Abstract
The crystal structures of three salts, namely N-(4-methoxyphenyl)piperazin-1-ium ethoxybenzoate monohydrate (I), N-(4-methoxyphenyl)piperazin-1-ium methoxybenzoate monohydrate (II) and N-(4-methoxyphenyl)piperazin-1-ium hydroxybenzoate monohydrate (III), have been determined and compared. In each of them, the ionic components and the water molecules are linked by a combination of N—H···O and O—H···O hydrogen bonds to form infinite chains of edge-fused centrosymmetric rings running parallel to the [100] direction. The C—H···O, C—H···π(arene) interactions and O—H···O in (III) are responsible for the further propagation of the aforementioned chains into di-periodic layers or tri-periodic networks. From an energetic point of view, all structures are primarily di-periodic; the very strong ionic interactions determine the periodicity. For comparison purposes, quantum chemical calculations were performed to show the difference between the ionic and neutral components. The energy of the hydrogen-bonded ring motifs was also estimated.
1. Introduction
Piperazine-based systems are key components for the development of drugs due to their broad spectrum of activities, viz. antibacterial, antifungal, antidepressant, antimycobacterial, etc. [1,2,3,4,5,6]. The pharmacological significance of piperazine derivatives has made them very important ligands for the synthesis of inclusion compounds [7,8,9] and hybrid materials [10,11,12]. Furthermore, the phenylpiperazine framework is one of special pharmaceutical interest because it has been known as active against many subtypes of serotonin receptors [13,14]. During the last two decades, N-(4-methoxyphenyl)piperazine (MeOPP) has emerged as a new addition to the range of designer recreational drugs, and considerable effort has been invested in the development of methods for the detection of both MeOPP itself and of its metabolites N-(4-hydroxyphenyl)piperazine and 4-hydroxyaniline [15] in human fluids [16,17]. MeOPP has euphoric stimulant properties, and its action on human physiology is similar to that of amphetamines [18,19] but has a significantly lower potential for abuse [20].
In view of the facts mentioned above and in continuation of our research work [21,22,23], in the present report, the synthesis and structural characterization of some novel heterocyclic compounds containing the phenylpiperazine moiety have been carried out. Here, the following salts of N-(4-methoxyphenyl)piperazine are considered, namely, N-(4-methoxyphenyl)piperazin-1-ium ethoxybenzoate monohydrate (I), N-(4-methoxyphenyl)piperazin-1-ium methoxybenzoate monohydrate (II) and N-(4-methoxyphenyl)piperazin-1-ium hydroxybenzoate monohydrate (III) (Scheme 1).
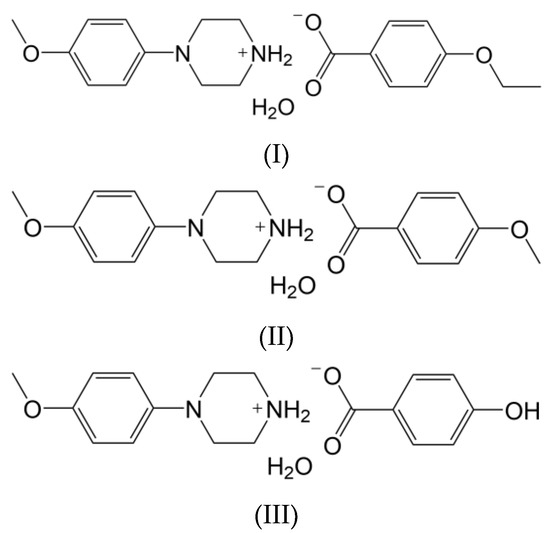
Scheme 1.
Chemical structures of I–III.
The numerous similarities and subtle differences in the supramolecular architecture of several N-(4-methoxyphenyl)piperazin-1-ium salts encouraged us to look more closely at the crystal-packing motifs from a geometrical and energetic point of view. For comparison purposes, some theoretical calculations are performed, and the results are discussed.
The proposed study is in line with the rational design of cocrystals and salts with the desired structural features [24,25,26]. A complete understanding of the supramolecular chemistry in terms of functional groups, synthons, building blocks, or structural frameworks is a prerequisite for crystal engineering [27,28,29]. Due to the complex and reversible nature of intermolecular interactions [30], predetermination of connectivity of molecules in the desired material is still a great challenge. Additional aspects, such as solvation or hydration, may also impact the final supramolecular structure [31,32].
2. Materials and Methods
2.1. Synthesis
All reagents were obtained commercially and were used as received. For the synthesis of the title compounds (Scheme 2), equimolar quantities (0.52 mmol of each component) of N-(4-methoxyphenyl)piperazine (from Sigma-Aldrich, St. Louis, MO, USA) and 4-ethoxybenzoic acid/4-methoxybenzoic acid/4-hydroxybenzoic acid were dissolved separately in methanol (10 mL), and the two solutions were mixed, stirred briefly, and then set aside to crystallize, giving the solid products (I), (II), and (III) after a few days. The products were collected by filtration and then dried in air [yield: 80%; m.p.: 421–423 K (I); 417–419 K (II); 423–425 K (III)].
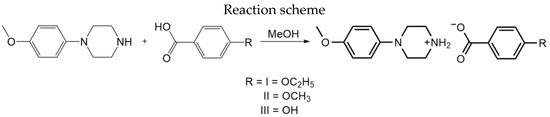
Scheme 2.
