Regarding the Nature of Charge Carriers Formed by UV or Visible Light Excitation of Carbon-Modified Titanium Dioxide



Abstract
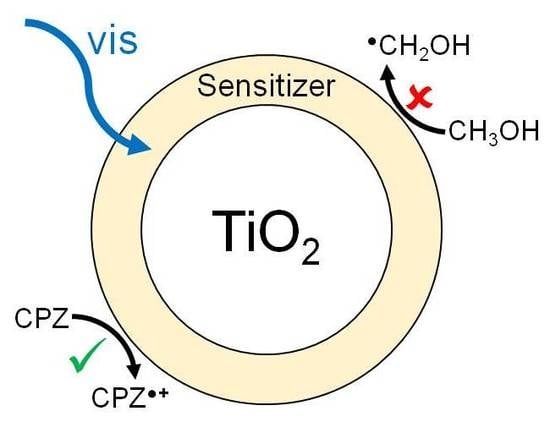
:
1. Introduction
2. Results
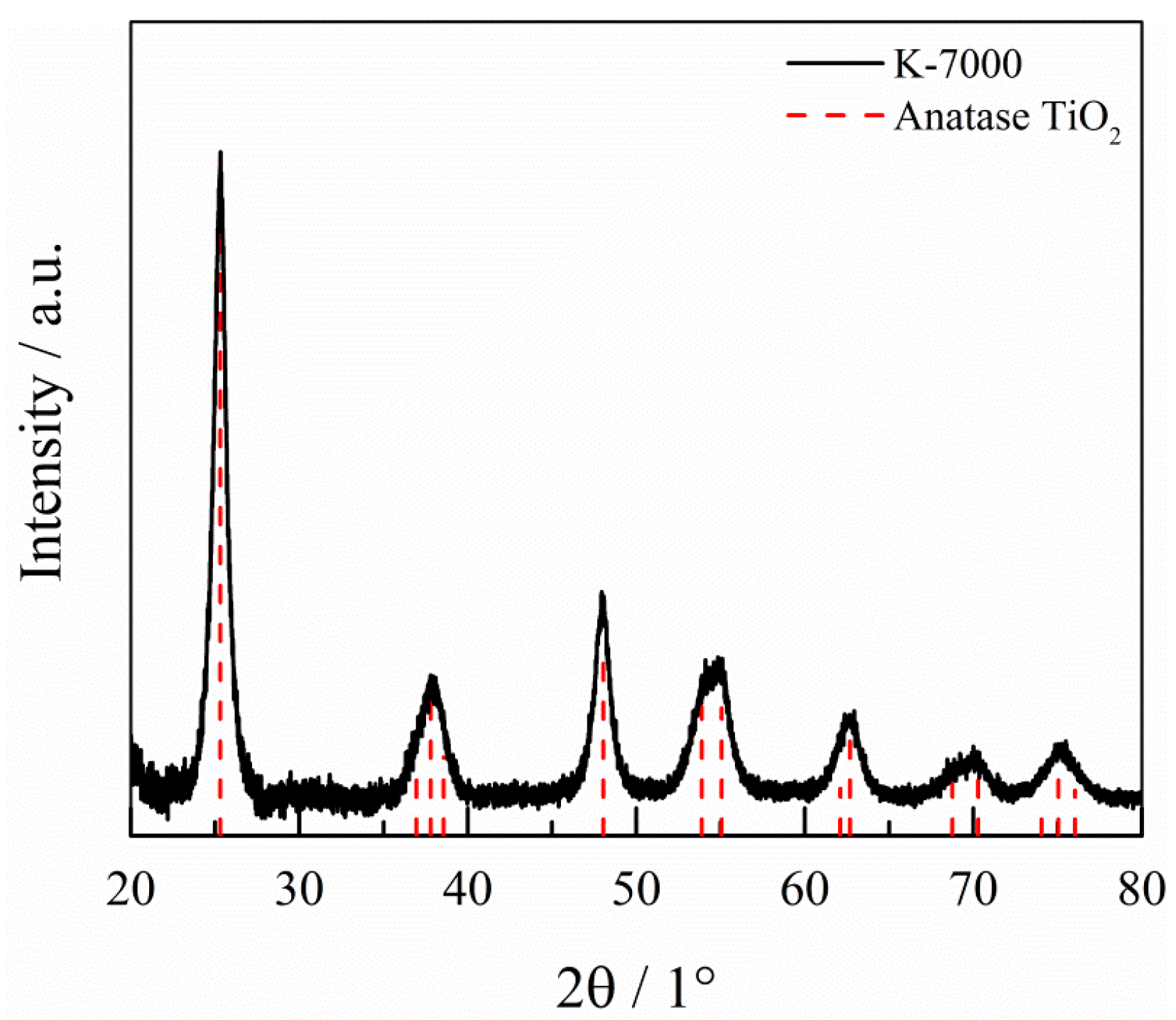
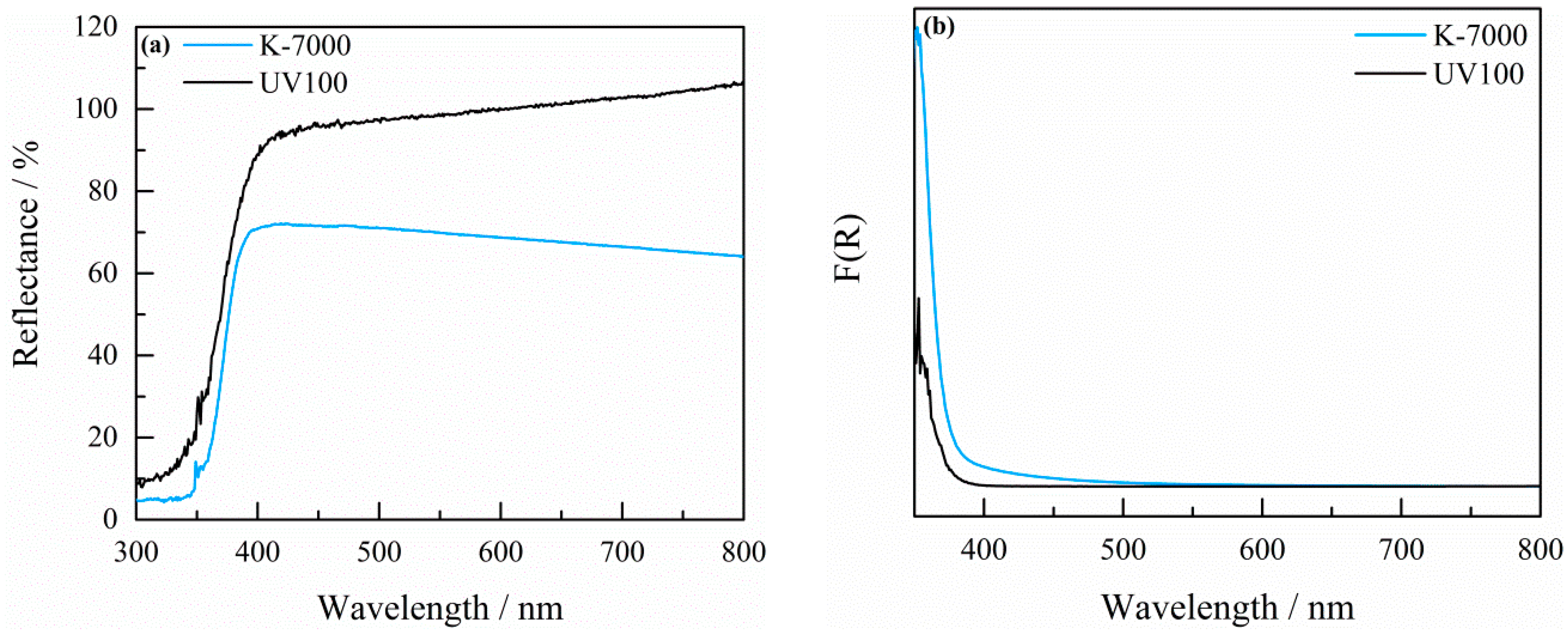
2.1. Characterization of the Photocatalyst
2.2. Mott–Schottky Measurements
