Structural, Spectroscopic, and Chemical Bonding Analysis of Zn(II) Complex [Zn(sal)](H2O): Combined Experimental and Theoretical (NBO, QTAIM, and ELF) Investigation

 ,
,
Abstract
:1. Introduction
2. Experimental
2.1. General Considerations
2.2. Synthesis
2.3. X-ray Diffraction Studies
2.4. Computational Details
3. Result and Discussion
3.1. Synthesis and Spectroscopic Characterization
3.2. X-ray Structure Investigation
3.3. Zn–Ligand Bonding and Zn···Zn Interaction
3.3.1. Reproducing the Structures
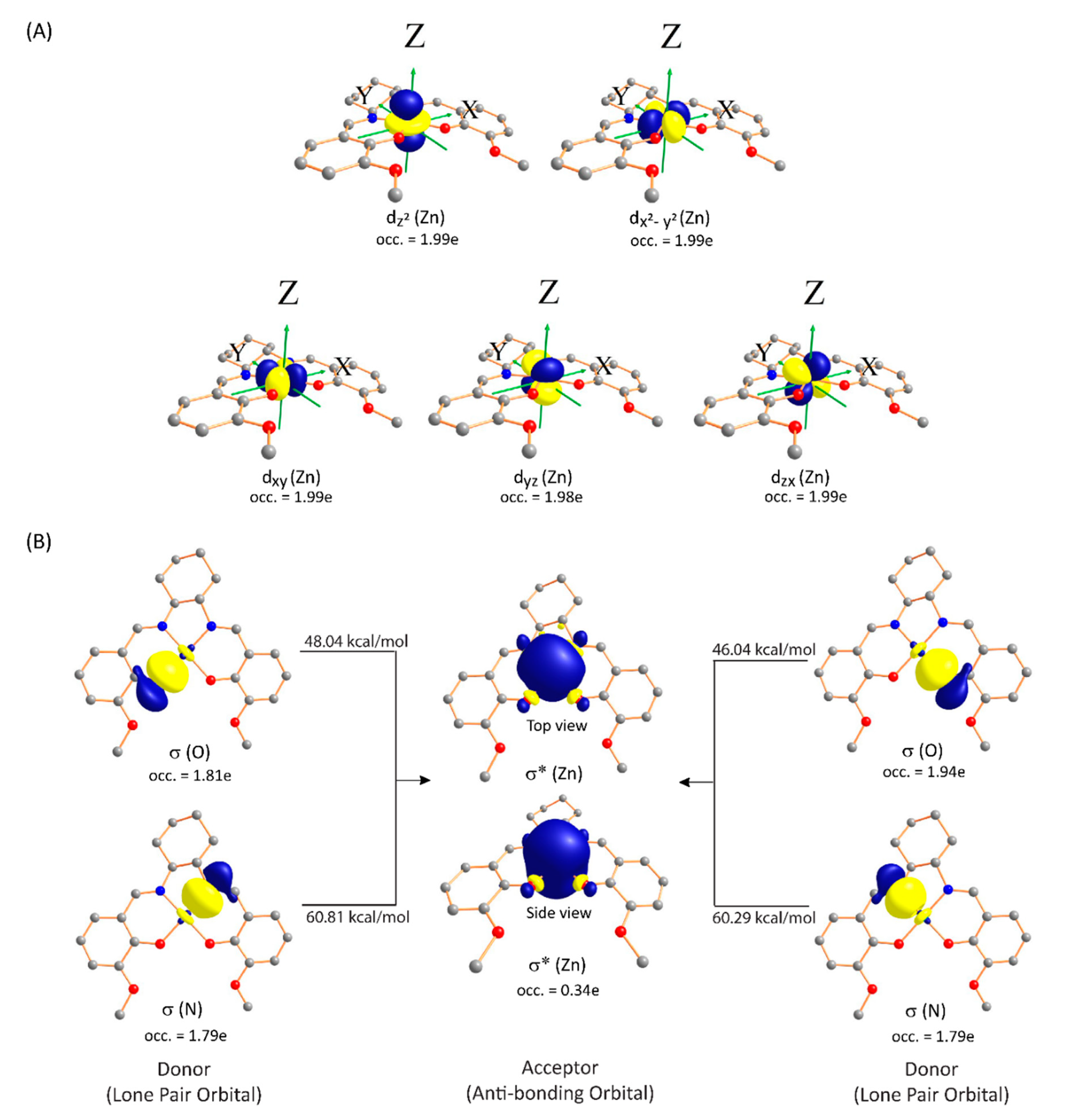
3.3.2. Natural Bond Orbital (NBO) Analysis
Natural Population Analysis
Second Order Perturbation Theory Analysis of Fock Matrix in NBO Basis
3.3.3. Quantum Theory of Atoms in Molecules (QTAIM) Analysis
3.3.4. Electron Localization Function (ELF) Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Werner, A. About the spatial change of position when converting space isomeric compounds. Ber. Dtsch. Chem. Ges. 1911, 44, 873–882. [Google Scholar] [CrossRef] [Green Version]
- Kauffman, G.B. Foundation of nitrogen stereochemistry: Alfred Werner’s inaugural dissertation. J. Chem. Educ. 1966, 43, 155. [Google Scholar] [CrossRef]
- Constable, E.C.; Housecroft, C.E. Coordination chemistry: The scientific legacy of Alfred Werner. Chem. Soc. Rev. 2013, 42, 1429–1439. [Google Scholar] [CrossRef] [PubMed]
- Werner, H. Alfred Werner: A Forerunner to Modern Inorganic Chemistry. Angew. Chem. Int. Ed. 2013, 52, 6146–6153. [Google Scholar] [CrossRef]
- Cotton, F.A.; Curtis, N.F.; Harris, C.B.; Johnson, B.F.G.; Lippard, S.J.; Mague, J.T.; Robinson, W.R.; Wood, J.S. Mononuclear and polynuclear chemistry of rhenium (III): Its pronounced homophilicity. Science 1964, 145, 1305–1307. [Google Scholar] [CrossRef]
- Dedieu, A.; Hoffmann, R. Platinum (0)-platinum (0) dimers. Bonding relationships in a d10-d10 system. J. Am. Chem. Soc. 1978, 100, 2074–2079. [Google Scholar] [CrossRef]
- Jiang, Y.; Alvarez, S.; Hoffmann, R. Binuclear and polymeric gold (I) complexes. Inorg. Chem. 1985, 24, 749–757. [Google Scholar] [CrossRef]
- Merz, K.M.; Hoffmann, R. d10-d10 Interactions: Multinuclear copper (I) complexes. Inorg. Chem. 1988, 27, 2120–2127. [Google Scholar] [CrossRef]
- Pyykkö, P.; Li, J.; Runeberg, N. Predicted ligand dependence of the Au(I)···Au(I) attraction in (XAuPH3)2. Chem. Phys. Lett. 1994, 218, 133–138. [Google Scholar] [CrossRef]
- Pyykkö, P.; Mendizabal, F. Theory of the d10–d10 Closed-Shell Attraction: 2. Long-Distance Behaviour and Nonadditive Effects in Dimers and Trimers of Type [(x-Au-L)n](n = 2, 3; X = Cl, I, H; L = PH3, PMe3,-N≡ CH). Chem. Eur. J. 1997, 3, 1458–1465. [Google Scholar]
- Pyykkö, P. Strong closed-shell interactions in inorganic chemistry. Chem. Rev. 1997, 97, 597–636. [Google Scholar] [CrossRef] [PubMed]
- Pyykko, P. Theoretical chemistry of gold. Angew. Chem., Int. Ed. 2004, 43, 4412–4456. [Google Scholar] [CrossRef] [PubMed]
- Pyykko, P. Theoretical chemistry of gold. II. Inorg. Chim. Acta 2005, 358, 4113–4130. [Google Scholar] [CrossRef]
