Abstract
The cocrystallization of trans-[PtI2(NCN(CH2)5)2] and iodoform (CHI3) yields crystalline adduct trans-[PtI2(NCN(CH2)5)2]∙2CHI3, the structure of which was studied via single-crystal X-ray diffractometry (XRD). In the XRD structure of trans-[PtI2(NCN(CH2)5)2]∙2CHI3, apart from rather predictable C–H∙∙∙I hydrogen bonds (HBs) and C−I∙∙∙I halogen bonds (XBs) with the iodide ligands, we identified C–I∙∙∙Pt metal-involving XBs, where the platinum center functions as an XB acceptor (that includes a metal dz2-orbital) toward the σ-holes of I atoms of CHI3. DFT calculations (PBE-D3/jorge-TZP-DKH with plane waves in the GAPW method) were carried out in the CP2K program for isolated molecules, complex–iodoform clusters, and crystal models with periodic boundary conditions, where the noncovalent nature and the existence of the interactions were confirmed using charge analysis, Wiberg bond indexes, and QTAIM topology analysis of electron density, whereas the philicities of the noncovalent partners were proved using charge analysis, electron localization function, electron density deformation, and one-electron potential projections, as well as electron density/electrostatic potential profiles for cluster models and electrostatic potential surfaces (ρ = 0.001 a.u.) for isolated molecules.
1. Introduction
Nowadays, halogen bonding (XB) [1,2] is the most studied σ-hole interaction due to its expressed directionality [3] and application as a useful tool for constructing supramolecular clusters, chains, layers, and even 3D scaffolds, which are important in material science, crystal engineering, and supramolecular chemistry [4,5,6,7]. These σ-hole interactions can be applied in synthetic, coordination, and organometallic chemistry [8], noncovalent catalysis [9,10,11,12], polymer science [13], and drug discovery [14,15].
Organic iodine-based species with electron-withdrawing substituents are the most popular σ-hole donors [16] (or, particularly, XB donors), whereas moieties featuring atoms with lone pairs (LPs) or electron-rich π-systems are commonly used as electron donors (XB acceptors). Metal centers with sterically available filled dz2-orbital can also form XBs as nucleophiles toward various σ-hole donors [17], despite their positive charges. To date, the nucleophilicity of metal centers in metal-involving XBs has been recognized for RhI [18], NiII [19,20,21], PdII [19,21,22], PtII [21,22,23,24,25,26], Au0 [27,28], AuI [29], and AuIII [30]. In a few instances, a metal center, together with the coordinated halogen atom, functions as an integrated two-centered nucleophile toward one σ-hole, forming metal-involving bifurcated R−X∙∙∙(Cl−M) (X = Br, I; M = PtII, AuI) [25,26,29] and R−I∙∙∙(I−MII) (M = PtII, PdII) possible XBs [31]. An important finding, in the context of the current work, was revealed when we studied the X-ray structure of trans-[PtCl2(NCN(CH2)5)2]∙2CHI3, where we detected the first example of metal-involving bifurcated XB, namely C−I∙∙∙(Cl−PtII) [25].
Halogen bonds can be investigated through both experimental and theoretical studies applied to the same objects. In the vast majority of reports on metal-involving XBs, theoretical calculations of intermolecular contacts were carried out for gas-phase model clusters based on atomic coordinates experimentally determined using X-ray diffractometry (XRD) [18,19,21,22,24,25,26,29], while studies using Kohn–Sham calculations with periodic boundary conditions are still quite rare—the only example is one of our previous studies presenting such calculations for three-center Br∙∙∙(Cl−PtII) XBs involving trans-[PtCl2(NCNR2)2] (R = Me2, Et2) complexes [26]. Inspired by the observation of these interactions and the results of the periodic calculations for the XBs, we decided to expand this area. We started with the identification of new metal-involving XB with dialkylcyanamide platinum(II) complexes.
In this study, we employed the trans-[PtI2(NCN(CH2)5)2] [32] dialkylcyanamide complex as an XB acceptor toward iodoform (CHI3) utilized as an efficient XB donor (Figure 1) [25]. The complex trans-[PtI2(NCN(CH2)5)2] was cocrystallized with CHI3 to yield cocrystal 1∙2CHI3, the structure of which was studied using single-crystal XRD. In trans-[PtI2(NCN(CH2)5)2]·2CHI3, apart from a rather predictable C–H∙∙∙I hydrogen bonds (HBs) and C−I∙∙∙I XBs with the iodide ligands, we identified C−I∙∙∙Pt XBs involving dz2-orbital-donating platinum(II) center.
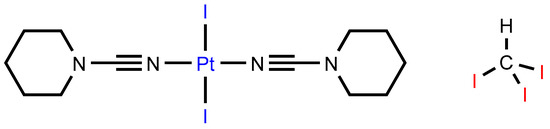
Figure 1.
Studied XB partners.
The CP2K software package [33,34,35,36,37,38,39] was chosen as a useful tool to investigate the nature of the detected intermolecular interactions in the crystals [40] since its results can be analyzed using the Multiwfn program [41]. For the comparison, the cluster models were also calculated in CP2K.
2. Materials and Methods
Analytically pure CHI3, K2[PtCl4], KI, piperidine-1-carbonitrile, and all solvents were obtained from Sigma-Aldrich (Merck, Germany) and used as received.
The NMR spectra were recorded on a Bruker AVANCE III 400 spectrometer at ambient temperature in acetone-d6 (at 400, 101, 86 MHz for 1H, 13C{1H}, and 195Pt NMR spectra, respectively) (Figures S1–S3). IR spectra were recorded on a Bruker TENSOR 27 FT-IR spectrometer (4000–200 cm–1, CsI pellets) (Figure S4). The CHN elemental analysis was carried out on a CHNS–analyzer LECO-932.
2.1. Synthesis of Complex trans-[PtI2(NCN(CH2)5)2]
A two-fold excess of KI (0.4 g, 2.4 mmol) was added to an aqueous solution of K2PtCl4 (0.5 g, 1.2 mmol, and 2.5 mL of H2O). The reaction mixture was left for 15 min until the darkening of the solution ceased. After that, a 10-fold excess of NCNC5H10 was added to the solution, and the reaction mixture was left for a week until the solution became clear. An orange precipitate was formed after one week. The resulting precipitate was filtered, washed with three portions of 3 mL of water and diethyl ether, and then dried in air at room temperature (RT). The substance was purified via column chromatography on silica gel (Merck 60 F254, CH2Cl2, first fraction). Yield: 76.7%. Anal. Calcd for C12H20N4I2Pt: C, 21.53; H, 3.01; N, 8.37. Found: C, 21.62; H, 2.94; N, 8.17%. TLC: Rf = 0.72 (eluent CH2Cl2:MeOH 50:1). IR (CsI, selected bands, cm−1): 2945 (m), 2923 (w), 2887 (w), ν(C−H); 1466 (w), 1452 (w), δ(CH2); 2288 (s), ν(C≡N); 1391 (m), ν(C−N). 1H NMR (acetone-d6, δ): 3.23 (m, 4H, NCH2), 1.58 (m, 4H, NCH2CH2), 1.48 (m, 2H, NCH2CH2CH2) ppm. 13C{1H} NMR (acetone-d6, δ): 119.10 (C≡N), 50.19 (NCH2), 25.11 (NCH2CH2), 22.87 (NCH2CH2CH2) ppm. 195Pt NMR (acetone-d6, δ): −3671.02 ppm.
