3.1. Characterization of the Vibrational Properties and Vibrational Splitting in LuVO4 Crystals
According to X-ray powder diffraction analysis [
11], the as-grown crystals had a single-phase zircon structure. All of the diffraction peaks were identified to result from LuVO
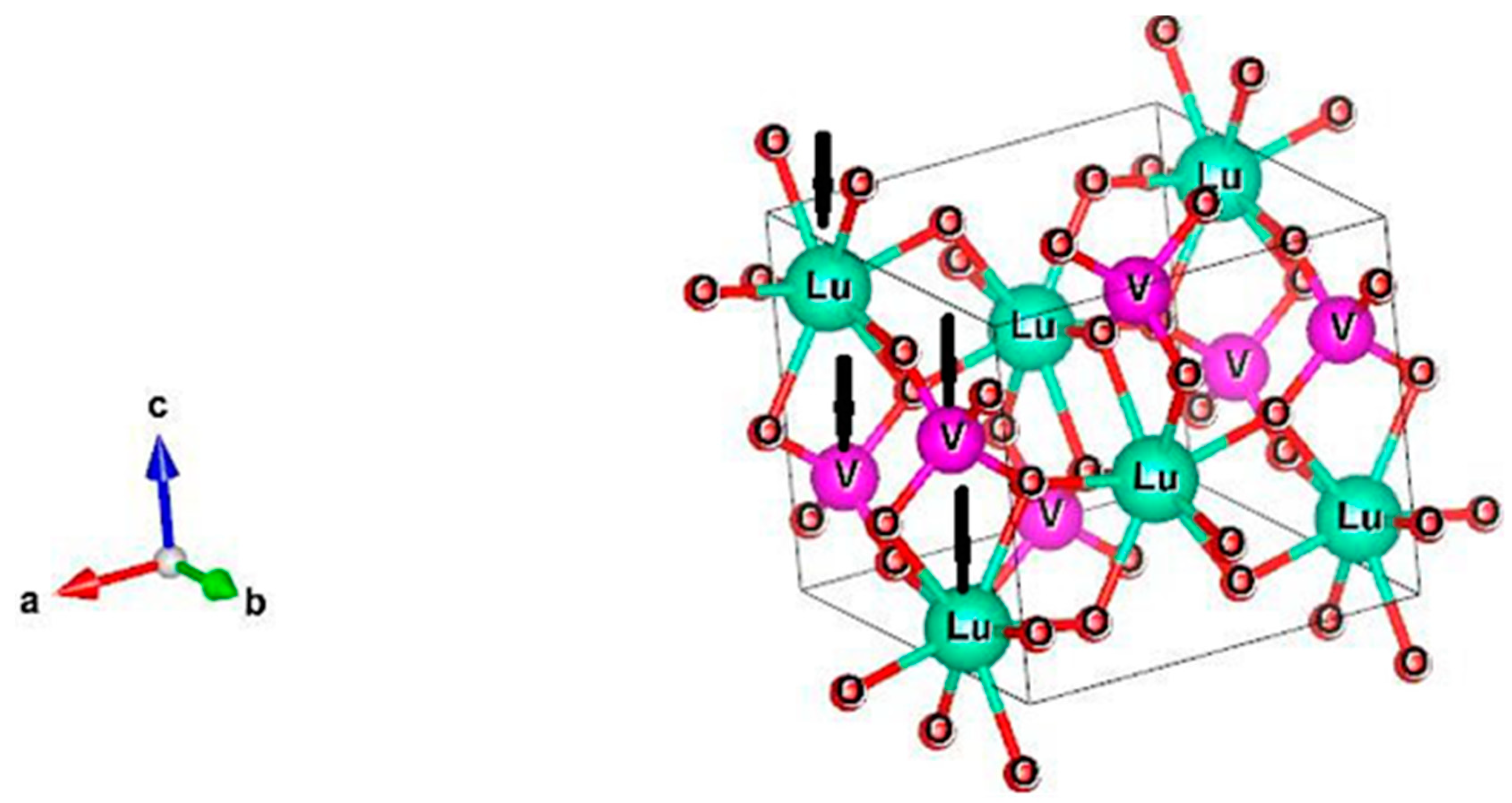
4 and no peaks of a second phase were found. The lattice structure of this crystal is depicted in
Figure 1. It can be considered as made up of chains formed by alternating LuO
8 polyhedra (bisdisphenoid) and VO
4 tetrahedra along the c-axis. These chains are joined together by sharing other edges of the LuO
8 polyhedra along the a- and b-directions to form a three-dimensional lattice with centrosymmetric tetragonal structure with the point group symmetry D
4h and space group I4
1/amd (D
4h19). Despite the different coordination environment, both Lu
3+ and V
5+ ions occupy sites of D
2d symmetry [
12]. The unit cell contains 4 formula units (2 formula units per primitive cell) with lattice parameters of a = b = 7.0236Å and c = 6.2293Å [
11]. Consequently, 12 atoms per primitive cell with 36 degrees of freedom should be taken into account.
