conversions around 80%.
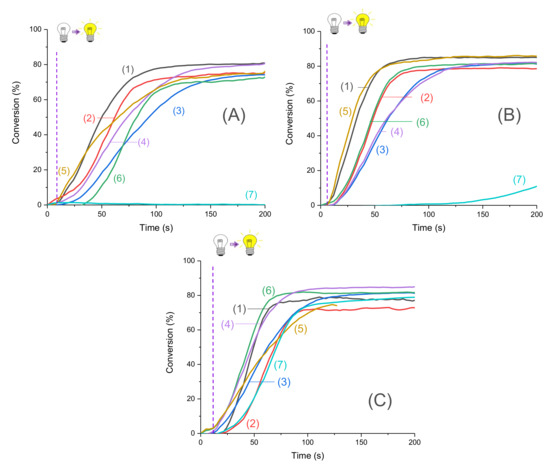
Figure 3.
Photopolymerization profiles of Mix-MA under air (methacrylates function conversion vs. irradiation time) in the presence of (1)–(6)/Ar2I+PF6− (3 wt%)/4-dppba (2 w%) with: (1) Napht-A1 (0.1 wt%); (2) Napht-A2 (0.1 wt%); (3) Napht-A3 (0.1 wt%); (4) Napht-B2 (0.1 wt%); (5) Napht-C (0.1 wt%); (6) Napht-B1 (0.1 wt%); (7) CQ (0.1 wt%) and EDB (1 wt%) upon irradiation by (A) LED@395 nm, (B) LED@405 nm, and (C) LED@470 nm; thickness = 1.4 mm under air, and irradiation starts after 10 s (see the dotted line).

Scheme 5.
Chemical structures of the reference photoinitiating system, camphorquinone/ethyl 4-(dimethylamino) benzoate (CQ/EDB).
Moreover, for the majority of proposed systems, photopolymerization started as soon as the light was turned on. The low oxygen inhibition observed is a significant advantage for many applications where photopolymerization under air is required. The polymers obtained were tack-free on the surface after only a few seconds of irradiation. Performance of each naphthalimide derivative to initiate free-radical polymerization of methacrylates depends on the chosen irradiation wavelength. Indeed, all compounds did not absorb in the same way at the three wavelengths of irradiation (as reported in Table 1). More particularly, it is interesting to compare the investigated compounds with camphorquinone, used as a reference. Under LED@395 nm (Figure 4A), was possible with this compound under LED@405 nm in our conditions of polymerization. In Figure 4C, we had a favorable case for the use of camphorquinone; however, polymerization was still less efficient than with our naphthalimide derivatives showing the interest of the proposed systems. Comparison of light absorption of these compounds reveals that at the investigated wavelengths, there was no direct correlation between absorption and kinetic/final conversion even if it influenced the reactivity of the system, as noted in Figure 3.
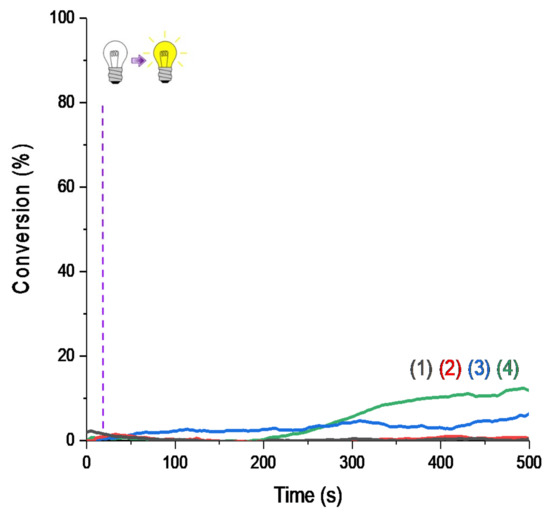
Figure 4.
RT-FTIR polymerization profiles (C=C conversion of Mix-MA resin vs. irradiation time upon LED@405 nm) in the presence of (1) monomer alone; (2) Ar2I+/PF6− (1.5 wt%); (3) 4-dppba (0.5 wt%); and (4) Ar2I+/PF6− (1.5 wt%) and 4-dppba (0.5 wt%) and LED@405 nm; thickness = 1.4 mm, under air, irradiation starts after 10 s.
Thus, we conclude that the mechanism involved in the free-radical polymerization reported here was not governed by light absorption properties alone, and it will be investigated in Section 3. We expect a redox mechanism from the excited state of the dye and the additives.
Thus, and in a way of conciseness, only Napht-B1 will be presented in the following section. This naphthalimide derivative was chosen regarding its kinetics of polymerization, final C=C conversion under the three wavelengths of irradiation tested, and its straightforward synthetic pathway (protocol, raw materials used, and yield). We focused on irradiation at 405 nm, a wavelength commonly used for photopolymerization, especially in the 3D printing field.
2.2.2. Blank Measurements
Photoinitiating systems (PISs) presented here are multicomponent systems. First of all, RT-FTIR follow-ups were performed on the system upon irradiation without any photosensitizer. As evidenced in Figure 4, presence of the photosensitizer is necessary for photopolymerization to be efficient. Only with the combination of iodonium salt/phosphine in the monomer was a slight polymerization observed after a long induction time, as shown with curve (4) in Figure 4. This was due to the formation of an absorbing charge transfer complex (as reported with this specific iodonium salt and phosphine in [25]).
Production of initiating radicals required an additive, here the iodonium salt, to produce aryl radicals. A third component was also necessary because of the oxygen in the atmosphere, which inhibited the reaction. Without one of the three components, polymerization was not possible, as shown in the Figure 5.
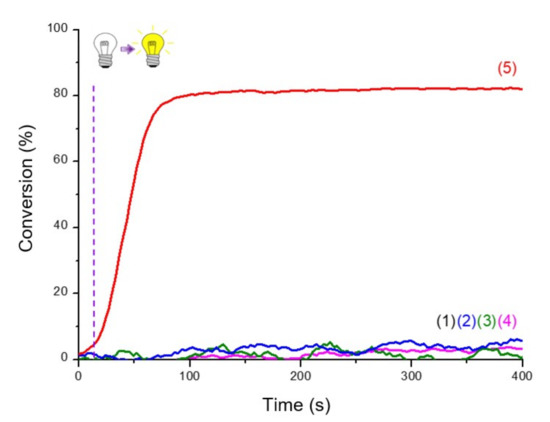
Figure 5.
RT-FTIR photopolymerization profiles (C=C conversion of Mix-MA resin vs. irradiation time upon LED@405 nm of (1) the resin Mix-MA alone; (2)–(5) in the presence of Napht-B1: (2) alone in the monomer; in combination with (3) Ar2I+PF6− (1.5 wt%); (4) 4-dppba (0.5 wt%) and (5) Ar2I+PF6− (1.5 wt%) and 4-dppba (0.5 wt%); LED@405 nm, thickness = 1.4 mm, under air, irradiation start after 10 s (dot line).
2.3. Chemical Mechanisms Associated with the Proposed Three-Component System
The different experiments presented in this section were performed to propose a full picture of the chemical mechanisms. Experiments included absorption and photolysis studied by UV-visible spectroscopy, characterization of the radical produced by ESR measurements, and determination of the redox properties by cyclic voltammetry.
