
Novel Non-Centrosymmetric NdSr4O(BO3)3 Borate and Nd(Ca1−xSrx)4O(BO3)3 Solid Solutions: Preparation, Crystal Structures, Thermal Expansion and Optical Properties

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis
2.2. Powder X-ray Diffraction
2.3. Single Crystal X-ray Diffraction
2.4. High-Temperature Single-Crystal X-ray Diffraction
2.5. High-Temperature Powder X-ray Diffraction
2.6. Thermal Analysis
2.7. Spectroscopy
2.8. Second Harmonic Generation (SHG) Measurements
3. Results
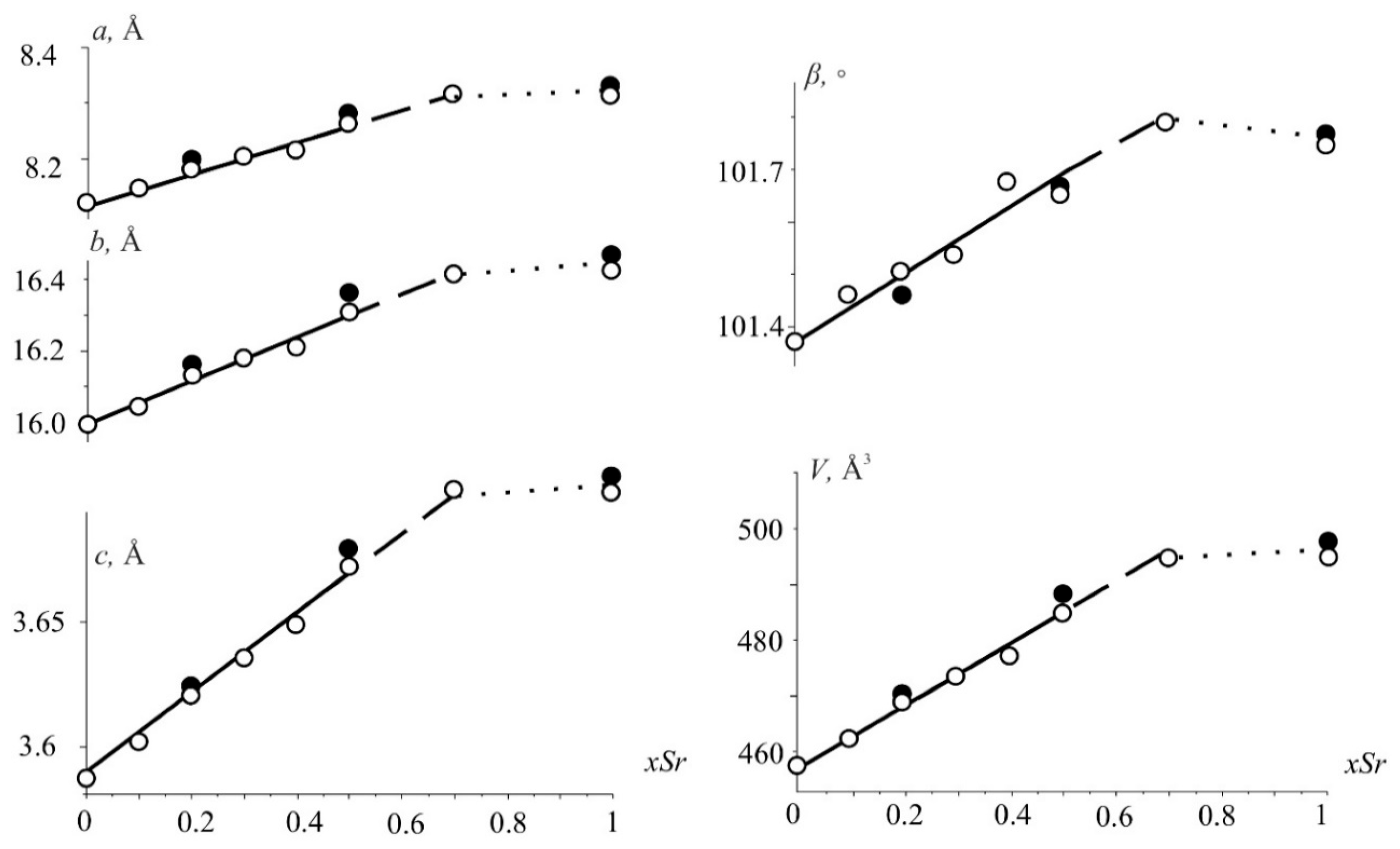