- Cotton, F.A.; Murillo, C.A.; Walton, R.A. (Eds.) Multiple Bonds between Metal Atoms, 3rd ed.; Springer: Berlin, Germany, 2005. [Google Scholar]
- Liddle, S.T. (Ed.) Molecular Metal-metal Bonds: Compounds, Synthesis, Properties; Wiley-VCH: Weinheim, Germany, 2015. [Google Scholar]
- Sculfort, S.; Braunstein, P. Intramolecular d10–d10 interactions in heterometallic clusters of the transition metals. Chem. Soc. Rev. 2011, 40, 2741–2760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.S.; Gabbai, F.P. Coordination-and redox-noninnocent behavior of ambiphilic ligands containing antimony. Acc. Chem. Res. 2016, 49, 857–867. [Google Scholar] [CrossRef]
- Tsipis, A.C. DFT challenge of intermetallic interactions: From metallophilicity and metallaromaticity to sextuple bonding. Coord. Chem. Rev. 2017, 345, 229–262. [Google Scholar] [CrossRef]
- Doyle, M.P.; Duffy, R.; Ratnikov, M.; Zhou, L. Catalytic carbene insertion into C− H bonds. Chem. Rev. 2010, 110, 704–724. [Google Scholar] [CrossRef]
- Georgiev, V.P.; Mohan, P.J.; DeBrincat, D.; McGrady, J.E. Low-symmetry distortions in Extended Metal Atom Chains (EMACs): Origins and consequences for electron transport. Coord. Chem. Rev. 2013, 257, 290–298. [Google Scholar] [CrossRef]
- Kampa, M.; Pandelia, M.E.; Lubitz, W.; van Gastel, M.; Neese, F. A metal–metal bond in the light-induced state of [NiFe] hydrogenases with relevance to hydrogen evolution. J. Am. Chem. Soc. 2013, 135, 3915–3925. [Google Scholar] [CrossRef]
- Xia, B.-H.; Zhang, H.-X.; Che, C.-M.; Leung, K.-H.; Phillips, D.L.; Zhu, N.; Shou, Z.-Y. Metal− Metal Interactions in Heterobimetallic d8−d10 Complexes. Structures and Spectroscopic Investigation of [M ‘M “(μ-dcpm)2 (CN)2]+(M‘ = Pt, Pd; M“ = Cu, Ag, Au) and Related Complexes by UV− vis Absorption and Resonance Raman Spectroscopy and ab Initio Calculations. J. Am. Chem. Soc. 2003, 125, 10362–10374. [Google Scholar]
- Lopez-de-Luzuriaga, J.M.; Monge, M.; Olmos, M.E.; Pascual, D. Experimental and Theoretical Comparison of the Metallophilicity between d10–d10 AuI–HgII and d8–d10 AuIII–HgII Interactions. Inorg. Chem. 2014, 53, 1275–1277. [Google Scholar] [CrossRef] [PubMed]
- Freitag, K.; Molon, M.; Jerabek, P.; Dilchert, K.; Rosler, C.; Seidel, R.W.; Gemel, C.; Frenking, G.; Fischer, R.A. Zn⋯ Zn interactions at nickel and palladium centers. Chem. Sci. 2016, 7, 6413–6421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, B.; Thakur, A.; Kumar, M.; Jasrotia, D. Metallophilic interactions and structure-stability relationship with secondary interactions in [ZnX]− based hybrid derivatives. Mater. Chem. Phys. 2017, 196, 52–61. [Google Scholar] [CrossRef]
- Katsuki, T. Unique asymmetric catalysis of cis-β metal complexes of salen and its related Schiff-base ligands. Chem. Soc. Rev. 2004, 33, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Baleizo, C.; Garcia, H. Chiral salen complexes: An overview to recoverable and reusable homogeneous and heterogeneous catalysts. Chem. Rev. 2006, 106, 3987–4043. [Google Scholar] [CrossRef]
