Abstract
The stoichiometric ratio 2:1 mix of 1-phenylpiperazine and oxalic acid dihydrate followed by slow evaporation results in a new material, bis(4-phenylpiperazin-1-ium) oxalate dihydrate, with the general chemical formula (C10H15N2)2(C2O4).2H2O, indicated by PPOXH. The title compound’s asymmetric unit and three-dimensional network have been determined by single crystal X-ray diffraction. Intermolecular O-H…O, N-H…O and C-H…O hydrogen bonding assist in maintaining and stabilization of the crystal structure of this new compound. Hirshfeld surface analysis and two-dimensional fingerprints have been performed to quantify the non-covalent interactions in the PPOXH structure. The vibrational modes of the different characteristic groups of the title chemical were identified using infrared spectrum analysis. The thermal characterization of this product was studied by a coupled TG/DTA analysis. The ultraviolet-visible absorption spectrum has been used to study the optical properties and the energy gap of this compound. DFT calculations were employed to evaluate the composition and properties of PPOXH. The analysis of HOMO-LUMO frontier orbitals analysis allows us to understand the chemical reactivity of this supramolecular compound and to determine the electrophilic and nucleophilic sites responsible for electron transfer. Topological analysis (AIM), reduced density gradient (RDG), molecular electrostatic potential surface (MEPS) and Mulliken population were analyzed to evaluate the types of non-covalent interactions, localization of electrons in space, atomic charges and molecular polarity in depth.
1. Introduction
Organic crystal engineering has become an area of intense research activity in recent years, with the objective that these designs lead to materials with interesting structures and properties. The complexation of organic molecules allows the development of supramolecular compounds with a more stable structure and more properties compared to the initial products. Supramolecular chemistry is the chemistry of non-covalent interactions. This branch has been developed by crystal engineering and is a very powerful tool to describe exactly how these interactions are established, which is the focus of recent papers [1,2,3]. It is an essential tool in the chemical and biological processes that control the elements of living systems [4]. Recently, the determination of crystal structures has influenced the evaluation in the biological field that allows a thorough understanding of the geometric study of intra- and intermolecular interactions in these molecules, which is directly related to the activity and reactivity of these systems [5]. The determination of the structures of biomolecules is highlighted by the leading role it has played in the awarding of numerous Nobel prizes in the disciplines of chemistry, physics and medicine [5].
The use of organic molecules for the synthesis of new materials is explained by the interest of the applications of these molecules in various fields such as pharmaceutical medicine [6], biology [7,8] and cosmetics [9]. The handling and processing of these materials greatly influence how drugs are developed [10].
Carboxylic acids constitute one of the most used organic molecules for the production of materials. The use of these organic acids for crystalline synthesis has a high probability of crystallization, which can be confirmed by the high interaction energy ∆E between the organic matrices to be complexed. This increases the possibility of crystal formation [11]. Recently, oxalic acid is among the carboxylic acids widely used for the complexation of new materials with interesting properties [12,13,14,15,16,17,18,19,20,21]. Specifically, oxalic acid is a dicarboxylic acid with the chemical formula C2H2O4. It has two hydroxyl groups on both molecular sides, and is a good reducing agent with an oxalate conjugate base.
Oxalic acid is an interesting organic matrix; it has great importance because of its interaction with the human body, animals and microorganisms [22]. It is an acid that is naturally produced in the human body when glycine and ascorbic acid are metabolized [22,23]. This acid is also present in many plants; it is produced by the metabolic conversion of ascorbic acid into oxalic acid [24]. The high content of oxalic acid in the human body may have a number of functions in the infection process, including chelation of calcium from cells and making pectic fractions more available to fungus. It is important to know that only plants are able to metabolize oxalic acid and oxalates [25,26]. The important roles and properties of oxalic acid provide the background for this work to pair it with 1-phenylpiperazine to create an anti-oxalate compound and for study of the interaction between these components.
Piperazines are well-studied heterocycles used in design for drug production and to improve the aqueous solubility of molecules. Currently, there are approximately 100 approved drugs that incorporate a piperazine cycle [27]. The piperazine families are used for the synthesis of crystalline compounds because of their interesting biological and pharmacological properties [28]. Recent research has shown the efficacy and interaction of one of the piperazine families against the COVID-19 virus [29], which confirms the usefulness of this amine against the most dangerous viruses.
In this work, we report on compounds resulting from the interaction of oxalic acid and 1-phenylpiperazine to create a new material that combines the characteristics of these two distinct entities. A new phase was prepared and characterized in experimental and quantum chemical studies.
2. Results and Discussion
2.1. Structure Description and Geometrical Parameters
The constituents of the structure of the title crystal are presented in Figure 1a. PPOXH is formed from two monoprotonated organic cations (C10H15N2)+, two water molecules and an oxalate anion (C2O4)2−. A Gaussian program was used to develop the optimized PPOXH structure shown in Figure 1b using the B3LYP/6-31G(d) level in comparison with the experimental data. The structure of PPOXH can be described by anion layers formed by oxalate anions and water molecules connected via OW-H…O hydrogen bonds that expand in the (100) plane (Figure 2). These layers are attached together by organic cations through N-H…O and C-H…O hydrogen bonds, thus forming a three-dimensional network.
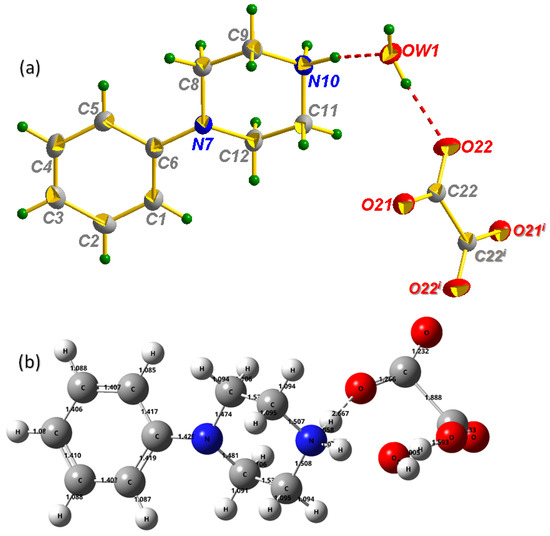
Figure 1.
Molecular structure of PPOXH with atom-labeling scheme, displacement ellipsoids are drawn at the 30% probability level (a), Optimized structure calculated by using B3LYP/6-31G(d) level (b). (i) −x; −y; −z. Intermolecular H-bonds are denoted by dotted lines.