2.2. Cocrystallization
Single crystals of trans-[PtI2(NCN(CH2)5)2]∙2CHI3 were obtained through the slow evaporation of a dichloromethane and ethylacetate solution (2 mL, 1:1) of a mixture of the corresponding trans-[PtI2(NCN(CH2)5)2] (0.007 mmol) and CHI3 taken in two-fold excess (0.014 mmol) at RT. The yellow crystals of trans-[PtI2(NCN(CH2)5)2]∙2CHI3 suitable for XRD were released after 3−4 d.
2.3. X-ray Structure Determination and Refinement
The suitable single crystals of trans-[PtI2(NCN(CH2)5)2] and trans-[PtI2(NCN(CH2)5)2]∙2CHI3 were studied on an Xcalibur Eos diffractometer (monochromated Mo Kα radiation, λ = 0.71073). The crystals were incubated at 100 K during data collection. Using Olex2 [42], the structures were solved with the ShelXT [43] structure solution program using intrinsic phasing and refined with the ShelXL [44] refinement package using least-square minimization. Hydrogen atoms in all structures were placed in ideally calculated positions according to neutron diffraction statistical data [45] and refined as colliding atoms with the parameters of relative isotropic displacement. The main data of crystallography and details of refinement are presented in Table S1 in Supporting Information. CCDC numbers 2252226–2252227 contain all supporting structural and refinement data.
2.4. Computational Details
Single-point DFT calculations based on experimentally determined coordinates with periodic boundary conditions for the crystal (1 × 1 × 1 cell) trans-[PtI2(NCN(CH2)5)2]∙2CHI3 model were performed in the CP2K-8.1 program [33,34,35,36,37,38,39], with a 350 Ry and a 50 Ry relative plane-wave cut-offs for the auxiliary grid using the PBE [46]-D3 [47,48] functional, with either (i) the Gaussian/augmented plane-wave (GAPW) method [49] with a full-electron jorge-TZP-DKH [50] mixed basis set with the Douglas–Kroll–Hess 2nd-order scalar relativistic calculations requested relativistic core Hamiltonian [51,52] or (ii) the mixed Gaussian/plane-wave (GPW) [53] basis set with the DZVP-MOLOPT-SR-GTH [54] basis in conjunction with the Goedecker—Teter—Hutter [55,56,57] pseudopotentials. The 1.0 × 10−6 Hartree convergence was achieved for the self-consistent field cycle in the Γ-point approximation. Similar methodologies were previously used for the investigation of related halogen-bonded systems [40,58,59,60]. For the restrained electrostatic potential (RESP) [61,62], atomic charges were calculated using the REPEAT [61] method, with constraints for all crystallographically dependent atoms to have the same RESP charges. The gas-phase studies for cluster models as well as isolated molecules were performed with experimentally determined coordinates in the same PBE-D3 [63] level of theory in CP2K with the same full-electron jorge-DZP-DKH bases with the Douglas–Kroll–Hess 2nd-order scalar relativistic calculations requested relativistic core Hamiltonian (the GAWP method) or with the DZVP-MOLOPT-SR-GTH basis in conjunction with the Goedecker—Teter—Hutter pseudopotentials (the GPW method), both in 20 × 20 × 20 Å3 boxes. The 0.500 rloc parameter was applied for Pt atoms in full-electron calculations. For pseudopotential calculations, the electron density functions (EDFs) [64] were applied for core electron modeling. The electron localization function (ELF) [65,66,67] and one-electron potential (OEP) [68,69] projection analysis, Bader [70,71,72] atoms-in-molecules topological analysis of electron density (QTAIM) [73], electron density difference (EDD) [74,75,76,77,78] projections, and electron density/electrostatic potential (ED/ESP) profile analysis [79] were performed and visualized in Multiwfn 3.8 [41,80,81]. The analysis of the electrostatic surface (ρ = 0.001 e/bohr3) [82] potentials [83,84,85] (ESP) was carried out for isolated molecules in Multiwfn 3.8 and visualized in VMD 1.9.3. [86]. In terms of natural population analysis (NPA) [87,88], atomic charges and Wiberg bond indexes [89,90,91] were calculated for cluster models using GENNBO utility in NBO 7.0 [92] based on .47 files generated in Multiwfn 3.8.
3. Results and Discussion
3.1. Electrostatic Surface Potentials
Electrostatic potentials (ESP) on the surface (ρ = 0.001 a.u.) and in projections based on the experimentally obtained coordinates (Tables S7 and S8) were calculated (PBE-D3/jorge-DZP-DKH, GAWP) for the isolated molecules trans-[PtI2(NCN(CH2)5)2] and CHI3. The σ-hole potential on CHI3 is positive (+22.9 kcal/mol) for the I sites that can be observed both on the ESP surface and ESP projection (Figure 2). In trans-[PtI2(NCN(CH2)5)2], both iodide ligands and the platinum center demonstrate significant negative potentials on all sides, with the smallest −23.8 kcal/mol and −32.8 kcal/mol values on Pt and I, respectively (Figure 2). The selected scale is optimal to convey as much information as possible about the molecular surface electrostatic potential. In this way, the opportunity of both C–I∙∙∙Pt and C–I∙∙∙I–Pt XB formation for their joint crystallization was predicted through ESP calculations.
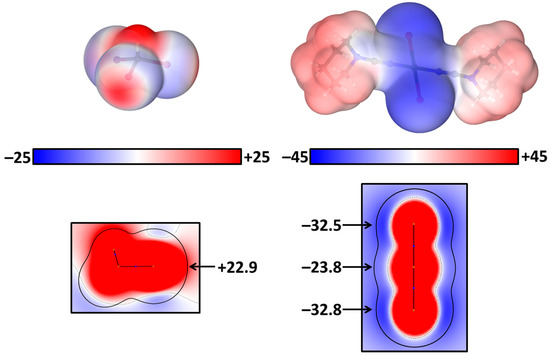
Figure 2.
ESP on surface (ρ = 0.001 a.u.) for CHI3 (left) and trans-[PtI2(NCN(CH2)5)2] (right) in kcal/mol.
3.2. Single-Crystal X-ray Diffraction Data
Complex trans-[PtI2(NCN(CH2)5)2] and iodoform were cocrystallized in a molar ratio of 1:2 via the slow evaporation of their dichloromethane/ethylacetate (1:1) mixture at RT. The cocrystallization yielded a crystalline adduct trans-[PtI2(NCN(CH2)5)2]∙2CHI3, the structure of which was studied via single-crystal XRD (Figure 3 and Table S1).
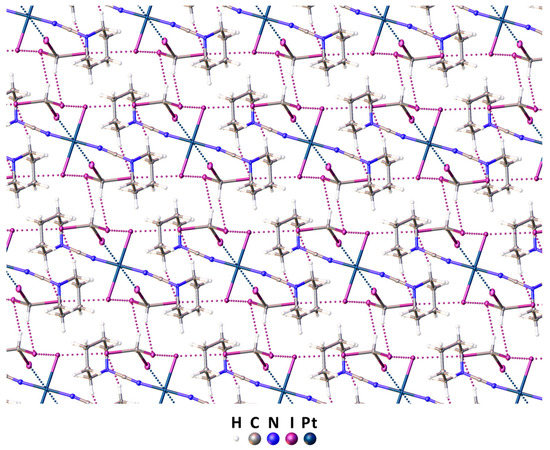
Figure 3.
The 3D network in the XRD structure of in trans-[PtI2(NCN(CH2)5)2]∙2CHI3. The contacts’ shorter Bondi vdW radii sums are presented by dotted lines. Hereinafter, thermal ellipsoids are shown with 50% probability.