2.3.1. Steady-State Photolysis
Absorption was measured on the three-component system investigated here with a particular interest for the properties at 405 nm, our wavelength of interest. Four experimental conditions were examined: Napht-B1 alone, Napht-B1 in combination with Ar2I+PF6−, Napht-B1 in combination with 4-dppba, and Napht-B1 in combination with both Ar2I+PF6− and 4-dppba.
First of all, absorption spectra for all freshly prepared solutions in acetonitrile and before any irradiation are presented in Figure 6. UV-Visible spectra of the phosphine and iodonium salts used here are available in Supporting Information (Figure S1). These compounds did not absorb at the wavelength of interest.
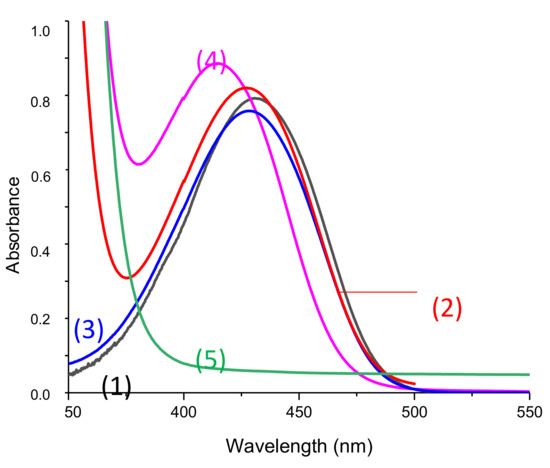
Figure 6.
UV-vis spectra of solutions used for photolysis measurement in acetonitrile before any irradiation and in presence of (1)–(4): Napht-B1 (1) alone; (2) with 4-dppba (5.10−2 M); (3) with Ar2I+/PF6− (4.10−2M); (4) with 4-dppba (5.10−2 M) and Ar2I+/PF6− (4.10−2 M); and Ar2I+/PF6− (4.10−2 M); and (5) 4-dppba (5.10−2 M) and Ar2I+/PF6− (4.10−2 M) in acetonitrile.
First, only a very slight difference in absorption was observed by adding iodonium salt to the solution containing the naphthalimide derivative (Curve (1) vs. (3)). However, the shape of the absorption spectra before any irradiation was influenced when phosphine was added to the system. With the three components in the same solution, this difference was even more pronounced. As shown in Supporting Information, phosphine did not absorb in the range of wavelength studied. We conclude that there were interactions between the chosen phosphine and the other components in solutions that influenced the absorption spectra. More particularly, we suppose that a charge transfer complex (CTC) formed. Garra et al. [25] previously studied the CTC obtained by the combination of a phosphine and the iodonium salt, and authors also observed a significant absorption of the CTC@405 nm.
The behaviors of the four solutions upon blue light irradiation were evaluated thanks to steady-state photolysis monitoring. Decay of the absorption at 405 nm (followed by UV-vis measurement) versus time of exposure is presented in Figure 7. UV-vis spectra of photolysis measurements are presented in Supporting Information (Figure S2).
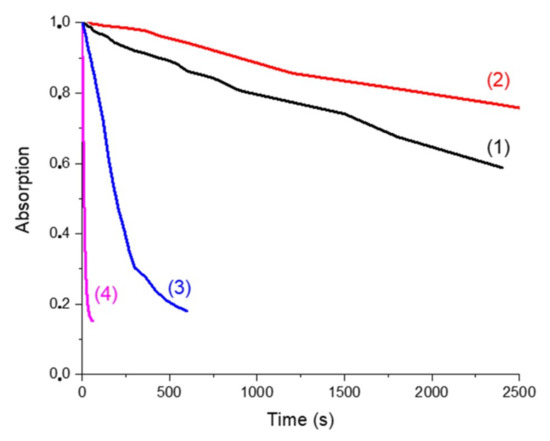
Figure 7.
Photolysis monitored by the decay of the absorption @405 nm (percentage of decay of the absorption peak at 405 nm vs. irradiation time of the solution upon LED exposure in the presence of Napht-B1: (1) alone; (2) with 4-dppba (5.10−2 M); (3) with Ar2I+PF6− (4.10−2 M) and (4) with 4-dppba (5.10−2 M) and Ar2I+PF6− (4.10−2 M) in acetonitrile.
In all cases, a decrease of the absorption peak at 405 nm was clearly observed when the samples were irradiated by the LED@405 nm. Curve (1) represents the solution with only the naphthalimide chromophore. It illustrates its direct photolysis. We compared it first with curve (2), which corresponds to the naphthalimide derivative in the presence of the phosphine. At the wavelength of interest (405 nm), the presence of the phosphine increased the absorption of the solution. The decay of absorption was influenced by the presence of this component. The CTC formed by the naphthalimide derivative and the phosphine was more stable than the naphthalimide alone, and consequently photolysis was slower.
Curves (1) and (3) correspond to the solution of Napht-B1 without and with Ar2I+PF6−, respectively. First, the iodonium salt only slightly influenced the shape of the absorption spectra. Photolysis of the solution upon irradiation at 405 nm was, however, different: decomposition of the derivative was accelerated by the presence of the iodonium salt. Thus, we supposed that the iodonium salt was able to quench the excited state of the photoinitiator through a redox process.
Finally, photolysis of the solution with the three components (curve (4)) clearly indicated that decomposition was the fastest when all components were present in solution. The CTC formed as reported in [25] not only absorbed at 405 nm but also was capable of reacting with the naphthalimide derivative, which increased its kinetic of degradation.
2.3.2. ESR-ST Experiments
In order to characterize the radicals obtained upon irradiation, ESR-ST measurements were performed in acetonitrile under air and in the presence of a spin-trap agent, the N-tert-butyl-α-phenylnitrone (PBN). No radicals were observed under air using only the naphthalimide derivative and the iodonium salt, as seen in Figure 8. This can be ascribed to the trapping of Ar• by O2. Addition of the phosphine (4-dppba) was mandatory in order to form the radicals and overcome this oxygen inhibition. Indeed, as shown in Figure 9A, formation of an aryl Ar•/PBN radical adduct was observed under air. The corresponding spectrum was simulated (Figure 9B) with the hyperfine coupling constants hfc (aN = 14.2 G; aH = 2.2 G) in full agreement with the reference values [27], which confirmed the formation of aryl radicals upon irradiation.
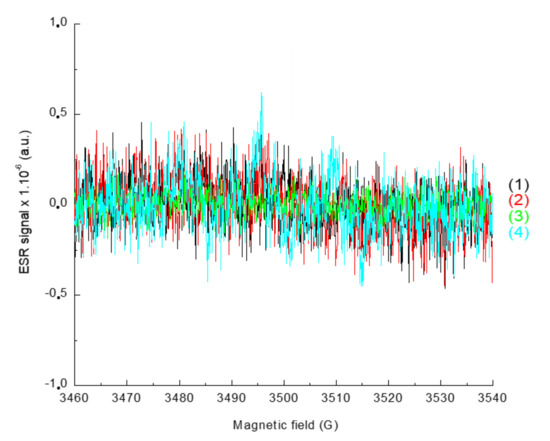
Figure 8.