- Reed, J.E.; Arnal, A.A.; Neidle, S.; Vilar, R. Stabilization of G-quadruplex DNA and inhibition of telomerase activity by square-planar nickel (II) complexes. J. Am. Chem. Soc. 2006, 128, 5992–5993. [Google Scholar] [CrossRef]
- Glasner, H.; Tshuva, E.Y. A marked synergistic effect in antitumor activity of salan titanium (IV) complexes bearing two differently substituted aromatic rings. J. Am. Chem. Soc. 2011, 133, 16812–16814. [Google Scholar] [CrossRef]
- Hitomi, Y.; Iwamoto, Y.; Kashida, A.; Kodera, M. Mononuclear nonheme iron (III) complexes that show superoxide dismutase-like activity and antioxidant effects against menadione-mediated oxidative stress. Chem. Commun. 2015, 51, 8702–8704. [Google Scholar] [CrossRef]
- Pessoa, J.C.; Correia, I. Salan vs. salen metal complexes in catalysis and medicinal applications: Virtues and pitfalls. Coord. Chem. Rev. 2019, 388, 227–247. [Google Scholar] [CrossRef]
- Hui, J.K.H.; Yu, Z.; MacLachlan, M.J. Supramolecular Assembly of Zinc Salphen Complexes: Access to Metal-Containing Gels and Nanofibers. Angew. Chem. Int. Ed. 2007, 46, 7980–7983. [Google Scholar] [CrossRef]
- Darensbourg, D.J. Making plastics from carbon dioxide: Salen metal complexes as catalysts for the production of polycarbonates from epoxides and CO2. Chem. Rev. 2007, 107, 2388–2410. [Google Scholar] [CrossRef] [PubMed]
- Kleij, A.W. Zinc-centred salen complexes: Versatile and accessible supramolecular building motifs. Dalton Trans. 2009, 24, 4635–4639. [Google Scholar] [CrossRef] [PubMed]
- Crane, A.K.; MacLachlan, M.J. Portraits of Porosity: Porous Structures Based on Metal Salen Complexes. Eur. J. Inorg. Chem. 2012, 17–30. [Google Scholar] [CrossRef]
- Wang, Y.D.; DuBois, J.L.; Hedman, B.; Hodgson, K.O.; Stack, T.D.P. Catalytic galactose oxidase models: Biomimetic Cu(II)-phenoxyl-radical reactivity. Science 1998, 279, 537–540. [Google Scholar] [CrossRef]
- Ueno, T.; Koshiyama, T.; Ohashi, M.; Kondo, K.; Kono, M.; Suzuki, A.; Yamane, T.; Watanabe, Y. Coordinated design of cofactor and active site structures in development of new protein catalysts. J. Am. Chem. Soc. 2005, 127, 6556–6562. [Google Scholar] [CrossRef]
- Laskin, J.; Yang, Z.; Chu, I.K. Energetics and dynamics of electron transfer and proton transfer in dissociation of metalIII (salen)− peptide complexes in the gas phase. J. Am. Chem. Soc. 2008, 130, 3218–3230. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Valency and Bonding: A Natural Bond Orbital Donor-acceptor Perspective; Cambridge UK Press: Cambridge, UK, 2005. [Google Scholar]
- Pauling, L. The Application of the Quantum Mechanics to the Structure of the Hydrogen Molecule and Hydrogen Molecule-Ion and to Related Problems. Chem. Rev. 1928, 28, 173–213. [Google Scholar] [CrossRef]
- Pauling, L. The nature of the chemical bond. Application of results obtained from the quantum mechanics and from a theory of paramagnetic susceptibility to the structure of molecules. J. Am. Chem. Soc. 1931, 53, 1367–1400. [Google Scholar] [CrossRef]
