Abstract
This study focuses on the synthesis, characterization, and properties of a yellowish, prism-shaped ligand, N,N′-(naphthalene-1,5-diyl) bis(1-(pyridin-2-yl) methanimine). The ligand was synthesized through refluxing 1,5-diaminonaphthalene and pyridine-2-carbaldehyde in extra-pure ethanol, employing X-ray diffraction on single crystal. The crystal is structured with two pyridylimine-binding units linked to a naphthalene. The crystal has a P21/c space group in a monoclinic system. The structure was confirmed through an infrared examination. Computational spectroscopy and theoretical methods were used to investigate the ligand HOMO, LUMO, and charge distribution. Additionally, a Hirshfeld analysis was performed to investigate noncovalent interactions in the crystalline form. The results showed that dispersion forces (H···H) were the primary factor contributing to the arrangement of the ligand molecule, accounting for 45.3% of the total interactions in the absence of hydrogen bonding. Overall, this study provides valuable insights into the synthesis, characterization, and properties of this unique ligand.
1. Introduction
Various fields benefit from a diverse array of practical implementations of structures based on naphthalene [1,2]. Imine systems can also be utilized to assemble discrete cyclophanes, double and triple helicates, dimers, trimmers, and grids [3,4,5,6,7,8,9,10,11,12,13]. One of the factors, which could affect these structure properties can be short contact. The formation of three-dimensional structures in biological and chemical systems relies heavily on short-range interactions, which can also aid in the creation of new materials with beneficial properties [14,15,16,17]. The other importance of short contacts is that these contacts a considerable impact on the physical and chemical properties of a compound, including its melting point, boiling point, and reactivity [18]. The study of short contacts in organic structures has gained increasing attention in recent years. This interest has been driven by advances in computational chemistry, which have made it possible to study the properties and behavior of organic compounds at the molecular level. Here we synthesized and characterized a structure that has two parallel pyridylimine binding units on both sides. These kinds of structures, which look rigid, can support grid structures such as Oborn and Youinou [19] or Lehn [20]. A similar structure on both sides of naphthalene has been observed in the works of other researchers, such as Piontek [21]. In this article, we will explore the synthesis, characterization, and DFT calculation of naphthalene-based crystal structures with pyridylimine-binding units.
2. Experimental
2.1. Synthesis
The ligand was made by refluxing 1,5-diaminonaphthalene (1 mol, 158.20 g) and pyridine-2-carbaldehyde (2 mol, 214.22 g) for 10 h in extra-pure ethanol. The crystal was washed with an ethanol/acetone solution and dried at 60 °C. Analysis calculated for C22H16N4: C, 78.55; H, 4.79; N, 16.66. Found: C, 79.00; H, 5.00; N, 16.00. Characteristic FT-IR data (KBr, cm−1): 519, 628, 996, 1063, 1108, 1448, 1359, 1592, 3261, 3421. Scheme 1 demonstrates the ligand preparation.
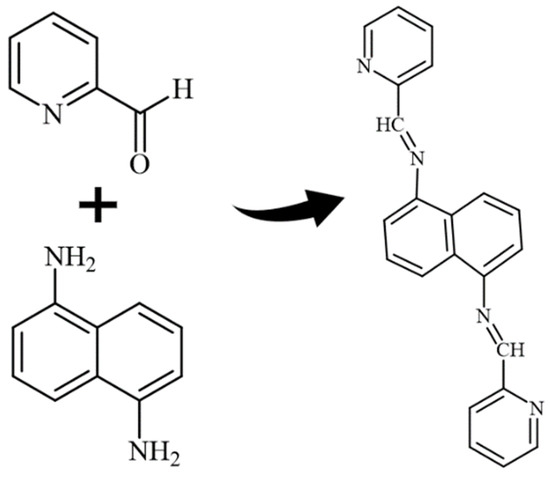
Scheme 1.
Ligand preparation.