Reaction scheme of I–III.
The products were characterized by IR and 1H NMR spectroscopies.
Compound (I):
FTIR: (KBr, cm−1): 2832 (O—CH3), 1644 (C=O), 1246 (C—N).
1H NMR: CD3OD (400 MHz, δ ppm): 1.408–1.373 (t, 3H, J = 7.2 Hz), 3.318–3.194 (q, 4H, J = 3.6 Hz), 3.318–3.194 (q, 4H, J = 2.8 Hz), 3.742 (s, 3H), 4.092–4.039 (q, 2H, J = 7.2 Hz), 6.890–6.843 (t, 4H) 6.974–6.952 (dd, 2H), 7.915–7.893 (dd, 2H).
Compound (II):
FTIR (KBr, cm−1): 2838 (O—CH3), 1642 (C=O), 1252 (C—N).
1H NMR: CD3OD (400 MHz, δ ppm): 3.31–3.263 (t, 4H, J = 4 Hz) 3.226–3.201 (t, 4H, J = 6.0 Hz) 3.74 (s, 3H), 3.82 (s, 3H), 6.908–6.842 (q, 4H, J = 9.2 Hz), 6.972–6.950 (d, 2H, J = 8.8 Hz), 7.930–7.908 (d, 2H, J = 8.8 Hz).
Compound (III):
FTIR: (KBr, cm−1) 3320 (—OH), 2860 (O—CH3), 1639 (C=O), 1244 (C—N).
1H NMR: DMSO-d6 (400 MHz, δ ppm): 2.91–2.89 (m, 4H), 2.98–2.96 (m, 4H), 3.678 (s, 3H), 6.87–6.78 (m, 4H), 6.89–6.87 (d, 2H, J = 8.0 Hz), 7.78–7.58 (d, 2H, J = 8.8 Hz).
2.2. X-ray Single-Crystal Diffraction
The X-ray diffraction data for (I)–(III) were collected at 90 K, with a Bruker D8 Venture diffractometer using Mo Kα radiation. Data collections, cell refinements, data reductions and analyses were carried out with APEX3 [33]. The multi-scan absorption correction was also applied [34]. The structures were solved with SHELXT [35] and refined using least-squares minimization with anisotropic displacement parameters for non-H atoms with SHELXL-2019/2 [36].
All H atoms were found in difference Fourier maps. Subsequently, the H atoms bonded to C atoms were treated as riding atoms, with constrained C—H distances of 0.95 Å (aromatic), 0.98 Å (methyl) or 0.99 Å (methylene), and with Uiso(H) = kUeq(C), where k = 1.5 for the methyl group and 1.2 for all other H atoms bonded to C atoms. For the H atoms bonded to heteroatoms (N, O), the atomic coordinates were refined with Uiso(H) = 1.2Ueq(N) or Uiso(H) = 1.5Ueq(O), respectively. Additionally, geometrical similarity restraints were applied to N—H bonds using the SADI instruction in SHELXL [36]. In (III), a riding model was also used for the Nsp3H2 donor group; the N—H bond lengths were constrained to 0.91 Å.
The geometries of molecular structures and intermolecular interactions were analysed using PLATON [37] and MERCURY [38] programs. Figures presenting the structures were made using MERCURY [38].
Crystal data for (I) (M = 376.44 g/mol): triclinic, space group P (no. 2), a = 6.1089(3) Å, b = 7.4876(4) Å, c = 20.7603(12) Å, α = 94.777(2)°, β = 91.948(2)°, γ = 95.954(2)°, V = 940.32(9) Å3, Z = 2, T = 90 K, μ(MoKα) = 0.10 mm−1, Dcalc = 1.330 g/cm3, 31,916 reflections measured (2.8° ≤ Θ ≤ 27.5°), 4301 unique (Rint = 0.040) which were used in all calculations. The final R1 was 0.037 [I > 2σ(I)] and wR2 was 0.081 (all data).
Crystal data for (II) (M = 362.42 g/mol): monoclinic, space group P21/c (no. 14), a = 6.0418(8) Å, b = 40.135(4) Å, c = 7.4851(10) Å, β = 95.929(2)°, V = 1805.3(4) Å3, Z = 4, T = 90 K, μ(MoKα) = 0.10 mm−1, Dcalc = 1.333 g/cm3, 26,791 reflections measured (2.8° ≤ Θ ≤ 27.5°), 4120 unique (Rint = 0.032) which were used in all calculations. The final R1 was 0.042 [I > 2σ(I)] and wR2 was 0.083 (all data).
Crystal data for (III) (M = 348.39 g/mol): triclinic, space group P (no. 2), a = 6.1734(7) Å, b = 7.405(1) Å, c = 18.737(3) Å, α = 81.519(4)°, β = 85.271(4)°, γ = 83.560(5)°, V = 839.93(19) Å3, Z = 2, T = 90 K, μ(MoKα) = 0.10 mm−1, Dcalc = 1.378 g/cm3, 34,608 reflections measured (2.2° ≤ Θ ≤ 27.5°), 3844 unique (Rint = 0.044) which were used in all calculations. The final R1 was 0.036 [I > 2σ(I)] and wR2 was 0.094 (all data).
2.3. Hirshfeld Surface Analysis and Energy Frameworks
Hirshfeld surface analysis [39] was performed using CrystalExplorer [40].