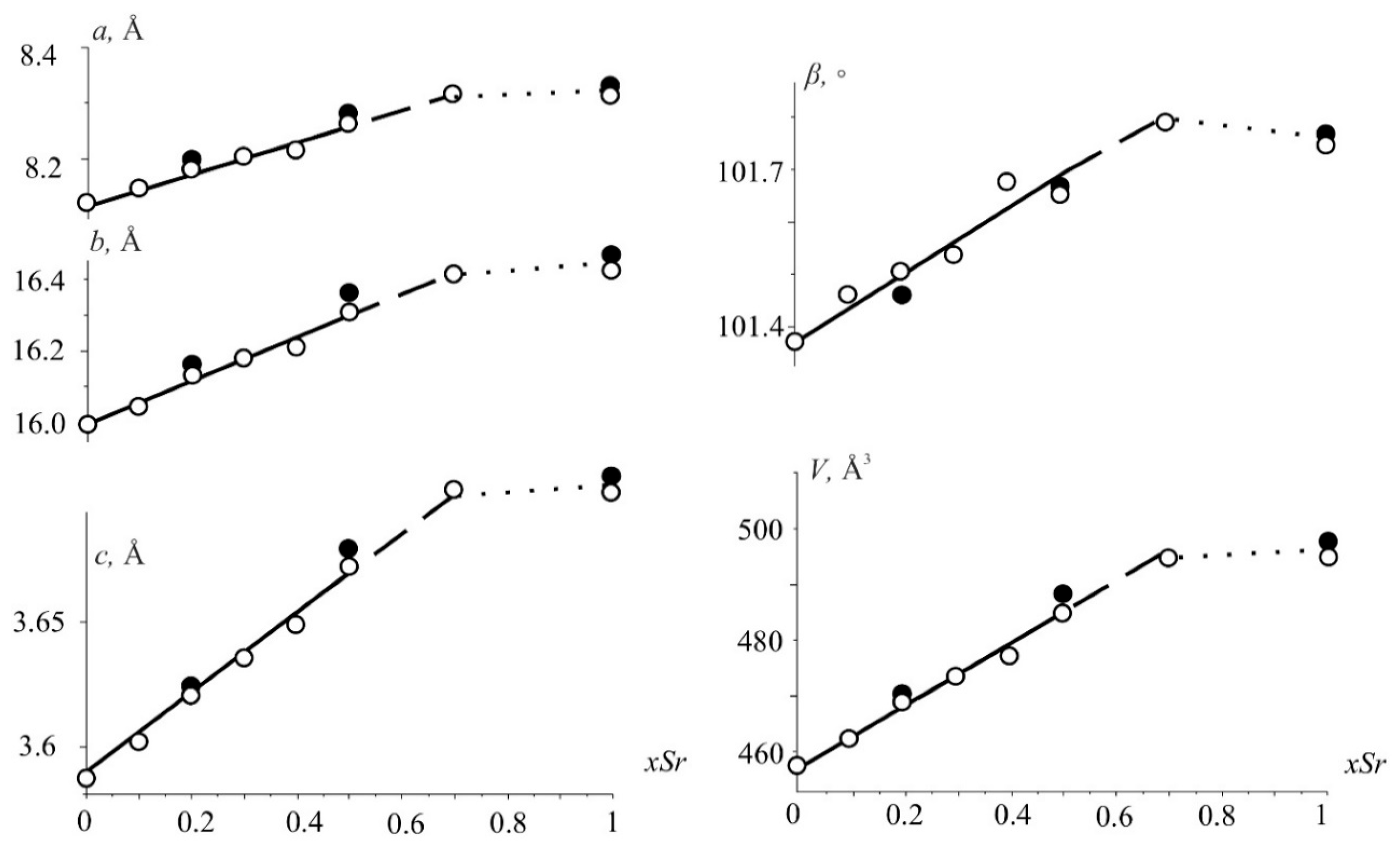
3.1. Powder X-ray Diffraction Studies
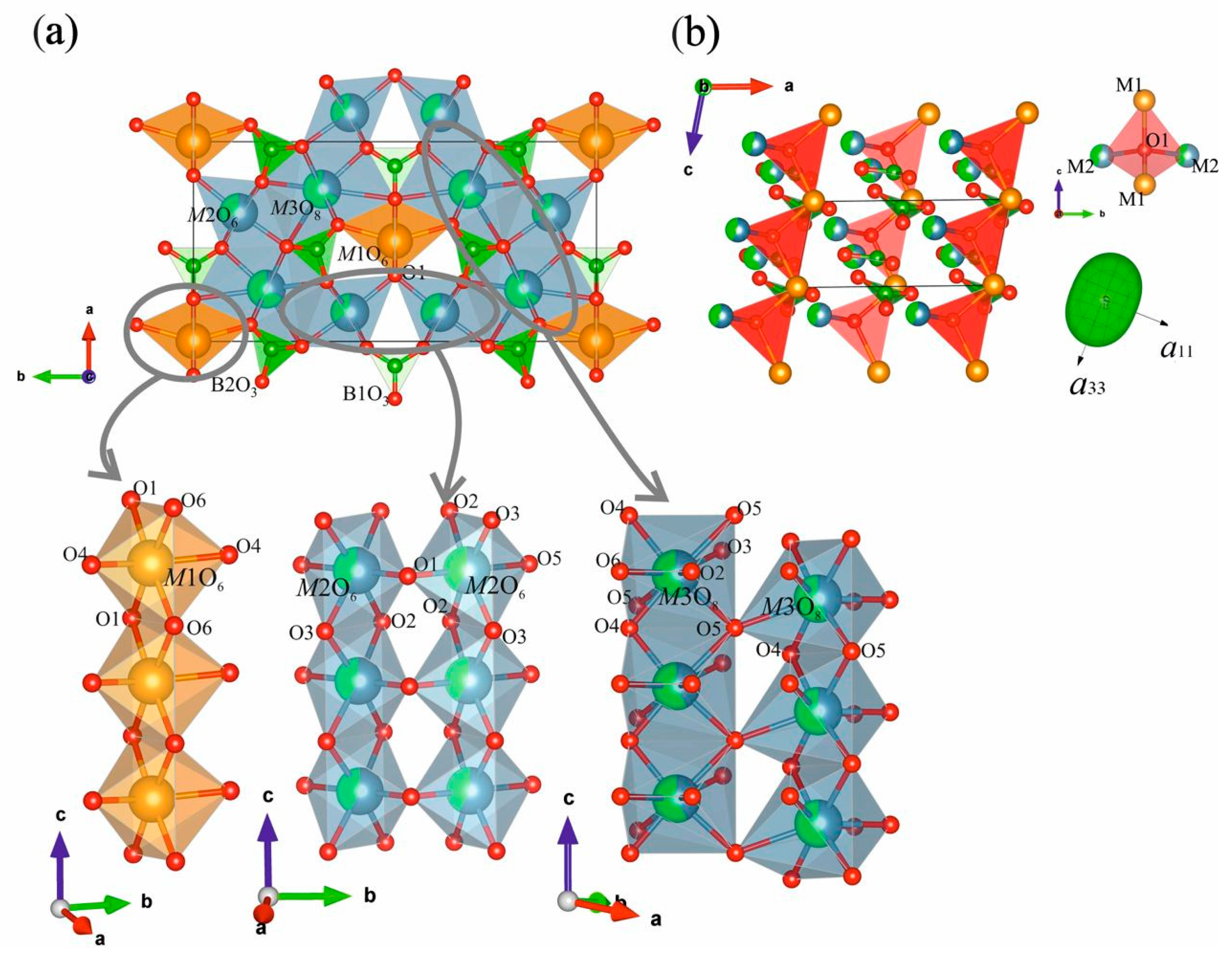
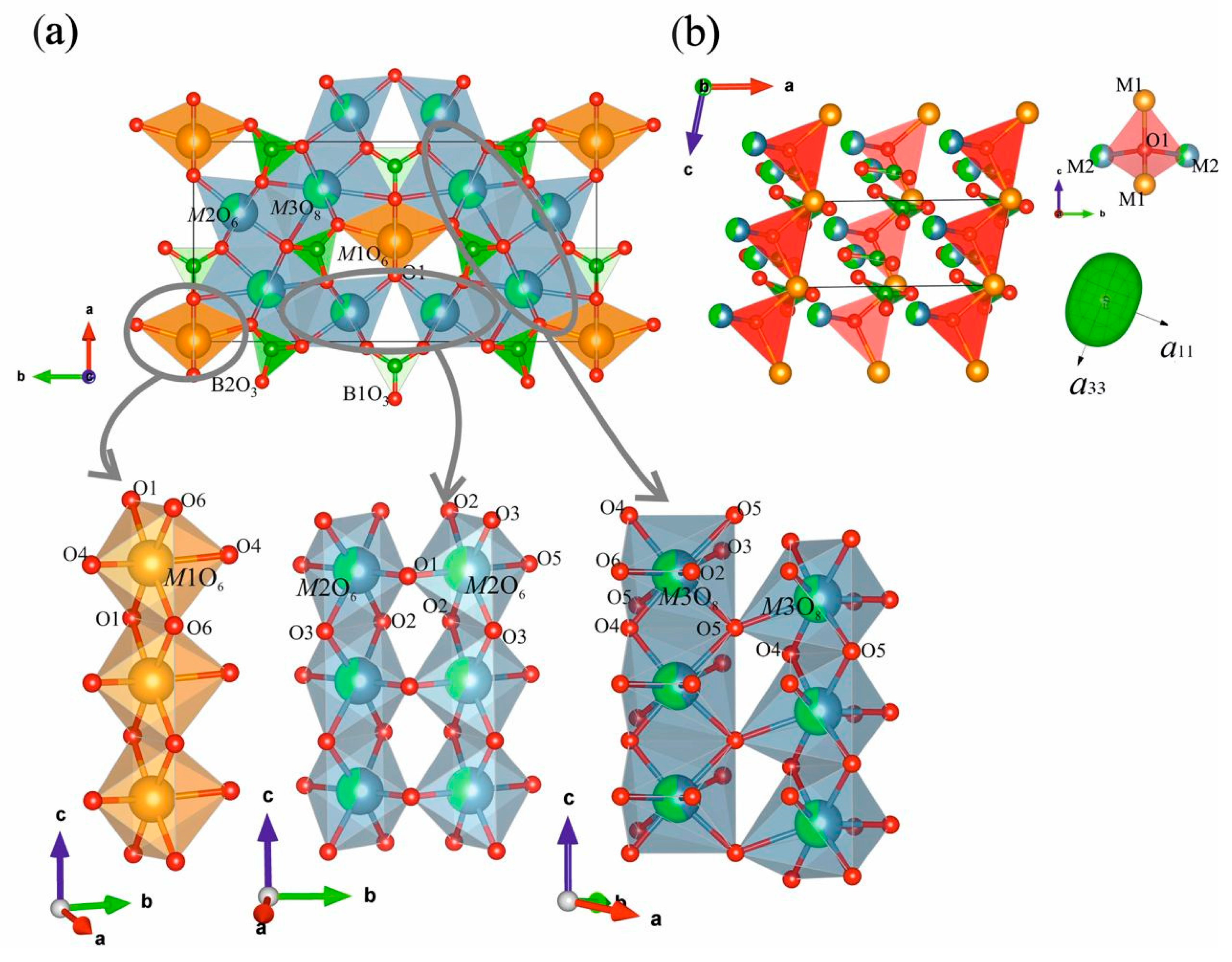
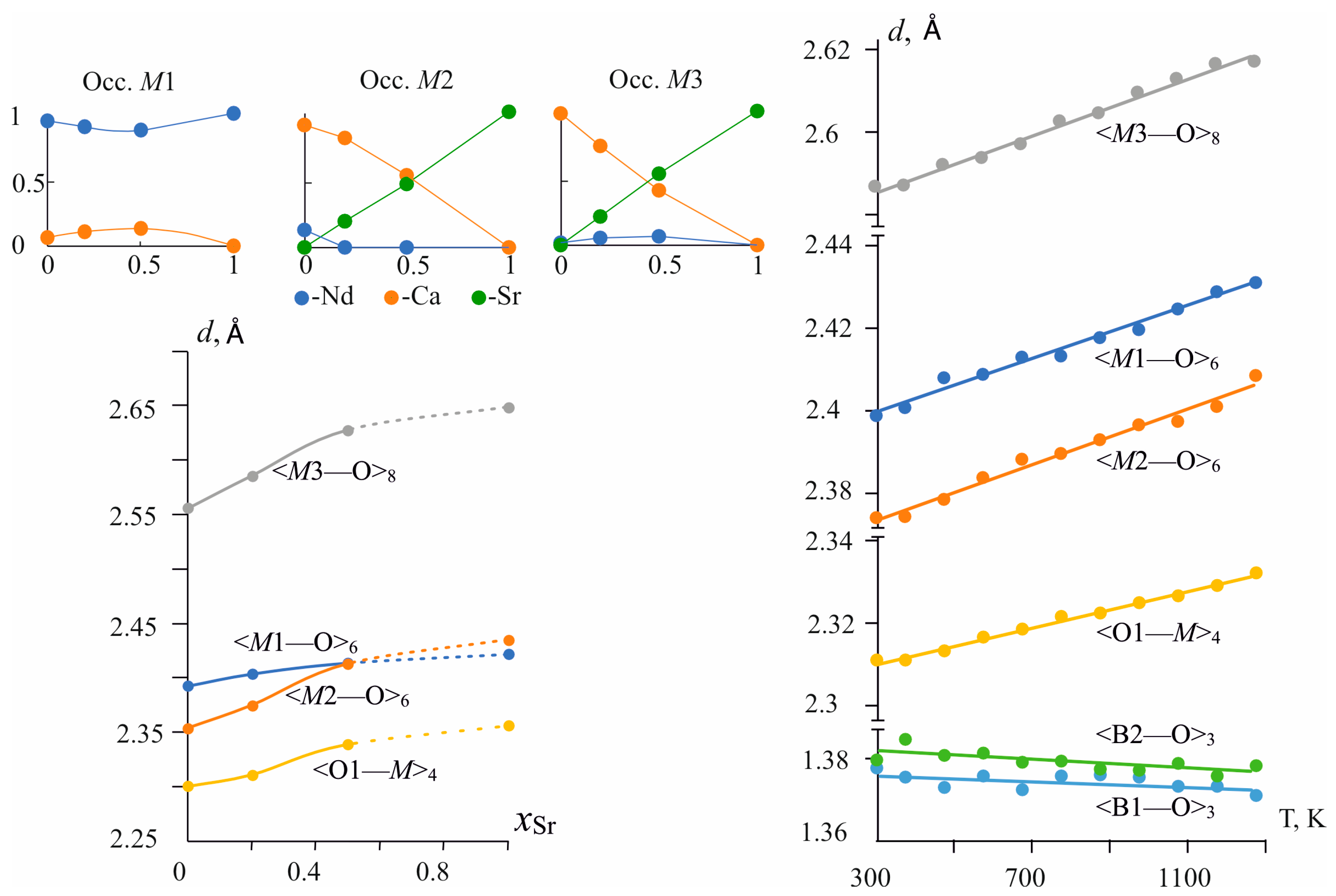