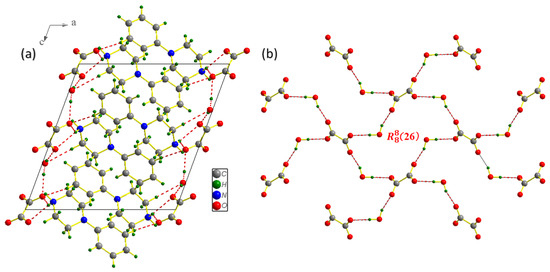
Figure 2.
Projection of the PPOXH structure along the (, ) plane (a) and the attachment of oxalate anions and water molecules by hydrogen bonds O-H…O in the (, ) plane (b).
The crystallographic data, structure refinement and measurement conditions for the collection are presented in Table 1. All bond angles and interatomic distances of different groups in PPOXH are recorded in Table 2.

Table 1.
Crystal data and experimental parameters used for the intensity data collection strategy and final results of the structure determination of PPOXH.
Table 2.
Principal distances (Å) and bond angles (°) in PPOXH.
Considering the oxalate anion, the lengths of the C-O bonds are found in the range 1.232–1.33 Å, theoretically, which is in agreement with that determined experimentally dC-O = 1.2621 (17) Å. The C-C bonds have the corresponding bond lengths: dexp = 1.565 (3) Å and dcalculated = 1.888 Å. The C-C-O interatomic bond angles determined by X-ray diffraction are in the following range [116.12(15)–116.76(15)°], while the O-C-O angle is 127.12 (13)°. The geometric data of the organic cation are given as follows; the length of the C-C and N-C bonds in the piperazine ring ranges from 1.513 (2) to 1.517 (2) Å and 1.4641 (18) to 1.4916 (19) Å, respectively. These values are calculated by the DFT method, with dC-C = 1.35 Å and dN-C = [1.474–1.508 Å]. The N-C bond length between the piperazine ring and the C6H5 group is 1.4176 (18) Å experimentally and 1.429 Å theoretically, which indicates a good agreement between the two results. The optimized length of the aromatic C-C bond of the phenyl ring group varies from 1.403 to 1.419 Å; these values are proportional to the empirically determined ones [1.384 (2)–1.4076 (19) Å]. The projection of the PPOXH structure along the (, ) plane (Figure 2a) shows that the structural cohesion of this compound is ensured by H-bonds of type N-H…O, O-H…O and C-H…O with donor-acceptor distances varying from 2.6781 (18) to 3.447 (2) Å.
The contributions of hydrogen bonds to the construction of three-dimensional networks are classified as follows: N-H…O bonds with distance donor…acceptr ranging from 2.6781 (18) to 2.9386 (17) Å, O-H…O bonds with ddonor…acceptor varying from 2.6989 (17) to 2.7040 (16) Å and C-H…O bond ddonor…acceptor = 3.447 (2) Å. According to Brown’s criterion, O-H…O bonds between oxalate anions and water molecules are considered as strong H-bonds (ddonor(O)…acceptor(O) < 2.73 Å) [30]. The geometry of the different existing H-bonds for the synthesized compound is given in Table 3. Oxalate anions and water molecules are attached by O-H…O hydrogen bonds (Figure 2b); this phenomenon will be repeated until a new supramolecular unit of type (26) is formed [31], which plays an important role in maintaining the crystal structure. With a distance value of 3.56 Å, Figure 3 allowed us to show the existence of C-H⋯π interactions between the organic groups, which contribute to the stability of the crystal structure; thus, the conditions for the existence of this type of interaction are verified [32].
Table 3.
Geometry of hydrogen bonds (Å, °) in PPOXH.
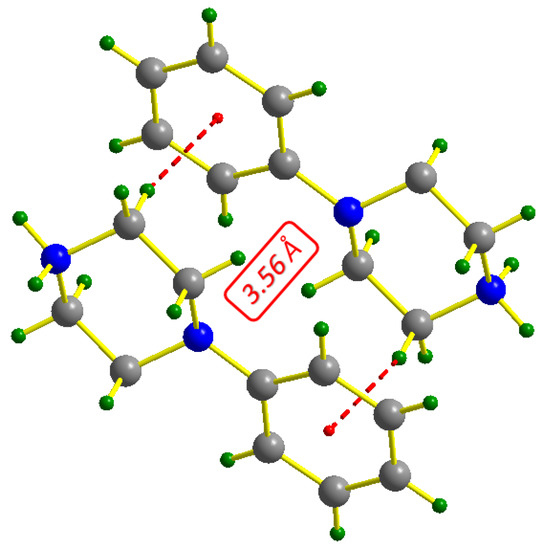
Figure 3.
Representation of C-H…π interactions in PPOXH.
2.2. Hirshfeld Surface Analysis
Non-covalent interactions in molecular crystals can be visualized by Hirshfeld surface analysis using the Crystal Explorer program [33], in which, the surface is determined by both the enclosed molecule and its nearest neighbors. The surface property can be determined by the dnorm types (Figure 4a), which is mapped according to a set of colors, red-white-blue, which represent interatomic contacts shorter than, equal to and longer than the van der Waals radius (contact due to hydrogen bonds), respectively. The two-dimensional fingerprints of the Hirshfeld surface show the proportionality, giving de as a function of di [34]. The latter provides quantitative information on the individual contribution of all interactions in the crystal stack. It illustrates the two-dimensional fingerprint of all the contacts contributing to the Hirshfeld surface. The 2D fingerprints (Figure 4b) show that the repulsive contacts of type H…H possess the most significant contribution, 44% of the total Hirshfeld surface, and appear as a predominantly blue area on the dnorm surface. The H…O/O…H type contacts appear as the second largest region of the fingerprint, representing 33.5% of the total area. This justifies the existence of strong hydrogen bonds and their important participation in the construction of the crystal system. H…C/C…H type contacts have a percentage representing 20.6 % of the global surface which verifies the presence of C-H…π type interactions and their efficiency in the stability of the PPOXH structure.
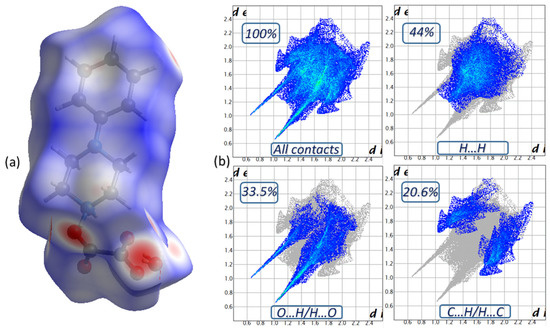
Figure 4.