2.2. X-ray Diffraction
The compound formed a yellowish, prism-shaped crystal measuring 0.06 × 0.06 × 0.30 mm3. The crystal was placed on a glass fiber and cooled to −93 °C using a nitrogen gas stream controlled by the Cryostream Controller 700. X-ray diffraction data were collected using a Bruker SMART APEX II instrument (Madison, WI, USA). The instrument used graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The diffractometer operated at 50 kV and 30 mA. The data was collected over 2θ ranges of 8.08~52.00°. The data collection process did not show any significant decay.
The data processing steps were performed on a PC using the Bruker AXS Crystal Structure Analysis Package [22,23,24,25]: The following steps were taken: The data were collected using APEX2, and cell refinement and data reduction were performed with SAINT. Absorption correction was carried out using SADABS, while the structure solution involved the use of XPREP and SHELXS-97. Structure refinement was performed with SHELXL-97, and molecular graphics and publication materials were generated using SHELXTL [26]. Cromer and Waber were the sources of the neutral atom scattering factors [27]. The monoclinic space group P21/c was identified through E statistics, systematic absences, and successful structure refinement. Direct methods were used to solve the structure. The compound was refined using full-matrix least-squares, with a minimization function of ∑w (Fo2 − Fc2)2. All non-hydrogen atoms underwent anisotropic refinement. Hydrogen atoms were positioned geometrically with a C-H distance of 0.95 Å and refined as riding atoms with a Ui-so(H) value of 1.2 UeqC. For 1204 independent reflections with I > 2σ(I), the final R1 value was 0.0410 and wR2 was 0.0872. For all 1599 independent reflections with R(int) = 0.0250, the final R1 value was 0.0606 and wR2 was 0.1021. The refinement involved 118 parameters and 0 restraints (R1 = ∑||Fo| − |Fc||/∑|Fo|; wR2 = {∑[w (Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2; (w = 1/[σ2(Fo2) + (0.0364P)2 + 0.2981P], where P = [Max (Fo2, 0) + 2Fc2]/3)). The largest residual peak was 0.200 e/Å3, and the largest residual hole was −0.231 e/Å3. The crystallographic data for compound 1 has been deposited with the Cambridge Crystallographic Data Centre as Supplementary Publication CCDC-2252233. The crystal characteristics and structure refinement were presented in Table 1. Table 2, Table 3, Table 4, Table 5 and Table 6 provide additional crystallographic information, including atomic coordinates (Table 2), bond lengths (Table 3), angles (Table 3), anisotropic displacement parameters (Table 4), hydrogen coordinates (Table 5), and torsion angles (Table 6).

Table 1.
Crystal data and structure refinement.
Table 2.
The atomic coordinates are expressed in units of 10−4, while the equivalent isotropic displacement parameters are expressed in units of 10−3 Å2. U (eq) is defined as one-third of the trace of the orthogonalized Uij tensor.
Table 3.
Bond lengths [Å] and angles [°].(Symmetry transformations used to generate equivalent atoms: #1 − x + 1, −y + 1, −z + 2).
Table 4.
The anisotropic displacement parameters expressed in units of Å2 × 103. The exponent of the anisotropic displacement factor is given by the formula: −2π2 [h2a*2U11 + ... + 2 h k a* b* U12].
Table 5.
Units of Measurement for Hydrogen Coordinates (×104) and Isotropic Displacement Parameters (Å2 × 103).
Table 6.
Torsion angle [°].(Symmetry transformations used to generate equivalent atoms: #1 − x + 1, −y + 1, −z + 2).
It might be interesting to present crystallography data in a visual format to provide a clear understanding of these pieces of information. Thus, we provided some Figure 1.
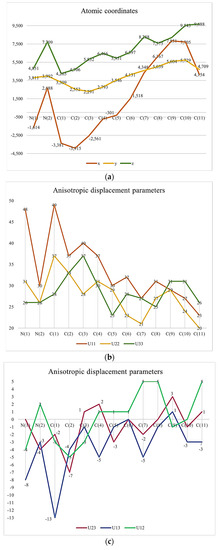
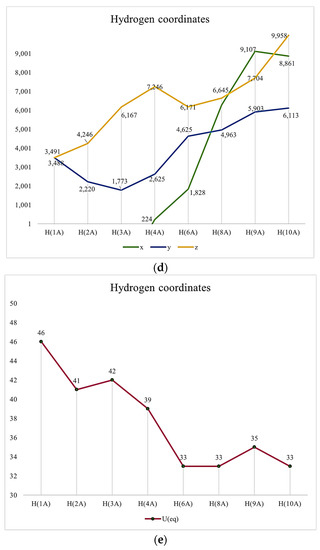
Figure 1.