- Pauling, L.; Wheland, G.W. The nature of the chemical bond. V. The quantum-mechanical calculation of the resonance energy of benzene and naphthalene and the hydrocarbon free radicals. J. Chem. Phys. 1933, 1, 362–374. [Google Scholar] [CrossRef] [Green Version]
- Wheland, G.W.; Pauling, L. A quantum mechanical discussion of orientation of substituents in aromatic molecules. J. Am. Chem. Soc. 1935, 57, 2086–2095. [Google Scholar] [CrossRef]
- Heitler, W.; London, F. Interaction of neutral atoms and homeopolar bonding according to quantum mechanics. Z. Phys. 1927, 44, 455–472. [Google Scholar] [CrossRef]
- Hund, F. To interpret some phenomena in the molecular spectra. Z. Phys. 1926, 36, 657–674. [Google Scholar] [CrossRef]
- Mulliken, R.S. The assignment of quantum numbers for electrons in molecules. I. Phys. Rev. 1927, 32, 186. [Google Scholar] [CrossRef]
- Lennard-Jones, J.E. The electronic structure of some diatomic molecules. Trans. Faraday Soc. 1929, 25, 668–686. [Google Scholar] [CrossRef]
- Bohr, N. On the constitution of atoms and molecules. Philos. Mag. 1913, 26, 476–502. [Google Scholar] [CrossRef] [Green Version]
- Bohr, N. Theory of Spectra and Atomic Constitution, Essay 3; Cambridge U. Press: London, UK, 1922. [Google Scholar]
- Lewis, G.N. The atom and the molecule. J. Am. Chem. Soc. 1916, 38, 762–785. [Google Scholar] [CrossRef] [Green Version]
- Lewis, G.N. Valence and the Structure of Atom and Molecules; Chemical Catalog Co.: New York, NY, USA, 1923. [Google Scholar]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1994. [Google Scholar]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Savin, A.; Silvi, B.; Coionna, F. Topological analysis of the electron localization function applied to delocalized bonds. Can. J. Chem. 1996, 74, 1088–1096. [Google Scholar] [CrossRef]
- Silvi, B. The synaptic order: A key concept to understand multicenter bonding. J. Mol. Struct. (THEOCHEM) 2002, 614, 3–10. [Google Scholar] [CrossRef]
- Matito, E.; Solà, M. The role of electronic delocalization in transition metal complexes from the electron localization function and the quantum theory of atoms in molecules viewpoints. Coord. Chem. Rev. 2009, 253, 647–665. [Google Scholar] [CrossRef]
- Lepetit, C.; Vabre, B.; Canac, Y.; Alikhani, M.E.; Zargarian, D. Pentacoordinated, square pyramidal cationic PCP Ni(II) pincer complexes: ELF and QTAIM topological analyses of nickel–triflate interactions. Theor. Chem. Acc. 2018, 137, 141. [Google Scholar] [CrossRef]
- Corté-Guzmán, F.; Bader, R.F.W. Complementarity of QTAIM and MO theory in the study of bonding in donor–acceptor complexes. Coord. Chem. Rev. 2005, 249, 633–662. [Google Scholar]
- Cromer, D.T.; Waber, J.T. International Tables for X-ray Crystallography, Kynoch. Birm. Engl. 1952, 3, 257–269. [Google Scholar]
- SAINT, Version 6.02, Bruker AXS: Madison, WI, USA, 1999.