ESR-Spin trapping spectrum of Napht-B1: (1) before irradiation; (2) after 180 s of irradiation with LED@405 nm (I ≈ 110 mW·cm−2) exposure; (3) and (4) in combination with Ar2I+PF6− (3) before irradiation and (4) after 180 s of irradiation with LED@405 nm (I ≈ 110 mW·cm−2); under air; phenyl-N-tert-butylnitrone (PBN) is used as a spin trap, in acetonitrile.
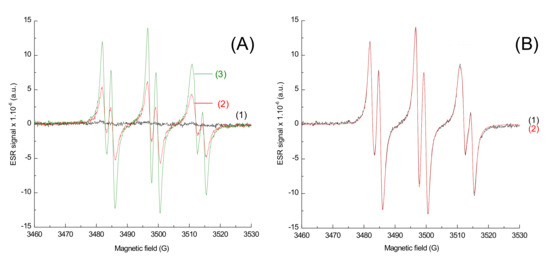
Figure 9.
ESR-Spin trapping spectrum of Napht-B1 in combination with Ar2I+PF6− and 4-dppba in acetonitrile; under air; phenyl-N-tert-butylnitrone (PBN) is used as a spin trap. (A) Experimental spectrum (1) before irradiation; after (2) 40 s and (3) 80 s of irradiation with LED@405 nm (I ≈ 110 mW·cm−2) exposure; (B) after 80 s of irradiation with LED@405 nm (I ≈ 110 mW·cm−2) exposure (1) experimental and (2) simulated with the hyperfine coupling constants hfc (aN = 14.2 G; aH = 2.2 G).
From the control experiments (Section 2.2), it was determined that no polymerization was possible using only the iodonium salt in combination with the naphthalimide derivative. Experiments conducted by ESR were in full agreement with this observation: the phosphine was essential for the system to produce enough aryl radicals, which are able to initiate the polymerization. ESR measurements were performed under air, and phosphine was essential to overcome oxygen inhibition. Indeed, aryl radicals, required for photopolymerization of (meth)acrylate, were able to react with O2. This reaction led to the formation of a peroxyl radical ROO•, too stable to continue polymerization. However, these radicals capable of reacting with a compound, such as a phosphine, to generate RO• radical were less stable and, therefore, were able to initiate free-radical polymerization. The role of phosphine as an oxygen scavenger has been already reported in the literature in [28]. The mechanism is illustrated in Scheme 6.
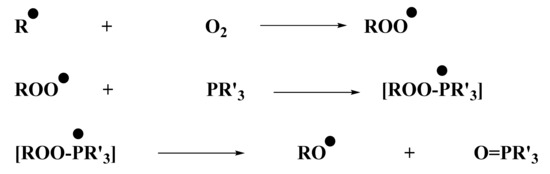
Scheme 6.
Chemical mechanism of peroxyl scavenging by the phosphine (abbreviated PR’3).
2.3.3. Redox Experiments
Photoinitiation with iodonium salts often relies on redox interactions with an appropriate reductant. In order to estimate if a redox reaction is possible, the free energy change ΔGet is calculated thanks to the free energy change equation (detailed in the experimental part above). For this purpose, fluorescence and cyclic voltammetry measurements were carried out, and the results are presented in the following section.
To determine the first singlet excited-state energy (ES1) of the investigated naphthalimide, absorption and emission spectra were recorded in acetonitrile (Figure 10). The crossing point of absorption and emission spectra allowed the determination of ES1. λ was measured at 480 nm. Thanks to the Einstein–Planck relation, we concluded that the ES1 = 2.58 eV. As represented in Figure 11, voltammetry measurements were performed on the Napht-B1 derivative. We estimated its oxidation potential as Eox = 0.57 V.
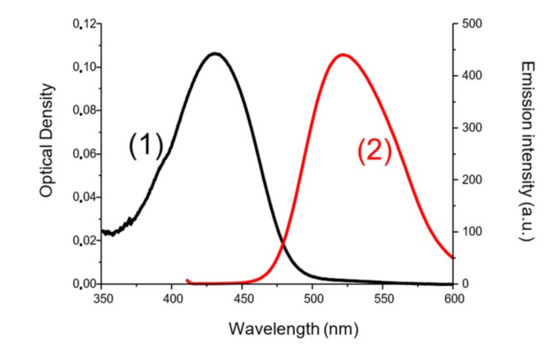
Figure 10.
ES1 determination for Napht-B1 (1) absorption spectra and (2) emission spectra in acetonitrile.
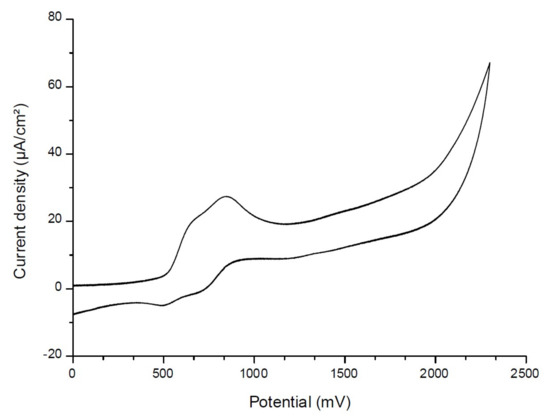
Figure 11.
Cyclic voltammetry of Napht-B1 dye in acetonitrile, in the presence of tetrabutylammonium hexafluorophosphate 0.1 M as a supporting electrolyte, under nitrogen.
Concerning the iodonium salt, its reduction potential has already been reported in [4] with a value of Ered = 0.2 eV. The feasibility of a redox reaction between the naphthalimide derivative and the iodonium salt can be calculated. Indeed, ΔGet was estimated at −2.21 eV for the redox reaction, which means that the reaction was highly favorable. Thus, we propose the reaction presented in Scheme 7 to be responsible for the polymerization process.
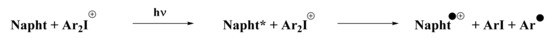
Scheme 7.
Redox reaction predicted between the iodonium salt and the naphthalimide derivative.
According to the photopolymerization results presented in Figure 3, Figure 4 and Figure 5, and thanks to experiments presented above, a general mechanism for the photoinitiation of methacrylate resins with 1,8-naphthalimide derivatives is presented in Scheme 8. Upon irradiation with blue light, the naphthalimide derivative absorbed light and reached its excited state. This excited state was able to react with the iodonium salt through an oxidative pathway to generate aryl radicals, capable of initiating the free-radical polymerization of methacrylate resins. Potential oxygen inhibition of these radicals was counter-balanced by the addition of phosphine (see Scheme 8). This particular phosphine in combination with iodonium salt has already been reported in [25] for the creation of a CTC able to generate aryl radicals upon blue light irradiation. These aryl radicals are able to initiate free-radical polymerization.
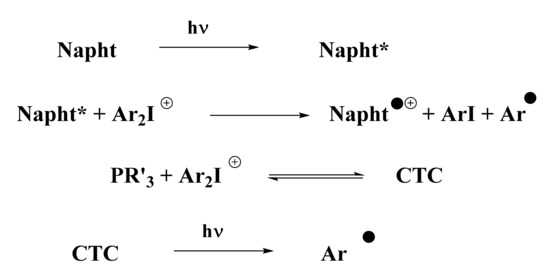
Scheme 8.