(a) Atomic Coordinates (×104). (b) Anisotropic displacement parameters (Å2 × 103) U11, U22, U33. (c) Anisotropic displacement parameters (Å2 × 103) U23, U13, U12. (d) Hydrogen Coordinates (×104). (e) Isotropic displacement parameters (Å2 × 103).
2.3. IR Characterization
Confirmation of the successful synthesis of the ligand was obtained through analysis of the IR. The spectrum displays a strong peak at 1288 cm−1, indicating the stretching frequency due to aromatic nitrogen. Additionally, there are two medium peaks at 1384 cm−1 and 1414 cm−1, which correspond to the deformation of CH2 and CH3, respectively. The medium peak at 1472 cm−1 is attributed to the symmetric bending vibration of the CH2 group. The weak peak at 1514 cm−1 in the IR spectrum is caused by the bending vibration of the aromatic ring in the compound. Figure 2 demonstrates IR spectrum.
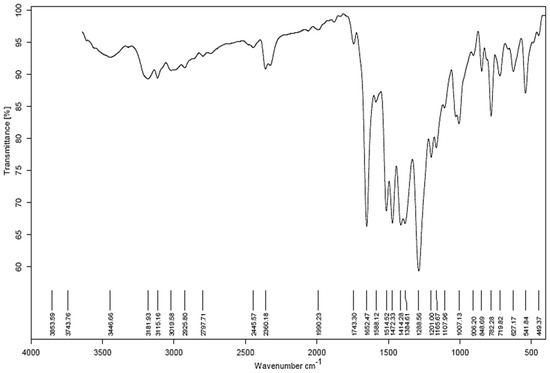
Figure 2.
IR spectrum.
2.4. Computational Method
For this research, we utilized GaussView 6.0 and Gaussian 09W software on a Windows operating system. To make the Gaussian work more manageable, we broke it down into two steps and completed them separately. Initially, we optimized the structure, and subsequently, we used the output file from the previous step to compute the Raman, NMR, HUMO, and LUMO structures.
2.5. Hirshfeld Surface Analysis
To perform an analysis of intermolecular contacts in the crystals under investigation, the Crystal Explorer Ver. 3.1 program package was used [28]. This involved utilizing Hirshfeld surface analyses, 2D fingerprint plots, and calculating the percentage contributions.
3. Results and Discussion
X-ray diffraction analysis was used to determine the structure of the ligand, revealing that it has a symmetrical structure with naphthalene in the center and two pyridines on the sides and belongs to the P21/c space group in a monoclinic system. C (7) on naphthalene and N (2) on the sides, connected naphthalene and pyridine with bond length 1.418 (2) to form a molecule structure (Figure 3a). Figure 3b depicts the unit cell packing. The multi-layer structure shows empty spaces in the unit cell in Figure 3b. Another structure, Figure 3c, is created by the direct growth of the compound structure and bears a striking resemblance to the Z shape when viewed from a specific orientation. Figure 3: The molecular structure: (a) The molecular structure (50% probability); (b) Unit cell packing; (c) The Z shape structure.