- Sheldrick, G.M. SAINT (Version 6.02), SADABS (Version 2.03); Bruker AXS: Madison, WI, USA, 2002. [Google Scholar]
- Sheldrick, G.M. SAINT and XPREP, Version 5.1; Siemens Industrial Automation Inc.: Madison, WI, USA, 1995. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. he anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment–Olex2 dissected. Acta Cryst. A 2015, 71, 59–75. [Google Scholar] [CrossRef] [Green Version]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO Version 3.1; Gaussian, Inc.: Wallingford, CT, USA, 2009; included in Gaussian 09. [Google Scholar]
- Frisch, M.J.T.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.C.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, G.A.N.B.; Petersson, H.; et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Chen, F.-W. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Usman, M.; Tabassum, S.; Arjmand, F.; Khan, R.A.; Ali, M.S.; Al-Lohedan, H.A.; Alsalme, A.; Farah, M.A.; Al-Anazi, K.M.; Ahmad, M. CuII-NaI heteronuclear complex as anticancer entity against human breast cancer cell lines: DNA binding, cleavage, and Computational studies. Inorg. Chim. Acta 2018, 479, 229–239. [Google Scholar] [CrossRef]
- Usman, M.; Arjmand, F.; Ahmad, M.; Khan, M.S.; Ahmad, I.; Tabassum, S. A comparative analyses of bioactive Cu (II) complexes using Hirshfeld surface and density functional theory (DFT) methods: DNA binding studies, cleavage and antibiofilm activities. Inorg. Chim. Acta 2016, 453, 193–201. [Google Scholar] [CrossRef]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds, 4th ed.; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Arif, M.; Nazir, S.; Iqbal, M.S.; Anjum, S. Synthesis and characterization of transition metal fluoride complexes with imidazole: X-ray crystal structure reveals short hydrogen bonds between lattice water and lattice fluoride. Inorg. Chim. Acta 2009, 362, 1624–1628. [Google Scholar] [CrossRef]
- Dumur, F.; Contal, E.; Wantz, G.; Gigmes, D. Photoluminescence of Zinc complexes: Easily tunable optical properties by variation of the bridge between the imido groups of schiff base ligands. Eur. J. Inorg. Chem. 2014, 25, 4186–4198. [Google Scholar] [CrossRef]
- Houjou, H.; Motoyama, T.; Araki, K. Electronic spectra of mono-and dinuclear complexes of fully π-conjugated salphen Ligands synthesized by using 2, 6-Dihydroxynaphthalene carbaldehydes. Eur. J. Inorg. Chem. 2009, 533–538. [Google Scholar] [CrossRef]
- Houjou, H.; Ito, M.; Arak, K. Comparison of the spectroscopic properties of π-conjugated, fused salphen triads embedded with Zn-homo-, Ni-homo-, and Ni/Zn-heteronuclei. Inorg. Chem. 2011, 50, 5298–5306. [Google Scholar] [CrossRef]
- Consiglio, G.; Failla, S.; Finocchiaro, P.; Oliveri, I.P.; Bella, S.D. An unprecedented structural interconversion in solution of aggregate zinc (II) salen Schiff-base complexes. Inorg. Chem. 2012, 51, 8409–8418. [Google Scholar] [CrossRef]
- Rogachev, A.Y.; Hoffmann, R. Iodine (I2) as a Janus-faced ligand in organometallics. J. Am. Chem. Soc. 2013, 135, 3262–3275. [Google Scholar] [CrossRef]
- Gericke, R.; Bennett, M.A.; Priver, S.H.; Bhargava, S.K. Formation of Heterobimetallic Complexes by Addition of d10-Metal Ions to cis-[(dppe) M (κ C-2-C6F4PPh2) 2](M = Ni, Pd, and Pt). Organometallics 2017, 36, 3178–3188. [Google Scholar] [CrossRef]
- Paenurk, E.; Poranne, R.G.; Chen, P. Trends in metallophilic bonding in Pd–Zn and Pd–Cu complexes. Organometallics 2017, 36, 4854–4863. [Google Scholar] [CrossRef]
- Kim, M.; Taylor, T.J.; Gabbai, F.P. Hg (II)··· Pd (II) metallophilic interactions. J. Am. Chem. Soc. 2008, 130, 6332–6333. [Google Scholar] [CrossRef] [PubMed]
- Runeberg, N.; Schutz, M.; Werner, H.-J. The aurophilic attraction as interpreted by local correlation methods. J. Chem. Phys. 1999, 110, 7210–7215. [Google Scholar] [CrossRef]