Pairwise model energies [41] for (I)–(III) were estimated and visualized [42,43] between molecules within a cluster of radius 25 Å (recommended for ionic structures), using CrystalExplorer [40]. The computational approach uses a B3LYP/6-31G(d,p) molecular wavefunction calculated for the respective molecular arrangement in the crystal. The total interaction energy between any nearest-neighbour molecular pairs was estimated in terms of four components: electrostatic, polarization, dispersion, and exchange-repulsion with scale factors of 1.057, 0.740, 0.871 and 0.618, respectively.
2.4. Theoretical Calculations
The density functional theory (DFT) single-point calculations to estimate energies of hydrogen-bonded ring systems [ and ] and hydrogen-bonded cation and anion pairs found in the crystalline state of (I)–(III) were carried out using a functional with correction for long range and dispersion effects, ωB97XD [44], combined with the basis set of 6-311++G(d,p). At the same level of theory, the full geometry optimization of neutral molecular pairs for (I)–(III) was performed. Since all the obtained interaction energies were not corrected for the basis-set superposition error (BSSE), they are not discussed in detail; the approximate set of energies is used to show the energetic trends. All calculations were carried out with GAUSSIAN09 [45]. H-atom positions for systems taken directly from crystal structures and subsequently used in single-point calculations were normalized according to mean X—H distances (X = C, N, O) [46].
3. Results and Discussion
3.1. Molecular Structures
All investigated compounds are 1:1 salts of the N-(4-methoxyphenyl)piperazin-1-ium cation (MeOPP) with ethoxybenzoate (I), methoxybenzoate (II), and hydroxybenzoate (III) anions, respectively (Figure 1). All crystallize as monohydrates.
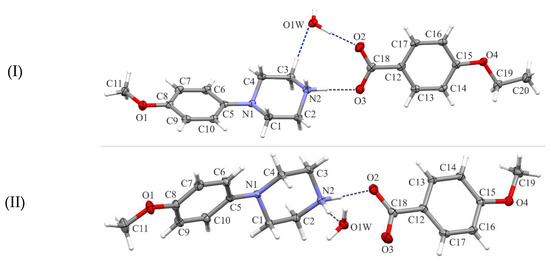
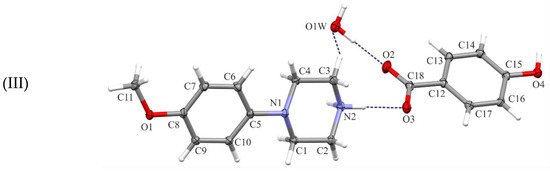
Figure 1.
Views of the asymmetric units of the title structures (I)–(III), with the atom-numbering schemes. Displacement ellipsoids are drawn at the 30% probability level. H atoms are shown as spheres of arbitrary radius.
In each of the compounds, the cations have very similar molecular conformation (Figure 2); since they are conformationally chiral molecules, they exist as a racemic mixture in the centrosymmetric space groups. The piperazine ring (N1/C1/C2/N2/C3/C4) adopts a chair conformation: the asymmetry parameters ΔC2 and ΔCs [37,47] do not exceed 5.44(13)° as the highest observed in (II). The methoxyphenyl substituent occupies an equatorial site on the piperazine ring. Moreover, the methoxy C atom (C11) lies close to the plane of the aryl ring (C5/C6/C7/C8/C9/C10): the two torsion angles, C11—O1—C8—C7 and C11—O1—C8—C9, defining this orientation are the most divergent from 180° and 0° in structure (III), −170.86(11)° and 7.94(18)°, respectively.
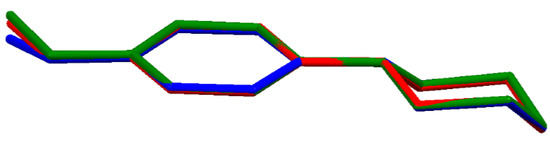
Figure 2.
Superimposed molecular structures of cations: (I)-red, (II)-green and (III)-blue.
The anions are simply benzoate substituted at position 4 by the ethoxy (I), methoxy (II) and hydroxy (III) groups, respectively. In all cases, the C—O bond lengths within the carboxylate group differ from each other by more than 3σ (roughly 1.24 Å vs. 1.27 Å), which clearly indicates that the negative charge is weakly delocalized.
The similar mutual orientation of cation and anion in the asymmetric units of the investigated salts can be described by the dihedral angle between the least-squares planes of aryl rings in both moieties: 68.4(1)° in (I), 65.9(1)° in (II) and 62.3(1)° in (III), respectively.
3.2. Supramolecular Features
In each of the three salts (I)–(III), the ionic components and the water molecules are linked by a combination of N—H···O and O—H···O hydrogen bonds (Table 1) to form infinite chains of edge-fused centrosymmetric rings running parallel to the [100] direction. Within these chain motifs, two types of hydrogen-bonded rings can be distinguished: according to graph-set notation [48,49,50], they can be described as and . In space group P for (I) and (III), the rings are centered at (n, 1, ½) and (½ +n, 1, ½), where n is an integer in each case, whereas in the space group P21/c for (II), they are centered at (n, ½, 0) and (½ +n, ½, 0), respectively.

Table 1.
Hydrogen-bond geometry for (I)–(III).