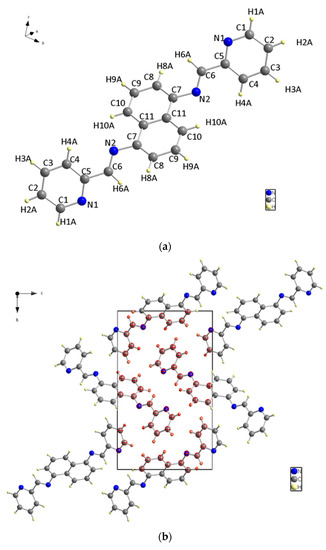
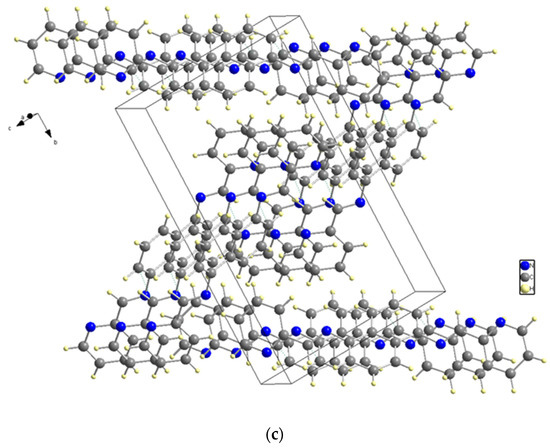
Figure 3.
(a) The molecular structure. (b) Unit cell packing. (c) The Z shape structure.
The bond between C (5) and C (6) has the longest bond length at 1.470 (2) Å. N (2) is bonded to C (7) on one side and C (6) on the other, with bonding distances of 1.418 (2) Å and 1.272 (2) Å, respectively. Additionally, the bond between N (2) and C (6) on both sides is the shortest non-hydrogen ligand bond.
The nitrogen and carbon bonds mentioned have slight differences compared to other nitrogen (N (1)) and carbon bonds within the ligand. At the ligand ends, we find N (1)-C (1) and N (1)-C (5), which measure 1.338 (2) Å and 1.348 (2) Å, respectively. The title compound molecule has a linker section (C (7)-N (2)-C (6)-C (5)) with an angle of −178.12° (15). There are no hydrogen bonding interactions present in the structure. The structure shown in Figure 4 is a result of the short contacts caused by C (6) and C (8), which attract neighboring molecules.
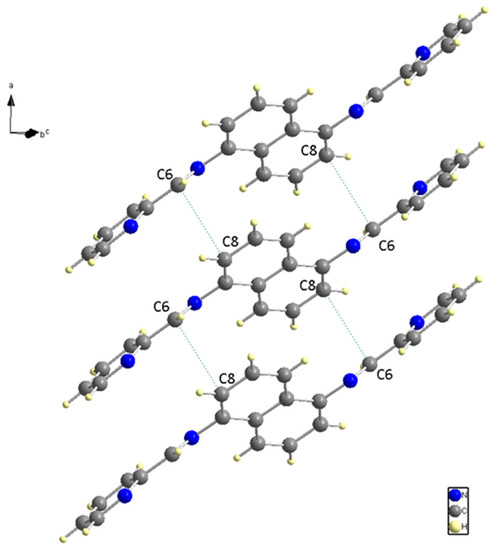
Figure 4.
Neighboring Molecules Short contacts.
Computational studies with Gaussian were conducted in this research, and Table 7 provides a summary of the ligand calculation obtained in the 6-311+(2d, p) basis set. The results suggest that the molecule is nonpolar or only slightly polar, which is supported by the very small dipole moment of 0.000039 D. This value indicates that there is minimal charge separation within the molecule. These findings align with previous discussions.
Table 7.
Gaussian calculation summary.
The charge distribution on the molecule in Figure 5 is symmetrical, and the level of polarity is low.
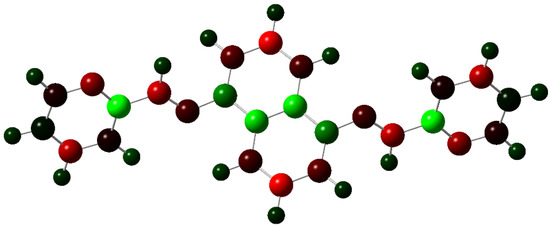
Figure 5.
Charge distribution with a color range from −0.614 to 0.614 Mulliken (red for negative, green for positive).