- Mendizabal, F.; Pyykko, P.; Runeberg, N. Aurophilic attraction: The additivity and the combination with hydrogen bonds. Chem. Phys. Lett. 2003, 370, 733–740. [Google Scholar] [CrossRef]
- Grimme, S.; Djukic, J.-P. Cation− cation “attraction”: When London dispersion attraction wins over Coulomb repulsion. Inorg. Chem. 2011, 50, 2619–2628. [Google Scholar] [CrossRef] [PubMed]
- Murray, L.J.; Weare, W.W.; Shearer, J.; Mitchell, A.D.; Abboud, K.A. Isolation of a (dinitrogen) tricopper (I) complex. J. Am. Chem. Soc. 2014, 136, 13502–13505. [Google Scholar] [CrossRef]
- -Kanssen, I.F.; Austin, J.; Kerridge, A. Topological study of bonding in aquo and bis (triazinyl) pyridine complexes of trivalent lanthanides and actinides: Does covalency imply stability? Inorg. Chem. 2016, 55, 10034–10042. [Google Scholar] [CrossRef]
- Teixeira, F.; Mosquera, R.; Melo, A.; Freire, C.; Cordeiro, M.N.D.S. Roots of Acetate-Vanadium Linkage Isomerism: A QTAIM Study. Inorg. Chem. 2016, 55, 3653–3662. [Google Scholar] [CrossRef]
- Wu, L.C.; Thomsen, M.K.; Madsen, S.R.; Schmoekel, M.; Jørgensen, M.R.V.; Cheng, M.C.; Peng, S.M.; Chen, Y.S.; Overgaard, J.; Iversen, B.B. Chemical Bonding in a Linear Chromium Metal String Complex. Inorg. Chem. 2014, 53, 12489–12498. [Google Scholar] [CrossRef]
- Duan, M.; Li, P.; Zhao, H.; Xie, F.; Ma, J. Organic Compounds of Actinyls: Systematic Computational Assessment of Structural and Topological Properties in [AnO2 (C2O4) n](2 n− 2)–(An = U, Np, Pu, Am; n = 1–3) Complexes. Inorg. Chem. 2019, 58, 3425–3434. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Matta, C.F. Bonding to titanium. Inorg. Chem. 2001, 40, 5603–5611. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Essen, H. The characterization of atomic interactions. J. Chem. Phys. 1984, 80, 1943–1960. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. Chemical bonds without bonding electron density—does the difference electron-density analysis suffice for a description of the chemical bond? Angew. Chem. Int. Ed. Engl. 1984, 23, 627–628. [Google Scholar] [CrossRef]
- Bianchi, R.; Gervasio, G.; Marabello, D. Experimental electron density analysis of Mn2 (CO) 10: Metal− metal and metal− Ligand bond characterization. Inorg Chem. 2000, 39, 2360–2366. [Google Scholar] [CrossRef] [PubMed]
- Macchi, P.; Proserpio, D.M.; Sironi, A. Experimental electron density in a transition metal dimer: Metal− metal and metal− ligand bonds. J. Am. Chem. Soc. 1998, 120, 13429–13435. [Google Scholar] [CrossRef]
- Hosseinnejad, T.; Dehghanpour, S.; Hosseinjani, A. Quantum chemical investigation on the structural and electronic properties of zinc–salphen complex: DFT and QTAIM analysis. Comput. Theor. Chem. 2013, 1004, 31–37. [Google Scholar] [CrossRef]
- Pitchumani, C.; Mary, V.; Shankar, R.; Vijayakumar, S. Theoretical insights into the metal chelating and antimicrobial properties of the chalcone based Schiff bases. Mol. Simul. 2019, 45, 636–645. [Google Scholar] [CrossRef]
- Gusev, A.N.; Kiskin, M.A.; Braga, E.V.; Chapran, M.; Wiosna-Salyga, G.; Baryshnikov, G.V.; Minaeva, V.A.; Minaev, B.F.; Ivaniuk, K.; Stakhira, P.; et al. Novel Zinc Complex with an Ethylenediamine Schiff Base for High-Luminance Blue Fluorescent OLED Applications. J. Phys. Chem. C 2019, 123, 11850–11859. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. DFT-B3LYP, NPA-, and QTAIM-based study of the physical properties of [M(II)(H2O)2 (15-crown-5)](M = Mn, Fe, Co, Ni, Cu, Zn) complexes. J. Phys. Chem. A 2011, 115, 5592–5601. [Google Scholar] [CrossRef]
- Li, X.; Huo, S.; Zeng, Y.; Sun, Z.; Zheng, S.; Meng, L. Metal–Metal and Metal–Ligand Bonds in (η5-C5H5) 2M2 (M = Be, Mg, Ca, Ni, Cu, Zn). Organometallics 2013, 32, 1060–1066. [Google Scholar] [CrossRef]