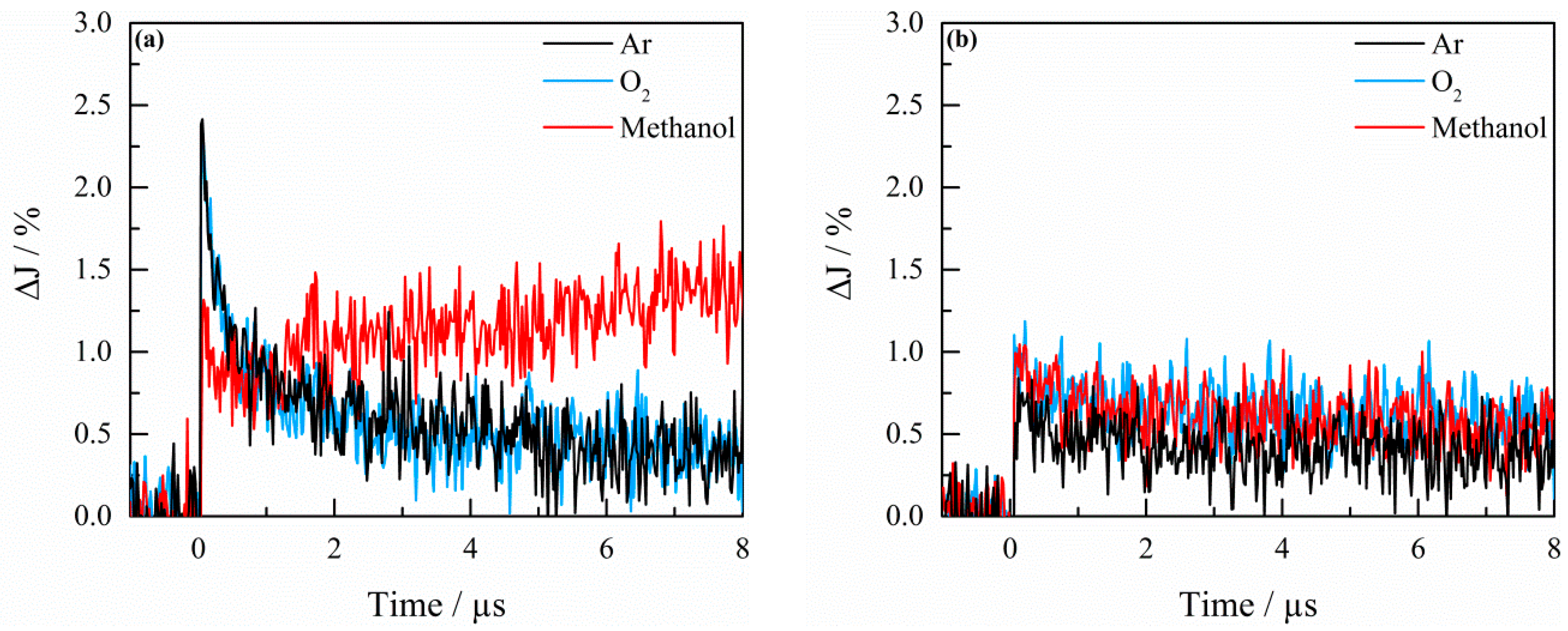
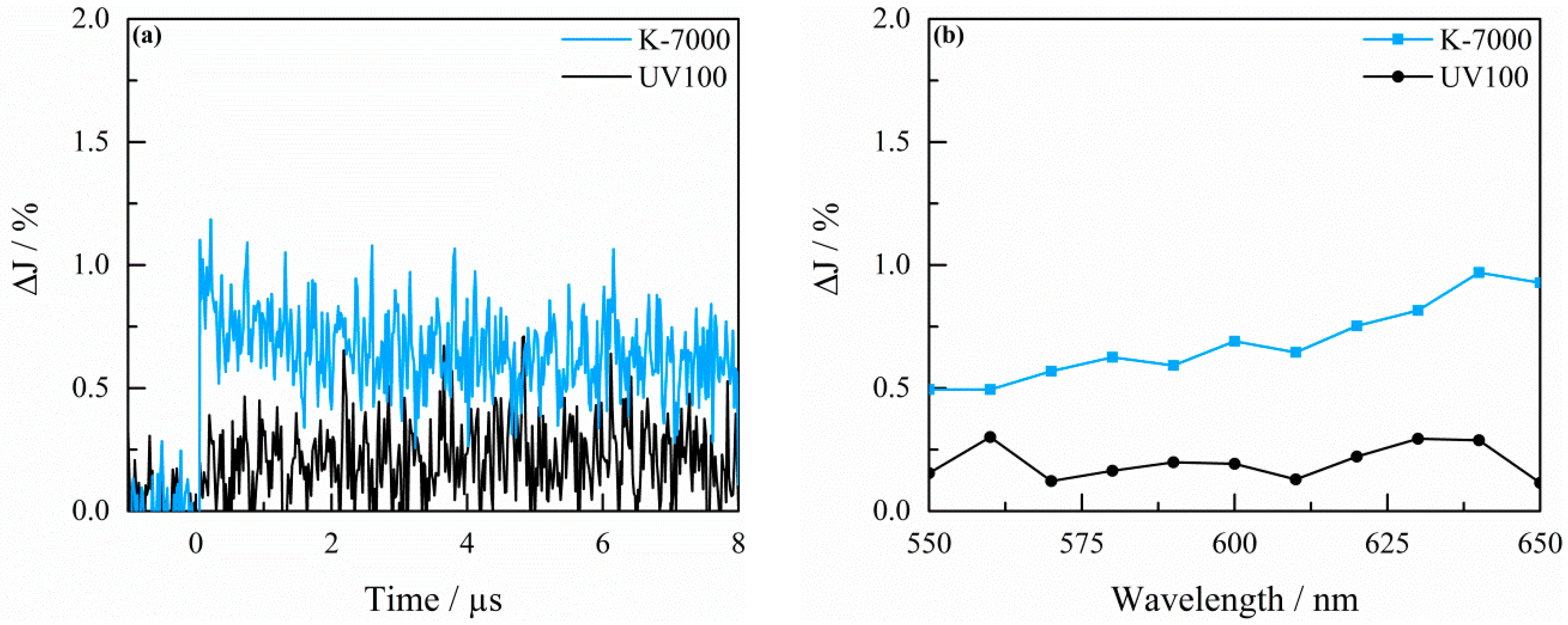
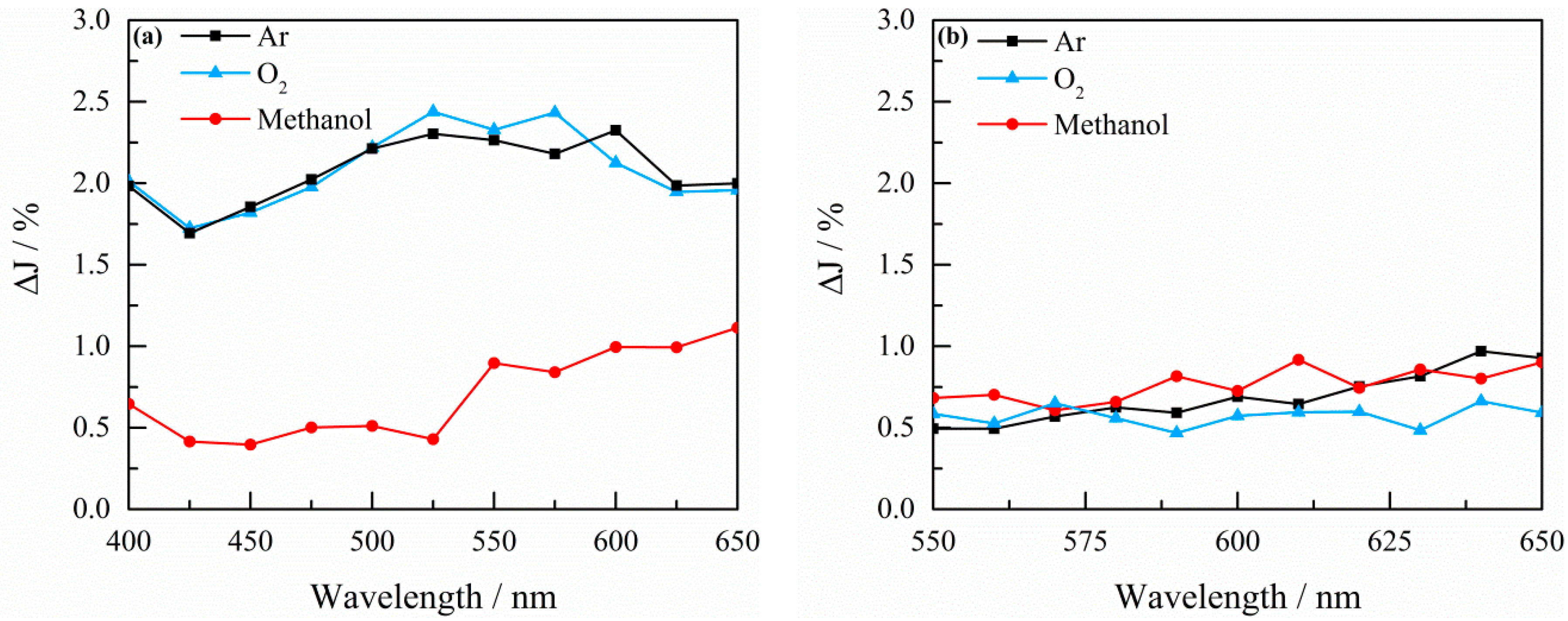
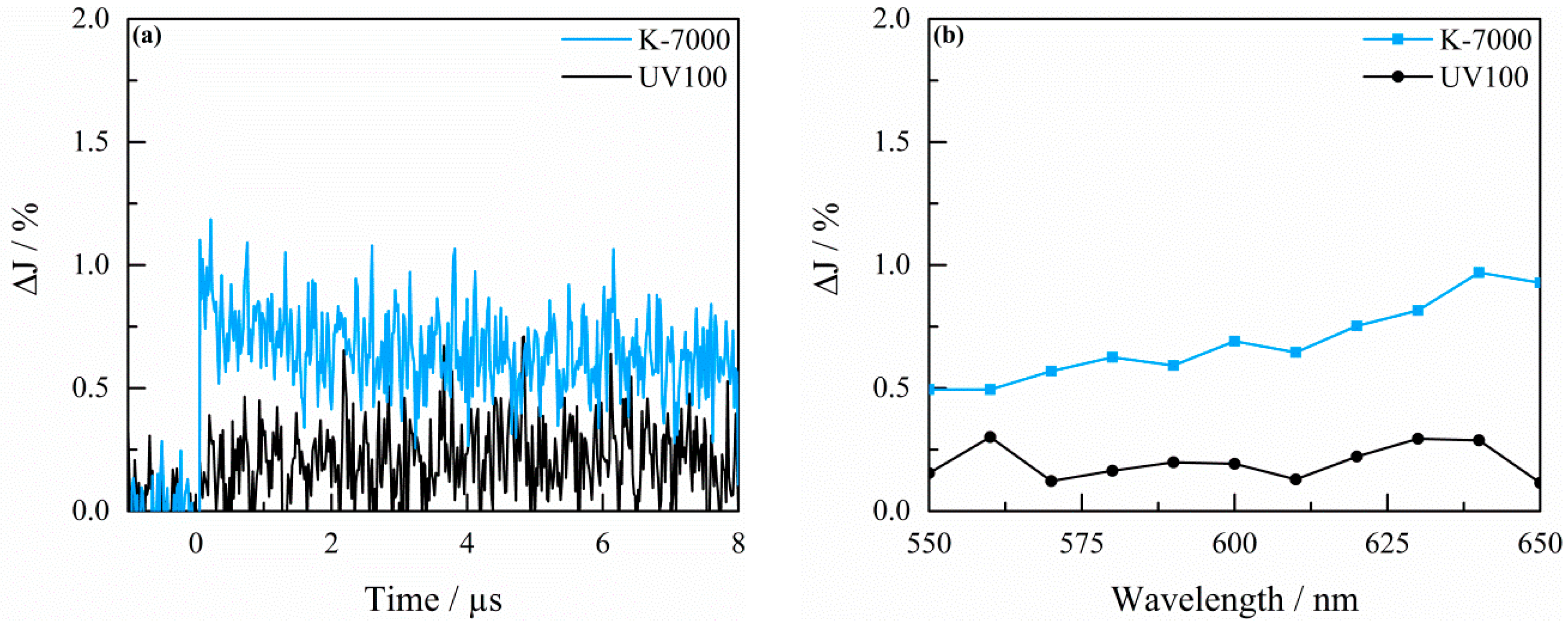
2.3. Transient Absorption Spectroscopy
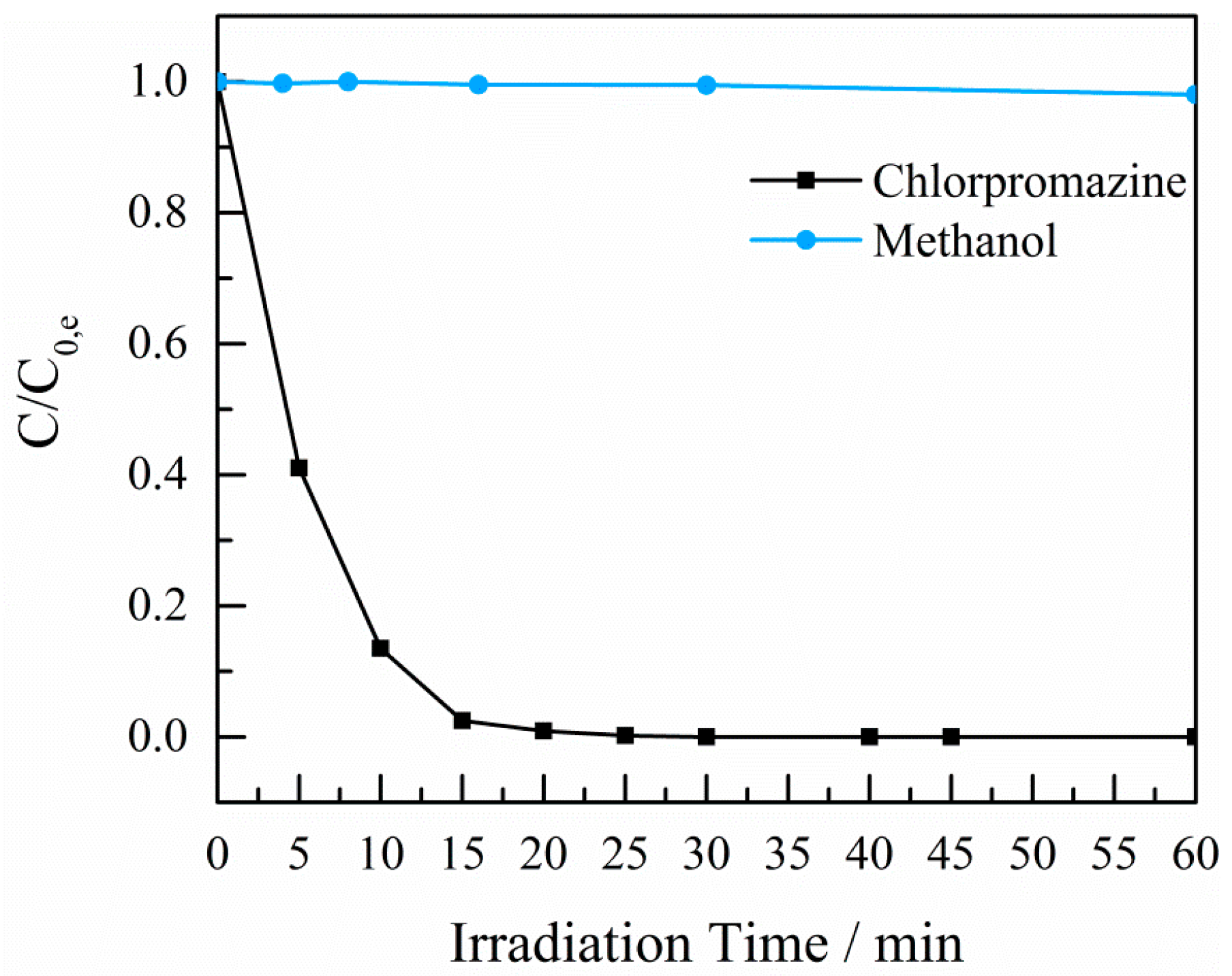
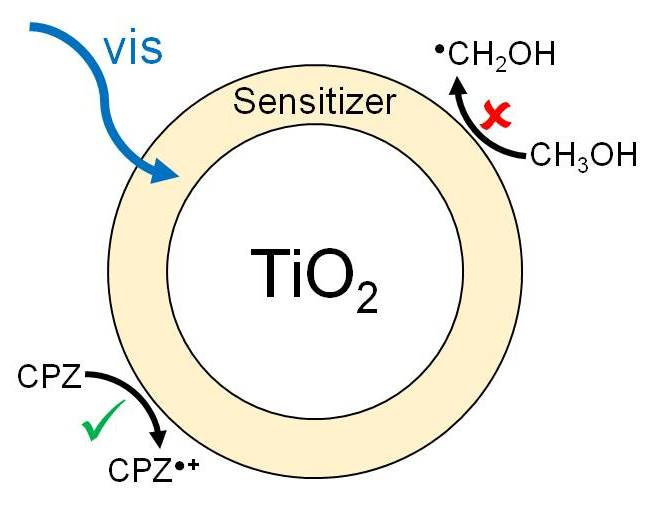
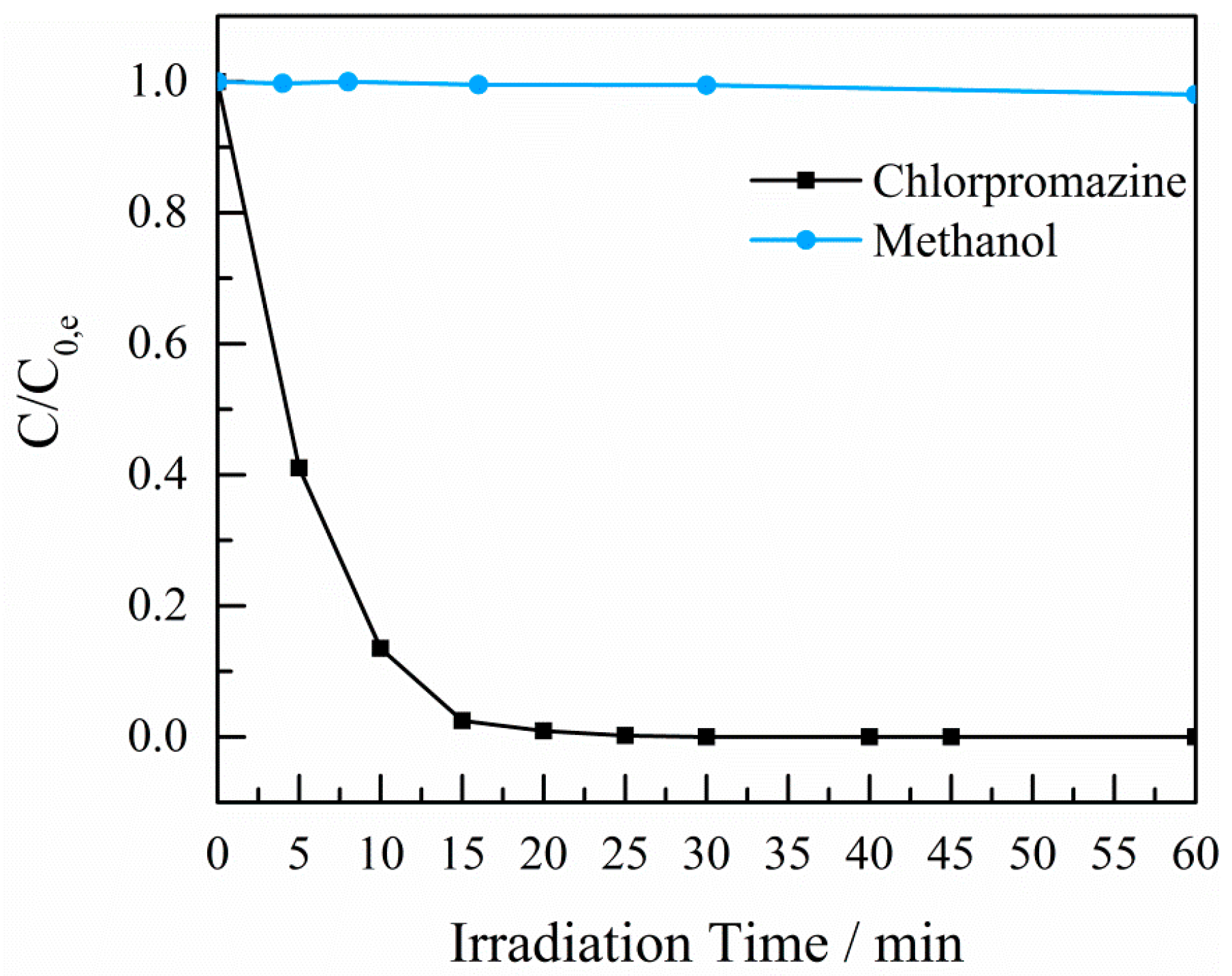
2.4. Photocatalytic Experiments
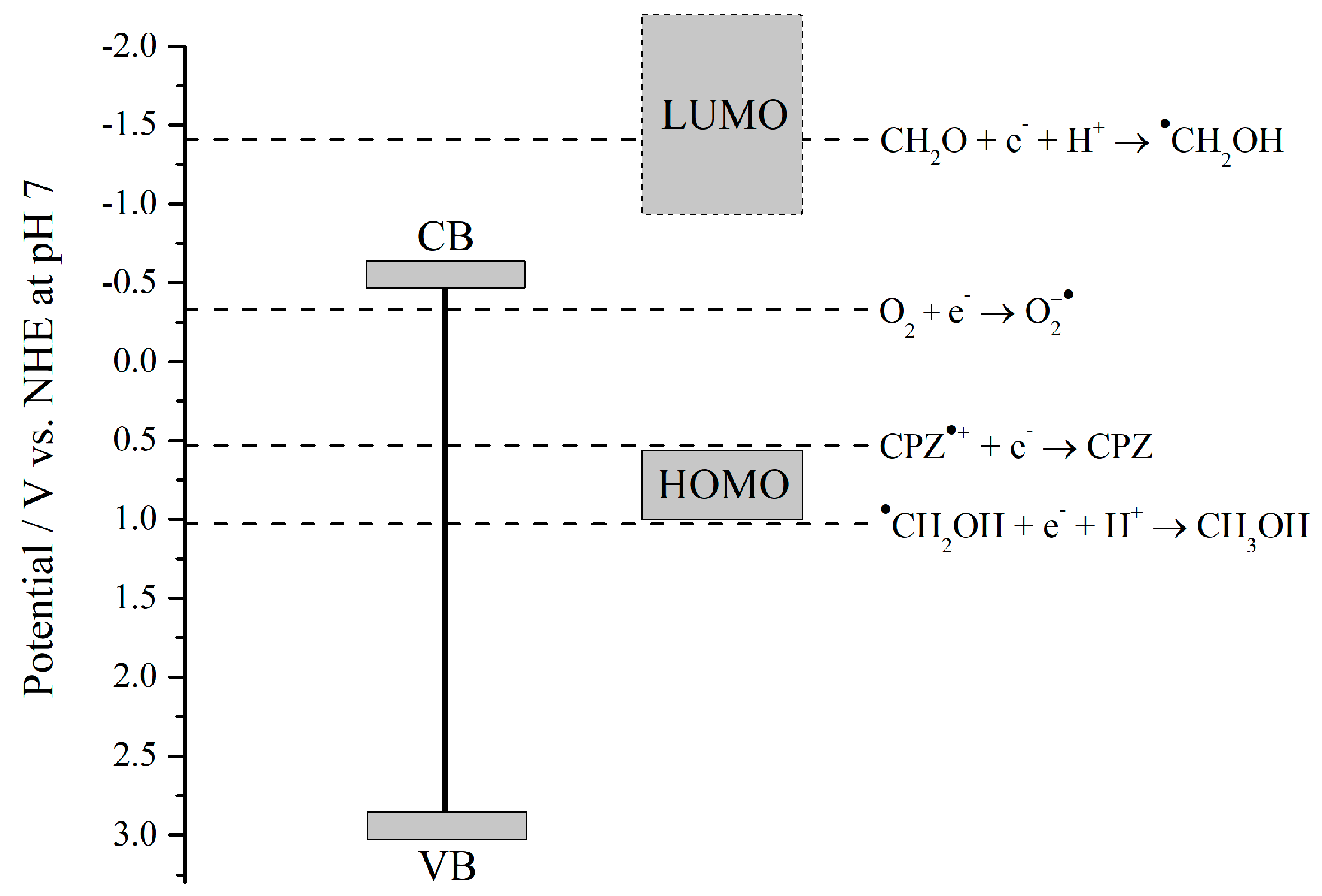