Proposed mechanism for the photoinitiation with the three-component system presented (abbreviations: Napht for the new naphthalimide derivatives, Ar2I+ for the iodonium salt, PR’3 for the phosphine, and CTC for the charge-transfer complex).
3. Materials and Methods
3.1. Chemical Compounds
All commercially available products in the present report were used without further purifications. Common solvents used (acetonitrile, acetone) were purchased from Sigma-Aldrich (Sigma-Aldrich, Saint Louis, MO, USA). The blend of monomers used in this study, referred to as “Mix-MA” (Scheme 3), was a mixture of 33.3 wt% of (hydroxypropyl) methacrylate (HPMA), 33.3 wt% of 1,4-butanediol dimethacrylate (1,4-BDDMA), and 33.3 wt% of a urethane dimethacrylate monomer. This resin has a low viscosity of 0.053 Pa·s and will be used as a benchmarked resin in photopolymerization reactions. These monomers were purchased from Sigma-Aldrich.
The iodonium salt bis(4-tert-butylphenyl)iodonium hexafluorophosphate (noted Iod or Ar2I+/PF6− or Speedcure 938, Scheme 4) was gifted from Lambson. 4-(Diphenylphosphino) benzoic acid (4-dppba, Scheme 4) was purchased from TCI Chemicals (TCI Chemicals, Tokyo, Japan). The camphorquinone (CQ, Scheme 5) and ethyl 4-(dimethylamino)benzoate (EDB, Scheme 5) were purchased from TCI Chemicals. CQ/EDB is used as a well-established reference for photopolymerization of methacrylates in the near-UV or visible range [4,29].
All reagents and solvents were purchased from Sigma-Aldrich or Alfa Aesar Chemicals (Alfa Aesar Chemicals, Ward Hill, MA, USA) and used as received without further purification. Mass spectroscopy was performed by the Spectropole at Aix-Marseille University. ESI (electron spray ionization) mass spectral analyses were recorded with a 3200 QTRAP (Applied Biosystems SCIEX) mass spectrometer. The HRMS (high resolution mass spectroscopy) mass spectral analysis was performed with a QStar Elite (Applied Biosystems SCIEX) mass spectrometer. Elemental analyses were recorded with a Thermo Finnigan EA 1112 elemental analysis apparatus driven by the Eager 300 software. 1H and 13C NMR spectra were determined at room temperature in 5 mm o.d. (outer diameter) tubes on a Bruker Avance 400 spectrometer of the Spectropole (Bruker, Billerica, MA, USA): 1H (400 MHz) and 13C (100 MHz). The 1H chemical shifts were referenced to the solvent peak DMSO (dimethylsulfoxide) (2.49 ppm), and the 13C chemical shifts were referenced to the solvent peak DMSO (49.5 ppm). All photoinitiators were prepared with analytical purity up to accepted standards for new organic compounds (>98%), which was checked by high-field NMR analysis. 5,8-Diaminobenzo[de]isochromene-1,3-dione, 9-aminobenzo[4,5]isochromeno[7,8-d]thiazole-1,3-dione and 9-aminobenzo[4,5] isochromeno[7,6-d]thiazole-4,6-dione were synthesized as previously reported [30]. They were used as precursors for the synthesis of the different naphthalimide derivatives presented here, as reported in Scheme 9.
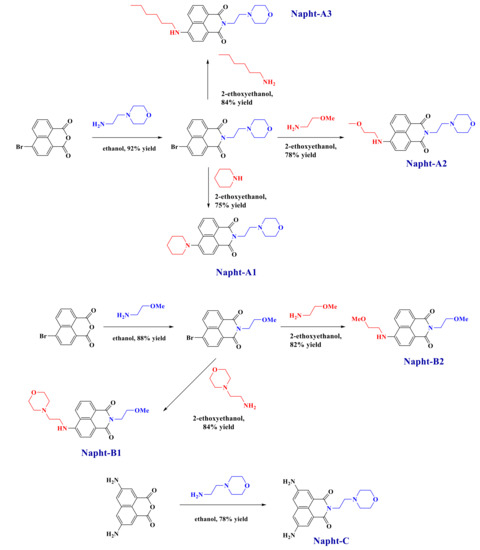
Scheme 9.
Synthetic routes giving access to the different naphthalimide derivatives reported in this work.
3.1.1. Synthesis of 6-bromo-2-(2-morpholinoethyl)-1H-benzo[de]isoquinoline-1,3(2H)-dione
A mixture of 6-bromobenzo[de]isochromene-1,3-dione (1.60 g, 5.77 mmol) and 4-(2-aminoethyl)morpholine (0.78 g, 0.79 mL, 6.04 mmol, 1.05 eq.) was suspended in absolute ethanol (50 mL), and the solution was refluxed overnight. Upon cooling, a precipitate formed. It was filtered off, washed with ethanol, and dried under reduced pressure (2.07 g, 92% yield). 1H NMR (CDCl3) δ 2.57–2.60 (m, 4H), 2.71 (t, 2H, J = 6.7 Hz), 3.65–3.68 (m, 4H), 4.34 (t, 2H, J = 6.7 Hz), 7.86 (t, 1H, J = 7.5 Hz), 8.05 (d, 1H, J = 7.9 Hz), 8.41 (d, 1H, J = 7.8 Hz), 8.58 (d, 1H, J = 8.5 Hz), 8.66 (d, 1H, J = 7.2 Hz); 13C NMR (CDCl3) δ 37.3, 53.8, 56.1, 67.0, 122.2, 123.0, 128.0, 128.9, 130.2, 130.6, 131.1, 131.2, 132.0, 133.2, 153.51, 163.53; HRMS (ESI MS) m/z: theoretical: 388.0423, found: 388.0421 ([M]+. detected).
3.1.2. Synthesis of 6-bromo-2-(2-methoxyethyl)-1H-benzo[de]isoquinoline-1,3(2H)-dione
A mixture of 6-bromobenzo[de]isochromene-1,3-dione (1.60 g, 5.77 mmol) and 2-methoxyethylamine (0.45 g, 0.53 mL, 6.06 mmol, 1.05 eq.) was suspended in absolute ethanol (50 mL), and the solution was refluxed overnight. Upon cooling, a precipitate formed. It was filtered off, washed with ethanol, and dried under reduced pressure (1.70 g, 88% yield). 1H NMR (DMSO d6) δ 3.27 (s, 3H, OMe), 3.61 (t, 2H, J = 6.1 Hz), 4.26 (t, 2H, J = 6.1 Hz), 8.02 (t, 1H, J = 7.4 Hz), 8.24 (d, 1H, J = 7.9 Hz), 8.35 (d, 1H, J = 7.9 Hz), 8.59 (t, 2H, J = 6.1 Hz); 1H NMR (CDCl3) δ 3.37 (s, 3H, OMe), 3.72 (t, 2H, J = 5.8 Hz), 4.42 (t, 2H, J = 5.8 Hz), 7.81 (t, 1H, J = 7.4 Hz), 8.01 (d, 1H, J = 7.9 Hz), 8.38 (d, 1H, J = 7.9 Hz), 8.52 (dd, 1H, J = 8.5 Hz, J = 1.0 Hz), 8.63 (dd, 1H, J = 8.5 Hz, J = 1.0 Hz); 13C NMR (CDCl3) δ 39.4, 58.8, 69.6, 122.2, 123.0, 128.0, 129.0, 130.2, 130.6, 131.0, 131.3, 132.1, 133.2, 163.63, 163.65; HRMS (ESI MS) m/z: theoretical: 333.0001, found: 333.0004 ([M]+. detected).