3.2. Crystal Structures of Nd(Ca1−xSrx)4O(BO3)3 Solid Solutions
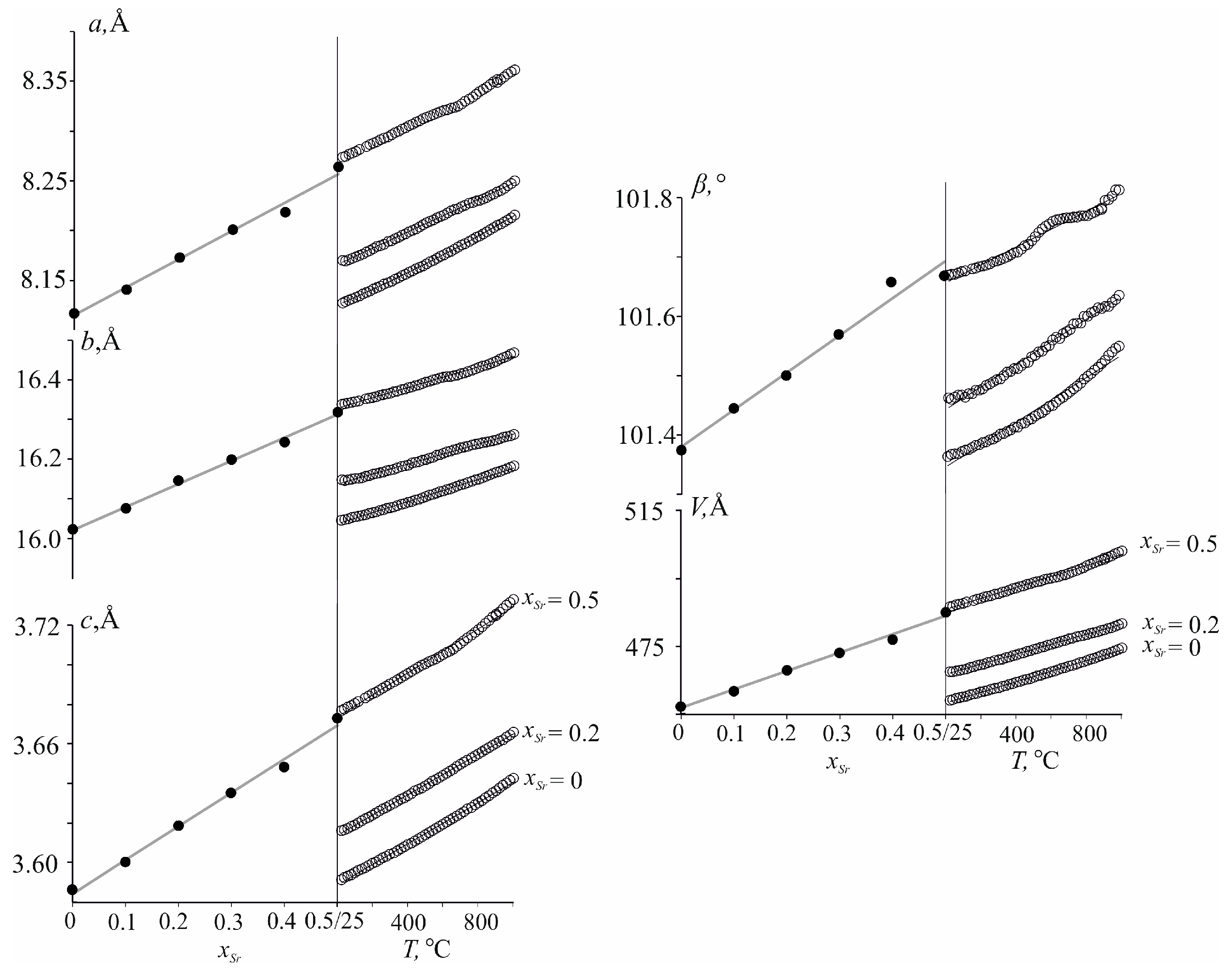
3.3. Compositional versus Thermal Properties in Nd(Ca1−xSrx)4O(BO3)3 Solid Solutions