3. Discussion




4. Materials and Methods
4.1. Chemicals
4.2. Characterization of K-7000
4.3. Transient Absorption Spectroscopy
4.4. Photocatalytic Procedure
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D.W. Understanding TiO2 Photocatalysis: Mechanisms and Materials. Chem. Rev. 2014, 114, 9919–9986. [Google Scholar] [CrossRef] [PubMed]
- Pelaez, M.; Nolan, N.T.; Pillai, S.C.; Seery, M.K.; Falaras, P.; Kontos, A.G.; Dunlop, P.S.M.; Hamilton, J.W.J.; Byrne, J.A.; O’Shea, K.; et al. A review on the visible light active titanium dioxide photocatalysts for environmental applications. Appl. Catal. B Environ. 2012, 125, 331–349. [Google Scholar] [CrossRef] [Green Version]
- Günnemann, C.; Haisch, C.; Fleisch, M.; Schneider, J.; Emeline, A.V.; Bahnemann, D.W. Insights into Different Photocatalytic Oxidation Activities of Anatase, Brookite, and Rutile Single-Crystal Facets. ACS Catal. 2019, 9, 1001–1012. [Google Scholar] [CrossRef]
- Tayade, R.J.; Surolia, P.K.; Kulkarni, R.G.; Jasra, R.V. Photocatalytic degradation of dyes and organic contaminants in water using nanocrystalline anatase and rutile TiO2. Sci. Technol. Adv. Mater. 2007, 8, 455–462. [Google Scholar] [CrossRef]
- Grätzel, M.; Rotzinger, F.P. The Influence of the Crystal Lattice Structure on the Conduction Band Energy of Oxides of Titanium(IV). Chem. Phys. Lett. 1985, 118, 474–477. [Google Scholar] [CrossRef]
- Reference Solar Spectral Irradiance: ASTM G-173. 2012.