According to the determined crystal structure [
7,
11,
12] and the data available in the International Tables for Crystallography for the space group D
4h19, the occupied Wyckoff positions are as follows: 4a (Lu
3+ ions), 4b (vanadium) and 16h (oxygen). As the mirror plane σ
v is the common symmetry element of the point group of the crystal D
4h and the site symmetry group C
s, C
s(σ
v) is identified as the correct site symmetry for the oxygen atoms, thus removing the ambiguity in the 16h positions for this structure. By carrying out the nuclear site group analysis [
14], shown in detail in
Table 1, one ultimately obtains the representation of the Raman active modes as follows:
It is also instructive to classify the vibrational modes with the molecular site group analysis, which is suitable for treating isolated ionic or molecular species within a crystal matrix [
14,
15]. The orthovanadate structure consists of heavy rare-earth ions and VO
43− tetrahedra that can be approximately regarded as separate units due to the strong internal bonds within each tetrahedron. Therefore, the lowest-frequency vibrations of LuVO
4 should comprise of translations and librations of the VO
43− tetrahedra as rigid units against the rare-earth ions, while the higher-frequency phonons should be almost entirely due to internal vibrations of the tetrahedra [
16]. The isolated VO
43- complex has the relatively high symmetry T
d and its degrees of freedom can be classified in terms of irreducible representations of the T
d group as follows:
The pure rotation and translation comprise zero-frequency motions of the complex as rigid structure that transform into librations and external translational modes, respectively, in the LuVO
4 crystal structure [
16]. The remaining four terms represent the four fundamental internal vibrations of the VO
43− tetrahedron. The first publications analyzing the Raman phonons in zircon-type structure and dividing them into external and internal modes according to the vibrating species were made by Miller et al. in 1968 [
16] and Elliot et al. in 1972 [
17]. Since then, these modes have been extensively studied by many authors for a number of orthovanadates. On lattice sites with D
2d symmetry (see
Table 1), the vibrational states undergo partial (crystal-field-induced) splitting according to the T
d/D
2d group-subgroup correlation rules. Then one should take into account the interaction between the two formula units in the primitive cell, which is equivalent to induction from the representations of the D
2d group to those of the D
4h group. In this way, a doubling of the vibrational states to the real number of degrees of freedom per primitive cell takes place and this doubling indicates the Davidov or correlation-field splitting, which is a measure of the interaction between non-equivalent tetrahedra in the primitive cell and also of the magnitude of the phonon dispersion in the Brillouin zone [
15].
From this analysis, the same total representation of the Raman active modes (Equation (1)) can be constructed with the valuable additional information that there will be 2
A1g + 2
B1g +
B2g + 2
Eg internal (higher-frequency) VO
43− modes; 2
B1g + 2
Eg external (translational) VO
43− modes and a librational
Eg mode of the VO
43− tetrahedron [
16]. The infrared active vibrations are:
A2u +
Eu translational, an
Eu librational and 2
A2u + 2
Eu internal VO
43- modes. The distribution of the optically active internal modes shall be as follows:
A1 (ν
1) →
A1g;
E (ν
2) →
A1g +
B2g and for both ν
3 and ν
4 from Equation (2):
F2 (ν
i) →
B1g +
Eg +
A2u +
Eu. This picture is of course an approximation of the real situation, where there are no purely internal or external modes, but still very helpful for the Raman mode assignment [
16,
17]. It also becomes clear that no Davidov splitting can be established from Raman measurements because the Davidov counterpart of each Raman mode is an odd-parity one. Combined Raman and IR measurements can detect the Davidov doublet
Eg +
Eu of the librational mode and the doublets
A2u +
B1g and
Eg +
Eu resulting from the
F2(ν
3) and
F2(ν
4) internal vibrations of the VO
43− tetrahedron.
The pertinent Raman tensors for the D
4h point group are the following [
15]:
A1g: ; B1g: ; B2g:; and Eg,1: ; Eg,2: .
The scattering intensity of a particular mode with Raman tensor
α is given by:
where the unit vectors
ei and
es denote the polarization of the incident and scattered light, respectively. For the polarized Raman measurements, we use the notations
X (100),
Y (010) and
Z (001) for the main crystal axes, and
X’(110) and
Y’(−110) for diagonal directions in the XY plane. The applied scattering configurations are labelled by Porto notations. Raman selection rules may suffer from depolarisation effects caused by the extremely strong birefringence of the LuVO
4 crystal. These effects lead to partial mixing of allowed and forbidden intensity between different Raman modes, which, nevertheless, can be minimized by using focusing/collecting optics with small numerical aperture (N.A.), while keeping the beam path within the sample short [
18]. The above two requirements were satisfactorily met by using a 20× microscope objective with N.A. = 0.4. Special care was also taken to achieve as perfect as possible back-scattering conditions on samples with smooth and optically defect-free surfaces. The main crystallographic planes of our samples were identified by X-ray diffractometry [
11].
The spectra also contain features that do not obey the Raman selection rules and obviously do not pertain to the first-order vibrational spectrum of the LuVO
4 crystal. Their intensity turns out to be dependent on the investigated sample spot and the excitation wavelength λ with strongest intensity appearing upon red (λ = 632.8 nm) excitation. Our earlier experience with similar polarized Raman measurements on LuVO
4 at 632.8 nm excitation [
11] shows that these features also display various polarization behaviour. As no significant impurities or second-phase inclusions were detected by XRD, we attribute these features to photoluminescence lines from electronic transitions of other rare-earth ions that are unavoidably present as substitutional defects in LuVO
4. In the spectra excited at 514.5 nm photoluminescence features are largely suppressed and upon blue excitation (488 nm) they are virtually missing. Besides, the birefringence of LuVO
4 is minimal in the blue spectral region [
7]. The 488 nm laser thus appears to be the obvious choice of excitation wavelength. We, nevertheless, used the 514.5 nm laser line in the present study because of indications that the generally missing Raman active E
g(4) mode might be resonantly enhanced in the green spectral region, and carefully selected sample regions with minimum and non-variable intensity of impurity features. For some spectra when the luminescence lines lie far from the studied Raman modes, the He-Ne laser excitation (632.8 nm) was used.
The measured Raman spectra are presented in
Figure 2. Although Raman selection rules are clearly discernible, in view of the known birefringence, the question may arise if the mode intensities in different scattering configurations are directly comparable. To address this question, we recall that besides the scattering cross section (given by the magnitude of the tensor elements), the Raman signal is influenced by the reflectivity
R at the sample surface, the possible absorption within the sample, and the size of the volume, in which the detected Raman scattering effectively occurs (scattering volume). The latter is determined by the focus spot depending entirely on the focusing optics and the depth of focus. The correction factor resulting from reflection losses has the form (1 −
R)
2 because both the incident and the scattered radiation undergo reflection. By means of the Fresnel equation for normal-incidence reflectivity, we obtain (1 −
R)
2 = 16
n2/(
n + 1)
4. We shall inspect that factor for the spectra labelled X(YY)
and X(ZZ)
representing the two limiting cases: ordinary wave (
n =
no = 2.1) and extraordinary wave (
n =
ne = 2.3), respectively. Using the results for the refraction index
n from [
7], we obtain a value of 0.764 for spectrum X(YY)
and for spectrum X(ZZ)
(1 −
R)
2 = 0.714. According to Gaussian beam theory [
19], the depth of focus is of the order λ/(N.A.)