3.1.3. Synthesis of 2-(2-morpholinoethyl)-6-(piperidin-1-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (Napht-A1)
6-Bromo-2-(2-morpholinoethyl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (0.95 g, 2.45 mmol) and piperidine (0.62 g, 0.72 mL, 7.34 mmol, 3 eq.) in 2-ethoxyethanol (10 mL) were stirred at reflux for 16 h. After cooling to room temperature, the solvent was removed under reduced pressure using a rotary evaporator. The crude product was purified by column chromatography on silica gel (CH2Cl2/MeOH: 9/1) to provide an orange oil. Dissolution in a minimum of DCM (dichloromethane), addition of pentane, and cooling in the fridge for one night afforded a yellow solid, which was filtered off, washed with pentane, and dried under vacuum (723 mg, 75% yield). 1H NMR (CDCl3) δ 1.66–1.75 (m, 2H), 1.84–1.97 (m, 4H), 2.57–2.60 (m, 4H), 2.69 (t, 2H, J = 7.1 Hz), 3.20–3.24 (m, 4H),3.67–3.69 (m, 4H), 4.32 (t, 2H, J = 7.1 Hz), 7.17 (d, 1H, J = 8.1 Hz), 7.66 (t, 1H, J = 7.5 Hz), 8.38 (d, 1H, J = 8.4 Hz), 8.47 (d, 1H, J = 8.1 Hz), 8.55 (d, 1H, J = 7.3 Hz); 13C NMR (CDCl3) δ 22.3, 22.4, 24.3, 26.2, 37.0, 44.6, 53.8, 54.5, 56.2, 67.1, 114.7, 115.9, 123.1, 125.3, 126.3, 130.0, 131.0, 132.6, 157.3, 164.1, 164.6; HRMS (ESI MS) m/z: theoretical: 393.2052, found: 393.2055 ([M]+. detected).
3.1.4. Synthesis of 6-((2-methoxyethyl)amino)-2-(2-morpholinoethyl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (Napht-A2)
6-Bromo-2-(2-morpholinoethyl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (950 mg, 2.45 mmol) and 2-methoxyethylamine (0.55 g, 0.64 mL, 7.34 mmol, 3 eq.) in 2-ethoxyethanol (10 mL) were stirred at reflux for 16 h. After cooling to room temperature, the solvent was removed under reduced pressure using a rotary evaporator. The crude product was purified by column chromatography on silica gel (CH2Cl2/MeOH: 9/1) to provide an orange oil. Dissolution in a minimum of DCM, addition of pentane, and cooling in the fridge for one night afforded a yellow solid, which was filtered off, washed with pentane, and dried under vacuum (732 mg, 78% yield). 1H NMR (CDCl3) δ 2.66–2.69 (m, 2H), 2.76 (t, 2H, J = 7.2 Hz), 3.46 (s, 3H, OMe), 3.56 (t, 2H, J = 5.2 Hz), 3.66–3.79 (m, 8H), 4.35 (t, 2H, J = 7.0 Hz), 5.68 (brs, 1H, NH), 6.70 (d, 1H, J = 8.4 Hz), 7.62 (t, 1H, J = 7.5 Hz), 8.13 (d, 1H, J = 8.4 Hz), 8.43 (d, 1H, J = 8.4 Hz), 8.56 (d, 1H, J = 7.5 Hz); 13C NMR (CDCl3) δ 36.7, 39.8, 40.0, 53.7, 56.2, 58.9, 66.8, 67.7, 70.0, 104.5, 110.6, 120.5, 123.0, 124.8, 126.2, 129.8, 131.2, 134.4, 149.5, 164.1, 164.7; HRMS (ESI MS) m/z: theoretical: 383.1845, found: 383.1847 ([M]+. detected).
3.1.5. Synthesis of 6-(hexylamino)-2-(2-morpholinoethyl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (Napht-A3)
6-Bromo-2-(2-morpholinoethyl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (0.95 g, 2.45 mmol) and hexylamine (0.74 g, 0.97 mL, 7.34 mmol, 3 eq.) in 2-ethoxyethanol (10 mL) were stirred at reflux for 16 h. After cooling to room temperature, the solvent was removed under reduced pressure using a rotary evaporator. The crude product was purified by column chromatography on silica gel (CH2Cl2/MeOH: 9/1) to provide an orange oil. Dissolution in a minimum of DCM, addition of pentane, and cooling in the fridge for one night afforded a yellow solid, which was filtered off, washed with pentane, and dried under vacuum (842 mg, 84% yield). 1H NMR (CDCl3) δ 0.84 (t, 3H, J = 6.9 Hz), 1.15–1.35 (m, 4H), 1.43 (qt, 2H, J = 7.2 Hz), 1.73 (qt, 2H, J = 7.4 Hz), 2.52–2.54 (m, 4H), 2.63 (t, 2H, J = 7.2 Hz), 3.33 (t, 2H, J = 7.2 Hz), 3.61–3.63 (m, 4H), 4.25 (t, 2H, J = 7.2 Hz), 5.28 (t, 1H, J = 5.0 Hz), 6.64 (d, 1H, J = 8.5 Hz), 7.53 (t, 1H, J = 7.5 Hz), 8.03 (d, 1H, J = 7.9 Hz), 8.37 (d, 1H, J= 8.4 Hz), 8.48 (dd, 1H, J = 7.3 Hz, J = 0.8 Hz); 13C NMR (CDCl3) δ 14.0, 22.6, 26.8, 29.0, 31.5, 37.0, 43.8, 53.8, 56.3, 67.1, 104.4, 110.2, 120.2, 123.2, 124.7, 125.7, 129.9, 131.1, 134.5, 149.5, 164.1, 164.7; HRMS (ESI MS) m/z: theoretical: 409.2365, found: 409.2366 ([M]+. detected).