The Hirshfeld surface of PPOXH in dnorm mode (a) and two-dimensional fingerprint plots of intermolecular contacts (b).
The weak interactions of H…N/N…H, N…C/C…N and N…N contribute to the packing of the title compound, which cover 1.1%, 0.6% and 0.2% of the total surface area, respectively. Crystal Explorer offers details on the percentage of interactions between one (XX) or two (XY) chemical elements in a crystal stacking, which can be used to compute enrichment ratios indirectly [35]. The enrichment ratio (ERXY) of different inter-contacts existing in the PPOXH structure are illustrated in Table 4. The list of enrichment ratios shows that the O…H/H…O contacts (ER(O…H/H…O) = 1.40) are the most numerous in the crystal packing with the formation of hydrogen bonds of type N-H…O, C-H…O and O-H…O. The C…H/H…C contacts are the second most important contacts for the packaging of the crystal, with an ER(C…H/H…C) value of 1.36, which confirms the effective presence of C-H…π type interactions in the crystal structure. The pairs H…H and H…N/N…H have enrichment ratios of 0.86 and 0.73,respectively, which are lower than unity and, therefore, their participation is lower than the previously noted contacts.
Table 4.
Enrichment ratio (ER(XY)) of different inter-contact and percentage of each atom on the surface Hirshfeld in PPOXH.
2.3. Thermal Characterization
Thermal behavior characterization was performed on PPOXH crystals using TG/DTA analysis, in order to evaluate their decomposition temperature, purity, melting point and crystallinity. The curve presented in Figure 5 shows that the thermal decomposition behavior of PPOXH has been examined in an air atmosphere from room temperature to 500 °C.
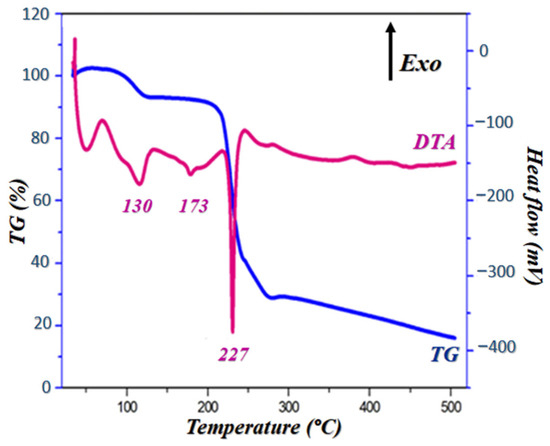
Figure 5.
DTA and TG curves of PPOXH at rising temperature.
Notably, the TG curve shows that this compound is stable up to 90 °C and it undergoes two stages of mass loss, one of which is 9% at 130 °C, which corresponds to the release of two crystallization water molecules (dehydration), and the other loss of mass is 51% which extends from 220 °C to 250 °C. The DTA curve gives three endothermic peaks; a first peak located at 130 °C that describes a dehydration accompanied by a loss of mass as it is presented in the TG curve. The second peak, at around 173 °C, corresponds to a PPOXH melting, justified by the absence of a mass loss on the TG curve. At the end, the third peak which reflects the decomposition of the rest of the product around 227 °C, coupled with a loss of mass which is confirmed by TG.
2.4. Vibrational IR Spectral Analysis
Infrared spectroscopy (IR) allows us to identify the functional groups in the structure of PPOXH. Figure 6 shows the experimental and theoretical FT-IR spectrum of PPOXH, where the observed bands are recorded between 4000 and 400 cm−1. In this way, our synthesized compound is in agreement with other referenced IR studies [36,37]. The presence of a small disagreement between the two spectra is explained by the fact that the FTIR spectrum was expressed in the crystalline state while the theoretical spectrum was analyzed in the gas phase.
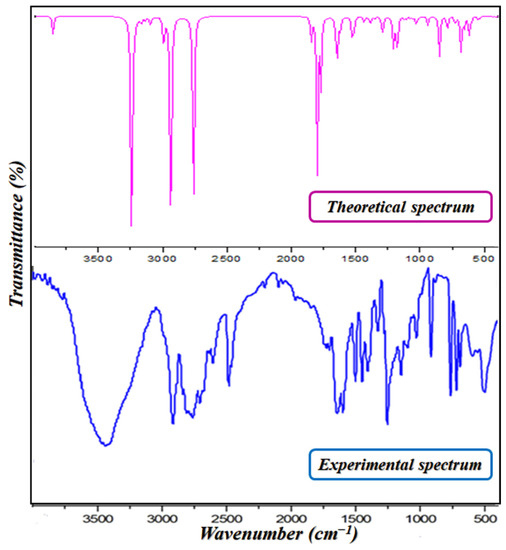
Figure 6.
The experimental and theoretical infrared spectra of PPOXH.
2.4.1. Vibration Modes of the 4-Phenylpiperazin-1-ium Cation
The 4-phenylpiperazinium cation contains the NH2+ group which is characterized by asymmetric and symmetric stretching modes. The band around 2931 cm−1 refers to this group’s vibrational modes, and, according to the theoretical calculation, is located around 2940 cm−1. The range extending from 2850 to 2750 cm−1 corresponds to the vibration of the CH2 group, which is calculated, theoretically, at around 2755 cm−1. The bands detected in the vicinity of 1326 to 1252 cm−1 are referred to the stretching modes of the C-N and C-C; these bands are calculated by DFT between 1290 and 1180 cm−1.
The bands from 1052 to 1000 cm−1 are related to the deformation modes of the C-H bond of phenyl group, and they are calculated around 1032 cm−1. In addition, the frequency region that extends from 1550 to 1600 cm−1 is assigned to the experimental deformation vibration of the C=C group and the calculated region extends from 1511 to 1542 cm−1.
2.4.2. Vibration Modes of Oxalate Anion
The oxalate anion C2O42− is characterized by a strong band around 1740 and 1700 cm−1, which is related to the elongation vibration of the C=O group, and another band which extends from 1028 to 1144 cm−1, which corresponds to the elongation vibration of the C-C group. These vibrations are calculated at 1796 cm−1–1769 cm−1 and from 1132 to 1124 cm−1, respectively, which presents good agreement between the two respective sets of ranges.