Figure 3 shows an exemplary scheme of intermolecular interactions forming the aforementioned chain of rings for structure (I). In the remaining structures (II)–(III), analogous chain-of-rings motifs also appear (Figure 4); however, the accompanying C—H···O interactions [and exceptionally the additional O—H···O hydrogen-bond in (III)] differentiate their crystal packings. In (I), two C—H···O interactions are observed within the basic chain motif (Figure 4a) in contrast to structure (II) (Figure 4b) where adjacent chains are linked by C3—H3A···O2(−x, −y + 1, −z + 1) along the [001] direction and by C11—H11C···O4(−x + 1, y + 1/2, −z + 1/2) along the [010] direction, respectively. In structure (III), the C2—H2A···O3(−x, −y + 1, −z + 1) intermolecular interaction connects the chain motifs along the [010] direction, whereas the O4—H4···O1(x, y, z − 1) hydrogen bond links them along the [001] direction (Figure 4c).
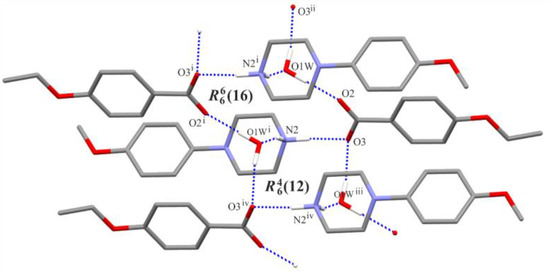
Figure 3.
Part of the crystal structure of (I), showing the formation of an infinite chain of rings. For the sake of clarity, (C)—H atoms not involved in the motif shown have been omitted. [Symmetry codes: (i) −x + 1, −y + 2, −z + 1; (ii) x−1, y, z; (iii) x + 1, y, z; (iv) −x + 2, −y + 2, −z + 1.].
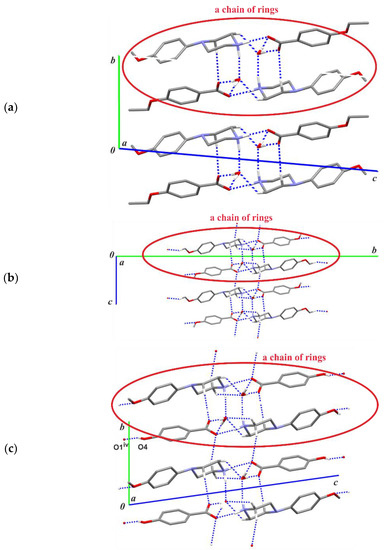
Figure 4.
Part of the crystal structures (a) of compound (I), (b) (II) and (c) (III) showing the basic chain-of-rings motifs parallel to the [100] direction, including the C—H···O intermolecular interactions. For the sake of clarity, the H atoms bonded to C atoms not involved in the motif shown have been omitted. [Symmetry code: (iv) x, y, z − 1.].
Finally, in (II)–(III) the combination of all chain sub-structures generates a tri-periodic supramolecular structure. In the case of (I), the presence of di-periodic complex sheets lying parallel to (001) can be found if one considers the C—H···π(arene) intermolecular interactions: C6—H6···Cg3, C10—H10···Cg3 and C19—H19A···Cg2 (Figure 5, Table S1). This type of interaction also occurs in (II) and (III), though the latter one vanishes in (III) due to the lack of a terminal alkoxy group in the anion. The behaviour of C−H···π (arene) interactions in the reported structures (I)–(III) agrees with observations of Umezawa et al. [51] and Nishio [52] that C−H···π contacts are much more frequently observed in organic crystals than that of O−H···π or N−H···π interactions because the CH group is more abundant than the OH or NH groups; in addition, the two latter groups prefer O or N acceptors to form ordinary hydrogen bonds.
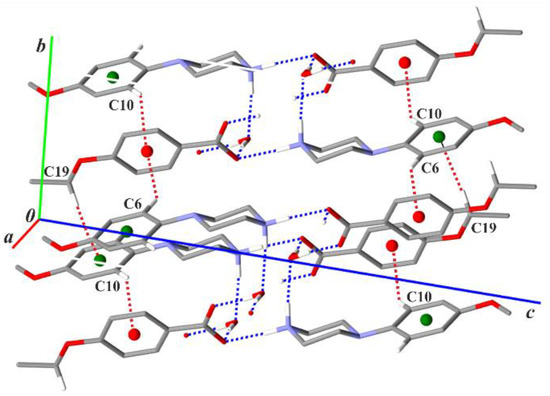
Figure 5.
Part of the crystal structure of compound (I) showing a scheme of the C—H···π(arene) intermolecular interactions (red dotted lines). Green and red balls represent the Cg2 and Cg3 centroids of the arene rings in the cations and anions, respectively. For the sake of clarity, the H atoms bonded to C atoms not involved in the motifs shown have been omitted.
3.3. Theoretical Considerations
The cation-anion pairs in the studied salts were further investigated by quantum chemical calculations in order to obtain more information about their binding interactions (Figure 6). At first, the single-point calculations for geometries taken directly from the crystals were performed (only H atom positions were normalized). Obtained energies are very comparable for the three analysed pairs; the descending order is −470.29 (II), −472.34 (I) and −473.41 (III) (all energies are given in kJ mol−1; BSSE uncorrected). The results show that the effect of the different substituent (ethoxy, methoxy, hydroxyl groups, respectively) in the anion molecule is negligible compared to the total binding energy of such pairs.