Figure 6 displays the structures of HOMO and LUMO, with EHOMO and ELUMO values of 0.20847 (eV) and 0.08374 (eV), respectively. To investigate noncovalent interactions in the solid state, we utilized Hirshfeld Surfaces analysis on the ligand. Figure 7 displays all the Hirshfeld properties. When the contact distance between atoms inside and outside the surface is greater than the sum of their respective van der Waals radii, the areas in blue on the dnorm property are indicated. The dnorm property has white areas that correspond to a contact distance equal to the sum of the van der Waals radii, and small amounts of red areas where the contact distance between atoms inside and outside the surface is less than the sum of their respective van der Waals radii [29].
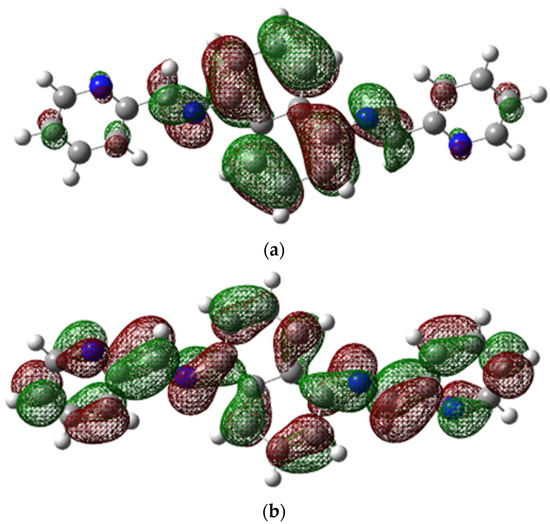
Figure 6.
(a) HOMO and (b) LUMO structures.
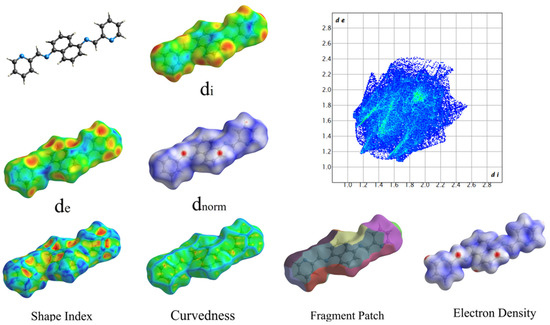
Figure 7.
Hershfield Surfaces mapper with all properties (a: −0.97, b: 0.08, c: 0.21).
The red areas on the plot represent non-covalent regions, and they are primarily located on the C6 and C8 atoms, which we identified in Figure 4 as being associated with these interactions. Figure 8 displays the fingerprint plot, which provides a visual representation of the interactions involved and their respective percentage contributions to the total interactions.
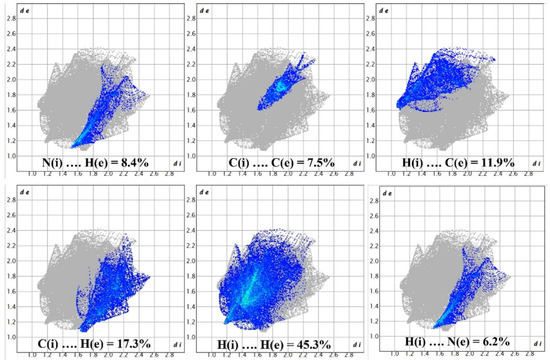
Figure 8.
Demonstration of percentage contributions in the total interactions.
According to Figure 8, the primary interaction throughout the surface is dispersion forces (H···H), which account for over 45% of the total interactions. The combined percentage of H and C in the structure is 29.2%, which includes contributions from both H(i)···C(e) and C(i)···H(e) interactions. Additionally, the total contribution of N and H is 14.6%. Therefore, carbon and hydrogen, with a contribution of 29.2%, along with the C8 and C6 interaction and dispersion forces (H···H), as depicted in Figure 9, are the forces that govern the stacking arrangement of the ligand molecules.
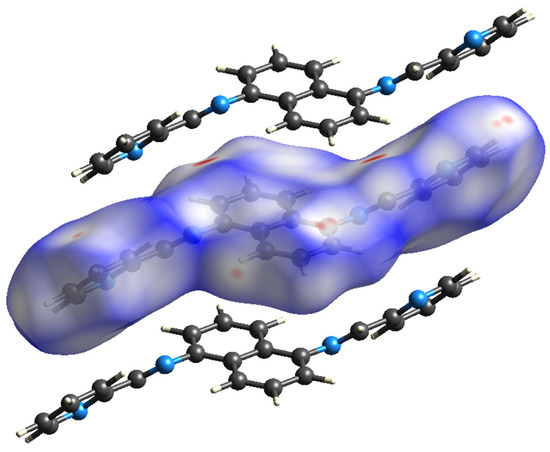
Figure 9.