- Arimi, A.; Megatif, L.; Granone, L.I.L.I.; Dillert, R.; Bahnemann, D.W.D.W. Visible-light photocatalytic activity of zinc ferrites. J. Photochem. Photobiol. A Chem. 2018, 366, 118–126. [Google Scholar] [CrossRef]
- Curti, M.; Kirsch, A.; Granone, L.I.; Tarasi, F.; López-Robledo, G.; Bahnemann, D.W.; Murshed, M.M.; Gesing, T.M.; Mendive, C.B. Visible-Light Photocatalysis with Mullite-Type Bi2(Al1– xFex)4O9: Striking the Balance between Bandgap Narrowing and Conduction Band Lowering. ACS Catal. 2018, 8, 8844–8855. [Google Scholar] [CrossRef]
- Xu, A.; Gao, Y.; Liu, H. The Preparation, Characterization, and their Photocatalytic Activities of Rare-Earth-Doped TiO2 Nanoparticles. J. Catal. 2002, 207, 151–157. [Google Scholar] [CrossRef]
- Klosek, S.; Raftery, D. Visible Light Driven V-Doped TiO2 Photocatalyst and Its Photooxidation of Ethanol. J. Phys. Chem. B 2001, 105, 2815–2819. [Google Scholar] [CrossRef]
- O’Regan, B.; Grätzel, M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 1991, 353, 737–740. [Google Scholar] [CrossRef]
- Cho, Y.; Choi, W.; Lee, C.; Hyeon, T.; Lee, H. Visible Light-Induced Degradation of Carbon Tetrachloride on Dye-Sensitized TiO2. Environ. Sci. Technol. 2001, 35, 966–970. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, P.; Moreira, J.; Gomaa, H.; Ray, A.K. Visible-Solar-Light-Driven Photocatalytic Degradation of Phenol with Dye-Sensitized TiO2: Parametric and Kinetic Study. Ind. Eng. Chem. Res. 2012, 51, 4523–4532. [Google Scholar] [CrossRef]
- Za<monospace>̧</monospace>bek, P.; Eberl, J.; Kisch, H. On the origin of visible light activity in carbon-modified titania. Photochem. Photobiol. Sci. 2009, 8, 264–269. [Google Scholar]
- Sankova, N.; Semeykina, V.; Selishchev, D.; Glazneva, T.; Parkhomchuk, E.; Larichev, Y.; Uvarov, N. Influence of Tetraalkylammonium Compounds on Photocatalytic and Physical Properties of TiO2. Catal. Lett. 2018, 148, 2391–2407. [Google Scholar] [CrossRef]
- Tobaldi, D.M.; Seabra, M.P.; Otero-Irurueta, G.; de Miguel, Y.R.; Ball, R.J.; Singh, M.K.; Pullar, R.C.; Labrincha, J.A. Quantitative XRD characterisation and gas-phase photocatalytic activity testing for visible-light (indoor applications) of KRONOClean 7000®. RSC Adv. 2015, 5, 102911–102918. [Google Scholar] [CrossRef]
- Ilan, Y.A.; Czapski, G.; Meisel, D. The one-electron transfer redox potentials of free radical I. The oxygen/superoxide system. Biochmica er Biophys. Acta 1976, 430, 209–224. [Google Scholar] [CrossRef]
- Yamakata, A.; Ishibashi, T.; Onishi, H. Water- and Oxygen-Induced Decay Kinetics of Photogenerated Electrons in TiO2 and Pt/TiO2: A Time-Resolved Infrared Absorption Study. J. Phys. Chem. B 2001, 105, 7258–7262. [Google Scholar] [CrossRef]
- Schneider, J.; Bahnemann, D.W. Undesired Role of Sacrificial Reagents in Photocatalysis. J. Phys. Chem. Lett. 2013, 4, 3479–3483. [Google Scholar] [CrossRef]
- Memming, R. Photoinduced Charge Transfer Processes at Semiconductor Electrodes and Particles. In Electron Transfer I. Topics in Current Chemistry; Mattay, J., Ed.; Springer: Berlin/Heidelberg, Germany, 1994; Volume 169, pp. 105–181. [Google Scholar]
- Wang, C.; Pagel, R.; Bahnemann, D.W.; Dohrmann, J.K. Quantum Yield of Formaldehyde Formation in the Presence of Colloidal TiO2-Based Photocatalysts: Effect of Intermittent Illumination, Platinization, and Deoxygenation. J. Phys. Chem. B 2004, 108, 14082–14092. [Google Scholar] [CrossRef]
- Iorio, Y.D.; Aguirre, M.E.; Brusa, M.A.; Grela, M.A. Surface Chemistry Determines Electron Storage Capabilities in Alcoholic Sols of Titanium Dioxide Nanoparticles. A Combined FTIR and Room Temperature EPR Investigation. J. Phys. Chem. C 2012, 116, 9646–9652. [Google Scholar] [CrossRef]
- Bahnemann, D.; Henglein, A.; Lilie, J.; Spanhel, L. Flash photolysis observation of the absorption spectra of trapped positive holes and electrons in colloidal titanium dioxide. J. Phys. Chem. 1984, 88, 709–711. [Google Scholar] [CrossRef]
- Morikawa, T.; Asahi, R.; Ohwaki, T.; Aoki, K.; Taga, Y. Band-Gap Narrowing of Titanium Dioxide by Nitrogen Doping. Jpn. J. Appl. Phys. 2001, 40, L561–L563. [Google Scholar] [CrossRef]