2.4.3. Vibration Modes of the Water Molecule
The O-H groups of the water molecule present a strong stretching band located experimentally at 3438 cm−1, and theoretically this band appears at 3244 cm−1. We also notice the appearance of another band related to the deformation vibration of H-O-H which stretches from 1670 to 1700 cm−1; this band is calculated by the DFT method at 1796 cm−1.
2.5. UV-Visible Spectroscopy
In the solid state, the experimental UV-visible spectrum of PPOXH was collected (Figure 7a).This analysis allowed us to analyze the charge transfer between the atoms constituting the structure of PPOXH. We observed the appearance of two absorption bands. At 350 nm, an intense band is observed that is relative to the π-π* transition, relative to aromatic conjugation in the organic cation. The second band appears with an absorption maximum located at 407 nm, attributed to the charge transfer n-π* between the oxalate anion and 4-phenylpiperazin-1-ium.
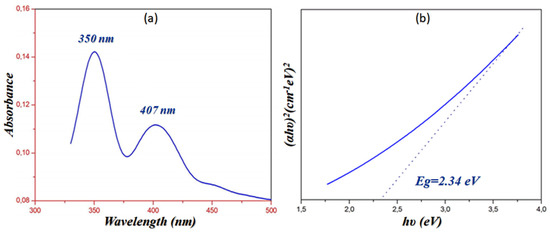
Figure 7.
Solid state Ultraviolet absorption spectrum of PPOXH (a) and determination of the energy gap obtained via the Tauc model (b).
The UV-visible spectroscopy also allows us measure the energy gap of PPOXH. Tauc proposed an extrapolation method that allows us to determine the Eg from the variation of (αhν)2 as a function of hν [38], as shown in Figure 7b, which indicates that PPOXH, with its value of Eg = 2.34 eV, can act as a semiconductor.
2.6. HOMO-LUMO Analysis
HOMO represents the highest occupied molecular orbital and LUMO, the lowest unoccupied molecular orbital. HOMO orbital acts as an electron donor and the LUMO orbital as an electron acceptor, so that the energy of the HOMO-LUMO orbitals is related to the nucleophilic and electrophilic character, respectively [39,40]. The energy gap between the boundary orbitals (Eg = |EHOMO − ELUMO|) is the most important parameter for the molecular reactivity and kinetic stability [41]. It is directly related to electricity conduction and the increase of the current is the result of the low significance of the HOMO-LUMO gap and vice-versa [42].
Using the B3LYP/6-31G(d) method, analytical calculations were performed. The energies, electronic affinity(A), global electrophilicity (ω), ionization potential (I), electronegativity (χ), chemical potential (μ), hardness (η) and softness (S) are presented in Table 5. These values illustrate some properties related to PPOXH: ionization potential (I) and electronic affinity (A) are directly related to HOMO and LUMO energy; (I) is the amount of free energy when the electron is extra to a neutral atom; hardness (η) is the opposite of softness (S), where softness measures the stability and reactivity of molecules; chemical potential (μ) is the opposite of electronegativity; (χ) is the ability of the molecule to attract electron pairs to it and global electrophilicity (ω) measures stabilization after a molecule accepts an extra-normal number of electrons [43].
Table 5.
Parameters of the HOMO-LUMO boundary molecular orbitals of PPOXH.
The energy level diagram with the HOMO-LUMO boundary of PPOXH is shown in Figure 8, which illustrates our observation that HOMOs are located on the organic cation while the LUMO ones are located on the oxalate anion. We can then deduce that the oxalate anion acts as an electron acceptor while the 4-phenylpiperazin-1-ium acts as an electron donor.
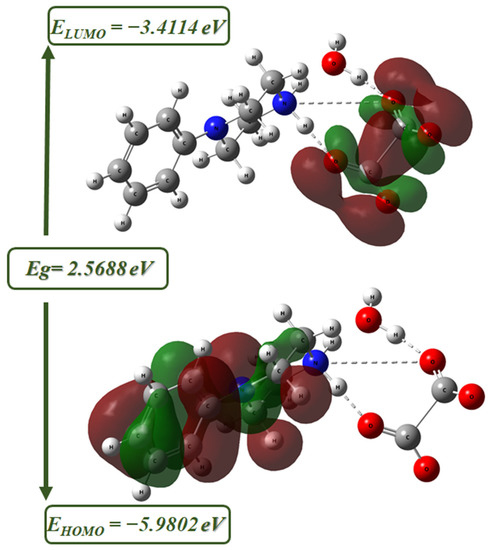
Figure 8.
The molecular frontier orbitals of PPOXH computed using the B3LYP/6-31G(d) level.
The values of the HOMO-LUMO frontier orbitals are −5.9802 and −3.4114 eV, respectively. The difference between these values gives the energy gap, Eg = 2.5688 eV, and this value allows this compound to be a semi-conductor. By contrasting the energy gap calculated for the HOMO-LUMO boundary (Eg = 2.36 eV) and that evaluated by the Tauc method (Eg = 3.16 eV), there is a good agreement between the two types of calculations since both procedures produce close energy values.
2.7. Non-Covalent Interactions (NCIs) Analysis
2.7.1. Topological Analysis (AIM)
The quantum theory of atoms in molecules (AIM) is one of the most efficient tools in the characterization of atomic and molecular interactions. It allows for the determination the existence of non-covalent bonds based on the critical bonding points (CBP), which appear when two neighboring atoms are chemically bonded or when there is a non-covalent interaction between them [44]. In particular, this analysis allows us to determine the strength of hydrogen bonds: strong, moderate or weak, based on topological parameters (Table S1), from which we can classify them. This classification is as follows: ∇2ρ(r) > 0 and H(r) > 0: Weak H-bonds (i.e., two topological parameters have a positive sign); ∇2ρ(r) > 0 and H(r) < 0: Moderate H-bonds (i.e., two topological parameters have opposite signs); ∇2ρ(r) < 0 and H(r) < 0: Strong H-bonds (i.e., two topological parameters have negative signs) [45].
We notice that the Figure 9 shows that PPOXH is stabilized by four non-covalent interactions, of which, three H-bonds N11-H36…O5, N11-H35…O7and O7-H8…O1 are considered as moderate hydrogen bonds since they have a Laplacian ∇2ρ(r) positive and an Hameltonian kinetic energy H(r) negative. We also notice the appearance of the non-covalent interaction O1…O7, which is worth an interaction energy Eint = 0.1694415 kJ·mol−1 and which also participates in the stability of the crystal system.