- Savin, A.; Nesper, R.; Wengert, S.; Fassler, T.F. ELF: The electron localization function. Angew. Chem. Int. Ed. Engl. 1997, 36, 1808–1832. [Google Scholar] [CrossRef]
- Jerabek, P.; von der Esch, B.; Schmidbaur, H.; Schwerdtfeger, P. Influence of Relativistic Effects on Bonding Modes in M (II) Dinuclear Complexes (M = Au, Ag, and Cu). Inorg. Chem. 2017, 56, 14624–14631. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identification Code | [Zn(sal)](H2O) |
|---|---|
| Empirical formula | C22H26N2O5Zn |
| Formula weight | 463.86 |
| Temperature/K | 100(2) |
| Crystal system | Monoclinic |
| Space group | P21/n |
| a/Å | 8.6332(6) |
| b/Å | 21.0131(15) |
| c/Å | 22.2375(16) |
| α/° | 90 |
| β/° | 97.101(2) |
| γ/° | 90 |
| Volume/Å3 | 4003.2(5) |
| Z | 8 |
| ρcalcg/cm3 | 1.5392 |
| μ/mm−1 | 1.266 |
| F(000) | 1939.3 |
| Crystal size/mm3 | 0.31 × 0.22 × 0.15 |
| Radiation | Mo Kα (λ = 0.71073) |
| 2θ range for data collection/° | 4.88 to 50.1 |
| Index ranges | −11 ≤ h ≤ 11, −27 ≤ k ≤ 27, −29 ≤ l ≤ 29 |
| Reflections collected | 62,209 |
| Independent reflections | 7088 [Rint = 0.1165, Rsigma = 0.0873] |
| Data/restraints/parameters | 7088/0/551 |
| Goodness-of-fit on F2 | 1.084 |
| Final R indexes (I ≥ 2σ (I)) | R1 = 0.0666, wR2 = 0.1509 |
| Final R indexes (all data) | R1 = 0.0959, wR2 = 0.1705 |
| Largest diff. peak/hole/e Å−3 | 1.64/−0.84 |
| CCDC | The CCDC No. 1978769 (see Supplementary) |
| Bonds | Experimental (SCXRD) | Calculated (DFT) | Calculated (DFT) | Angles | Experimental (SCXRD) | Calculated (DFT) | Calculated (DFT) |
|---|---|---|---|---|---|---|---|
| Dimer | Monomer | Dimer | Monomer | ||||
| Zn1–N1 | 1.855(5) | 1.87927 | 2.02935 | N1–Zn1–O2 | 86.1(2) | 91.55826 | 94.59652 |
| Zn1–N2 | 1.856(5) | 1.88835 | 2.02604 | N1–Zn1–O3 | 174.62(19) | 162.62912 | 179.01430 |
| Zn1–O2 | 1.859(4) | 1.86004 | 2.00440 | N2–Zn1–O2 | 174.93(19) | 172.33079 | 179.33971 |
| Zn1–O3 | 1.851(4) | 1.86365 | 2.00188 | N2–Zn1–O3 | 94.69(19) | 88.59658 | 94.64959 |
| Zn2–N3 | 1.858(5) | 1.85823 | - | N1–Zn1–N2 | 86.1(2) | 80.79105 | 85.60376 |
| Zn2–N4 | 1.859(5) | 1.86134 | - | O2–Zn1–O3 | 85.07(16) | 98.95992 | 85.33979 |
| Zn2–O6 | 1.851(4) | 1.86370 | - | N3–Zn2–O6 | 94.73(19) | 91.32315 | - |
| Zn2–O7 | 1.861(3) | 1.87021 | - | N3–Zn2–O7 | 175.77(19) | 161.50932 | - |
| Zn1···Zn2 | 3.539 | 3.54991 | - | N4–Zn2–O6 | 175.70(19) | 162.16173 | - |
| - | - | - | - | N4–Zn2–O7 | 94.46(18) | 91.48183 | - |
| - | - | - | - | N3–Zn2–N4 | 85.7(2) | 83.75503 | - |
| - | - | - | - | O6–Zn2–O7 | 85.42(16) | 98.23381 | - |
| Atom | Charge | TNP | Natural Electron Configuration | Bond | WBI |
|---|---|---|---|---|---|
| Monomer | |||||
| Zn1 | 1.23501 | 28.76499 | Zn1[core]4s0.373d9.944p0.45 | - | - |
| N1 | −0.58352 | 7.58352 | N1[core]2s1.352p4.203S0.013p0.02 | Zn1–N1 | 0.3191 |
| N2 | −0.58084 | 7.58084 | N2[core]2s1.352p4.203S0.013p0.02 | Zn1–N2 | 0.3171 |
| O2 | −0.70141 | 8.70141 | O2[core]2s1.672p5.013p0.01 | Zn1–O2 | 0.3051 |
| O3 | −0.70962 | 8.70962 | O3[core]2s1.682p5.013p0.01 | Zn1–O3 | 0.3024 |
| Dimer | |||||
| Zn1 | 1.36908 | 28.63092 | Zn1[core]4s0.293d9.964p0.37 | Zn1···Zn2 | 0.0221 |
| N1 | −0.64894 | 7.64894 | N1[core]2s1.352p4.283p0.01 | Zn1–N1 | 0.2349 |
| N2 | −0.64039 | 7.64039 | N2[core]2s1.352p4.273p0.01 | Zn1–N2 | 0.2321 |
| O2 | −0.79317 | 8.53347 | O2[core]2s1.672p5.113p0.01 | Zn1–O2 | 0.2288 |
| O3 | −0.78575 | 8.78575 | O3[core]2s1.672p5.113p0.01 | Zn1–O3 | 0.2221 |
| Zn2 | 1.38118 | 28.61882 | Zn2[core]4s0.293d9.964p0.36 | - | - |
| N3 | −0.64202 | 7.64202 | N3[core]2s1.352p4.283p0.01 | Zn2–N3 | 0.2275 |
| N4 | −0.64680 | 7.64680 | N4[core]2s1.352p4.293p0.01 | Zn2–N4 | 0.2269 |
| O6 | −0.79987 | 8.79987 | O6[core]2s1.672p5.123p0.01 | Zn2–O6 | 0.2127 |
| O7 | −0.81127 | 8.81127 | O7[core]2s1.682p5.123p0.01 | Zn2–O7 | 0.2190 |