The solid phase of the parent complex demonstrates the P21/c space group, whereas the P-1 space group is realized in the cocrystal (Table S1). In the XRD structure of trans-[PtI2(NCN(CH2)5)2]∙2CHI3, the bond angles around the PtII center are very close to 90° (88.78(10)° and 91.22(10)°; Table S2). The Pt1–N1–C1 and N1–C1–N2 fragments incline from the linearity, with the bond angles of 170.6(5)° and 174.9(6)°, correspondingly. The bond distances Pt1–N1, N1–C1, and C1–N2 are equal, within 3σ, to those of the parent unassociated complex trans-[PtI2(NCN(CH2)5)2] (Figure S5). The Pt1–I1 distances (2.6068(4)Å) are slightly shorter than similar distances in trans-[PtI2(NCN(CH2)5)2] (2.6216(3)Å) but typical for Pt–I bonds [93]. In trans-[PtI2(NCN(CH2)5)2]∙2CHI3, the plane of the piperidine ring has a typical chair conformation, but in contrast to the unassociated complex, it strongly deviates from the plane of the linear fragments Pt1–N1–C1 and N1–C1–N2 (Figure S6).
The molecular structure of trans-[PtI2(NCN(CH2)5)2] is represented by 2D layers (Figure S7), where the complex molecules are linked to each other via intermolecular C–H∙∙∙X (X = I, Pt) hydrogen bonds (HBs) between the Hs of the piperidine rings and the iodide ligands or the metal center of trans-[PtI2(NCN(CH2)5)2] (Table S3). In contrast to the parent unassociated complex, the crystal structure of trans-[PtI2(NCN(CH2)5)2]∙2CHI3 exhibit 3D networks (Figure 3) comprising the complexes and CHI3 molecules, which are linked to each other via intermolecular C–I∙∙∙X (X = I, Pt) contacts (Table 1) and C–H∙∙∙I HBs between the H atoms of the piperidine ring and I centers of the iodoform (Table 2). At the same time, CHI3 molecules form intermolecular C1S–H1S···I3S HBs between each other.

Table 1.
Parameters of XBs in trans-[PtI2(NCN(CH2)5)2]∙2CHI3.
Table 2.
Parameters of HBs in trans-[PtI2(NCN(CH2)5)2]∙2CHI3.
The crystal structure of trans-[PtI2(NCN(CH2)5)2]∙2CHI3 features two molecules of CHI3 per one molecule of trans-[PtI2(NCN(CH2)5)2], where each complex molecule is surrounded by six CHI3 molecules (Figure 4). The molecular structure of trans-[PtI2(NCN(CH2)5)2]∙2CHI3 includes C–I∙∙∙I–Pt short contacts formed between two iodide ligands of 1 and I centers of CHI3, which comprise from 89% to 90% of the vdW radii sum defined by Bondi [94] (ΣvdW; 2RvdW(I) = 3.96 Å) (Figure 4 and Table 1). The angles around the iodine centers of CHI3 are close to 180° and are far from linear around the iodide ligands of 1. In turn, an inspection of the calculated ESP data for trans-[PtI2(NCN(CH2)5)2] (Figure 2) reveals that the minimal ESP (−32.8 kcal/mol) is located on the I atoms of the Pt–I bonds. All these observations indicate the C–I∙∙∙I–Pt short contacts should be treated as XBs defined by IUPAC or as “type II” halogen–halogen interactions [1,95], where the iodide ligands act as nucleophilic centers toward iodoform σ-holes.
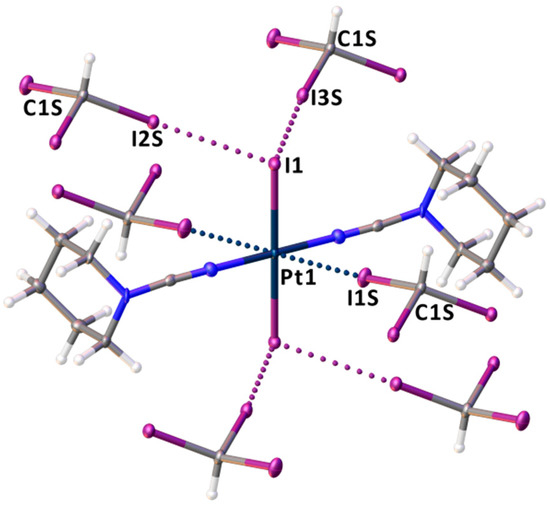
Figure 4.
The environment of trans-[PtI2(NCN(CH2)5)2] in trans-[PtI2(NCN(CH2)5)2]∙2CHI3, XBs are presented by dotted lines.
Besides C–I∙∙∙I–Pt XBs, the XRD structure of trans-[PtI2(NCN(CH2)5)2]·2CHI3 exhibits C–I∙∙∙Pt short contacts between the I centers of CHI3 and the metal center of trans-[PtI2(NCN(CH2)5)2] (Figure 3). The I1S∙∙∙Pt1 distances (3.5131(5) Å) are less than ΣvdW(I + Pt) = 3.73 Å, and the corresponding angles around I1S are close to linear (176.64(13)°). Thus, the observed interactions can be treated as C–I∙∙∙[dz2-PtII] metal-involving XBs, where PtII functions as a dz2-orbital-centered nucleophile (dz2-nucleophile). The nucleophilicity of metal complexes in similar X∙∙∙[dz2-M] XBs (X = I, Br) has been previously revealed for such metal centers as RhI [18], NiII [19,20,21], PdII [19,21,22], and PtII [21,22,23,24,25,26].
Other types of noncovalent interactions in trans-[PtI2(NCN(CH2)5)2]∙2CHI3 are represented by C–H∙∙∙I hydrogen bonds (HBs) between the H atoms of the piperidine ring and the I centers of the iodoform (Table S3).
3.3. Theoretical Consideration
More information about the nature of interactions in cocrystals can be obtained through DFT calculations, which were performed in this work using two methodologies. The 1 × 1 × 1 cell (Figure 5 and Table S4 for Cartesian coordinates) containing 49 atoms was used for the calculations with periodic boundary conditions (the crystal model). Since the CP2K software package was chosen for the implementation of the calculations, no symmetry elements were applied in this system.
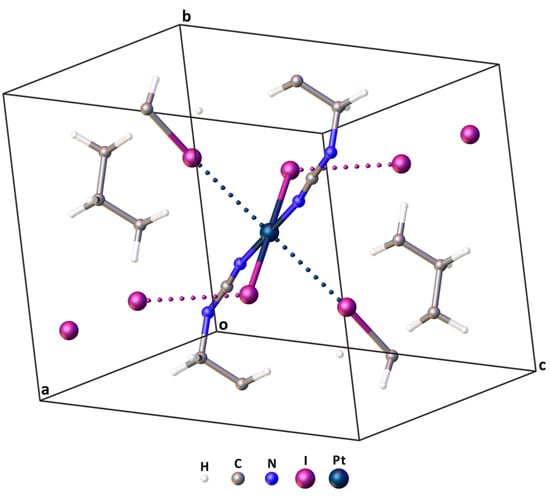
Figure 5.
The 1 × 1 × 1 cell applied for DFT calculations with periodic boundary conditions. XBs within the cell are presented by dotted lines.
For gas-phase cluster calculations, the closest environment of trans-[PtI2(NCN(CH2)5)2] in the adduct, or the so-called second sphere [96], was used for cluster construction (Figure 4; for Cartesian coordinates, see Table S9). The cluster center, i.e., the Pt atom, was placed in the center of a 20 × 20 × 20 Å3 box. Note that both the crystal and cluster models were based on the experimentally obtained atomic coordinates.
The CP2K software package was chosen for the implementation of the calculations under periodic boundary conditions using a PBE-D3 with a jorge-DZP-DKH basis set using the Douglas–Kroll–Hess second-order (DKH2) scalar relativistic calculations requested relativistic core Hamiltonian (Gaussian and augmented plane waves) or with DZVP-MOLOPT-SR-GTH with the Goedecker—Teter—Hutter pseudopotentials (Gaussian and plane waves). For the comparison, the same functional and basis sets were applied in the cluster model.