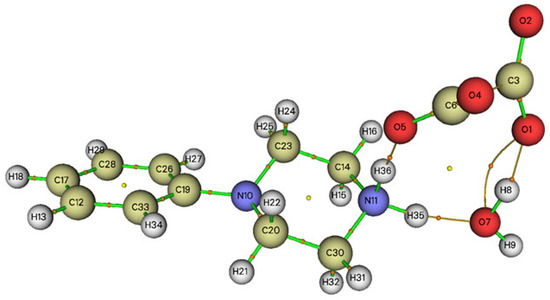
Figure 9.
Graphical representation of the AIM analysis of PPOXH (the critical binding points (BCP) and the connection paths are represented respectively by small balls and orange lines).
2.7.2. Reduced Density Gradient (RDG)
The reduced density gradient (RDG) analysis developed by Johnson et al. [46] was used to assess the type of non-covalent interactions (NCIs) that exist in PPOXH. As it is presented in Figure 10, we can evaluate the type and the strength of the NCIs by analyzing the amount of electron density as a function of the (sign (λ2)*ρ) and of the iso-surface density. The RDG analysis is based on the sign of λ2 (Figure 10a) and a set of colors; blue, green and red (Figure 10b), which gives information about the nature and strength of NCIs as follows: (sign (λ2)*ρ) < 0: H-bonding interactions (blue colors); (sign (λ2)*ρ) close to zero: van der Waals interactions (green colors); (sign (λ2)*ρ) > 0: steric effect (red colors) [47].
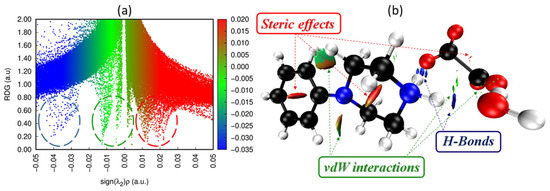
Figure 10.
Graphical display of RDG (a) and iso-surface density (b) of PPOXH to show the non-covalent interactions.
As shown in Figure 10b, the blue spots located between the hydrogen atoms of 4-phenylpiperazin-1-ium, the oxygen atoms of the oxalate anion and the water molecule attributed to the hydrogen bonds strongly participate in the crystalline stability of PPOXH. The green spots located between the amine atoms are attributed to van der Waals interactions, while the repulsion between the atoms due to the steric effect is represented by the red color.
2.8. Molecular Electrostatic Potential Surface (MEPS)
MEPS provides a method for mapping the surface of the molecular electrostatic potential, which allows analysis of the relative polarity to molecules. It is a method to evaluate the electrophilic and nucleophilic sites that are related to the reactivity and charge transfer [48]. DFT theory was used to determine the MEPS for the optimized geometry and identify the reactive sites in the molecule, using a variety of colors. The different ranges of electrostatic potential on the surface of PPOXH were mapped, and the potential is presented by this set of colors in an increasing manner, from negative to positive, as follows: red ‣ orange ‣ yellow ‣ green ‣ blue [49].
Figure S1 shows that the negative regions (red), rich in electrons, are located near oxygen atoms corresponding to nucleophilic sites, while the electrophilic sites, represented by the positive regions (blue), are located on the hydrogen atoms. These results confirm the charge transfer and the formation of N-H…O and O-H…O H-bonds.
2.9. Mulliken Population Analysis
The Mulliken population is a theoretical analysis method that determines the distribution of atomic charges, which is related to many properties of molecular structures, such as vibrational properties, dipole moment and polarizability [50]. It has an important role in the detection of electrophilic and nucleophilic attacks which allows the understanding of the reactivity of molecules. Mulliken atomic charges of PPOXH calculated at the B3LYP/6-311G are shown in Figure S2.
We notice that the carbon atoms C8, C9, C11 and C12 of the piperazine ring have negative charges because each of them is linked to two positively charged hydrogen atoms. The carbon atoms of the phenyl group other than C6 are negatively charged because each of them is linked to a positively charged hydrogen atom. They are more electronegative than the piperazine ring carbon; this is due to the delocalization of electrons around the aromatic ring. C6 is positively charged because it is attached to a nitrogen atom N7 and two carbon atoms C1 and C5 that are negatively charged. The nitrogen atom N10 is more electronegative than N7; this is due to the protonation of this nitrogen by another hydrogen atom during its reaction with oxalic acid. The carbon atoms C22 and C22i (i: −x, −y, −z + 1) of the oxalate anion are positively charged because each of them is linked to two negatively charged oxygen atoms.
It can be concluded that the electronegativity of an element increases by agreement with another positively charged element and, in contrast, this well-defined charge distribution causes hydrogen bonds of types C-H…O, O-H…O and N-H…O which contribute to the attachment of the molecules forming the PPOXH.
3. Experimental
3.1. Chemical Preparation
The new bis(4-phenylpiperazin-1-ium) oxalate dihydrate, (C10H15N2)2(C2O4).2H2O, was synthesized by the 1:2 reaction between oxalic acid and 1-phenylpiperazine. An amount of 1 mmol of oxalic acid was dissolved in 8 mL of water and 2 mmol of 1-phenylpiperazine in 8 mL of ethanol; both solutions are subjected to magnetic stirring for a few minutes. The mixture is then subjected to slow evaporation in petri dishes at room temperature. Single crystals suitable for structural studies were observed after 7 days (Yield 75%). The chemical reaction can be written as in Scheme 1.
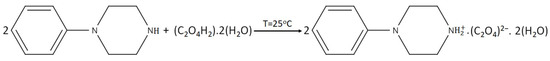
Scheme 1.
The chemical reaction scheme of PPOXH.
3.2. Characterization Techniques
3.2.1. Single-Crystal X-ray Diffraction
APEXII-Bruker-AXS is the diffractometer used for single crystal X-ray diffraction connected to a CCD detector whose X-ray source is a molybdenum Kα (Mo Kα) radiation with monochromatic wavelength λ = 0.71073 Å. The measurements were made between 3.1 and 27.4 degrees. Absorption corrections were performed by the multi-scan technique using the SADABS program [51]. A total of 6397 reflections were measured, of which, 2596 were independent and 1895 had an intensity I > 2σ(I). The structure was solved directly using SHELXS-97, which provided the positions of all atoms, save hydrogen. Full-matrix least-square methods based on F2 (SHELXL-97) [52] included in the WINGX program were then used to refine the result [51]. The hydrogen atoms bonded to N and OW were located from a difference map and were allowed to refine. The rest of the H atoms were treated as riding, with C-H = 0.95 Å (aromatic), C-H = 0.99 Å (methylene) and Uiso(H) = 1.2Ueq(C).