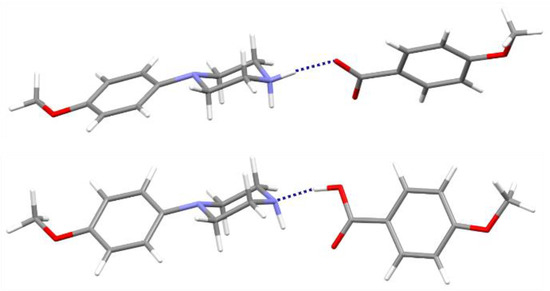
Figure 6.
The cation-anion pair linked by the N—H···O hydrogen from the analyzed salt (above) and corresponding neutral pair with the O—H···N interactions optimized in the gas-phase (below); components of structure (II) are taken as representatives.
Secondly, the molecular geometries for the discussed pairs were fully optimized. As a result, neutral molecular pairs were obtained (Figure 6) instead of ionic pairs, with the O—H···N hydrogen bond as the binding interaction (dH···N = 1.70 Å, ∠ O—H···N = 167°). It should be mentioned that the cocrystal structures of the analyzed components have not yet been determined experimentally. Energies of neutral molecular pairs are nearly eight times lower than for corresponding ionic pairs: −61.10 (I), −61.37 (II) and −61.62 (III), respectively. Interestingly, the molecular conformation of N-(4-methoxyphenyl)piperazine (MeOPP) differs both from its corresponding ionic moiety in the crystal studied and from the neutral one determined in the solid state (IHILOD refcode; [21]). Figure 7 shows the superposition of two neutral MeOPP molecules, and the main differences are observed in the mutual orientation of rings and the position of the H atom at the N2 atom. In the MeOPP crystal, the (N)—H atom occupies the equatorial position, while in the gas-phase pair with derivatives of benzoic acids it prefers the axial position.
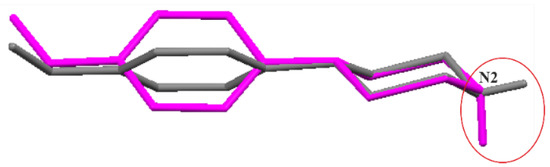
Figure 7.
Superimposed molecular structures of N-(4-methoxyphenyl)piperazine (MeOPP): a component from the optimized pair of (II) (magenta) and IHILOD (grey) [21].
Encouraged by the good agreement of the energy values for the pairs in the crystal obtained from the proposed single-point calculations and those implemented in CrystalExplorer (see Section 3.5.), we also estimated the binding energy of the graph-set ring motifs, and , which play a dominant role in supramolecular architecture of many salts with the N-(4-methoxyphenyl)piperazin-1-ium cation. The same principal chain-of-rings motifs have been found in the five structures of salts of the mentioned cations with 4-amino-, 4-chloro-, 4-fluoro-, 4-bromo- and unsubstituted benzoate monohydrate [21,22]. Each motif consists of six molecules: two cations, two anions and two water molecules related by the inversion centre, but the combination of hydrogen bonds differs (Figure 3). The binding energies of such systems are: −1275.14, −1267.18, −1264.96 for and −1274.65, −1269.39, −1267.97 for , for (I)–(III), respectively (all energies are given in kJ mol−1; BSSE uncorrected). The comparable values of energy seem to stabilize the chain motif constructed from fused ring motifs.
3.4. Hirshfeld Surface Analysis
As described in Section 3.2, the X—H···O (X = O, N, C) hydrogen bonds and the C—H···π(arene) interactions play a dominant role in the crystal packing of the investigated salts. Therefore, a fingerprint plot analysis for the cation-anion pairs as a basic building block of these salts was performed to estimate the contribution of close contacts to the Hirshfeld surface (please note that, in this way, the N—H···O interaction binding the Hirshfeld surface components is not included in this analysis). The images of the 2D fingerprint plots (Figure S1) and the Hirshfeld fingerprint breakdown with the most common contacts of (I)–(III) (Figure S2) are given in the supporting information. It can be seen that H···H close contacts dominate the Hirshfeld surface of all salts: 52.6% (I), 49.6% (II) and 46% (III). The next most dominant contact types are only C···H [24.9% (I), 25.9% (II), 27.7% (III)] and O···H [20.8% (I), 22.7% (II), 23.9% (III)]. This reveals the following relationship: a slight decrease in the contribution of H···H contacts in structures (II) and (III) compensates for a slight increase in the other two types of contacts.
3.5. Energy Frameworks
To gain additional insight into the crystal structures of the three title salts, energy calculations and visualizations of the corresponding energy frameworks were carried out with CrystalExplorer [40]. The pairwise energies calculated for the cation–anion pair from the asymmetric unit are in descending order: −473.7 (II), −474.7 (I) and −479.9 (III) (all energies are given in kJ mol−1); which is in a good agreement with energies estimated using quantum chemical calculations described in Section 2.4 and Section 3.3 The analysis of pairwise energies for salts is very complicated, since the interactions between ionic components demonstrate a long-range nature. In view of this, an energy framework approach is only used to visualize the energetic trends in the investigated salts. Figure 8 shows that all three structures are energetically di-periodic if one considers both the smaller number of interactions between layers and the difference in the magnitude of energy values between these within a layer and between layers. The energies between adjacent layers do not exceed +/−130 kJ mol−1; thus, they are roughly five times lower than within layers (please note that, in Figure 8, the energy threshold is 100 kJ mol−1; all interactions are depicted in Figure S3 in the supporting information). This may lead to the conclusion that the di-periodic supramolecular assembly involving a chain-of-rings motif stabilizes the investigated salts.