2 but it also linearly depends on
n. Thus, the larger value of
ne effectively increases the scattering volume by about 10%, entirely compensating the higher reflection losses. Even if we neglect one of these counter-acting effects and consider only the other, the expected change in Raman signal does not exceed 10%. The depth of focus is of the order 10 μm and the crystal exhibits high transparency above 500 nm [
11]; therefore, possible absorption losses should be negligible. Consequently, the different spectra in
Figure 2 are perfectly comparable for the purpose of Raman mode assignment and even for establishing relations between the Raman tensor elements
a and
b for the A
1g modes. The relative peak intensity of the A
1g(2) line in spectra X(YY)
and X(ZZ)
is ≈ 17000 and ≈ 19900 (arb. u.), respectively. This implies |
a| ≈ |
b| for the A
1g(2) mode. However, the intensity of the A
1g(1) line in spectrum X(YY)
is about four times smaller than in spectrum X(ZZ)
. The A
1g(1) mode thus has a strongly anisotropic Raman tensor (|
b| ≈ 2|
a|).
Concerning the depolarization effect addressed in [
18], we point out that the allowed A
1g(2) intensity in spectrum X(YY)
is more than 10 times higher than the corresponding forbidden intensity in spectrum X(ZY)
. Similar relations are valid for all higher-intensity lines in
Figure 2, i.e., their assignment is not threatened by birefringence effects.
Based on the polarization behavior of the different spectral lines, most of them could be unambiguously assigned to predicted Raman active phonon modes of the LuVO
4 crystal. Besides phonon lines, there are several photoluminescence lines within the spectral region of 300–450 cm
−1. The mode assignment, along with some new results on problematic modes that will be outlined in detail in
Section 3.2, is given in
Table 2 with juxtaposition with the vibrational species of the isolated VO
43− tetrahedron [
20,
21]. We also included in
Table 2 the peak-intensities and the linewidths in order to highlight the SRS-promoting phonons by means of their two most important properties: high Raman efficiency and long lifetime. As noted in [
22], the steady-state Raman gain coefficient of a certain phonon mode in SRS depends linearly on the ratio of its scattering cross-section (integral intensity) and its linewidth in spontaneous Raman scattering. This ratio is proportional to the peak intensity [
22]. Indeed, for phonons that were identified as SRS-promoters [
10], the peak intensity (given in bold font in
Table 2) is many times higher than for most of the other normal vibrations in LuVO
4, and the linewidth is small.
Table 2 contains some new results on the LuVO
4 Raman mode assignment, which will be discussed in detail in the next subsection. Here, we will briefly review the crystal-field splitting Δω
CF and Davidov splitting Δω
D within the manifolds arising from the fundamentals ν
3 and ν
4, all components of which are optically active. Within those manifolds, the frequency difference between the E
g(E
u) and the B
1g(A
2u) mode provides an estimate for the crystal-field splitting while the separation in the E
g/E
u or B
1g/A
2u doublet serves as a measure for the Davidov splitting. Although IR measurements on LuVO
4 [
6,
23] are scarce, its IR active modes are long known [
23], except for the E
u in the ν
4 manifold. Nevertheless, this mode can be credibly assumed to lie between 300 and 350 cm
−1 by analogy to the well-studied internal vibrations of YVO
4 [
16,
27,
28]. Additionally, only Δω
CF can be estimated for ν
2 by the separation A
1g(1)–B
2g for their odd-parity counterparts A
1u and B
2u are not optically active.
Table 3 represents the estimated splitting using the first column of
Table 2. Three observations can be made from
Table 3, which should be more or less valid for all members of the orthovanadate family: (i) Δω
CF decreases monotonically in going from bond-bending to bond-stretching vibrations. (ii) For the mixed bending/stretching vibrations of the ν
4 manifold, Δω
CF and Δω
D are comparable with slight prevalence of Δω
CF. (iii) In the ν
3 manifold, Δω
D for the E-modes dominates over Δω
CF and is eight times stronger than Δω
D for the B
1g(4)/A
2u doublet. The fundamental frequency ν
3 = 825 cm
−1 of isolated VO
43− [
21] appears like a “common center of gravity” of both doublets. For this effect, we propose a simple qualitative explanation. In crystals of complex ions as VO
43−, the interaction causing Davidov splitting is mainly governed by short-range repulsion forces [
15]. Therefore, Δω
D should primarily depend on the relative motion of the vibrating ions for the modes of a particular doublet. In the rectangular unit cell, the difference in the Z-coordinates of the two non-equivalent tetrahedra of the primitive cell is more than twice smaller than their horizontal distance [
17] (see also
Figure 1). The E
g(5) mode has equally charged ions of the two tetrahedra vibrating horizontally against each other, while the corresponding E
u mode has these ions executing the same vibration in phase. Due to the proximity of their Z-coordinates, a considerable additional repulsive force should arise between the two tetrahedra during the E
g(5) vibration, unlike the corresponding E
u motion, which completely lacks such a force. In the analogous case with the B
1g(4) mode and its Davidov counterpart A
2u, the vibrational motion takes place along the vertical Z-axis. Due to the larger horizontal distance between the two non-equivalent tetrahedral, the mutual influence between motions of equally charged ions during the B
1g(4) vibration should be much weaker, which might explain the small Δω
D of only 7 cm
−1 in the B
1g(4)/A
2u doublet. This splitting resembles only a slight perturbation of the fundamental
F2(ν
3).