Hirshfeld surface.
The ligand being discussed can potentially form coordination complexes with multiple metals. Some of the metals that have been utilized to create coordination complexes with Schiff base ligands, which have structural similarities to our synthesized ligand, are Co (II), Ni (II), Cu (II), Zn (II), gold (III), and copper (II) [30,31,32,33]. Furthermore, the ligand has the potential to create coordination complexes with other transition metals, including Pd (II) [34]. The coordination geometry of metal complexes may differ based on the specific metal involved, although octahedral coordination is preferred for certain metals [30]. The crystal structures of metal complexes may differ based on the metal used and the coordination geometry formed. As an example, in a previous study [31], the author reported on gold (III) complexes with 1,1-dimethylbiguanide. In this case, the gold atom is coordinated by two chloride ligands and two nitrogen atoms from the biguanide ligand, resulting in a square planar coordination geometry.
4. Conclusions
In conclusion, we successfully synthesized and characterized a symmetrical ligand, N, N′-(naphthalene-1,5-diyl) bis(1-(pyridin-2-yl) methanimine), through refluxing 1,6-diaminonaphthalene and pyridine-2-carbaldehyde in extra-pure ethanol. The ligand was found to have a structure containing central naphthalene and two parallel parts on the sides, with two pyridylimine-binding units connected to a 1,5-naphthalene structure. The structure was confirmed through infrared examination, and computational spectroscopy and theoretical methods were used to show the ligand HOMO, LUMO, and charge distribution. Additionally, we conducted a Hirshfeld analysis and demonstrated all of its properties. Our findings suggest that dispersion forces (H···H) were the primary factor contributing to the arrangement of the ligand molecule, accounting for 45.3% of the total interactions in the absence of hydrogen bonding.
Supplementary Materials
Crystallographic data for the structure reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication CCDC-2252233 for C22H16N4 (1). Copies of the data can be obtained by request from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (Fax: +44-1223-336033, e-Mail: de-posit@ccdc.cam.ac.uk).
Author Contributions
B.M., writing—review and editing; A.K., validation, formal analyses, writing—original draft preparation and editing; N.R., writing—original draft preparation; Y.H., supervision, writing—review and editing and S.W.J., project administration and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by grant NRF-2019R1A5A8080290 of the National Research Foundation of Korea.
Data Availability Statement
Not applicable.
Acknowledgments
We thank the University of Qom for their assistance with this project.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Altintop, M.D.; Sever, B.; Özdemir, A.; Kuş, G.; Oztopcu-Vatan, P.; Kabadere, S.; Kaplancikli, Z.A. Synthesis and Evaluation of Naphthalene-Based Thiosemicarbazone Derivatives as New Anticancer Agents against LNCaP Prostate Cancer Cells. J. Enzym. Inhib. Med. Chem. 2016, 31, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.; Tashkandi, N.; Hadjichristidis, N.; Alkayal, N.S. Synthesis of Naphthalene-Based Polyaminal-Linked Porous Polymers for Highly Effective Uptake of CO2 and Heavy Metals. Polymers 2022, 14, 1136. [Google Scholar] [CrossRef]