| Bonds | ρ(r) | ∇2ρ(r) | V(r) | G(r) | H(r) |
|---|---|---|---|---|---|
| Free Ligand | |||||
| O3–C15 | 0.33472 | −0.43827 | −0.95688 | 0.42365 | −0.53322 |
| O1–C28 | 0.32705 | −0.47176 | −0.91170 | 0.39688 | −0.51482 |
| N5–C21 | 0.38331 | −0.96352 | −0.97264 | 0.36588 | −0.60676 |
| N5–C51 | 0.24651 | −0.54978 | −0.32827 | 0.95413 | −0.23285 |
| N6–C43 | 0.24854 | −0.56114 | −0.33363 | 0.96675 | −0.23696 |
| N6–C29 | 0.38390 | −0.96397 | −0.97602 | 0.36751 | −0.60851 |
| C29–C27 | 0.29863 | −0.93736 | −0.39924 | 0.82453 | −0.31679 |
| C27–C15 | 0.31448 | −0.99291 | −0.45569 | 0.10373 | −0.35195 |
| C28–C31 | 0.31422 | −0.99078 | −0.45492 | 0.10361 | −0.35131 |
| C31–C21 | 0.30494 | −0.98150 | −0.41673 | 0.85678 | −0.33105 |
| Monomer | |||||
| Zn1–N1 | 0.12489 | 0.54129 | −0.22762 | 0.18147 | −0.04614 |
| Zn1–N2 | 0.12417 | 0.53805 | −0.22583 | 0.18017 | −0.04566 |
| Zn1–O2 | 0.11318 | 0.60032 | −0.21993 | 0.18500 | −0.03492 |
| Zn1–O3 | 0.11081 | 0.58531 | −0.21328 | 0.17980 | −0.03347 |
| O4–C24 | 0.33012 | −0.64531 | −0.85801 | 0.34834 | −0.50967 |
| O4–C12 | 0.33759 | −0.62583 | −0.90178 | 0.37266 | −0.52912 |
| C24–C22 | 0.32137 | −0.10575 | −0.47080 | 0.10320 | −0.36760 |
| C12–C23 | 0.31374 | −1.1002 | −0.44824 | 0.09876 | −0.34948 |
| C22–C20 | 0.29922 | −0.92077 | −0.41241 | 0.09111 | −0.32130 |
| C23–C18 | 0.29562 | −0.89616 | −0.40226 | 0.08911 | −0.31315 |
| C20–N7 | 0.12417 | 0.53805 | −0.22583 | 0.18017 | −0.04566 |
| C18–N6 | 0.37738 | −0.90728 | −0.98151 | 0.37734 | −0.60416 |
| N7–C46 | 0.25215 | −0.60040 | −0.35784 | 0.10387 | −0.25397 |
| N6–C34 | 0.25276 | −0.60561 | −0.36068 | 0.10463 | −0.25604 |
| C46–C34 | 0.26776 | −0.73000 | −0.31228 | 0.64893 | −0.24739 |
| Dimer | |||||
| Zn1···Zn2 | 0.00479 | 0.01073 | −0.00196 | 0.00232 | 0.00035 |
| Zn1–N1 | 0.12433 | 0.53525 | −0.22559 | 0.17970 | −0.04588 |
| Zn1–N2 | 0.12369 | 0.53327 | −0.22426 | 0.17879 | −0.04547 |
| Zn1–O2 | 0.11306 | 0.60029 | −0.21990 | 0.18498 | −0.03491 |
| Zn1–O3 | 0.11048 | 0.58281 | −0.21241 | 0.17905 | −0.03335 |
| Zn2–N3 | 0.12512 | 0.54051 | −0.22797 | 0.18154 | −0.04642 |
| Zn2–N4 | 0.12447 | 0.53619 | −0.22601 | 0.18003 | −0.04598 |
| Zn2–O6 | 0.11075 | 0.58587 | −0.21336 | 0.17991 | −0.03344 |
| Zn2–O7 | 0.11315 | 0.60117 | −0.22015 | 0.18522 | −0.34930 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Usman, M.; Khan, R.A.; Alsalme, A.; Alharbi, W.; Alharbi, K.H.; Jaafar, M.H.; Abu Khanjer, M.; Tabassum, S. Structural, Spectroscopic, and Chemical Bonding Analysis of Zn(II) Complex [Zn(sal)](H2O): Combined Experimental and Theoretical (NBO, QTAIM, and ELF) Investigation. Crystals 2020, 10, 259. https://doi.org/10.3390/cryst10040259
Usman M, Khan RA, Alsalme A, Alharbi W, Alharbi KH, Jaafar MH, Abu Khanjer M, Tabassum S. Structural, Spectroscopic, and Chemical Bonding Analysis of Zn(II) Complex [Zn(sal)](H2O): Combined Experimental and Theoretical (NBO, QTAIM, and ELF) Investigation. Crystals. 2020; 10(4):259. https://doi.org/10.3390/cryst10040259
Chicago/Turabian StyleUsman, Mohammad, Rais Ahmad Khan, Ali Alsalme, Walaa Alharbi, Khadijah H. Alharbi, Mohammad H. Jaafar, Maymonah Abu Khanjer, and Sartaj Tabassum. 2020. "Structural, Spectroscopic, and Chemical Bonding Analysis of Zn(II) Complex [Zn(sal)](H2O): Combined Experimental and Theoretical (NBO, QTAIM, and ELF) Investigation" Crystals 10, no. 4: 259. https://doi.org/10.3390/cryst10040259
APA StyleUsman, M., Khan, R. A., Alsalme, A., Alharbi, W., Alharbi, K. H., Jaafar, M. H., Abu Khanjer, M., & Tabassum, S. (2020). Structural, Spectroscopic, and Chemical Bonding Analysis of Zn(II) Complex [Zn(sal)](H2O): Combined Experimental and Theoretical (NBO, QTAIM, and ELF) Investigation. Crystals, 10(4), 259. https://doi.org/10.3390/cryst10040259