3.2. Assignment of Problematic Raman Active Modes Using Their Rotational Transformation Properties
Thanks to several detailed group-theoretical and spectroscopic investigations of orthovanadate crystals, the symmetry properties of the stronger lines in their Raman spectra are well-established and their assignment has been known for decades [
16,
17,
18,
24,
29]. However, the modes E
g(1), B
1g(2) and E
g(4) are known to have exceptionally weak scattering intensity in almost all orthovanadates of the zircon-family [
28]. Due to their low intensity and complications from birefringence effects, these modes are frequently missing [
24] or wrongly assigned [
10,
25,
26] in some recent Raman studies of orthovanadates. Therefore, to complete the mode assignment for the LuVO
4 crystal, i.e., to obtain the results shown in
Table 2, we measured Raman spectra as a function of the rotation angle φ by rotating the sample around the exciting beam in its polarization plane. This approach utilizing the differences in the transformation properties of phonons of different symmetry has proved to be helpful in many instances. Beside its conventional use for checking phonon symmetry, it has also been applied for determination of crystal structure [
30] and detection of lattice defects [
31].
For the present experiments, φ is defined as the angle between the X-axis and the actual polarization direction of the exciting laser beam. For rotating scattering configurations in perpendicular polarization x
φy
φ and x
φzφ are orthogonal φ-dependent directions in the XY and the XZ plane, respectively. For rotation in parallel polarization, the transition from the initial to the final polarization is denoted. For instance, Y(XX→ZZ)Ȳ means gradual transition from Y(XX)Ȳ to Y(ZZ)Ȳ through rotation about the Y axis. Vertically stacked rotation-angle dependent spectra are shown in
Figure 3,
Figure 4,
Figure 5 and
Figure 6 for various frequency regions and scattering configurations. The theoretically predicted dependence of the Raman intensity (Equation (3)) on the angle φ for the examined configurations is given in
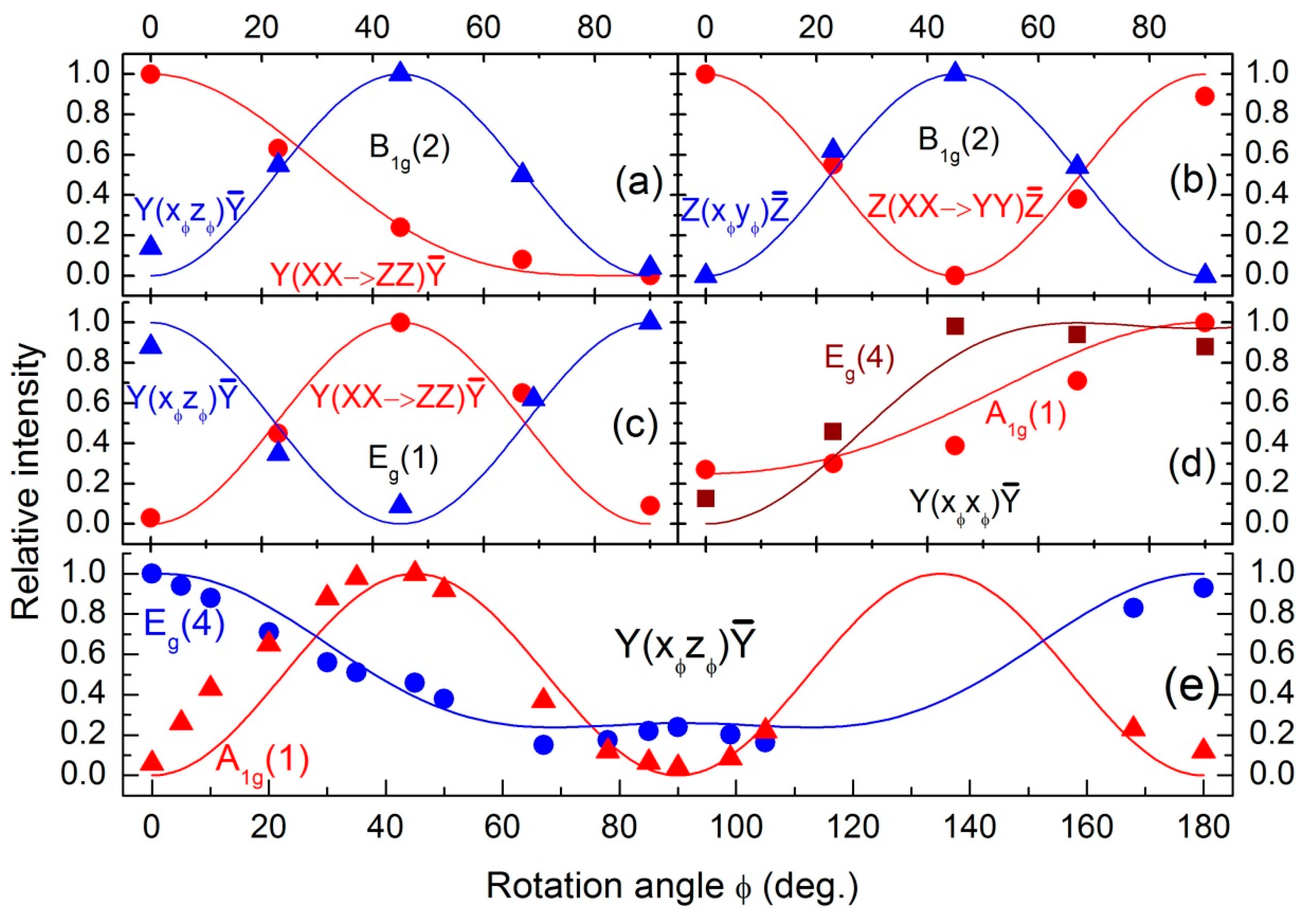
Table 4. The angular dependences of the measured mode intensities normalized to the maximum intensity for each line of interest are plotted in
Figure 7. Theoretical curves of the corresponding functions in
Table 4 are also plotted in
Figure 7 for comparison.