The Bader quantum theory of atoms-in-molecules (QTAIM) method allows for the confirmation of the formation of the observed interactions and their noncovalent nature. The QTAIM topological analysis was performed for both the crystal and cluster models and revealed the presence of bond critical points (3, −1) (BCP) for the metal-involving and iodine∙∙∙iodine short contacts (Table 3). Small and negative values of sign(λ2)ρ at the BCPs can be used as evidence for the noncovalent and attractive nature of the XBs, correspondingly. Close to zero and positive values of energy density H(r) (0.000–0.002 Hartrees) and the relation of the potential energy density module |V(r)| and the Lagrangian kinetic energy G(r) (|V(r)|/G(r) ≤ 1) [72] on the BCPs allow for the identification of these interactions as typically noncovalent.
Table 3.
Sign(λ2)ρ (in e/bohr3), Laplacian ∇2ρ (in e/bohr5), potential energy density V(r), Lagrangian kinetic energy G(r), and energy density H(r) (in Hartree) at the bond critical points (3, −1), corresponding to different noncovalent interactions in trans-[PtI2(NCN(CH2)5)2]·2CHI3.
Another way to confirm the existence and noncovalent nature of the interactions under consideration is the Wiberg bond indexes (WBIs) [89,90,91] in the natural atomic orbital partitioning scheme [87,88], which was calculated for the (trans-[PtI2(NCN(CH2)5)2])∙(CHI3)6 cluster model in both relativistic and pseudopotential calculations (Table 4). In both calculation schemes, the WBIs for halogen bonds are in the 0.04–0.10 range, which is less than the typical indexes for coordination [97] or coordinative [98] halogen bonds [99].
Table 4.
Sums of NPA charges in e for CHI3 molecules, forming different interactions in (trans-[PtI2(NCN(CH2)5)2])∙(CHI3)6.
In the (trans-[PtI2(NCN(CH2)5)2])∙(CHI3)6 cluster model, the charge transfer from the complex to iodoform molecules was also calculated using the summation of the atomic charges in the natural atomic partitioning scheme (natural population analysis, NPA). The sums of NPA atomic charges are negative for all three types of CHI3 molecules (Figure 4), corresponding to the three types of XBs (Table 5). The I2CH–I∙∙∙I–Pt halogen bonds were also confirmed with the corresponding charge transfer.
Table 5.
Sums of NPA charges in e for CHI3 molecules, forming different interactions in (trans-[PtI2(NCN(CH2)5)2])∙(CHI3)6.
In the crystal model, the charge sums were also calculated as restricted electrostatic potential charges using the REPEAT methodology. In both models (with DKH2 or pseudopotentials) the charge sums per iodoform molecule are negative and almost the same (−0.230 and −0.219 e, respectively), which confirms the total electrophilicity of CHI3 toward trans-[PtI2(NCN(CH2)5)2] in trans-[PtI2(NCN(CH2)5)2]∙2CHI3.
The electron localization function (ELF), which is a derivative of the electron wavefunctions, electron density, and its gradient, allows for the determination of the areas where shared and unshared electron pairs are located. A conjunction of the ELF projections and QTAIM critical points, bond, and interbasin paths is represented in the upper part of Figure 6. The OEP is steric potential with a negative value, which, like the ELF, locates shared and unshared electron pair areas but depends only on electron density and its derivatives. The same OEP + QTAIM combination can also be found in the lower part of Figure 6.
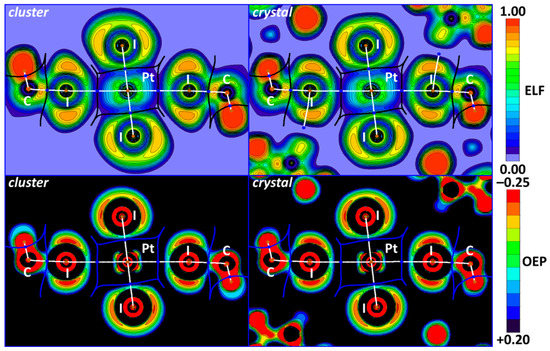
Figure 6.
OEP (upper) and ELF (lower) projections for the I2HC−I1S∙∙∙Pt∙∙∙I1S−CHI2 XBs in cluster and crystal models and QTAIM white bond paths, brown nuclear, blue bond, and orange ring critical points, and blue or black interbasin paths.
In the cluster and crystal models, the I∙∙∙Pt bond paths (Figure 6) pass through the depletion I ELF areas in the iodoform. These observations confirm the electrophilicity of iodine atoms toward the metal center in the I∙∙∙Pt interactions. The same analysis can be performed for the OEP projections. Notably, the location of critical bond points and the bond paths is the same in both the cluster and crystal models, which indicates the similarity of the nature of noncovalent interactions.
The nucleophilicity of the Pt center toward iodine atoms in CHI3 was also proved through a comparison of the electron density (ED) and electrostatic potential (ESP) profiles along the I∙∙∙Pt bond paths in the cluster models. This analysis [79,100,101], which has already been applied for halogen bonds including metals [18,21,29,30], shows the roles of noncovalent partners since the ED minimum is closer to the electrophile nucleus, the ESP minimum is near the nucleophile nucleus, and the area between the minima corresponds to nucleophile lone pair. Accordingly, in both relativistic and pseudopotential calculations (Figure 7), the ESP minimum along the I∙∙∙Pt bond path is closer to the Pt nucleus; therefore, the metal center can be treated as a nucleophile in the XB.
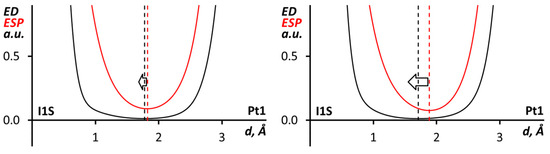
Figure 7.
ED and ESP profiles along the I∙∙∙Pt bond path in (trans-[PtI2(NCN(CH2)5)2])∙(CHI3)6 cluster model with relativistic (left) or pseudopotential (right) calculations.
Another way to confirm the Pt nucleophilicity toward iodine in iodoform is the electron density difference (EDD, also known as electron density shift) calculations [74,75,76,77,78]. This method is well known for cluster calculations (Figure 8A), when the electron density of the isolated molecules is subtracted from the cluster electron density, showing electron gain and loss under cluster formation with preserved geometries. In this work, we first performed analogous calculations for the crystal model (Figure 8B), where the electron densities of hypothetical cells with atoms from only the first (Table S5) or second (Table S6) type of molecules were used as subtrahends for the electron density from an original crystal model (Table S4).
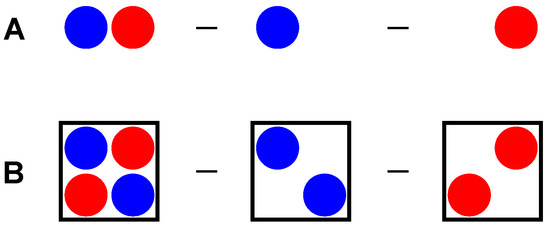
Figure 8.
Principal scheme of EDD calculations for cluster (A) and crystal (B) models.