3.2.2. Materials and Physical Measurements
Using a Fourier transform infrared spectrometer (Nicolet IR 200, FT-IR), at room temperature, the IR spectrum of sample in KBr pellets in the frequency range of 4000–400 cm−1 was collected. Solid state ultraviolet spectroscopy in the 200–800 nm range was measured using a Perkin-Elmer Lambda 35 Spectrophotometer. With 11.5 mg, the TG-DTA analysis was performed using a Setaram 92 multimodule analyzer. The sample was heated from room temperature to 500 °C in an exposed platinum crucible; an empty crucible was used as a reference.
3.2.3. Computational Details
With the GaussView program, the structure of PPOXH was modeled [53] and then optimized in the gas phase with the Gaussian 09 package [54].The B3LYP/6-31G(d) method was used for all quantum chemistry computations of the electronic structure and optimized geometry of PPOXH [55]. We used the molecular electrostatic potential analysis (MEPS) and the Mulliken population to fully understand the polarity, distribution and charge transfer of PPOXH [39,56].The intermolecular interactions of the compound were evaluated using various methods, such as topological analysis (AIM, RDG) [57]. The electrophilic and nucleophilic sites responsible for the chemical reactivity in PPOXH and the energy gap were quantified by HOMO-LUMO analysis [49].
The ability of theoretical chemists to quantitatively model non-covalent interactions between organic molecules was exploited to predict their crystal structures and estimate their physical properties. This explains the growth in the number of recent research papers that are mainly based on this theoretical method [58,59,60].
4. Conclusions
The work presented in this manuscript was devoted to the synthesis of a new organic crystal, bis(4-phenylpiperazin-1-ium) oxalate dehydrate, in order to create an anti-oxalate compound in which the oxalate anion is strongly bound to the organic cation by different non-covalent interactions. The structural study of PPOXH was carried out based on single crystal X-ray diffraction; PPOXH crystallizes in a monoclinic system with the space group P21/c.
The non-covalent intermolecular interactions were visualized using the dnorm Hirshfeld surface and two-dimensional fingerprints, which confirmed the predominance of hydrogen bond and H…C/C…H type interactions. DTA/TG analysis was used to study the thermal behavior of PPOXH crystals. Spectroscopic and vibrational properties were determined using IR and UV-visible spectroscopy, which showed characteristic PPOXH-related peaks and the existence of π-π* and n-π* transitions.
Quantum chemistry studies were performed to investigate and confirm the properties and reactivity of PPOXH. HOMO-LUMO boundary analysis, non-covalent interactions (AIM and RDG), molecular electrostatic potential surface (MEPS) and Mulliken population analysis were performed to optimize the geometry of this synthesized supramolecular compound and approximate the molecular conformation in order to obtain comparable predictions between these results and those of the experimental study.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cryst13060875/s1, Figure S1: Molecular Electrostatic Potential Surface (MEPS) of PPOXH; Figure S2: Mulliken charges populations calculated for the PPOXH; Table S1: Topological parameters of PPOXH.
Author Contributions
Conceptualization, M.J., M.K., T.R., H.M. and N.I.; methodology, H.M. and N.I.; software, M.J., M.K., T.R., H.M. and N.I.; validation, M.J., M.K., T.R., H.M., N.I., A.S.K. (Aleksandr S. Kazachenko), Y.N.M. and A.S.K. (Aleksandr S. Kazachenko); formal analysis, M.J., M.K., T.R., H.M., N.I., O.A.-D. and A.S.K. (Anna S. Kazachenko); investigation, M.J., M.K., T.R., H.M., N.I. and A.S.K. (Aleksandr S. Kazachenko); resources, H.M., N.I. and O.A.-D.; data curation, N.I.,M.J., M.K., T.R. and H.M.; writing—original draft preparation, N.I., M.J., M.K., T.R., H.M., A.S.K. (Aleksandr S. Kazachenko) and A.S.K. (Anna S. Kazachenko); writing—review and editing, M.J., M.K., T.R. and H.M.; visualization, M.J., M.K., T.R., H.M. and N.I.; supervision, N.I. and H.M.; project administration, N.I. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Researchers Supporting Project no. RSP2023R61 of King Saud University, Riyadh, Saudi Arabia.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
This study was carried out within the state assignment no. 0287-2021-0012 for the Institute of Chemistry and Chemical Technology, Siberian Branch of the Russian Academy of Sciences.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sahoo, P.R.; Kumar, A.; Kumar, A.; Kumar, S. Experimental and computational investigation of polymorphism in methyl 3-hydroxy-4-(piperidin-1-ylmethyl)-2-naphthoate. J. Mol. Struct. 2020, 1219, 128619. [Google Scholar] [CrossRef]
- Sahoo, P.R.; Kathuria, I.; Kumar, S. The structural arrangement of the ligand-metal complex with centered zinc and nickel atoms and their optical features. J. Mol. Struct. 2022, 1262, 133010. [Google Scholar] [CrossRef]
- Sahoo, P.R.; Kumar, A.; Kumar, A.; Kumar, S. Synthesis and optical properties of copper(II) and nickel(II) complexes of a highly fluorescent morpholine-derivative. Polyhedron 2019, 171, 559–570. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Kopylovich, M.N.; Silva, M.F.C.G.; Pombeiro, A.J.L. Non-covalent interactions in the synthesis of coordination compounds: Recent advances. Coord. Chem. Rev. 2017, 314, 54–72. [Google Scholar] [CrossRef]
- Brammer, L. Developments in inorganic crystal engineering. Chem. Soc. Rev. 2004, 33, 476–489. [Google Scholar] [CrossRef]