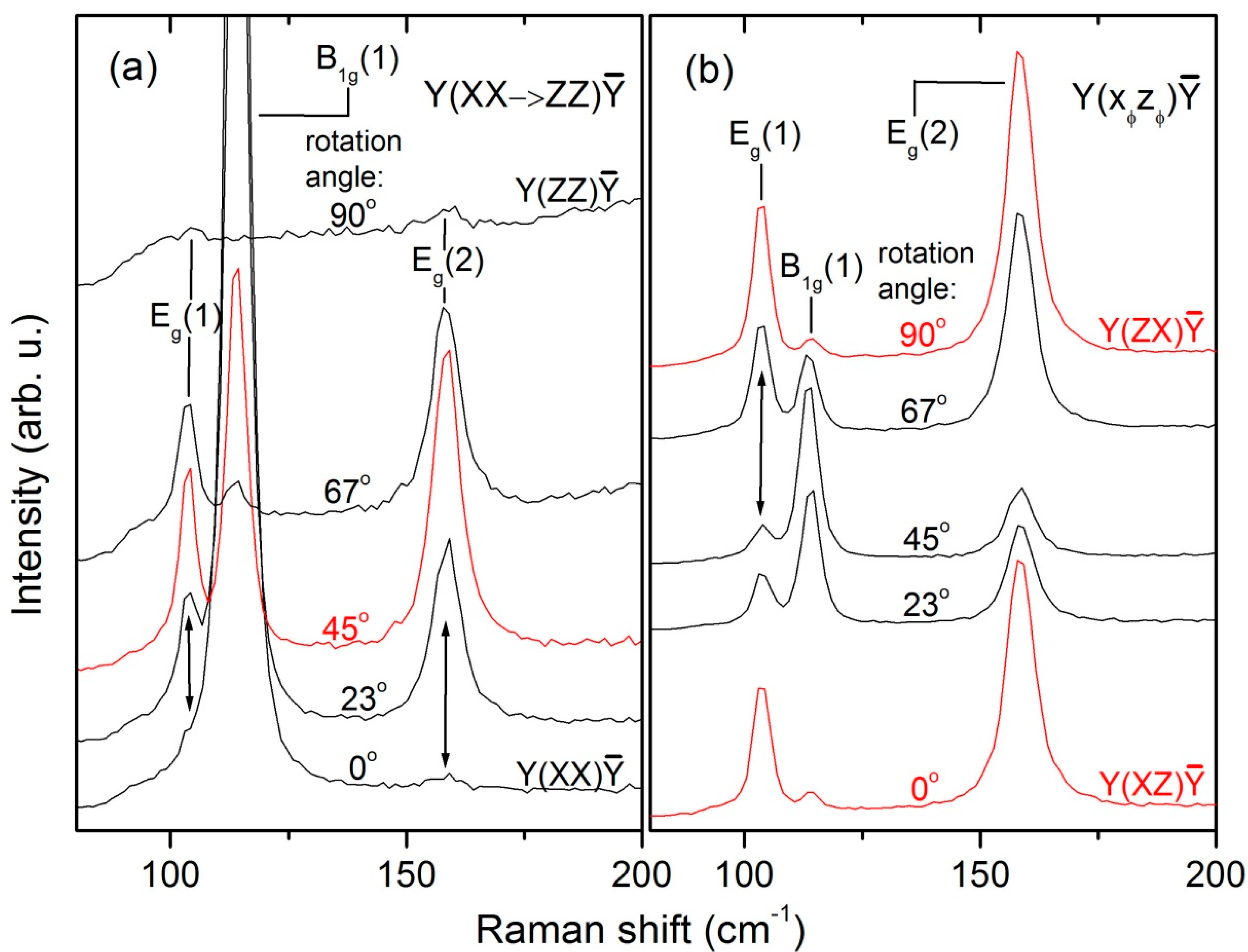
The rotation-angle dependent spectra of the lowest-energy translational modes of the LuVO
4 crystal are displayed in
Figure 3. From
Figure 7c and the observed behavior of the line at 103 cm
−1, it is evident that its φ-dependent intensity oscillation is in excellent agreement with the formulae listed in the last row of
Table 4. Consequently, we can unambiguously assign the E
g(1) mode to the line at 103 cm
−1.
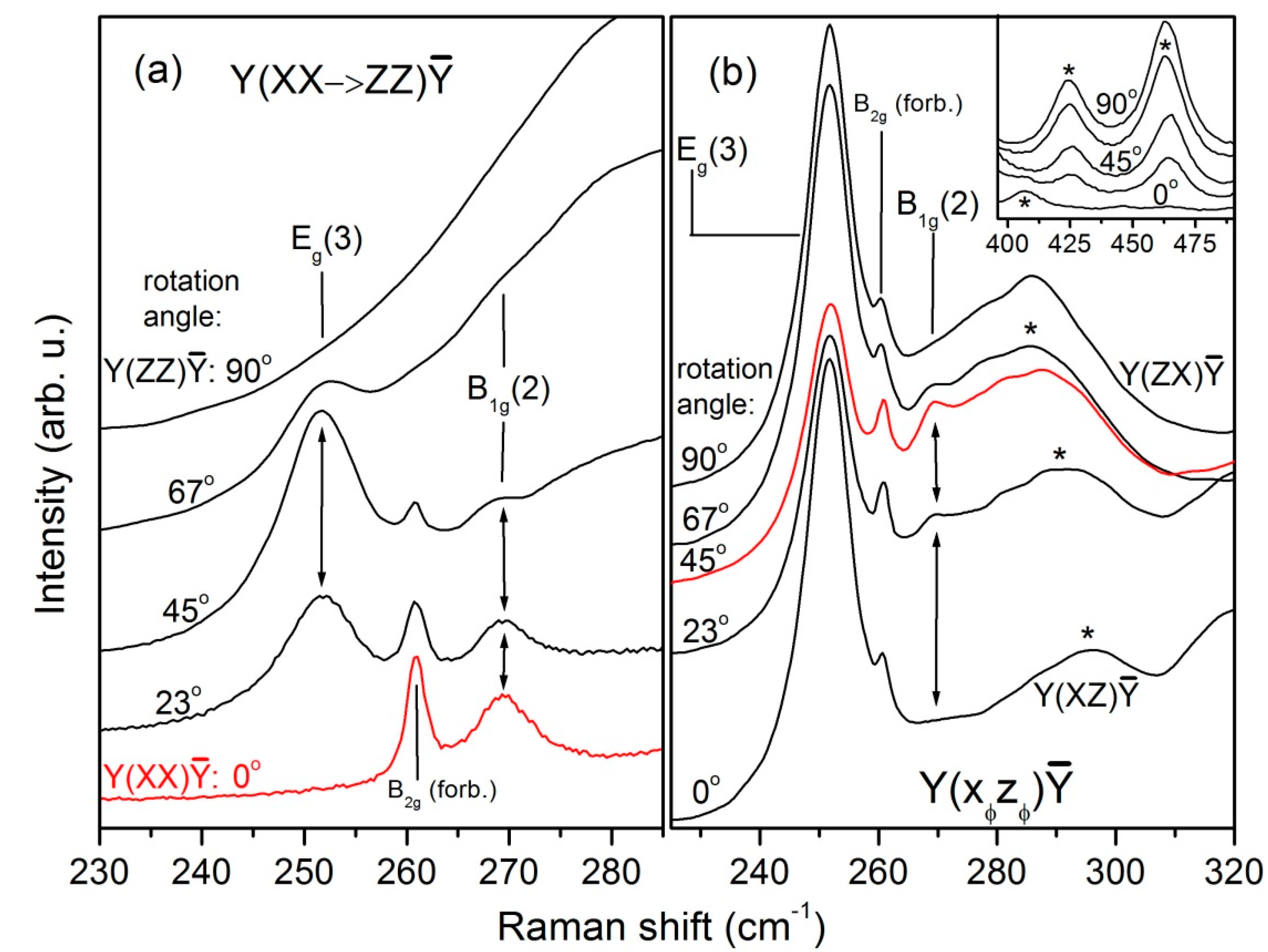
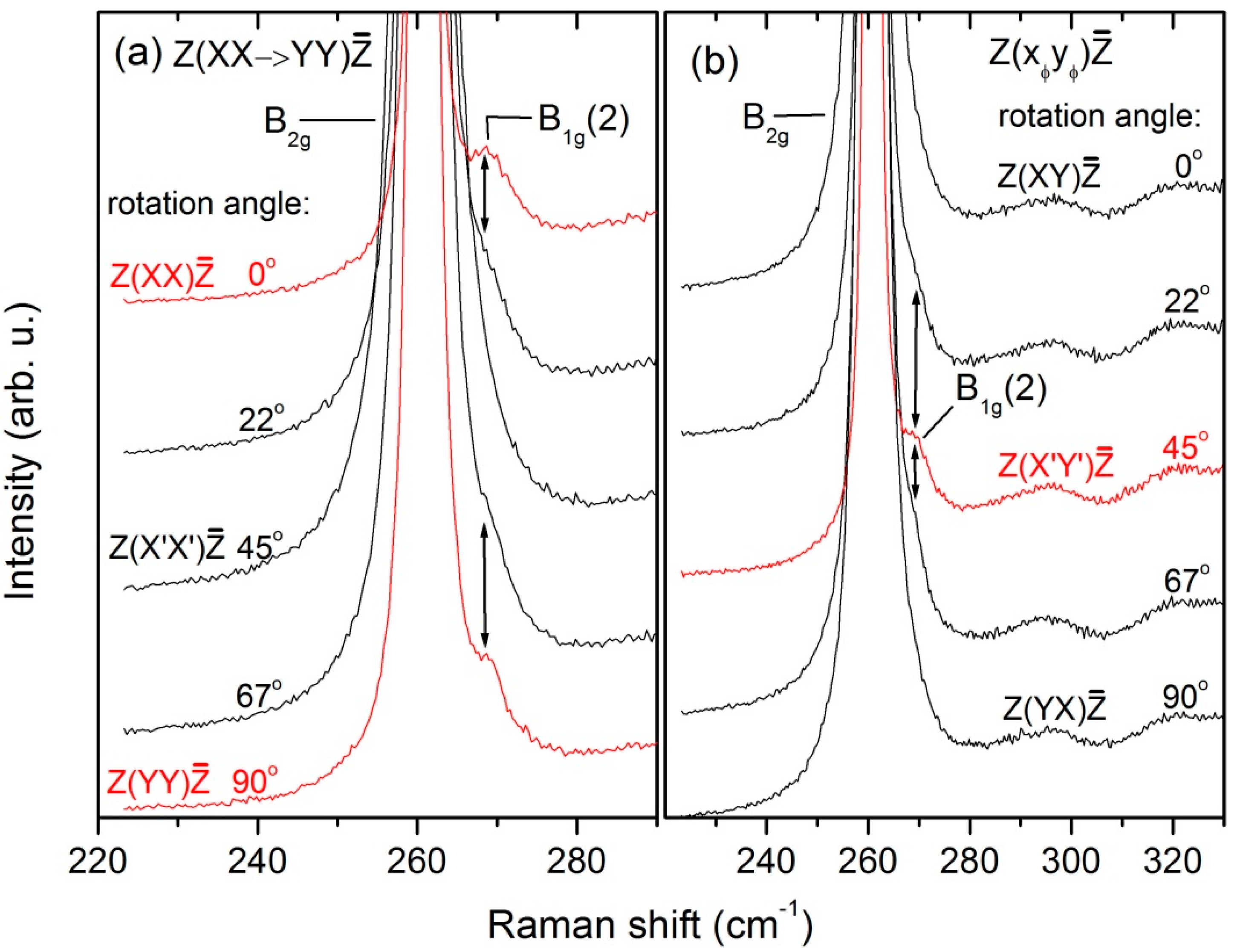
To confirm the assignment of the B
1g(2) mode, which has extremely weak intensity, we performed thorough investigations by rotating in two perpendicular planes: XZ (excitation along Y) and XY (excitation along Z) using the high-resolution diffraction grating (1800 gr./mm). The corresponding sets of Raman spectra are shown in
Figure 4 and
Figure 5, respectively. Because of interference fringes, the spectra in
Figure 4 were smoothed out by means of fast Fourier Transform (FFT) filtering. The first impression from
Figure 4 is that both 261.5 cm
−1 and 269 cm
−1 lines satisfy the theoretical φ-dependence of the intensity of the B
1g mode. Thus, if only the polarizations used in
Figure 4 are considered, one can easily declare that the stronger 261.5 cm
−1 line is associated with the B
1g(2) mode. However, due to its high intensity in XY polarization (
Figure 2) and its φ-dependent behavior (
Figure 5), the 261.5 cm
−1 line is undoubtedly recognized as the only B
2g mode of the LuVO
4 crystal. In
Figure 7a,b, the φ-dependence of the 269 cm
−1 line intensity is presented together with theoretical curves according to the formulae listed in the second row of
Table 4.