EDD projections (Figure 9) were drawn for both pseudopotential and full-electron relativistic calculations for the cluster and crystal models, and they were performed together with the topological analysis of electron density (QTAIM). In all cases, sufficient electron lost areas (red) can be found around Pt nuclear points, whereas the electron gain areas (blue) are mostly around the C atoms of iodoform molecules. These observations can be interpreted as electron charge transfer from Pt dz2-orbital to LUMOs of iodoform molecules, in accordance with NPA and RESP charge sums for the cluster and crystal models, respectively. In the case of the cluster models, the electron concentration on the C atoms of CHI3 is caused only by the C−I∙∙∙Pt halogen bonds. Thus, the EDD projections can also be viewed as the last evidence for XB formation and the nucleophilicity of the PtII center toward iodoform molecules.
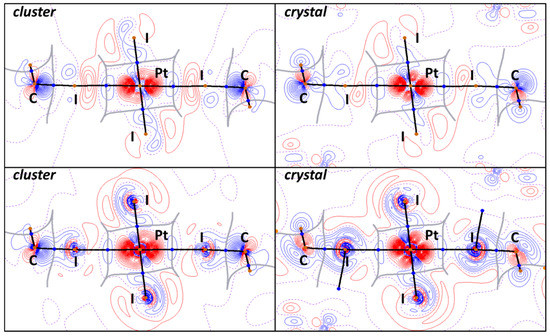
Figure 9.
EDD projections for the I2HC−I1S∙∙∙Pt∙∙∙I1S−CHI2 XBs in cluster and crystal models with QTAIM black bond paths, brown nuclear, blue bond, orange ring critical points, and gray interbasin paths for calculations performed with GTH pseudopotentials (upper) or full-electron bases with relativism (lower). Contour lines were drawn from −0.02 to 0.02 e with 0.0005 steps; lines with negative values are red, lines with positive values are blue, and zero lines are purple and dotted.
4. Conclusions
In this work, we demonstrated that the platinum(II) iodide dialkylcyanamide complex trans-[PtI2(NCN(CH2)5)2] can be cocrystallized with iodoform, forming the cocrystal trans-[PtI2(NCN(CH2)5)2]∙2CHI3 (Figure 4). Upon the analysis of noncovalent forces in the XRD structure of trans-[PtI2(NCN(CH2)5)2]∙2CHI3, in addition to a rather conventional C–H∙∙∙I HBs and C−I∙∙∙I XBs, we recognized C–I∙∙∙PtII metal-involving XBs, where the platinum(II) center functions as a dz2-orbital-donating nucleophile.
The nature of the observed contacts was theoretically confirmed using DFT calculations performed with two complementary methodologies: (i) single-point calculations (cluster model) and (ii) calculations with periodic boundary conditions (crystal model), both performed using the CP2K program with pseudopotential or full-electron relativistic bases. DFT calculations (PBE-D3/jorge-DZP-DKH, GAWP) for the isolated molecules revealed the σ-holes (ρ = 0.001 e/bohr3) on the I atoms of CHI3 and negative electrostatic potentials on the Pt center and the iodide ligands of trans-[PtI2(NCN(CH2)5)2]. The noncovalent nature and the existence of the C–I∙∙∙I–PtII and C–I∙∙∙PtII XBs were verified via the topological analysis of electron density within the QTAIM method and Wiberg bond indexes, whereas natural charges for the cluster models and RESP charges for the crystal models, as well as ELF, OEP, EDD projections, and ED/ESP profiles, confirmed the nucleophilicity of the platinum(II) center. Although CP2K has already been used to confirm σ-hole interactions, in this work, it was applied for the first time to investigate metal-involving XBs. The EDD methodology was also used, for the first time, in calculations with periodic boundary conditions.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cryst13050712/s1, Figure S1: 1H NMR spectrum of trans-[PtI2(NCN(CH2)5)2] (400 MHz, acetone-d6, 298 K); Figure S2: 13C NMR spectrum of trans-[PtI2(NCN(CH2)5)2] (101 MHz, acetone-d6, 298 K); Figure S3: 195Pt NMR spectrum of trans-[PtI2(NCN(CH2)5)2] (86 MHz, acetone-d6, 298 K); Figure S4: IR spectrum of trans-[PtI2(NCN(CH2)5)2] (CsI); Figure S5: XRD structure of trans-[PtI2(NCN(CH2)5)2]. Thermal ellipsoids are shown with 50% probability; Figure S6: Conformation of the complexes in trans-[PtI2(NCN(CH2)5)2] (top) and trans-[PtI2(NCN(CH2)5)2]·2CHI3 (bottom); Figure S7: The 2D layers in the XRD structure of trans-[PtI2(NCN(CH2)5)2]: (a) view along the a-axis; (b) view along the b-axis; (c) ) view along the c-axis; Table S1: Crystal data and structure refinement for trans-[PtI2(NCN(CH2)5)2]∙2CHI3; Table S2: Selected bond distances and angles in the XRD structures of trans-[PtI2(NCN(CH2)5)2] and trans-[PtI2(NCN(CH2)5)2]·2CHI3; Table S3: Parameters of HBs in the XRD structure of trans-[PtI2(NCN(CH2)5)2]; Tables S4–S10: Cartesian coordinates for models.
Author Contributions
Conceptualization, A.A.E., D.M.I. and S.A.B.; methodology, A.A.E. and D.M.I.; investigation, M.A.K., A.M.C. and I.S.A.; writing—original draft preparation, R.I.K., O.A.M., A.A.E. and D.M.I.; writing—review and editing, A.A.E. and D.M.I.; visualization, A.M.C., R.I.K., E.S.O., A.V.R., O.A.M. and D.M.I.; supervision, A.A.E. and D.M.I.; project administration, A.A.E., D.M.I. and S.A.B. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Russian Science Foundation (project 22-73-10021, synthetic and crystal engineering studies; project 21-73-00059, theoretical calculations).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data underlying the results presented in this paper are not publicly available at this time but may be obtained from the authors upon reasonable request.