- Zhou, Z.; Vázquez-Gonzáleza, M.; Willner, I. Stimuli-responsive metal–organic framework nanoparticles for controlled drug delivery and medical applications. Chem. Soc. Rev. 2021, 50, 4541–4563. [Google Scholar] [CrossRef]
- Giliopoulos, D.; Zamboulis, A.; Giannakoudakis, D.; Bikiaris, D.; Triantafyllidis, K. Polymer/Metal Organic Framework (MOF) Nanocomposites for Biomedical Applications. Molecules 2020, 25, 185. [Google Scholar] [CrossRef]
- Nasrollahzadeh, M.; Sajjadi, M.; Tahsili, M.R. High efficiency treatment of organic/inorganic pollutants using recyclable magnetic N-heterocyclic copper(II) complex and its antimicrobial applications. Sep. Purif. Technol. 2020, 238, 116403. [Google Scholar] [CrossRef]
- Iravani, S.; Varma, R.S. Greener synthesis of lignin nanoparticles and their applications. Green Chem. 2020, 22, 612–636. [Google Scholar] [CrossRef]
- Li, T.; Mattei, A. Pharmaceutical Crystals: Science and Engineering; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar]
- Barbas, R.; Fael, H.; Lee, S.; Ruiz, R.; Hunter, C.A.; Fuguet, E.; Ràfols, C.; Prohens, R. Virtual Cocrystal Screening of Adefovir Dipivoxyl: Identification of New Solid Forms with Improved Dissolution and Permeation Profiles. Pharmaceutics 2022, 14, 2310. [Google Scholar] [CrossRef]
- Senthil, R.; Vijayaragavan, G.; Ayeshamariam, A.; Kaviyarasu, K. Nonlinear optical properties of single crystal of L-OOMHCL incorporation with Glycine Oxalic Acid (GOA) with high chemical stability for optoelectronic applications. Surf. Interfaces 2020, 18, 18100417. [Google Scholar] [CrossRef]
- Khan, I.M.; Alam, K.; Alamb, M.J.; Ahmad, M. Spectrophotometric and photocatalytic studies of H-bonded charge transfer complex of oxalic acid with imidazole: Single crystal XRD, experimental and DFT/TD-DFT studies. New J. Chem. 2019, 43, 9039–9051. [Google Scholar] [CrossRef]
- Khan, I.M.; Shakya, S.; Akhtar, R.; Alam, K.; Islam, M.; Alam, N. Exploring interaction dynamics of designed organic cocrystal charge transfer complex of 2-hydroxypyridine and oxalic acid with human serum albumin: Single crystal, spectrophotometric, theoretical and antimicrobial studies. Bioorg. Chem. 2020, 100, 103872. [Google Scholar] [CrossRef] [PubMed]
- Mahendra, K.; Udayashankar, N.K. Investigation on mechanical and temperature dependent electrical properties of potassium hydrogen oxalate oxalic acid dihydrate single crystal. Phys. Lett. A 2020, 384, 126475. [Google Scholar] [CrossRef]
- Naseema, K.; Ravi, S.; Sreedharan, R. Studies on a novel organic NLO single crystal: L-asparaginium oxalate. Chin. J. Phys. 2019, 60, 612–622. [Google Scholar] [CrossRef]
- Senthil, K.; Elangovan, K.; Senthil, A.; Vinitha, G. Growth and characterization of N-methylurea oxalic (NMUO) acid single crystal. Rasayan J. Chem. 2019, 12, 1262–1268. [Google Scholar] [CrossRef]
- Sasaki, T.; Sakamoto, S.; Takamizawa, S. Organoferroelastic Crystal Prepared by Supramolecular Synthesis. Cryst. Growth Des. 2020, 20, 1935–1939. [Google Scholar] [CrossRef]
- Rohith, P.S.; Jagannatha, N.; Kumar, K.V.P. Growth and Characterization of Pure and Magnesium Doped Copper Cadmium Oxalate Single Crystals. Mater. Today 2019, 8, 85–93. [Google Scholar] [CrossRef]
- Owoyemi, B.C.D.; Silva, C.C.P.D.; Diniz, L.F.; Souza, M.S.; Ellena, J.; Carneiro, R.L. Fluconazolium oxalate: Synthesis and structural characterization of a highly soluble crystalline form. CrystEngComm 2019, 21, 1114–1121. [Google Scholar] [CrossRef]
- Essid, M.; Muhammad, S.; Marouani, H.; Saeed, A.; Aloui, Z.; Al-Sehem, A.G. Synthesis, characterization, Hirshfeld surface analysis and computational studies of 1-methylpiperazine-1,4-diium bis(hydrogen oxalate): [C5H14N2](HC2O4)2. J. Mol. Struct. 2020, 1211, 128075. [Google Scholar] [CrossRef]
- Mahendra, K.; Udayashankar, N.K. Growth and comparative studies on oxalic acid dihydrate, potassium oxalate hydrate and potassium hydrogen oxalate oxalic acid dihydrate single crystals. J. Phys. Chem. Solids 2020, 138, 109263. [Google Scholar] [CrossRef]
- Stewart, R.D. The function of oxalic acid in the human metabolism. Clin. Chem. Lab. Med. 2011, 49, 1405–1412. [Google Scholar]
- Yang, J.C.; Loewus, F.A. Metabolic Conversion of L-Ascorbic Acid to Oxalic Acid in Oxalate-accumulating Plants. Plant Physiol. 1975, 56, 283–285. [Google Scholar] [CrossRef]
- Çalişkan, M. The Metabolism of Oxalic Acid. Turk. J. Zool. 2000, 24, 103–106. [Google Scholar]
- Singh, P.P.; Kothari, L.K.; Sharma, D.C.; Saxena, S.N. Nutritional value of foods in relation to their oxalic acid content. Am. J. Clin. Nutr. 1972, 25, 1147–1152. [Google Scholar] [CrossRef]
- Meanwell, N.A.; Loiseleur, O. Applications of Isosteres of Piperazine in the Design of Biologically Active Compounds: Part 1. J. Agric. Food Chem. 2022, 70, 10942–10971. [Google Scholar] [CrossRef]
- Gatfaoui, S.; Mezni, A.; Roisnel, T.; Marouani, H. Synthesis, characterization, Hirshfeld surface analysis and antioxidant activity of anovel organic-inorganic hybrid material 1-methylpiperazine-1,4-diium bis(nitrate). J. Mol. Struct. 2017, 1139, 52–59. [Google Scholar] [CrossRef]
- Gatfaoui, S.; Sagaama, A.; Issaoui, N.; Roisnel, T.; Marouani, H. Synthesis, experimental, theoretical study and molecular docking of 1-ethylpiperazine-1,4-diium bis(nitrate). Solid State Sci. 2020, 106, 106326. [Google Scholar] [CrossRef]
- Brown, I.D. On the geometry of O-H⋯O hydrogen bonds. ActaCryst. 1976, 32, 24–31. [Google Scholar] [CrossRef]