3.3.1. Thermal Expansion
3.3.2. Compositional Properties
3.3.3. Temperature-Dependent Structural Evolution of Nd(Ca0.8Sr0.2)4O(BO3)3 Solid Solution from Single-Crystal XRD Data
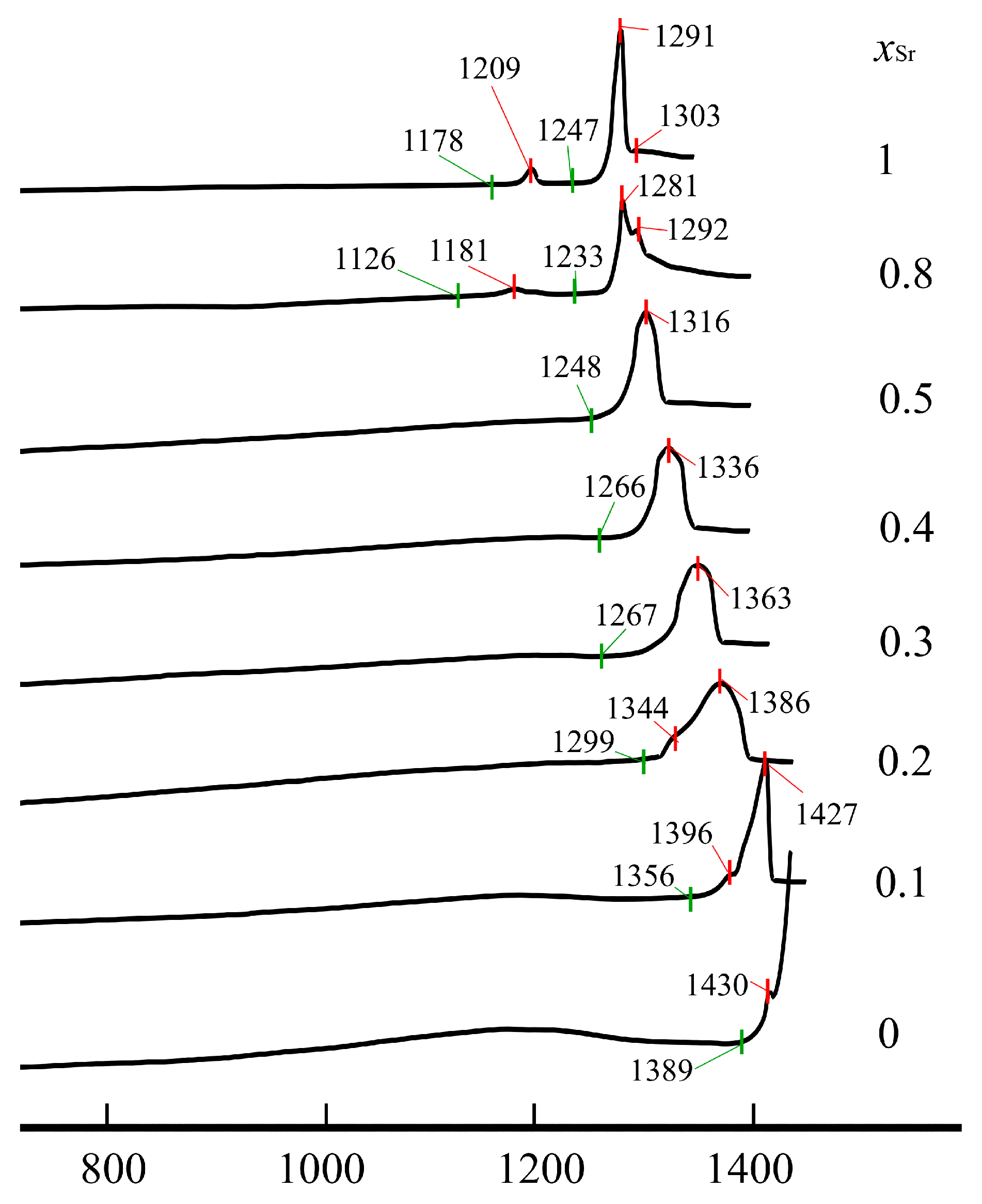
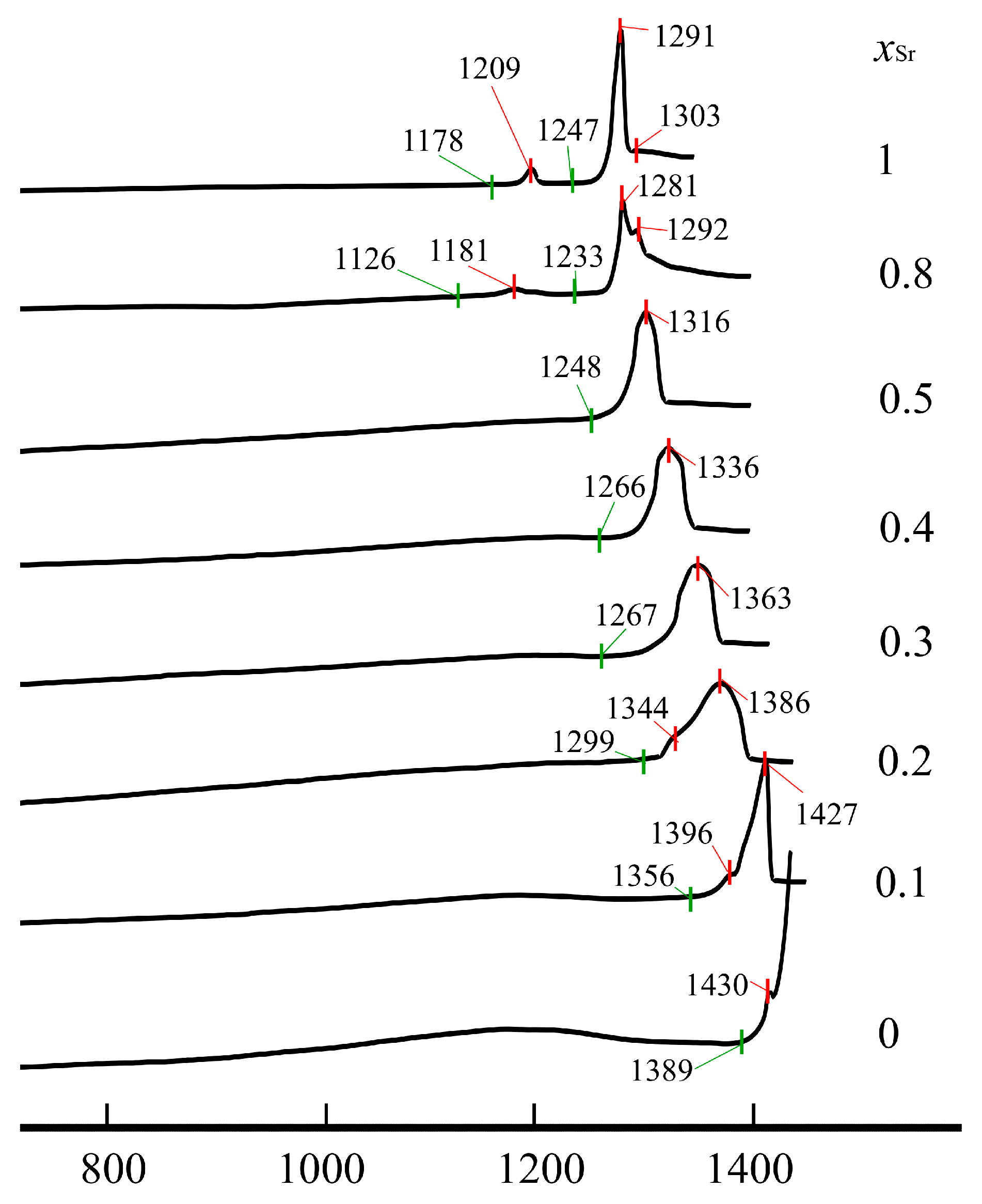
3.4. Melting Processes of Nd(Ca1−xSrx)4O(BO3)3 Solid Solutions from DCS Data
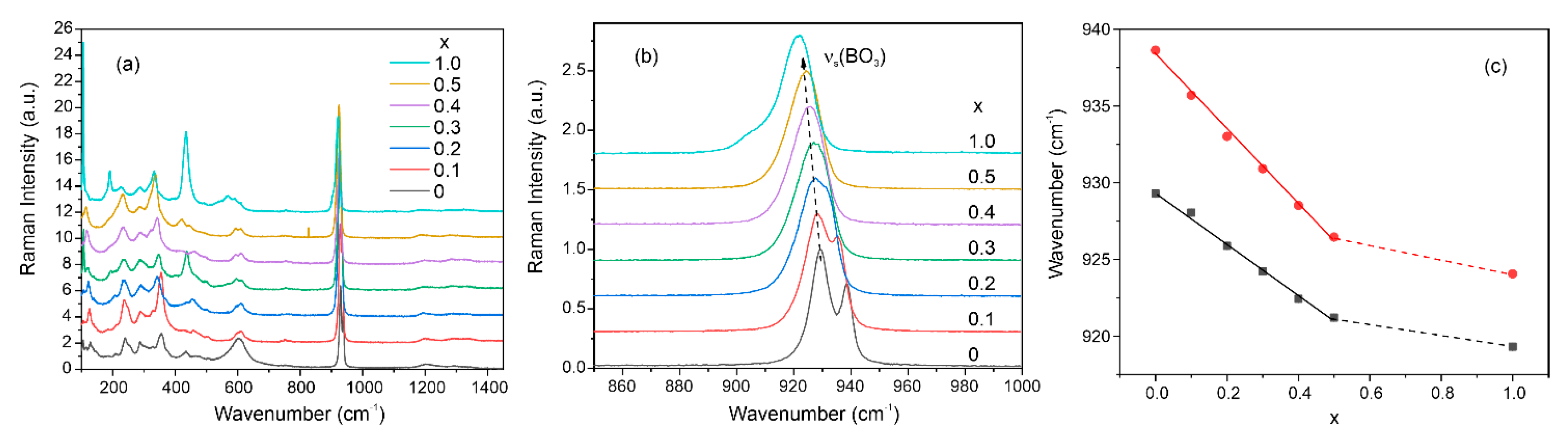