Acknowledgments
Physicochemical measurements were performed at the Center for XRD Studies, Center for Magnetic Resonance, and Center for Chemical Analysis and Materials Research (all belonging to St Petersburg State University).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the Halogen Bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J. An Overview of Strengths and Directionalities of Noncovalent Interactions: σ-Holes and π-Holes. Crystals 2019, 9, 165. [Google Scholar] [CrossRef]
- Wang, H.; Bisoyi, H.K.; Urbas, A.M.; Bunning, T.J.; Li, Q. The Halogen Bond: An Emerging Supramolecular Tool in the Design of Functional Mesomorphic Materials. Chem. A Eur. J. 2019, 25, 1369–1378. [Google Scholar] [CrossRef]
- Mukherjee, A.; Sanz-Matias, A.; Velpula, G.; Waghray, D.; Ivasenko, O.; Bilbao, N.; Harvey, J.N.; Mali, K.S.; De Feyter, S. Halogenated Building Blocks for 2D Crystal Engineering on Solid Surfaces: Lessons from Hydrogen Bonding. Chem. Sci. 2019, 10, 3881–3891. [Google Scholar] [CrossRef] [PubMed]
- Gilday, L.C.; Robinson, S.W.; Barendt, T.A.; Langton, M.J.; Mullaney, B.R.; Beer, P.D. Halogen Bonding in Supramolecular Chemistry. Chem. Rev. 2015, 115, 7118–7195. [Google Scholar] [CrossRef]
- Usoltsev, A.N.; Adonin, S.A.; Novikov, A.S.; Abramov, P.A.; Sokolov, M.N.; Fedin, V.P. Chlorotellurate(iv) Supramolecular Associates with “Trapped” Br2: Features of Non-Covalent Halogen⋯halogen Interactions in Crystalline Phases. CrystEngComm 2020, 22, 1985–1990. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Kopylovich, M.N.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Non-Covalent Interactions in the Synthesis of Coordination Compounds: Recent Advances. Coord. Chem. Rev. 2017, 345, 54–72. [Google Scholar] [CrossRef]
- Tepper, R.; Schubert, U.S. Halogen Bonding in Solution: Anion Recognition, Templated Self-Assembly, and Organocatalysis. Angew. Chem. Int. Ed. 2018, 57, 6004–6016. [Google Scholar] [CrossRef]
- Bulfield, D.; Huber, S.M. Halogen Bonding in Organic Synthesis and Organocatalysis. Chem. A Eur. J. 2016, 22, 14434–14450. [Google Scholar] [CrossRef] [PubMed]
- Mahmudov, K.T.; Gurbanov, A.V.; Guseinov, F.I.; Guedes da Silva, M.F.C. Noncovalent Interactions in Metal Complex Catalysis. Coord. Chem. Rev. 2019, 387, 32–46. [Google Scholar] [CrossRef]
- Benz, S.; Poblador-Bahamonde, A.I.; Low-Ders, N.; Matile, S. Catalysis with Pnictogen, Chalcogen, and Halogen Bonds. Angew. Chem. Int. Ed. 2018, 57, 5408–5412. [Google Scholar] [CrossRef]
- Berger, G.; Soubhye, J.; Meyer, F. Halogen Bonding in Polymer Science: From Crystal Engineering to Functional Supramolecular Polymers and Materials. Polym. Chem. 2015, 6, 3559–3580. [Google Scholar] [CrossRef]
- Dalpiaz, A.; Pavan, B.; Ferretti, V. Can Pharmaceutical Co-Crystals Provide an Opportunity to Modify the Biological Properties of Drugs? Drug Discov. Today 2017, 22, 1134–1138. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.S. Halogen Bonding in Medicinal Chemistry: From Observation to Prediction. Fut. Med. Chem. 2017, 9, 637–640. [Google Scholar] [CrossRef]
- Gorokh, I.D.; Adonin, S.A.; Novikov, A.S.; Usoltsev, A.N.; Plyusnin, P.E.; Korolkov, I.V.; Sokolov, M.N.; Fedin, V.P. Halobismuthates with 3-Iodopyridinium Cations: Halogen Bonding-Assisted Crystal Packing. Polyhedron 2019, 166, 137–140. [Google Scholar] [CrossRef]
- Ivanov, D.M.; Bokach, N.A.; Kukushkin, V.Y.; Frontera, A. Metal Centers as Nucleophiles: Oxymoron of Halogen Bond-Involving Crystal Engineering. Chem. A Eur. J. 2022, 28, e202103173. [Google Scholar] [CrossRef]
- Eliseeva, A.A.; Ivanov, D.M.; Rozhkov, A.V.; Ananyev, I.V.; Frontera, A.; Kukushkin, V.Y. Bifurcated Halogen Bonding Involving Two Rhodium(I) Centers as an Integrated σ-Hole Acceptor. JACS Au 2021, 1, 354–361. [Google Scholar] [CrossRef]
- Kucheriv, O.I.; Shylin, S.I.; Ksenofontov, V.; Dechert, S.; Haukka, M.; Fritsky, I.O.; Gural’skiy, I.A. Spin Crossover in Fe(II)–M(II) Cyanoheterobimetallic Frameworks (M = Ni, Pd, Pt) with 2-Substituted Pyrazines. Inorg. Chem. 2016, 55, 4906–4914. [Google Scholar] [CrossRef]
- Bikbaeva, Z.M.; Ivanov, D.M.; Novikov, A.S.; Ananyev, I.V.; Bokach, N.A.; Kukushkin, V.Y. Electrophilic–Nucleophilic Dualism of Nickel(II) toward Ni···I Noncovalent Interactions: Semicoordination of Iodine Centers via Electron Belt and Halogen Bonding via σ-Hole. Inorg. Chem. 2017, 56, 13562–13578. [Google Scholar] [CrossRef]
- Zelenkov, L.E.; Eliseeva, A.A.; Baykov, S.V.; Suslonov, V.V.; Galmés, B.; Frontera, A.; Kukushkin, V.Y.; Ivanov, D.M.; Bokach, N.A. Electron Belt-to-s-Hole Switch of Noncovalently Bound Iodine(I) Atoms in Dithiocarbamate Metal Complexes. Inorg. Chem. Front. 2021, 8, 2505–2517. [Google Scholar] [CrossRef]
- Baykov, S.V.; Dabranskaya, U.; Ivanov, D.M.; Novikov, A.S.; Boyarskiy, V.P. Pt/Pd and I/Br Isostructural Exchange Provides Formation of C–I···Pd, C–Br···Pt, and C–Br···Pd Metal-Involving Halogen Bonding. Cryst. Growth Des. 2018, 18, 5973–5980. [Google Scholar] [CrossRef]
- Rozhkov, A.V.; Ivanov, D.M.; Novikov, A.S.; Ananyev, I.V.; Bokach, N.A.; Kukushkin, V.Y. Metal-Involving Halogen Bond Ar–I⋯[Dz2PtII] in a Platinum Acetylacetonate Complex. CrystEngComm 2020, 22, 554–563. [Google Scholar] [CrossRef]
- Katlenok, E.A.; Haukka, M.; Levin, O.V.; Frontera, A.; Kukushkin, V.Y. Supramolecular Assembly of Metal Complexes by (Aryl)I⋅⋅⋅dz2[PtII] Halogen Bond. Chem. A Eur. J. 2020, 26, 7692–7701. [Google Scholar] [CrossRef]
- Ivanov, D.M.; Novikov, A.S.; Ananyev, I.V.; Kirina, Y.V.; Kukushkin, V.Y. Halogen Bonding Between Metal Center and Halocarbon. Chem. Commun. 2016, 52, 5565–5568. [Google Scholar] [CrossRef] [PubMed]
- Dabranskaya, U.; Ivanov, D.M.; Novikov, A.S.; Matveychuk, Y.V.; Bokach, N.A.; Kukushkin, V.Y. Metal-Involving Bifurcated Halogen Bonding C–Br···η2(Cl–Pt). Cryst. Growth Des. 2019, 19, 1364–1376. [Google Scholar] [CrossRef]
- Blakey, I.; Merican, Z.; Rintoul, L.; Chuang, Y.-M.; Jack, K.S.; Micallef, A.S. Interactions of Iodoperfluorobenzene Compounds with Gold Nanoparticles. Phys. Chem. Chem. Phys. 2012, 14, 3604–3611. [Google Scholar] [CrossRef] [PubMed]
- Komoto, Y.; Fujii, S.; Hara, K.; Kiguchi, M. Single Molecular Bridging of Au Nanogap Using Aryl Halide Molecules. J. Phys. Chem. C 2013, 117, 24277–24282. [Google Scholar] [CrossRef]
- Aliyarova, I.S.; Tupikina, E.Y.; Ivanov, D.M.; Kukushkin, V.Y. Metal-Involving Halogen Bonding Including Gold(I) as a Nucleophilic Partner. The Case of Isomorphic Dichloroaurate(I)·Halomethane Cocrystals. Inorg. Chem. 2022, 61, 2558–2567. [Google Scholar] [CrossRef] [PubMed]