- Bernstein, J. Polymorphism of L-glutamic acid: Decoding the α–β phase relationship via graph-set analysis. Acta Cryst. 1991, B47, 1004–1010. [Google Scholar] [CrossRef]
- Zhao, Z.; Parrish, R.M.; Smith, M.D.; Pellechia, P.J.; Sherrill, C.D.; Shimizu, K.D. Do DeuteriumsForm Stronger CH–π Interactions. J. Am. Chem. Soc. 2012, 134, 14306–14309. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystal. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Jelsch, C.; Ejsmont, K.; Huder, L. The enrichment ratio of atomic contacts in crystals, an indicator derived from the Hirshfeld surface analysis. IUCrJ 2014, 1, 119–128. [Google Scholar] [CrossRef]
- Noureddine, O.; Gatfaoui, S.; Antonia Brandán, S.; Marouani, H.; Issaoui, N. Structural, docking and spectroscopic studies of a new piperazine derivative, 1-phenylpiperazine-1,4-diium-bis (hydrogen sulfate). J. Mol. Struct. 2020, 1202, 127351. [Google Scholar] [CrossRef]
- Peterson, K.I.; Pullman, D.P. Determining the Structure of Oxalate Anion Using Infrared andRaman Spectroscopy Coupled with Gaussian Calculations. J. Chem. Educ. 2016, 93, 1130–1133. [Google Scholar] [CrossRef]
- Tauc, J. Optical properties and electronic structure of amorphous Ge and Si. Mater. Res. Bull. 1968, 3, 37–46. [Google Scholar] [CrossRef]
- Sidir, I.; Sidir, Y.G.; Kumalar, M.; Tasal, E. Ab initio Hartree–Fock and density functional theory investigations on the conformational stability, molecular structure and vibrational spectra of 7-acetoxy-6-(2,3-dibromopropyl)-4,8-dimethylcoumarin molecule. J. Mol. Struct. 2010, 964, 134–151. [Google Scholar] [CrossRef]
- Akman, F. A density functional theory study based on monolignols: Molecular structure, HOMO-LUMO analysis, molecular electrostatic potential. Cellul. Chem. Technol. 2019, 53, 243–250. [Google Scholar] [CrossRef]
- Daghar, C.; Issaoui, N.; Roisnel, T.; Dorcet, V.; Marouani, H. Empirical and computational studies on newly synthesiscyclohexylammonium perchlorate. J. Mol. Struct. 2021, 1230, 129820. [Google Scholar] [CrossRef]
- Staykov, A.; Nozaki, D.; Yoshizawa, K. Photoswitching of Conductivity through a Diarylperfluorocyclopentene Nanowire. J. Phys. Chem. C 2007, 111, 3517–3521. [Google Scholar] [CrossRef]
- Radhi, A.H.; Du, E.A.B.; Khazaal, F.A.; Abbas, Z.M.; Aljelawi, O.H.; Hamadan, S.D.; Almashhadani, H.A.; Kadhim, M.M. HOMO-LUMO Energies and Geometrical Structures Effecton Corrosion Inhibition for Organic Compounds Predict by DFT and PM3 Methods. NeuroQuantology 2020, 18, 37–45. [Google Scholar] [CrossRef]
- Gatfaoui, S.; Issaoui, N.; Roisnel, T.; Marouani, H. Synthesis, experimental and computational study of a non-centrosymmetricmaterial 3-methylbenzylammonium trioxonitrate. J. Mol. Struct. 2020, 1225, 129132. [Google Scholar] [CrossRef]
- Akman, F.; Issaoui, N.; Kazachenko, A.S. Intermolecular hydrogen bond interactions in the thiourea/water complexes (Thio-(H2O)n) (n = 1, …, 5): X-ray, DFT, NBO, AIM, and RDG analyses. J. Mol. Model. 2020, 26, 161. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Gatfaoui, S.; Issaoui, N.; Roisnel, T.; Marouani, H. A proton transfer compound template phenylethylamine: Synthesis, a collective experimental and theoretical investigations. J. Mol. Struct. 2019, 1191, 183–196. [Google Scholar] [CrossRef]
- Luque, F.J.; López, J.M.; Orozco, M. Perspective on Electrostatic interactions of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects. Theor. Chem. Acc. 2000, 103, 343–345. [Google Scholar] [CrossRef]
- Jmai, M.; Gatfaoui, S.; Issaoui, N.; Roisnel, T.; Kazachenko, A.S.; Al-Dossary, O.; Marouani, H.; Kazachenko, A.S. Synthesis, Empirical and Theoretical Investigations on New Histaminium Bis(Trioxonitrate) Compound. Molecules 2023, 28, 1931. [Google Scholar] [CrossRef]
- Govindasamy, P.; Gunasekaran, S.; Srinivasan, S. Molecular geometry, conformational, vibrational spectroscopic, molecular orbital and Mulliken charge analysis of 2-acetoxybenzoic acid. Spectrochim. Acta A Mol. 2014, 130, 329–336. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.; Millam, J. GaussView, version 5; Semichem, Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; revision C.01; Gaussian, Inc.: Wallingford, UK, 2009. [Google Scholar]
- Tirado-Rives, J.; Jorgensen, W.L. Performance of B3LYP Density Functional Methods for a Large Set of Organic Molecules. J. Chem. Theory Comput. 2008, 4, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Comparison of Various Means of Evaluating Molecular Electrostatic Potentials for Noncovalent Interactions. J. Comput. Chem. 2017, 39, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Noureddine, O.; Issaoui, N.; Medimagh, M.; Al-Dossary, O.; Marouani, H. Quantum chemical studies on molecular structure, AIM, ELF, RDG and antiviral activities of hybrid hydroxychloroquine in the treatment of COVID-19: Molecular docking and DFT calculations. J. King Saud Univ. Sci. 2021, 33, 101334. [Google Scholar] [CrossRef]
- Sahoo, P.R.; Kumar, S. Synthesis of an optically switchable salicylaldimine substituted naphthopyran for selective and reversible Cu2+ recognition in aqueous solution. RSC Adv. 2016, 6, 20145–20154. [Google Scholar] [CrossRef]
- Kumar, A.; Sahoo, P.R.; Kathuria, I.; Prakash, K.; Kumar, S. Oxazine as an efficient precursor for the development of photochromic spiropyrans. J. Photochem. Photobiol. A Chem. 2023, 438, 114541. [Google Scholar] [CrossRef]
- Sahoo, P.; Prakash, K.; Kumar, S. Experimental and theoretical investigations of cyanide detection using a photochromic naphthopyran. Supramol. Chem. 2017, 29, 183–192. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).