- Meistermann, I.; Moreno, V.; Prieto, M.J.; Moldrheim, E.; Sletten, E.; Khalid, S.; Rodger, P.M.; Peberdy, J.C.; Isaac, C.J.; Rodger, A.; et al. Intramolecular DNA Coiling Mediated by Metallo-Supramolecular Cylinders: Differential Binding of P and M Helical Enantiomers. Proc. Natl. Acad. Sci. USA 2002, 99, 5069–5074. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Wang, Y.; Lin, J.; You, L. A Novel Synthesis Route to Rare Earth Polyborates. Chem. Commun. 2001, 13, 1178–1179. [Google Scholar] [CrossRef]
- Janica, I.; Patroniak, V.; Samorì, P.; Ciesielski, A. Imine-Based Architectures at Surfaces and Interfaces: From Self-Assembly to Dynamic Covalent Chemistry in 2D. Chem. Asian J. 2018, 13, 465–481. [Google Scholar] [CrossRef]
- Hamblin, J.; Childs, L.J.; Alcock, N.W.; Hannon, M.J. Directed One-Pot Syntheses of Enantiopure Dinuclear Silver(i) and Copper(i) Metallo-Supramolecular Double Helicates. J. Chem. Soc. Dalton Trans. 2002, 2, 164–169. [Google Scholar] [CrossRef]
- Childs, L.J.; Alcock, N.W.; Hannon, M.J. Assembly of Nano-Scale Circular Supramolecular Arrays through π–π Aggregation of Arc-Shaped Helicate Units. Angew. Chem. Int. Ed. 2001, 113, 1079–1081. [Google Scholar] [CrossRef]
- Hamblin, J.; Jackson, A.; Alcock, N.W.; Hannon, M.J. Triple Helicates and Planar Dimers Arising from Silver(i) Coordination to Directly Linked Bis-Pyridylimine Ligands. J. Chem. Soc. Dalton Trans. 2002, 8, 1635. [Google Scholar] [CrossRef]
- Zaier, R.; Ayachi, S. Toward Designing New Cyclopentadithiophene-Naphthalene Derivatives Based Small Molecules for Organic Electronic Applications: A Theoretical Investigation. Mater. Today Commun. 2021, 27, 102370. [Google Scholar] [CrossRef]
- Gondia, N.K.; Sharma, S.K. Comparative Optical Studies of Naphthalene Based Schiff Base Complexes for Colour Tunable Application. Mater. Chem. Phys. 2019, 224, 314–319. [Google Scholar] [CrossRef]
- Li, Q.; Ma, L.; Li, J.; Wang, L.; Yu, L.; Zhao, Y.; Lv, Y. Study of a Fluorescent System Based on the Naphthalene Derivative Fluorescent Probe Bound to Al3+. Micromachines 2023, 14, 868. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Cao, P.; Wang, Y.; Wu, W.; Guo, J.; Sun, J.; Zou, X.; Wang, W.; Ruan, H. Effects of Naphthalene Application on Soil Fungal Community Structure in a Poplar Plantation in Northern Jiangsu, China. Appl. Sci. 2023, 13, 5794. [Google Scholar] [CrossRef]
- Sadek, O.; Bouhadir, G.; Bourissou, D. Lewis Pairing and Frustration of Group 13/15 Elements Geometrically Enforced by (Ace)Naphthalene, Biphenylene and (Thio)Xanthene Backbones. Chem. Soc. Rev. 2021, 50, 5777–5805. [Google Scholar] [CrossRef] [PubMed]
- Nishino, M.; Miyashita, S. Effect of the Short-Range Interaction on Critical Phenomena in Elastic Interaction Systems. Phys. Rev. B 2013, 88, 014108. [Google Scholar] [CrossRef]
- Melkikh, A.V.; Meijer, D.K.F. On a Generalized Levinthal’s Paradox: The Role of Long- and Short Range Interactions in Complex Bio-Molecular Reactions, Including Protein and DNA Folding. Prog. Biophys. Mol. Biol. 2018, 132, 57–79. [Google Scholar] [CrossRef]
- Claesson, P.M.; Kjellin, M.; Rojas, O.J.; Stubenrauch, C. Short-Range Interactions between Non-Ionic Surfactant Layers. Phys. Chem. Chem. Phys. 2006, 8, 5501. [Google Scholar] [CrossRef] [PubMed]