- Aliyarova, I.S.; Tupikina, E.Y.; Soldatova, N.S.; Ivanov, D.M.; Postnikov, P.S.; Yusubov, M.; Kukushkin, V.Y. Halogen Bonding Involving Gold Nucleophiles in Different Oxidation States. Inorg. Chem. 2022, 61, 15398–15407. [Google Scholar] [CrossRef] [PubMed]
- Bulatova, M.; Ivanov, D.M.; Rautiainen, J.M.; Kinzhalov, M.A.; Truong, K.-N.; Lahtinen, M.; Haukka, M. Studies of Nature of Uncommon Bifurcated I–I···(I–M) Metal-Involving Noncovalent Interaction in Palladium(II) and Platinum(II) Isocyanide Cocrystals. Inorg. Chem. 2021, 60, 13200–13211. [Google Scholar] [CrossRef] [PubMed]
- Mindich, A.L.; Haukka, M.; Bokach, N.A. Synthesis and Crystal Structure of Trans -Diiodobis(Piperidine-1-Carbonitrile)Platinum(II). J. Crystallogr. 2013, 2013, 312310. [Google Scholar] [CrossRef]
- Hutter, J.; Iannuzzi, M.; Schiffmann, F.; VandeVondele, J. cp2k: Atomistic simulations of condensed matter systems. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 15–25. [Google Scholar] [CrossRef]
- Kühne, T.D.; Iannuzzi, M.; Del Ben, M.; Rybkin, V.V.; Seewald, P.; Stein, F.; Laino, T.; Khaliullin, R.Z.; Schütt, O.; Schiffmann, F.; et al. CP2K: An Electronic Structure and Molecular Dynamics Software Package—Quickstep: Efficient and Accurate Electronic Structure Calculations. J. Chem. Phys. 2020, 152, 194103. [Google Scholar] [CrossRef] [PubMed]
- Frigo, M.; Johnson, S.G. The Design and Implementation of FFTW3. Proc. IEEE 2005, 93, 216–231. [Google Scholar] [CrossRef]
- Vandevondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Quickstep: Fast and Accurate Density Functional Calculations Using a Mixed Gaussian and Plane Waves Approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef]
- Borštnik, U.; VandeVondele, J.; Weber, V.; Hutter, J. Sparse Matrix Multiplication: The Distributed Block-Compressed Sparse Row Library. Parallel Comput. 2014, 40, 47–58. [Google Scholar] [CrossRef]
- Schütt, O.; Messmer, P.; Hutter, J.; VandeVondele, J. GPU-Accelerated Sparse Matrix-Matrix Multiplication for Linear Scaling Density Functional Theory. In Electronic Structure Calculations on Graphics Processing Units; John Wiley & Sons, Ltd.: Chichester, UK, 2016; pp. 173–190. [Google Scholar]
- Goerigk, L.; Hansen, A.; Bauer, C.; Ehrlich, S.; Najibi, A.; Grimme, S. A Look at the Density Functional Theory Zoo with the Advanced GMTKN55 Database for General Main Group Thermochemistry, Kinetics and Noncovalent Interactions. Phys. Chem. Chem. Phys. 2017, 19, 32184–32215. [Google Scholar] [CrossRef]
- Zelenkov, L.E.; Ivanov, D.M.; Tyumentsev, I.A.; Izotova, Y.A.; Kukushkin, V.Y.; Bokach, N.A. Structure-Directing Interplay between Tetrel and Halogen Bonding in Co-Crystal of Lead(II) Diethyldithiocarbamate with Tetraiodoethylene. Int. J. Mol. Sci. 2022, 23, 11870. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Allen, F.H.; Bruno, I.J. Bond Lengths in Organic and Metal-Organic Compounds Revisited: X–H Bond Lengths from Neutron Diffraction Data. Acta Crystallogr. Sect. B Struct. Sci. 2010, 66, 380–386. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Lippert, G.; Hutter, J.; Parrinello, M. The Gaussian and Augmented-Plane-Wave Density Functional Method for Ab Initio Molecular Dynamics Simulations. Theor. Chem. Acc. 1999, 103, 124–140. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Barysz, M.; Sadlej, A.J. Two-Component Methods of Relativistic Quantum Chemistry: From the Douglas-Kroll Approximation to the Exact Two-Component Formalism. J. Mol. Struct. Theochem. 2001, 573, 181–200. [Google Scholar] [CrossRef]
- Reiher, M. Relativistic Douglas-Kroll-Hess Theory. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 139–149. [Google Scholar] [CrossRef]
- Lippert, G.; Hutter, J.; Parrinello, M. A Hybrid Gaussian and Plane Wave Density Functional Scheme. Mol. Phys. 1997, 92, 477–488. [Google Scholar] [CrossRef]
- VandeVondele, J.; Hutter, J. Gaussian Basis Sets for Accurate Calculations on Molecular Systems in Gas and Condensed Phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef]
- Goedecker, S.; Teter, M.; Hutter, J. Separable Dual-Space Gaussian Pseudopotentials. Phys. Rev. B 1996, 54, 1703–1710. [Google Scholar] [CrossRef] [PubMed]
- Hartwigsen, C.; Goedecker, S.; Hutter, J. Relativistic Separable Dual-Space Gaussian Pseudopotentials from H to Rn. Phys. Rev. B 1998, 58, 3641–3662. [Google Scholar] [CrossRef]
- Krack, M. Pseudopotentials for H to Kr Optimized for Gradient-Corrected Exchange-Correlation Functionals. Theor. Chem. Acc. 2005, 114, 145–152. [Google Scholar] [CrossRef]
- Kinzhalov, M.A.; Ivanov, D.M.; Melekhova, A.A.; Bokach, N.A.; Gomila, R.M.; Frontera, A.; Kukushkin, V.Y. Chameleonic Metal-Bound Isocyanides: A π-Donating CuI-Center Imparts Nucleophilicity to the Isocyanide Carbon toward Halogen Bonding. Inorg. Chem. Front. 2022, 9, 1655–1665. [Google Scholar] [CrossRef]
- Nieland, E.; Komisarek, D.; Hohloch, S.; Wurst, K.; Vasylyeva, V.; Weingart, O.; Schmidt, B.M. Supramolecular Networks by Imine Halogen Bonding. Chem. Commun. 2022, 58, 5233–5236. [Google Scholar] [CrossRef]
- Sokolova, E.V.; Kinzhalov, M.A.; Smirnov, A.S.; Cheranyova, A.M.; Ivanov, D.M.; Kukushkin, V.Y.; Bokach, N.A. Polymorph-Dependent Phosphorescence of Cyclometalated Platinum(II) Complexes and Its Relation to Non-Covalent Interactions. ACS Omega 2022, 7, 34454–34462. [Google Scholar] [CrossRef] [PubMed]
- Campañá, C.; Mussard, B.; Woo, T.K. Electrostatic Potential Derived Atomic Charges for Periodic Systems Using a Modified Error Functional. J. Chem. Theory Comput. 2009, 5, 2866–2878. [Google Scholar] [CrossRef] [PubMed]
- Golze, D.; Hutter, J.; Iannuzzi, M. Wetting of Water on Hexagonal Boron Nitride@Rh(111): A QM/MM Model Based on Atomic Charges Derived for Nano-Structured Substrates. Phys. Chem. Chem. Phys. 2015, 17, 14307–14316. [Google Scholar] [CrossRef]
- Zhao, D.; Liu, S.; Rong, C.; Zhong, A.; Liu, S. Toward Understanding the Isomeric Stability of Fullerenes with Density Functional Theory and the Information-Theoretic Approach. ACS Omega 2018, 3, 17986–17990. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Cai, Z.; Wang, J.; Xin, K. An Open Library of Relativistic Core Electron Density Function for the QTAIM Analysis with Pseudopotentials. J. Comput. Chem. 2018, 39, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D.; Edgecombe, K.E. A Simple Measure of Electron Localization in Atomic and Molecular Systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of Chemical Bonds Based on Topological Analysis of Electron Localization Functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Savin, A.; Nesper, R.; Wengert, S.; Fässler, T.F. ELF: The Electron Localization Function. Angew. Chem. Int. Ed. Engl. 1997, 36, 1808–1832. [Google Scholar] [CrossRef]