- Umebayashi, T.; Yamaki, T.; Itoh, H.; Asai, K. Band gap narrowing of titanium dioxide by sulfur doping. Appl. Phys. Lett. 2003, 81, 2–5. [Google Scholar] [CrossRef]
- Qin, P.; Yang, X.; Chen, R.; Sun, L.; Marinado, T.; Edvinsson, T.; Boschloo, G.; Hagfeldt, A. Influence of π-Conjugation Units in Organic Dyes for Dye-Sensitized Solar Cells. J. Phys. Chem. C 2007, 111, 1853–1860. [Google Scholar] [CrossRef]
- Youngblood, W.J.; Lee, S.-H.A.; Kobayashi, Y.; Hernandez-Pagan, E.A.; Hoertz, P.G.; Moore, T.A.; Moore, A.L.; Gust, D.; Mallouk, T.E. Photoassisted Overall Water Splitting in a Visible Light-Absorbing Dye-Sensitized Photoelectrochemical Cell. J. Am. Chem. Soc. 2009, 131, 926–927. [Google Scholar] [CrossRef]
- Ahmed, A.Y.; Kandiel, T.A.; Ivanova, I.; Bahnemann, D. Photocatalytic and photoelectrochemical oxidation mechanisms of methanol on TiO2 in aqueous solution. Appl. Surf. Sci. 2014, 319, 44–49. [Google Scholar] [CrossRef]
- Somasundaram, N.; Srinivasan, C. Oxidation of aryl methyl sulfides and sulfoxides on irradiated TiO2. J. Photochem. Photobiol. A Chem. 1998, 115, 169–173. [Google Scholar] [CrossRef]
- Merkle, F.H.; Discher, C.A. Electrochemical Oxidation of Chlorpromazine Hydrochloride. J. Pharm. Sci. 1963, 53, 620–623. [Google Scholar] [CrossRef]
- Bahnemann, D.; Asmus, K.-D.; Willson, R.L. Phenothiazine Radical-cations: Electron Transfer Equilibria with Iodide Ions and the Determination of One-electron Redox Potentials by Pulse Radiolysis. J. Chem. Soc. Perkin Trans. II 1983, 1669–1673. [Google Scholar] [CrossRef]
- Feldt, S.M.; Lohse, P.W.; Kessler, F.; Nazeeruddin, M.K.; Grätzel, M.; Boschloo, G.; Hagfeldt, A. Regeneration and recombination kinetics in cobalt polypyridine based dye-sensitized solar cells, explained using Marcus theory. Phys. Chem. Chem. Phys. 2013, 15, 7087–7097. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.A. Chemical and Electrochemical Electron-Transfer Theory. Annu. Rev. Phys. Chem. 1964, 15, 155–196. [Google Scholar] [CrossRef]
- Maruthamuthu, P.; Sharma, D.K.; Serpone, N. Subnanosecond relaxation dynamics of 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonate) and chlorpromazine. Assessment of photosensitization of a wide band gap metal oxide semiconductor TiO2. J. Phys. Chem. 1995, 99, 3636–3642. [Google Scholar] [CrossRef]
- Arimi, A.; Dillert, R.; Dräger, G.; Bahnemann, D.W. Light-Induced Reactions of Chlorpromazine in the Presence of a Heterogeneous Photocatalyst: Formation of a Long-Lasting Sulfoxide. Catalysts 2019, 9, 627. [Google Scholar] [CrossRef]
- Lang, X.; Chen, X.; Zhao, J. Heterogeneous visible light photocatalysis for selective organic transformations. Chem. Soc. Rev. 2014, 43, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Chen, P.; Comte, P.; Nazeeruddin, M.K.; Liska, P.; Péchy, P.; Grätzel, M. Fabrication of screen-printing pastes from TiO2 powders for dye-sensitised solar cells. Prog. Photovoltaics Res. Appl. 2007, 15, 603–612. [Google Scholar] [CrossRef]
- Nash, T. The colorimetric estimation of formaldehyde by means of the Hantzsch reaction. Biochem. J. 1953, 55, 416–421. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arimi, A.; Günnemann, C.; Curti, M.; Bahnemann, D.W. Regarding the Nature of Charge Carriers Formed by UV or Visible Light Excitation of Carbon-Modified Titanium Dioxide. Catalysts 2019, 9, 697. https://doi.org/10.3390/catal9080697
Arimi A, Günnemann C, Curti M, Bahnemann DW. Regarding the Nature of Charge Carriers Formed by UV or Visible Light Excitation of Carbon-Modified Titanium Dioxide. Catalysts. 2019; 9(8):697. https://doi.org/10.3390/catal9080697
Chicago/Turabian StyleArimi, Arsou, Carsten Günnemann, Mariano Curti, and Detlef W. Bahnemann. 2019. "Regarding the Nature of Charge Carriers Formed by UV or Visible Light Excitation of Carbon-Modified Titanium Dioxide" Catalysts 9, no. 8: 697. https://doi.org/10.3390/catal9080697
APA StyleArimi, A., Günnemann, C., Curti, M., & Bahnemann, D. W. (2019). Regarding the Nature of Charge Carriers Formed by UV or Visible Light Excitation of Carbon-Modified Titanium Dioxide. Catalysts, 9(8), 697. https://doi.org/10.3390/catal9080697