The excellent agreement between theory and experiment manifested in
Figure 7a,b confirms the assignment of the 269 cm
−1 line to the
B1g(2) mode. This assignment is further corroborated by Figure B1 of [
32], where the lines E
g(3), B
2g and B
1g(2) of LuVO
4 are detected in the same sequence and at nearly the same frequencies. From that figure it is seen that the B
1g(2) mode crosses over the B
2g mode in going from NdVO
4 to LuVO
4, while the B
2g frequency remains nearly constant at 260–261 cm
−1. The same behaviour may be deduced from Table 1 of [
24], if the labels B
2g(ν
2) and B
1g(2) are interchanged. We therefore argue that it is the B
2g mode that remains relatively unchanged for all rare-earth vanadates while the B
1g(2) mode undergoes a slight hardening over the rare-earth vanadate series from NdVO
4 to LuVO
4. This is, however, contradictory to the mass-dependent frequency behavior ω~(µ)
−1/2 (here µ is the reduced mass of the rare-earth and the VO
43− ions) for its vibrational pattern (out-of-phase translation) expected from the simple model adopted in [
24]. This seemingly contradictory behavior is supposed to be due to the mixing of the three close-by lying modes E
g(3), B
2g and B
1g(2) of totally different character. B
2g as internal tetrahedral mode should be least susceptible to such mixing which supports the assumption for its nearly constant frequency. Additionally, the B
2g vibrational pattern having all 4 oxygens move perpendicular to the V-O bonds should also keep this phonon insensitive to a change in the rare earth ion radius. On the other hand, the mass-dependent frequency behavior of the in-phase translational B
1g(2) mode (113 cm
−1 for LuVO
4) agrees qualitatively with the analogue model expected for its vibrational pattern, namely,
ω~(
m)
−1/2 with
m being the total mass of the rare-earth and the VO
43− ions [
24].
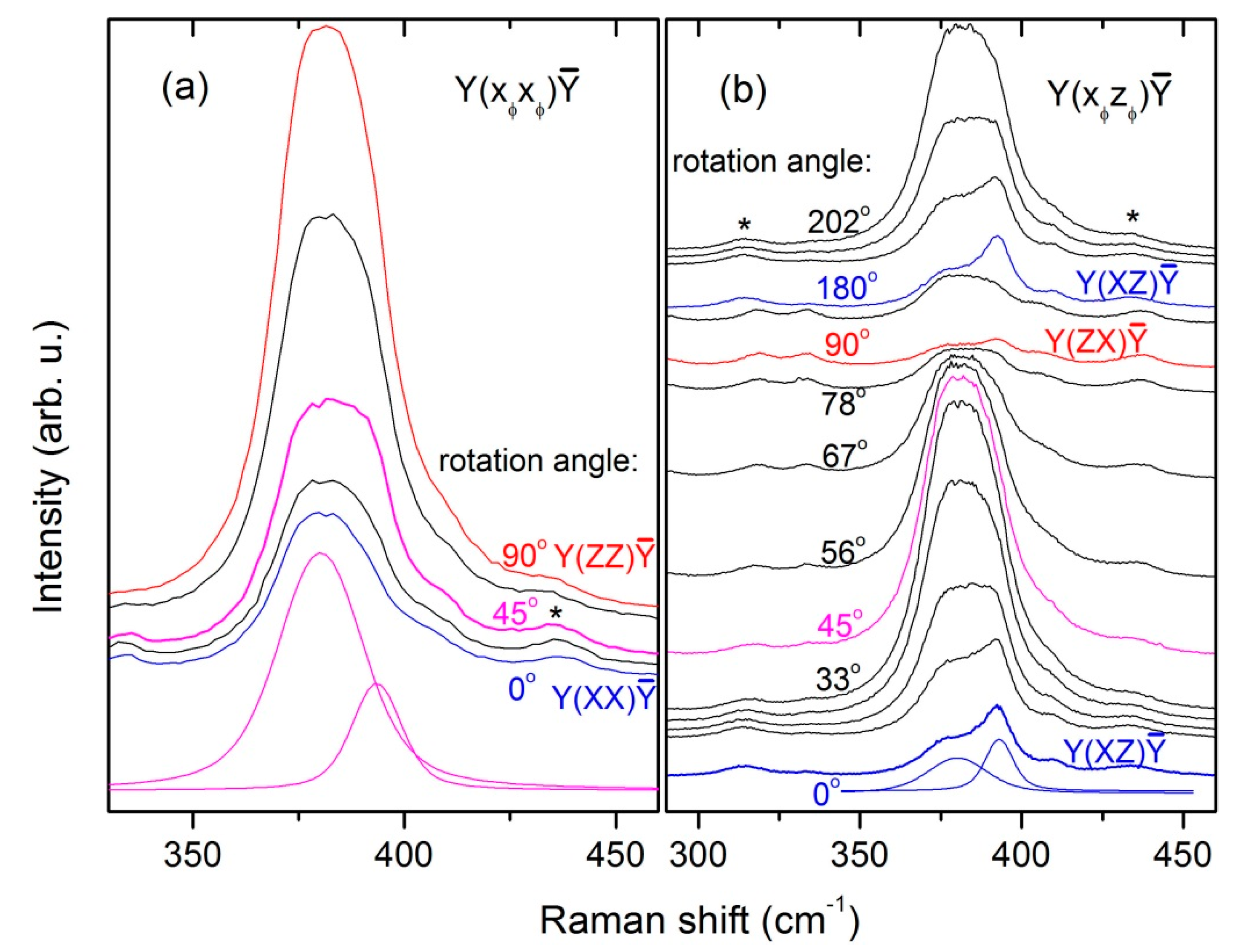
The E
g(4) mode is expected to lie in the vicinity of the A
1g(1) mode at 381 cm
−1 for all members of the orthovanadate family with a frequency nearly independent of the mass of rare-earth ion [
28]. It is the second lowest in energy of the internal Raman modes of the VO
43− complex and seems to have exceptionally low scattering intensity. The experimental detection of the E
g (4) mode in orthovanadates has almost never been reported, except for the study of Sanson et al. [
28] on YVO
4, where the E
g(4) mode was found at 387 cm
−1. Interestingly, in the present study, we find in the spectra excited at 514.5 nm a line at 393 cm
−1 with promising polarization behaviour (see
Figure 2 and
Figure 6a). In parallel polarization, the relative intensity of this line resembles more or less the φ-dependence for an E
g mode, as depicted in
Figure 7d. However, its intensity drops significantly when going from Y(XZ)Ȳ to Y(ZX)Ȳ scattering configuration (see
Figure 2), which is extremely unusual for a phonon mode. We therefore scanned its angular dependence in perpendicular polarisation in the full range from φ = 0° to 180° using the high-resolution diffraction grating (1800 gr./mm). The obtained spectra are shown in
Figure 6b and the resulting φ-dependences are plotted in
Figure 7e. It is seen that, despite the intensity drop towards 90
°, the relative intensity of this line tends to form a local minimum at about 45° and has a weak local maximum at 90°. It thus appears that there is an accidental coincidence of two lines: a weak one with a period of 90° and a stronger one with a period of 180° in the angular dependence of their intensities. The former one has the right angular dependence for an E
g mode, while the latter one has the right angular dependence for a polarized luminescence line. Although luminescence features normally appear in optical spectra in form of wider bands, rare-earth compounds are an exception. Sharp phonon-like luminescence lines are typical for rare-earth ions due to the radial confinement of their f-electrons [