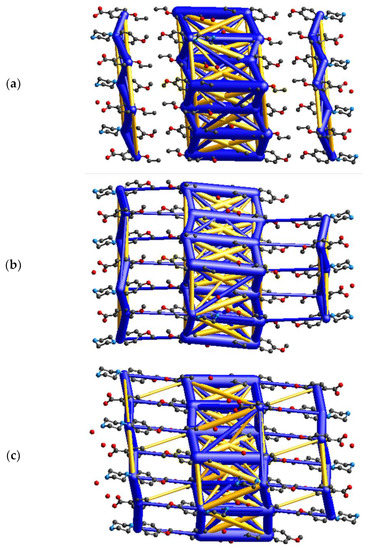
Figure 8.
Diagrams of the total energy frameworks for 10 Å cluster of (a) (I), (b) (II), and (c) (III), respectively; drawn along the crystallographic a axis. All diagrams use the same energy tube scale factor of 20 and the energy threshold of 100 kJ mol−1. The yellow cylinders represent destabilizing interactions.
4. Conclusions
Three salts with the N-(4-methoxyphenyl)piperazin-1-ium cation and benzoate anion substituted at position 4 by the ethoxy (I), methoxy (II) and hydroxy (III) groups were synthesized and characterized by FTIR, 1H NMR and X-ray single-crystal structure analysis. Their stability in the crystalline state is ensured by the combination of X—H···O (X = O, N, C) hydrogen bonds and the C—H···π(arene) interactions, which strongly determine the contribution of only three dominant types of intermolecular contacts (H···H, C···H, O···H) to the Hirshfeld surface of two ionic components.
Interestingly, the analysed structures are not typically isostructural; however, they exhibit some similar supramolecular features, e.g., the formation of a hydrogen-bonded chain of fused rings [ and ]. Furthermore, the energetic assemblies of the three salts are substantially the same; they are all di-periodic with respect to the strongest attractive and repulsive interactions within a layer built from the aforementioned chain motifs.
The role of the cation-anion pairs and hydrogen-bonded motifs has been highlighted by estimation of their binding energies using quantum chemical calculations and an energy framework approach. It was shown that, compared to salts, the neutral molecular pairs are expected to differ in molecular conformation of N-(4-methoxyphenyl)piperazine, and the donor N atom from piperazine moiety will be an acceptor of the O—H···N binding interaction.
In summary, our analysis might be useful in crystal engineering of new derivatives of N-(4-methoxyphenyl)piperazine or its analogues.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cryst12121807/s1, Table S1: Geometry of C—H···π(arene) intermolecular interactions for (I)–(III); Figure S1: Hirshfeld surface of two ionic components and corresponding fingerprint plot for (I)–(III); Figure S2: The percentage contributions of various intermolecular close contacts to the Hirshfeld surface area of two ionic components of (I)–(III); Figure S3: Diagrams of the total energy frameworks for 10 Å cluster of (I)–(III) (no energy threshold); Table S2: Final Cartesian coordinates for the gas-phase neutral pair of (I); Table S3: Final Cartesian coordinates for the gas-phase neutral pair of (II); Table S4: Final Cartesian coordinates for the gas-phase neutral pair of (III).
Author Contributions
Conceptualization, P.P. and H.S.Y.; methodology, G.P.S.; software, S.R.P. and L.C.; validation, L.C., S.R.P. and H.S.Y.; formal analysis, S.R.P. and T.R.D.; investigation, P.P. and B.K.J.; resources, V., Y.B.B. and T.R.D.; data curation, S.R.P. and L.C.; writing—original draft preparation, L.C.; writing—review and editing, L.C., H.S.Y. and S.R.P.; visualization, H.S.Y., S.R.P. and L.C.; supervision, H.S.Y. and S.R.P.; project administration, T.R.D.; funding acquisition, L.C., H.S.Y. and S.R.P. All authors have read and agreed to the published version of the manuscript.
Funding
The D8 Venture diffractometer was funded by the NSF (MRI CHE1625732), and by the University of Kentucky.
Data Availability Statement
Deposition numbers CCDC 2216869–2216871 for (I)–(III) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures (accessed on 1 November 2022).