3.1.6. Synthesis of 2-(2-methoxyethyl)-6-((2-morpholinoethyl)amino)-1H-benzo[de]isoquinoline-1,3(2H)-dione (Napht-B1)
6-Bromo-2-(2-methoxyethyl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (0.82 g, 2.45 mmol) and 4-(2-aminoethyl)morpholine (0.95 g, 0.96 mL, 7.34 mmol, 3 eq.) in 2-ethoxyethanol (10 mL) were stirred at reflux for 16 h. After cooling to room temperature, the solvent was removed under reduced pressure using a rotary evaporator. The crude product was purified by column chromatography on silica gel (CH2Cl2/MeOH: 9/1) to provide a yellow oil as the pure product. For a higher purity, the product was dissolved in a minimum of DCM followed by an addition of pentane. Cooling in the fridge for one night afforded a yellow precipitate that was filtered off, washed several times with pentane, and dried under vacuum (789 mg, 84% yield). 1H NMR (CDCl3) δ 2.56 (m, 4H), 2.84 (t, 2H, J = 5.7 Hz), 3.39 (s, 3H), 3.41–3.46 (m, 2H), 3.74 (t, 2H, J = 6.1 Hz), 3.77–3.80 (m, 4H), 4.42 (t, 2H, J = 6.1 Hz), 6.25 (t, 1H, J = 5.2 Hz), 6.68 (d, 1H, J = 8.4 Hz), 7.64 (t, 1H, J = 7.6 Hz), 8.10 (d, 1H, J = 8.0 Hz), 8.46 (d, 1H, J = 8.4 Hz), 8.59 (d, 1H, J = 6.7 Hz); 13C NMR (CDCl3) δ 36.3, 39.0, 53.1, 58.7, 66.8, 67.0, 69.8, 104.5, 110.3, 120.4, 123.1, 124.8, 126.0, 129.9, 131.3, 134.6, 149.4, 164.2, 164.8; HRMS (ESI MS) m/z: theoretical: 383.1845, found: 383.1848 ([M]+. detected).
3.1.7. Synthesis of 2-(2-methoxyethyl)-6-((2-methoxyethyl)amino)-1H-benzo[de]isoquinoline-1,3(2H)-dione (Napht-B2)
6-Bromo-2-(2-methoxyethyl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (0.82 g, 2.45 mmol) and 2-methoxyethylamine (0.55 g, 0.64 mL, 7.34 mmol, 3 eq.) in 2-ethoxyethanol (10 mL) were stirred at reflux for 16 h. After cooling to room temperature, the solvent was removed under reduced pressure using a rotary evaporator. The crude product was purified by column chromatography on silica gel (CH2Cl2/MeOH: 9/1) to provide a yellow oil as the pure product. For a higher purity, the product was dissolved in a minimum of DCM followed by an addition of pentane. Cooling in the fridge for one night afforded a yellow precipitate that was filtered off, washed several times with pentane, and dried under vacuum (659 mg, 82% yield). 1H NMR (CDCl3) δ 3.39 (s, 3H), 3.45 (s, 3H), 3.51–3.55 (m, 2H), 3.70–3.77 (m, 4H), 4.40 (t, 2H, J = 5.9 Hz), 5.70 (brs, 1H), 6.63 (d, 1H, J = 8.4 Hz), 7.53 (t, 1H, J = 7.6 Hz), 8.05 (d, 1H, J = 8.4 Hz), 8.37 (d, 1H, J = 8.4 Hz), 8.49 (d, 1H, J = 7.3 Hz); 13C NMR (CDCl3) δ 38.9, 40.0, 58.7, 58.9, 69.8, 70.1, 104.3, 110.4, 120.4, 122.9, 124.6, 126.1, 129.7, 131.1, 134.3, 149.4, 164.1, 164.7; HRMS (ESI MS) m/z: theoretical: 328.1423, found: 328.1422 ([M]+. detected).
3.1.8. Synthesis of 5,8-diamino-2-(2-morpholinoethyl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (Napht-C)
5,8-Diaminobenzo[de]isochromene-1,3-dione (0.92 g, 4.04 mmol) and 4-(2-aminoethyl)morpholine (0.78 g, 0.79 mL, 6.04 mmol, 1.5 eq.) were suspended in absolute ethanol (50 mL), and the solution was refluxed overnight. After cooling, the solvent was removed under reduced pressure. Dissolution in THF (tetrahydrofuran) and addition of pentane precipitated a brown solid that was filtered off, washed several times with pentane, and dried under vacuum (1.07 g, 78% yield). 1H NMR (DMSO d6) δ 2.42–2.48 (m, 4H), 2.54 (t, 2H, J = 6.7 Hz), 3.52–3.54 (m, 4H), 4.12 (t, 2H, J = 6.7 Hz), 5.68 (brs, 4H, NH2), 6.93 (d, 2H, J = 2.1 Hz), 7.57 (d, 2H, J = 2.1 Hz); 13C NMR (DMSO d6) δ 36.6, 53.4, 55.7, 66.2, 109.6, 114.5, 117.0, 122.3, 135.5, 147.6, 163.9; HRMS (ESI MS) m/z: theoretical: 340.1535, found: 340.1537 ([M]+. detected).
3.2. Irradiation Sources
The following light-emitting diodes (LEDs) were used as irradiation sources: (i) LED@395 nm (incident light intensity at the sample surface (I ≈ 110 mW·cm−2); (ii) LED@405 nm (I ≈ 110 mW·cm−2); and (iii) LED@470 nm (I ≈ 80 mW·cm−2), provided by Thorlabs.
3.3. Free-Radical Photopolymerization (FRP)
The three-component photoinitiating systems proposed in the present paper were mainly based on 1,8-naphthalimide derivatives/Iod/4-dppba (0.1 wt%/3 wt%/2 wt%). Other concentrations were also used (see text and Figure captions). The weight percent of the photoinitiating system was calculated from the monomer content. The photosensitive formulation (photoinitiating system mixed in the monomer) was deposited in a mold with a controlled thickness of 1.4 mm, on a polypropylene film. Free-radical polymerizations were performed under air upon irradiation with an LED. The conversion was followed by real-time Fourier transform infrared spectroscopy (RT-FTIR) using a JASCO 4600 spectrometer.
The double bond C=C content of Mix-MA was continuously followed at 6100–6220 cm−1 (see Figure 12), which corresponds to a methacrylate peak. For all experiments, irradiation of the photosensitive formulation started after 10 s. Measurements were carried out by transmission. The procedure has been described in detail in Reference [31].
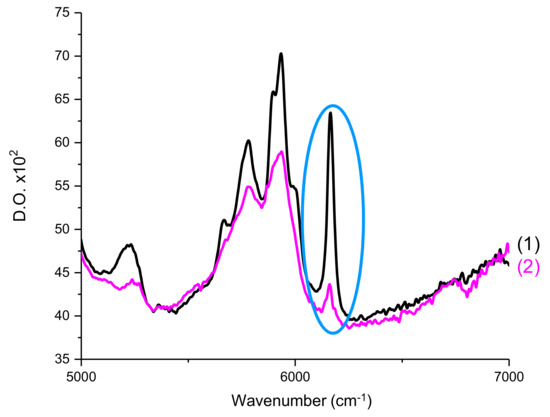
Figure 12.
RT-FTIR spectra of Mix-MA between 5000–7000 cm−1 (1) before polymerization and (2) after polymerization. Circled in blue is the peak used to calculate the double bond C=C conversions of the monomer.
3.4. Redox Potentials
Redox potentials (Eox: oxidation potential of the electron donor and Ered: reduction potential of the electron acceptor vs. saturated calomel electrode—SCE) were measured in acetonitrile by cyclic voltammetry using tetrabutylammonium hexafluorophosphate 0.1 M as a supporting electrolyte. To predict the electron transfer reaction, the free energy change ΔGet was calculated according to the classic equation ΔGet = Eox − Ered − ES1 + C, where ES1 and C stand for the excited-state energy and the coulombic term for the initially formed ion pair, respectively [32]. As the measurement was done in a polar solvent, the coulombic term C was neglected.