33]. Radiative discharge of some excited states in RE ions via dipole-allowed transitions was shown to produce polarized spectra [
34] having cos
2φ-like angular dependence (hence period of 180°), which is typical for radiation of an oscillating dipole with fixed orientation. We also established a cos
2φ-like angular dependence for the intensity of some luminescence features around 400 cm
−1 in the spectra from
Figure 4b, which we have depicted in the inset of
Figure 4b. Therefore, to provide theoretical curves for panels (d) and (e) of
Figure 7, we modelled the intensity behaviour of the 393 cm
−1 line with functions proportional to (sin
2φ + A sin
22φ) for parallel and (cos
2φ + A cos
22φ) for perpendicular polarization, respectively. The constant A was estimated to be ≈ 1/3 from comparison of the line’s intensity in crossed polarization at 0° where both components should be at maximum and 90° where the Raman line is again at maximum while the luminescence vanishes. The model shows satisfactory agreement with the measured data in both polarizations, especially in view of the fact that the region 300–450 cm
−1 is rich in luminescence lines and there might be more than one of them interfering with the Raman modes A
1g(1) and E
g(4). For instance, in [
35] luminescence spectra of rare-earth-doped LuVO
4 nanoleaves were reported containing lines around 526 nm, which corresponds to a Raman shift of ≈ 400 cm
−1 in a spectrum excited at 514.5 nm. We consider the presented arguments sufficient to assign the 393 cm
-1 line to the so far missing E
g(4) mode. This assignment is corroborated by the presence of a weak line in an E
g Raman spectrum of LuVO
4 at the same frequency in Figure B3 of [
32] for 514.5 nm excitation and at 386 cm
−1 in Figure 5 of [
25] for 532 nm excitation. In both publications, these faint features were not discussed probably due to their weak intensity. We should also mention the work of Voron’ko et al. [
36] who reported to have found the E
g(4) mode around 440 cm
−1 in X(ZY)
Raman spectra of YVO
4 and GdVO
4. In their temperature-dependent Raman study, these authors observed a double-peak structure for the A
1g(1) line, which they explained by thermally-activated population of higher local minima in the lattice potential through rotation of some VO
43− tetrahedra. The higher-energy component, whose A
1g character was reportedly confirmed by polarization studies [
36], nearly coincides in frequency with the line identified by us as the E
g(4) mode. Although we do not rule out a possible inhomogeneous broadening of the A
1g line in our LuVO
4 crystal due to VO
43− to rotation at higher temperatures, we emphasize that we do not detect any splitting in its lineshape despite the large linewidth. We also have confirmed the symmetry of the E
g(4) mode at 393 cm
−1 in the relevant polarization configurations, and additionally by demonstrating its rotational transformation properties. Furthermore, the E
g(4) mode is expected at a nearly constant position between 370 and 410 cm
−1 for all orthovanadates and has already been detected at 387 cm
−1 for YVO
4 in full agreement with theoretical model calculations [
28].
It should be pointed out, however, that no Raman feature at 393 cm
−1 is detectable upon blue excitation at 488 nm where the spectrum is almost free of luminescence lines. This implies a possible resonance character of the E
g(4) mode. A strong dependence of another E
g mode intensity on the excitation energy has already been established for YVO
4 [
28]. Unfortunately, no complete resonance excitation profile can be constructed for this mode because upon red excitation (633 nm) the region 350–500 cm
−1 is overshadowed by intense luminescence features stronger than the phonon lines. Still, measurements at other excitation wavelengths in the green region and, possibly at low temperatures, could provide ultimate confirmation of our E
g(4) mode assignment.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}