- Hunter, G. The Exact One-electron Model of Molecular Structure. Int. J. Quantum Chem. 1986, 29, 197–204. [Google Scholar] [CrossRef]
- Chan, W.T.; Hamilton, I.P. Valence Shell Structures in the Distributions of the Laplacian of the Electron Density and the One-Electron Potential for Diatomic Molecules. J. Chem. Phys. 1998, 108, 2473–2485. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Nguyen-Dang, T.T. Quantum Theory of Atoms in Molecules–Dalton Revisited. In Advances in Quantum Chemistry; Academic Press: Cambridge, MA, USA, 1981; pp. 63–124. [Google Scholar]
- Bader, R.F.W. A Quantum Theory of Molecular Structure and Its Applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From Weak to Strong Interactions: A Comprehensive Analysis of the Topological and Energetic Properties of the Electron Density Distribution Involving X–HF–Y Systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Ai-Guo, Z. Dissecting the Nature of Halogen Bonding Interactions from Energy Decomposition and Wavefunction Analysis. Monatshefte Chem. Chem. Mon. 2017, 148, 1259–1267. [Google Scholar] [CrossRef]
- Solimannejad, M.; Shirazi, S.G.; Scheiner, S. Analysis of Complexes Pairing Hydroperoxyl Radical with Peroxyformic Acid. J. Phys. Chem. A 2007, 111, 10717–10721. [Google Scholar] [CrossRef]
- Blanco, F.; Alkorta, I.; Rozas, I.; Solimannejad, M.; Elguero, J. A Theoretical Study of the Interactions of NF3 with Neutral Ambidentate Electron Donor and Acceptor Molecules. Phys. Chem. Chem. Phys. 2011, 13, 674–683. [Google Scholar] [CrossRef]
- Solimannejad, M.; Malekani, M.; Alkorta, I. Cooperativity between the Hydrogen Bonding and Halogen Bonding in F3CX⋯ NCH(CNH)⋯NCH(CNH) Complexes (X=Cl, Br). Mol. Phys. 2011, 109, 1641–1648. [Google Scholar] [CrossRef]
- Bartashevich, E.V.; Tsirelson, V.G. Interplay between Non-Covalent Interactions in Complexes and Crystals with Halogen Bonds. Russ. Chem. Rev. 2014, 83, 1181–1203. [Google Scholar] [CrossRef]
- Oliveira, V.; Kraka, E.; Cremer, D. The Intrinsic Strength of the Halogen Bond: Electrostatic and Covalent Contributions Described by Coupled Cluster Theory. Phys. Chem. Chem. Phys. 2016, 18, 33031–33046. [Google Scholar] [CrossRef] [PubMed]
- Mata, I.; Molins, E.; Alkorta, I.; Espinosa, E. Topological Properties of the Electrostatic Potential in Weak and Moderate N···H Hydrogen Bonds. J. Phys. Chem. A 2007, 111, 6425–6433. [Google Scholar] [CrossRef]
- Cao, X.; Rong, C.; Zhong, A.; Lu, T.; Liu, S. Molecular Acidity: An Accurate Description with Information-Theoretic Approach in Density Functional Reactivity Theory. J. Comput. Chem. 2018, 39, 117–129. [Google Scholar] [CrossRef]
- Wang, K.; He, X.; Rong, C.; Zhong, A.; Liu, S.; Zhao, D. On the Origin and Nature of Internal Methyl Rotation Barriers: An Information-Theoretic Approach Study. Theor. Chem. Acc. 2022, 141, 68. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Carroll, M.T.; Cheeseman, J.R.; Chang, C. Properties of Atoms in Molecules: Atomic Volumes. J. Am. Chem. Soc. 1987, 109, 7968–7979. [Google Scholar] [CrossRef]
- Scrocco, E.; Tomasi, J. Electronic Molecular Structure, Reactivity and Intermolecular Forces: An Euristic Interpretation by Means of Electrostatic Molecular Potentials; Academic Press: Cambridge, MA, USA, 1978; Volume 11, ISBN 012034811X. [Google Scholar]
- Scrocco, E.; Tomasi, J. The Electrostatic Molecular Potential as a Tool for the Interpretation of Molecular Properties. In New Concepts II; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2007; pp. 95–170. [Google Scholar]
- Brinck, T.; Murray, J.S.; Politzer, P. Surface Electrostatic Potentials of Halogenated Methanes as Indicators of Directional Intermolecular Interactions. Int. J. Quantum Chem. 1992, 44, 57–64. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural Population Analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Natural Bond Orbitals and Extensions of Localized Bonding Concepts. Chem. Educ. Res. Pract. 2001, 2, 91–104. [Google Scholar] [CrossRef]
- Wiberg, K.B. Application of the Pople-Santry-Segal CNDO Method to the Cyclopropylcarbinyl and Cyclobutyl Cation and to Bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Trindle, C. Bond Index Description of Delocalization. J. Am. Chem. Soc. 1969, 91, 219–220. [Google Scholar] [CrossRef]
- Trindle, C.; Sinanoglu, O. Local Orbital and Bond Index Characterization of Hybridization. J. Am. Chem. Soc. 1969, 91, 853–858. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Karafiloglou, P.; Landis, C.R.; Weinhold, F. NBO 7.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2018. [Google Scholar]
- Scaffidi-Domianello, Y.Y.; Meelich, K.; Jakupec, M.A.; Arion, V.B.; Kukushkin, V.Y.; Galanski, M.S.; Keppler, B.K. Novel Cis- and Trans-Configured Bis(Oxime)Platinum(II) Complexes: Synthesis, Characterization, and Cytotoxic Activity. Inorg. Chem. 2010, 49, 5669–5678. [Google Scholar] [CrossRef]
- Bondi, A. Van Der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G. Type II Halogen···halogen Contacts Are Halogen Bonds. IUCrJ 2014, 1, 5–7. [Google Scholar] [CrossRef]
- Brammer, L.; Mínguez Espallargas, G.; Adams, H. Involving Metals in Halogen–Halogen Interactions: Second-Sphere Lewis Acid Ligands for Perhalometallate Ions (M–X⋯X′–C). CrystEngComm 2003, 5, 343–345. [Google Scholar] [CrossRef]
- Georgiou, D.C.; Butler, P.; Browne, E.C.; Wilson, D.J.D.; Dutton, J.L. On the Bonding in Bis-Pyridine Iodonium Cations. Aust. J. Chem. 2013, 66, 1179. [Google Scholar] [CrossRef]
- Koskinen, L.; Hirva, P.; Kalenius, E.; Jääskeläinen, S.; Rissanen, K.; Haukka, M. Halogen Bonds with Coordinative Nature: Halogen Bonding in a S–I + –S Iodonium Complex. CrystEngComm 2015, 17, 1231–1236. [Google Scholar] [CrossRef]
- Frosch, J.; Koneczny, M.; Bannenberg, T.; Tamm, M. Halogen Complexes of Anionic N-Heterocyclic Carbenes. Chem.—A Eur. J. 2021, 27, 4349–4363. [Google Scholar] [CrossRef] [PubMed]
- Lamberts, K.; Handels, P.; Englert, U.; Aubert, E.; Espinosa, E. Stabilization of Polyiodide Chains via Anion⋯anion Interactions: Experiment and Theory. CrystEngComm 2016, 18, 3832–3841. [Google Scholar] [CrossRef]
- Bartashevich, E.V.; Matveychuk, Y.V.; Troitskaya, E.A.; Tsirelson, V.G. Characterizing the Multiple Non-Covalent Interactions in N, S-Heterocycles–Diiodine Complexes with Focus on Halogen Bonding. Comput. Theor. Chem. 2014, 1037, 53–62. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).