- Kotelchuck, D.; Scheraga, H.A. The Influence of Short-Range Interactions on Protein Conformation, ii. a Model for Predicting the α-Helical Regions of Proteins. Proc. Natl. Acad. Sci. USA 1969, 62, 14–21. [Google Scholar] [CrossRef]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The Role of Short-Chain Fatty Acids in the Interplay between Diet, Gut Microbiota, and Host Energy Metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef]
- Youinou, M.-T.; Rahmouni, N.; Fischer, J.; Osborn, J.A. Self-Assembly of a Cu4 Complex with Coplanar Copper(I) Ions: Synthesis, Structure, and Electrochemical Properties. Angew. Chem. Int. Ed. Engl. 1992, 31, 733–735. [Google Scholar] [CrossRef]
- Baxter, P.N.W.; Lehn, J.-M.; Fischer, J.; Youinou, M.-T. Self-Assembly and Structure of a 3 × 3 Inorganic Grid from Nine Silver Ions and Six Ligand Components. Angew. Chem. Int. Ed. Engl. 1994, 33, 2284–2287. [Google Scholar] [CrossRef]
- Piontek, A.; Siodłak, D.; Zarychta, B. Naphthalene-2,6-Diyl Bis(4-Methylbenzenesulfonate). IUCrdata 2018, 3, x180890. [Google Scholar] [CrossRef]
- Bruker. SADABS, Version 2008/1; Bruker AXS Inc.: Madison, WI, USA, 2008. [Google Scholar]
- Bruker. XPREP, Version 2008/2; Bruker AXS Inc.: Madison, WI, USA, 2008. [Google Scholar]
- Bruker. SAINT, Version 7.68A.; Bruker AXS Inc.: Madison, WI, USA, 2009. [Google Scholar]
- Bruker. APEX2, Version 2010.3-0; Bruker AXS Inc.: Madison, WI, USA, 2010. [Google Scholar]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Cromer, D.T.; Waber, J.T. International Tables for X-ray Crystallography; Kynoch Press: Birmingham, UK, 1974; Volume 4, Table 2.2 A. [Google Scholar]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A Program for Hirshfeld Surface Analysis, Visualization and Quantitative Analysis of Molecular Crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards Quantitative Analysis of Intermolecular Interactions with Hirshfeld Surfaces. Chem. Commun. 2007, 7, 3814. [Google Scholar] [CrossRef] [PubMed]
- Abouelatta, A.I.; Sonk, J.A.; Hammoud, M.M.; Zurcher, D.M.; McKamie, J.J.; Schlegel, H.B.; Kodanko, J.J. Synthesis, Characterization, and Theoretical Studies of Metal Complexes Derived the Chiral Tripyridyldiamine Ligand Bn-CDPy3. Inorg. Chem. 2010, 49, 5202–5211. [Google Scholar] [CrossRef]
- Makotchenko, E.V.; Kharlamova, V.Y.; Baidina, I.A.; Bardina, E.E.; Korolkov, I.V.; Mironov, I.V.; Gushchin, A.L. Synthesis, Crystal Structure and Solution Studies of Gold(III) Complexes with 1,1-Dimethylbiguanide. Inorganica Chim. Acta 2023, 552, 121496. [Google Scholar] [CrossRef]
- Murillo, J.; Goodwin, C.A.P.; Stevens, L.; Fortier, S.; Gaunt, A.J.; Scott, B.L. Synthesis and Comparison of Iso-Structural f-Block Metal Complexes (Ce, U, Np, Pu) Featuring η6-Arene Interactions. Chem. Sci. 2023, 14, 7438–7446. [Google Scholar] [CrossRef]
- Kollur, S.P.; Castro, J.O.; Frau, J.; Flores-Holgu’ın, N.; Shruthi, G.; Shivamallu, C.; Glossman-Mitnik, D. Preparation, Spectroscopic Investigations and Chemical Reactivity Properties of a New Schiff Base Ligand and Its Copper (II) Complexes. J. Mol. Struct. 2019, 1191, 17–23. [Google Scholar] [CrossRef]
- Kurpik, G.; Walczak, A.; Gołdyn, M.; Harrowfield, J.; Stefankiewicz, A.R. Pd(II) Complexes with Pyridine Ligands: Substituent Effects on the NMR Data, Crystal Structures, and Catalytic Activity. Inorg. Chem. 2022, 61, 14019–14029. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).