Acknowledgments
One of the authors (PP) is grateful to B.N.M. Institute of Technology for research facilities. HSY thanks UGC for a BSR Faculty fellowship for three years.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Brito, A.F.; Moreira, L.K.S.; Menegatti, R.; Costa, E.A. Piperazine derivatives with central pharmacological activity used as therapeutic tools. Fundam. Clin. Pharmacol. 2019, 33, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Shaquiquzzaman, M.; Verma, G.; Marella, A.; Akhter, M.; Akhtar, W.; Khan, M.F.; Tasneem, S.; Alam, M.M. Piperazine scaffold: A remarkable tool in generation of diverse pharmacological agents. Eur. J. Med. Chem. 2015, 102, 487–529. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghorbani, M.; Bushra, B.A.; Zabiulla, S.; Mamatha, S.V.; Khanum, S.A. Piperazine and morpholine: Synthetic preview and pharmaceutical applications. J. Chem. Pharm. Res. 2015, 7, 281–301. [Google Scholar] [CrossRef]
- Asif, M. Piperazine and pyrazine containing molecules and their diverse pharmacological activities. Int. J. Adv. Sci. Res. 2015, 1, 5–11. [Google Scholar] [CrossRef]
- Patel, R.V.; Park, S.W. An evolving role of piperazine moieties in drug design and discovery. Mini-Rev. Med. Chem. 2013, 13, 1579–1601. [Google Scholar] [CrossRef]
- Chaudhary, P.; Kumar, R.; Verma, A.K.; Singh, D.; Yadav, V.; Chhillar, A.K.; Sharma, G.L.; Chandra, R. Synthesis and antimicrobial activity of N-alkyl and N-aryl piperazine derivatives. Bioorg. Med. Chem. 2006, 14, 1819–1826. [Google Scholar] [CrossRef]
- Jin, Y.; Huang, T.; Zhao, W.; Yang, X.; Meng, Y.; Ma, P. A study on the self-assembly mode and supramolecular framework of complexes of cucurbit[6]urils and 1-(4-methoxyphenyl)piperazine. RSC Adv. 2020, 10, 37369–37373. [Google Scholar] [CrossRef]
- Zhang, Y.; Chao, J.; Zhao, S.; Xu, P.; Wang, H.; Guo, Z.; Liu, D. Investigation on the inclusion interaction of 4-sulfonatocalix[n]arenes with 1-(4-nitrophenyl)piperazine. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2014, 132, 44–51. [Google Scholar] [CrossRef]
- König, O.; Bürgi, H.-B.; Armbruster, T.; Hulliger, J.; Weber, T. A Study in crystal engineering: Structure, crystal growth, and physical properties of a polar perhydrotriphenylene inclusion compound. J. Am. Chem. Soc. 1997, 119, 10632–10640. [Google Scholar] [CrossRef]
- Gharbi, C.; Toumi, B.; Soudani, S.; Lefebvre, F.; Kaminsky, W.; Jelsch, C.; Nasr, C.B.; Khedhiri, L. Synthesis, structural characterization, antibacterial activity, DFT computational studies and thermal analysis of two new thiocyanate compounds based on 1-phenylpiperazine. J. Mol. Struct. 2022, 1257, 132620. [Google Scholar] [CrossRef]
- Bhati, S.; Kumar, V.; Singh, S.; Singh, J. Synthesis, biological activities and docking studies of piperazine incorporated 1,3,4-oxadiazole derivatives. J. Mol. Struct. 2019, 1191, 197–205. [Google Scholar] [CrossRef]
- Upadhayaya, R.S.; Sinha, N.; Jain, S.; Kishore, N.; Chandra, R.; Arora, S.K. Optically active antifungal azoles: Synthesis and antifungal activity of (2R,3S)-2-(2,4- difluorophenyl)-3-(5-{2-[4-aryl-piperazin-1-yl]-ethyl}-tetrazol-2-yl/1-yl)-1-[1,2,4]-triazol-1-yl-butan-2-ol. Bioorg. Med. Chem. 2004, 12, 2225–2238. [Google Scholar] [CrossRef]
- Horton, D.A.; Bourne, G.T.; Smythe, M.L. The combinatorial synthesis of bicyclic privileged structures or privileged substructures. Chem. Rev. 2003, 103, 893–930. [Google Scholar] [CrossRef]
- Perez, M.; Fourrier, C.; Sigogneau, I.; Pauwels, P.J.; Palmier, C.; John, G.W.; Valentin, J.-P.; Halazy, S. Synthesis and serotonergic activity of arylpiperazide derivatives of serotonin: Potent agonists for 5-HT1D receptors. J. Med. Chem. 1995, 38, 3602–3607. [Google Scholar] [CrossRef]
- Arbo, M.D.; Bastos, M.L.; Carmo, H.F. Piperazine compounds as drugs of abuse. Drug Alcohol Depend. 2012, 122, 174–185. [Google Scholar] [CrossRef]
- Staack, R.F.; Maurer, H.H. Toxicological detection of the new designer drug 1-(4-methoxyphenyl)piperazine and its metabolites in urine and differentiation from an intake of structurally related medicaments using gas chromatography–mass spectrometry. J. Chromatogr. B 2003, 798, 333–342. [Google Scholar] [CrossRef]
- Staack, R.F.; Theobald, D.S.; Paul, D.; Springer, D.; Kraemer, T.; Maurer, H.H. In vivo metabolism of the new designer drug 1-(4-methoxyphenyl)piperazine (MeOPP) in rat and identification of the human cytochrome P450 enzymes responsible for the major metabolic step. Xenobiotica 2004, 34, 179–192. [Google Scholar] [CrossRef]
- Staack, R.F.; Maurer, H.H. Metabolism of designer drugs of abuse. Curr. Drug Metab. 2005, 6, 259–274. [Google Scholar] [CrossRef]
- Wohlfarth, A.; Weinmann, W.; Dresen, S. LC-MS/MS screening method for designer amphetamines, tryptamines, and piperazines in serum. Anal. Bioanal. Chem. 2010, 396, 2403–2414. [Google Scholar] [CrossRef]