3.5. ESR Spin Trapping (ESR-ST) Experiments
The ESR-ST experiments were carried out using an X-Band spectrometer (Bruker EMX-Plus). Results presented were recorded in a solution of acetonitrile, and phenyl-N-tert-butylnitrone (PBN) was added as a spin-trapping agent. The radicals were created under air at room temperature upon exposure to the LED@405 nm (I ≈ 110 mW·cm−2) according to a procedure described in detail [33]. The ESR spectra simulations were carried out with the PEST WINSIM program [27].
3.6. Absorption Spectra
Absorption properties of the investigated compounds were investigated by UV-vis spectroscopy using a Varian Cary 3 spectrometer. Molar extinction coefficients were calculated thanks to the Beer–Lambert Law, A = ℓ·ε·c, with A representing the absorbance of the sample, ℓ the path length of the beam of light through the sample, and ε the molar extinction coefficient of the sample at the wavelength of choice. Measurements were performed in acetonitrile at room temperature using a quartz cell with 10 mm optical pathlength. Photolysis experiments were also performed using UV-vis spectroscopy in acetonitrile and at room temperature. Measurements were made at different times during irradiation by LED@405 nm (I ≈ 110 mW·cm−2). More details about the experimental procedure are given in the caption of the concerned figure.
3.7. Emission Spectra
Fluorescence properties of the compound were studied using a JASCO FP-6200 spectrometer. Experiments were made in acetonitrile at room temperature. Determination of the first singlet excited-state energy (ES1) was made using the crossing point of the absorption and fluorescence spectra [34].
3.8. Molecular Orbital Calculation
The electronic absorption spectra for the different compounds were calculated with the time-dependent density functional theory at the mPW1PW91/6-31G * level of theory on the relaxed geometries calculated at the UB3LYP/6-31G * level of theory. The computational procedure was carried out with the Gaussian 03 suite of programs, as presented in [35].
4. Conclusions
In this paper, new 1,8-naphthalimide derivatives have been proposed for the initiation of fast free-radical photopolymerizations under air upon excitation with violet to blue light delivered by LEDs. The different dyes have been examined in three-component photoinitiating systems in combination with two additives: iodonium salt and phosphine (4-dppba). As photopolymerization was performed under mild irradiation conditions (i.e., LED exposure with visible light range of irradiation, in air, and at room temperature), these new photoinitiators could be used for applications in 3D printing, for example. All 1,8-naphthalimides presented here worked better than the commercial camphorquinone at the three selected wavelengths. A study on the spatial resolution and optimization of the system for application in the 3D printing field is currently underway, and the results will be presented in a forthcoming paper.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4344/9/8/637/s1, Figure S1: UV-vis spectra of (1) 4-dppba and (2) Ar2I+/PF6− in acetonitrile, Figure S2: (A) Photolysis of Napht in ACN upon LED@405 nm: UVvis spectra for different irradiation times; (B) Photolysis of Napht + Ar2+/PF6− (3 w%) in ACN upon LED@405 nm: UV-vis spectra for different irradiation times; (C) Photolysis of Napht + 4-dppba (2 w%) in ACN upon LED@405 nm: UVvis spectra for different irradiation times; (D) Photolysis of Napht + Ar2+/PF6− (3 w%) + 4-dppba (2 w%) in ACN upon LED@405 nm: UVvis spectra for different irradiation times.
Author Contributions
J.L., C.D., F.D., N.H., and F.M. conceived and designed the experiments; A.-H.B. and S.Z. carried out most of the experiments; B.G. carried out the theoretical calculations; M.I.-O. and D.G. contributed to review the manuscript. F.D. and J.L. wrote the paper, which was improved by the rest of authors.
Funding
This research was funded by the ANR agency under the frame of the project “FastPrinting” (ANR-15-CE08-0012).
Acknowledgments
The authors thank the ANR agency for the financial support of the ANR project “FastPrinting”. Computations were partly achieved using high-performance computing (HPC) resources from the Mesocentre of the University of Strasbourg, and the authors thank them for that.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Iwata, T. Biodegradable and Bio-Based Polymers: Future Prospects of Eco-Friendly Plastics. Angew. Chem. Int. Ed. 2015, 54, 3210–3215. [Google Scholar] [CrossRef] [PubMed]
- Khare, A.; Deshmukh, S. Studies toward Producing Eco-Friendly Plastics. J. Plast. Film Sheet. 2006, 22, 193–211. [Google Scholar] [CrossRef]
- Jambeck, J.R.; Geyer, R.; Wilcox, C.; Siegler, T.R.; Perryman, M.; Andrady, A.; Narayan, R.; Law, K.L. Plastic Waste Inputs from Land into the Ocean. Science 2015, 347, 768–771. [Google Scholar] [CrossRef] [PubMed]
- Fouassier, J.P.; Lalevée, J. Photoinitiators for Polymer Synthesis; Wiley Online Library: Weinheim, Germany, 2012. [Google Scholar]
- Moseley, H. Ultraviolet and Laser Radiation Safety. Phys. Med. Biol. 1994, 39, 1765. [Google Scholar] [CrossRef] [PubMed]
- Ganster, B.; Fischer, U.K.; Moszner, N.; Liska, R. New Photocleavable Structures, 4. Macromol. Rapid Commun. 2008, 29, 57–62. [Google Scholar] [CrossRef]
- Lalevée, J.; Allonas, X.; Fouassier, J.P. Acylgermanes: Excited State Processes and Reactivity. Chem. Phys. Lett. 2009, 469, 298–303. [Google Scholar] [CrossRef]
- Mitterbauer, M.; Knaack, P.; Naumov, S.; Markovic, M.; Ovsianikov, A.; Moszner, N.; Liska, R. Acylstannanes: Cleavable and Highly Reactive Photoinitiators for Radical Photopolymerization at Wavelengths above 500 Nm with Excellent Photobleaching Behavior. Angew. Chem. Int. Ed. 2018, 57, 12146–12150. [Google Scholar] [CrossRef]
- Radebner, J.; Eibel, A.; Leypold, M.; Jungwirth, N.; Pickl, T.; Torvisco, A.; Fischer, R.; Fischer, U.K.; Moszner, N.; Gescheidt, G.; et al. Tetraacylstannanes as Long-Wavelength Visible-Light Photoinitiators with Intriguing Low Toxicity. Chem. Eur. J. 2018, 24, 8281–8285. [Google Scholar] [CrossRef]