- Nagai, F.; Nonaka, R.; Kamimura, K.S.H. The effects of non-medically used psychoactive drugs on monoamine neurotransmission in rat brain. Eur. J. Pharmacol. 2007, 559, 132–137. [Google Scholar] [CrossRef]
- Kiran Kumar, H.; Yathirajan, H.S.; Foro, S.; Glidewell, C. Crystal structures of the recreational drug N-(4-methoxyphenyl)piperazine (MeOPP) and three of its salts. Acta Crystallogr. Sect. E Crystallogr. Comm. 2020, 76, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Kiran Kumar, H.; Yathirajan, H.S.; Foro, S.; Glidewell, C. Twelve 4-(4-methoxyphenyl)piperazin-1-ium salts containing organic anions: Supra-molecular assembly in one, two and three dimensions. Acta Crystallogr. Sect. E Crystallogr. Comm. 2019, 75, 1494–1506. [Google Scholar] [CrossRef] [PubMed]
- Kiran Kumar, H.; Yathirajan, H.S.; Sagar, B.K.; Foro, S.; Glidewell, C. Six 1-aroyl-4-(4-methoxyphenyl)piperazines: Similar molecular structures but different patterns of supra molecular assembly Acta crystallogr. Sect. E Crystallogr. Comm. 2019, 75, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
- Budzikur, D.; Kinzhybalo, V.; Ślepokura, K. Crystal engineering and structural diversity of 2-aminopyridinium hypodiphosphates obtained by crystallization and dehydration. CrystEngComm 2022, 24, 4417–4429. [Google Scholar] [CrossRef]
- Barbas, R.; Portell, A.; Hunter, C.A.; Prohens, R.; Frontera, A. Combined computational/experimental investigation of new cocrystals of the drug bosentan. CrystEngComm 2022, 24, 5105–5111. [Google Scholar] [CrossRef]
- Pang, Y.; Xing, P.; Geng, X.; Zhu, Y.; Liu, F.; Wang, L. Supramolecular assemblies of 2-hydroxy-3-naphthoic acid and N-heterocycles via various strong hydrogen bonds and weak X···π (X=C–H, π) interactions. RSC Adv. 2015, 5, 40912–40923. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal engineering: From molecule to crystal. J. Am. Chem. Soc. 2013, 135, 9952–9967. [Google Scholar] [CrossRef]
- Zaworotko, M.J. Molecules to crystals, crystals to molecules... and back again? Cryst. Growth Des. 2007, 7, 4–9. [Google Scholar] [CrossRef]
- Desiraju, G.R. Supramolecular synthons in crystal engineering—A new organic synthesis. Angew. Chem. Int. Ed. Engl. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Müller-Dethlefs, K.; Hobza, P. Noncovalent interactions: A challenge for experiment and theory. Chem. Rev. 2000, 100, 143–168. [Google Scholar] [CrossRef]
- Infantes, L.; Chisholm, J.; Motherwell, S. Extended motifs from water and chemical functional groups in organicmolecular crystals. CrystEngComm 2003, 5, 480–486. [Google Scholar] [CrossRef]
- Görbitz, C.H.; Hersleth, H.-P. On the inclusion of solvent molecules in the crystal structures of organic compounds. Acta Crystallogr. B 2000, 56, 526–534. [Google Scholar] [CrossRef]
- Bruker-AXS. APEX3; Bruker-AXS Inc.: Madison, WI, USA, 2016. [Google Scholar]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Cryst. 2015, 48, 3–10. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. checkCIF validation ALERTS: What they mean and how to respond. Acta Crystallogr. Sect. E Crystallogr. Comm. 2020, E76, 1–11. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17.5; University of Western Australia: Crawley, Australia, 2017. [Google Scholar]
- Turner, M.J.; Grabowsky, S.; Jayatilaka, D.; Spackman, M.A. Accurate and efficient model energies for exploring intermolecular interactions in molecular crystals. J. Phys. Chem. Lett. 2014, 5, 4249–4255. [Google Scholar] [CrossRef]
- Turner, M.J.; Thomas, S.P.; Shi, M.W.; Jayatilaka, D.; Spackman, M.A. Energy frameworks: Insights into interaction anisotropy and the mechanical properties of molecular crystals. Chem. Commun. 2015, 51, 3735–3738. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Rev. D.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Allen, F.H.; Bruno, I.J. Bond lengths in organic and metal-organic compounds revisited: X—H bond lengths from neutron diffraction data. Acta Crystalogr. B 2010, 66, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Duax, L.; Norton, D.A. Atlas of Steroid Structure; IFI/Plenum: New York, NY, USA, 1975; Volume 1, pp. 16–22. [Google Scholar]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Etter, M.C.; MacDonald, J.C.; Bernstein, J. Graph-set analysis of hydrogen-bond patterns in organic crystals. Acta Crystallogr. B 1990, 46, 256–262. [Google Scholar] [CrossRef]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.-L. Patterns in hydrogen bonding: Functionality and graph set analysis in crystals. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Umezawa, Y.; Tsuboyama, S.; Takahashi, H.; Uzawa, J.; Nishio, M. CHπ interaction in the conformation of organic compounds. A database study. Tetrahedron 1999, 55, 10047–10056. [Google Scholar] [CrossRef]
- Nishio, M. The CH/π hydrogen bond in chemistry. Conformation, supramolecules, optical resolution and interactions involving carbohydrates. Phys. Chem. Chem. Phys. 2011, 13, 13873–13900. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).