- Bouzrati-Zerelli, M.; Kirschner, J.; Fik, C.P.; Maier, M.; Dietlin, C.; Morlet-Savary, F.; Fouassier, J.P.; Becht, J.-M.; Klee, J.E.; Lalevée, J. Silyl Glyoxylates as a New Class of High Performance Photoinitiators: Blue LED Induced Polymerization of Methacrylates in Thin and Thick Films. Macromolecules 2017, 50, 6911–6923. [Google Scholar] [CrossRef]
- Xiao, P.; Zhang, J.; Dumur, F.; Tehfe, M.A.; Morlet-Savary, F.; Graff, B.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Visible Light Sensitive Photoinitiating Systems: Recent Progress in Cationic and Radical Photopolymerization Reactions under Soft Conditions. Prog. Polym. Sci. 2015, 41, 32–66. [Google Scholar] [CrossRef]
- Lalevée, J.; Fouassier, J.-P. Dyes and Chromophores in Polymer Science; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Fouassier, J.; Allonas, X.; Burget, D. Photopolymerization Reactions under Visible Lights: Principle, Mechanisms and Examples of Applications. Prog. Org. Coat. 2003, 47, 16–36. [Google Scholar] [CrossRef]
- Jin, R.; Tang, S. Theoretical Investigation into Optical and Electronic Properties of 1, 8-Naphthalimide Derivatives. J. Mol. Model. 2013, 19, 1685–1693. [Google Scholar] [CrossRef]
- Bureš, F. Fundamental Aspects of Property Tuning in Push–Pull Molecules. RSC Adv. 2014, 4, 58826–58851. [Google Scholar] [CrossRef]
- Banerjee, S.; Veale, E.B.; Phelan, C.M.; Murphy, S.A.; Tocci, G.M.; Gillespie, L.J.; Frimannsson, D.O.; Kelly, J.M.; Gunnlaugsson, T. Recent Advances in the Development of 1, 8-Naphthalimide Based DNA Targeting Binders, Anticancer and Fluorescent Cellular Imaging Agents. Chem. Soc. Rev. 2013, 42, 1601–1618. [Google Scholar] [CrossRef]
- Bojinov, V.B.; Simeonov, D.B. Synthesis of Highly Photostable Blue-Emitting 1, 8-Naphthalimides and Their Acrylonitrile Copolymers. Polym. Degrad. Stab. 2010, 95, 43–52. [Google Scholar] [CrossRef]
- Grabchev, I. Photophysical Characteristics of Polymerizable 1, 8-Naphthalimide Dyes and Their Copolymers with Styrene or Methylmethacrylate. Dyes Pig. 1998, 38, 219–226. [Google Scholar] [CrossRef]
- Xiao, P.; Dumur, F.; Frigoli, M.; Tehfe, M.-A.; Graff, B.; Fouassier, J.P.; Gigmes, D.; Lalevée, J. Naphthalimide Based Methacrylated Photoinitiators in Radical and Cationic Photopolymerization under Visible Light. Polym. Chem. 2013, 4, 5440–5448. [Google Scholar] [CrossRef]
- Xiao, P.; Dumur, F.; Graff, B.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Blue Light Sensitive Dyes for Various Photopolymerization Reactions: Naphthalimide and Naphthalic Anhydride Derivatives. Macromolecules 2014, 47, 601–608. [Google Scholar] [CrossRef]
- Zhang, J.; Zivic, N.; Dumur, F.; Xiao, P.; Graff, B.; Fouassier, J.P.; Gigmes, D.; Lalevee, J. UV-Violet-Blue LED Induced Polymerizations: Specific Photoinitiating Systems at 365, 385, 395 and 405 Nm. Polymer 2014, 55, 6641–6648. [Google Scholar] [CrossRef]
- Zhang, J.; Dumur, F.; Xiao, P.; Graff, B.; Bardelang, D.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Structure Design of Naphthalimide Derivatives: Toward Versatile Photoinitiators for near-UV/Visible LEDs, 3D Printing, and Water-Soluble Photoinitiating Systems. Macromolecules 2015, 48, 2054–2063. [Google Scholar] [CrossRef]
- Xiao, P.; Dumur, F.; Zhang, J.; Graff, B.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Amino and Nitro Substituted 2-Amino-1H-Benzo[de]Isoquinoline-1,3(2H)-Diones: As Versatile Photoinitiators of Polymerization from Violet-Blue LED Absorption to a Panchromatic Behavior. Polym. Chem. 2015, 6, 1171–1179. [Google Scholar] [CrossRef]
- Xiao, P.; Dumur, F.; Zhang, J.; Graff, B.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Naphthalimide-Phthalimide Derivative Based Photoinitiating Systems for Polymerization Reactions under Blue Lights. J. Polym. Sci. A Polym. Chem. 2015, 53, 665–674. [Google Scholar] [CrossRef]
- Garra, P.; Graff, B.; Morlet-Savary, F.; Dietlin, C.; Becht, J.-M.; Fouassier, J.-P.; Lalevée, J. Charge Transfer Complexes as Pan-Scaled Photoinitiating Systems: From 50 Μm 3D Printed Polymers at 405 Nm to Extremely Deep Photopolymerization (31 Cm). Macromolecules 2017, 51, 57–70. [Google Scholar] [CrossRef]
- Morlet-Savary, F.; Klee, J.E.; Pfefferkorn, F.; Fouassier, J.P.; Lalevée, J. The Camphorquinone/Amine and Camphorquinone/Amine/Phosphine Oxide Derivative Photoinitiating Systems: Overview, Mechanistic Approach, and Role of the Excitation Light Source. Macromol. Chem. Phys. 2015, 216, 2161–2170. [Google Scholar] [CrossRef]
- Duling, D.R. Simulation of Multiple Isotropic Spin-Trap EPR Spectra. J. Magn. Reson. Ser. B 1994, 104, 105–110. [Google Scholar] [CrossRef]
- Bouzrati-Zerelli, M.; Maier, M.; Fik, C.P.; Dietlin, C.; Morlet-Savary, F.; Fouassier, J.P.; Klee, J.E.; Lalevée, J. A Low Migration Phosphine to Overcome the Oxygen Inhibition in New High-Performance Photoinitiating Systems for Photocurable Dental Type Resins. Polym. Int. 2017, 66, 504–511. [Google Scholar] [CrossRef]
- Cook, W.D. Photopolymerization Kinetics of Dimethacrylates Using the Camphorquinone/Amine Initiator System. Polymer 1992, 33, 600–609. [Google Scholar] [CrossRef]
- Xiao, P.; Dumur, F.; Zhang, J.; Graff, B.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. New Role of Aminothiazonaphthalimide Derivatives: Outstanding Photoinitiators for Cationic and Radical Photopolymerizations under Visible LEDs. RSC Adv. 2016, 6, 48684–48693. [Google Scholar] [CrossRef]
- Lalevée, J.; Blanchard, N.; Tehfe, M.-A.; Peter, M.; Morlet-Savary, F.; Fouassier, J.P. A Novel Photopolymerization Initiating System Based on an Iridium Complex Photocatalyst. Macromol. Rapid Commun. 2011, 32, 917–920. [Google Scholar] [CrossRef]
- Rehm, D.; Weller, A. Kinetics of Fluorescence Quenching by Electron and H-Atom Transfer. Isr. J. Chem. 1970, 8, 259–271. [Google Scholar] [CrossRef]
- Tehfe, M.-A.; Lalevee, J.; Gigmes, D.; Fouassier, J.P. Green Chemistry: Sunlight-Induced Cationic Polymerization of Renewable Epoxy Monomers under Air. Macromolecules 2010, 43, 1364–1370. [Google Scholar] [CrossRef]
- Atkins, P.; De Paula, J. Elements of Physical Chemistry; Oxford University Press: Oxford, MS, USA, 2013. [Google Scholar]
- Foresman, J.; Frish, E. Exploring Chemistry; Gaussian Inc.: Pittsburg, CA, USA, 1996. [Google Scholar]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).