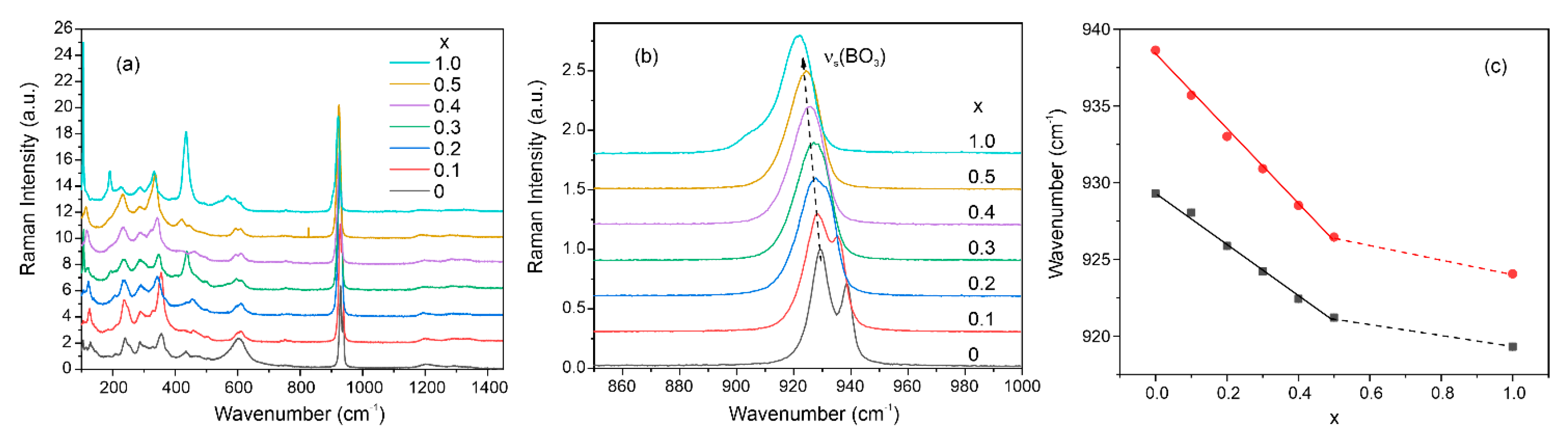
3.5. Raman Spectra
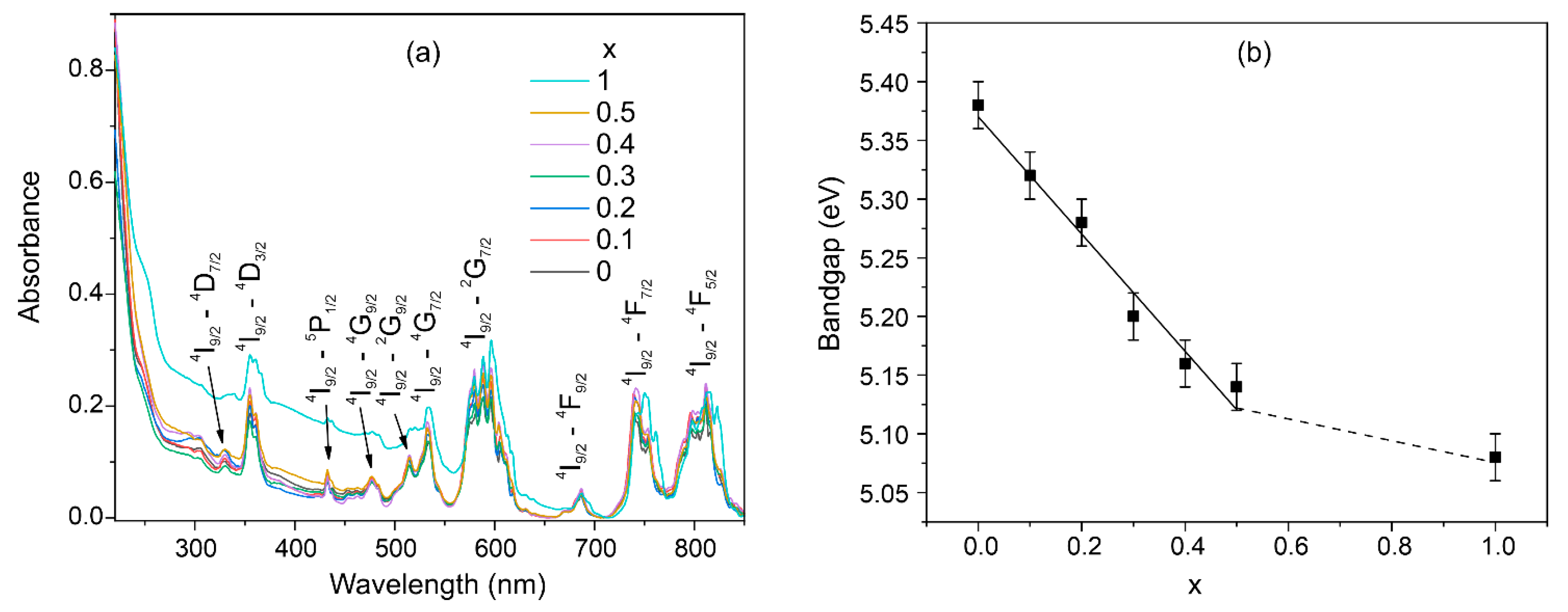
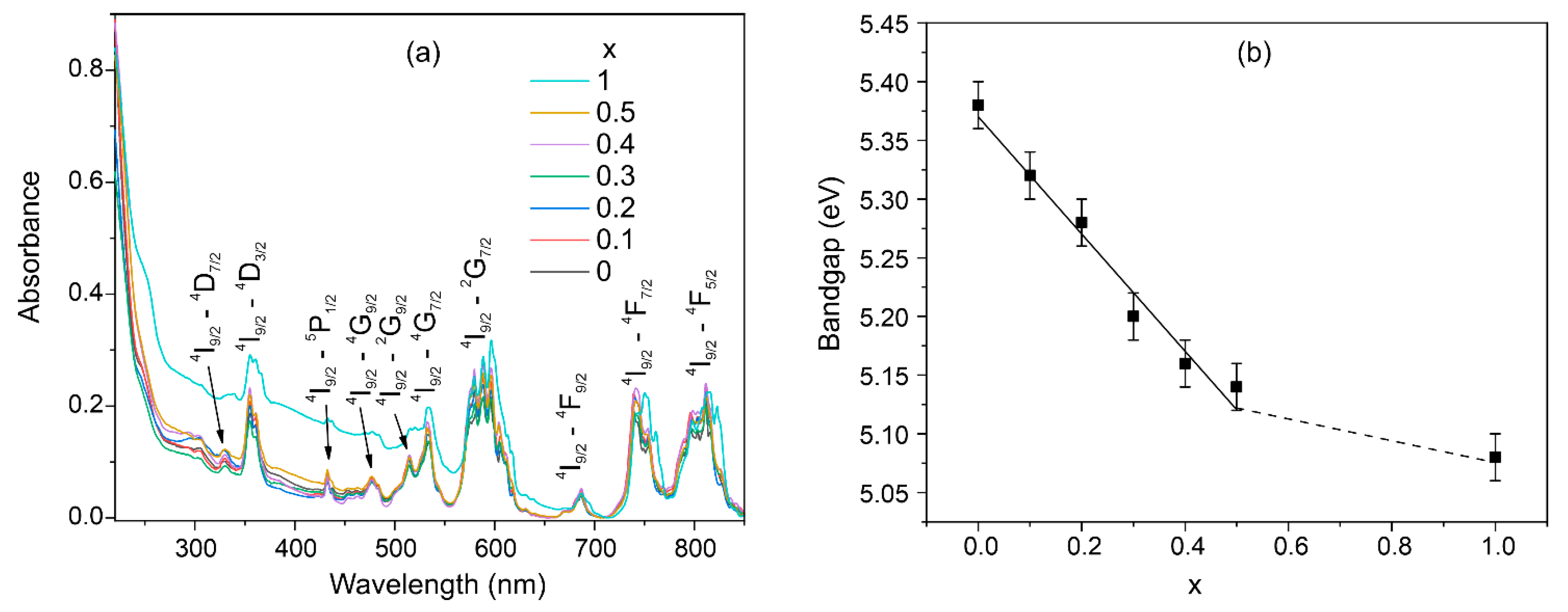
3.6. Optical Bandgap
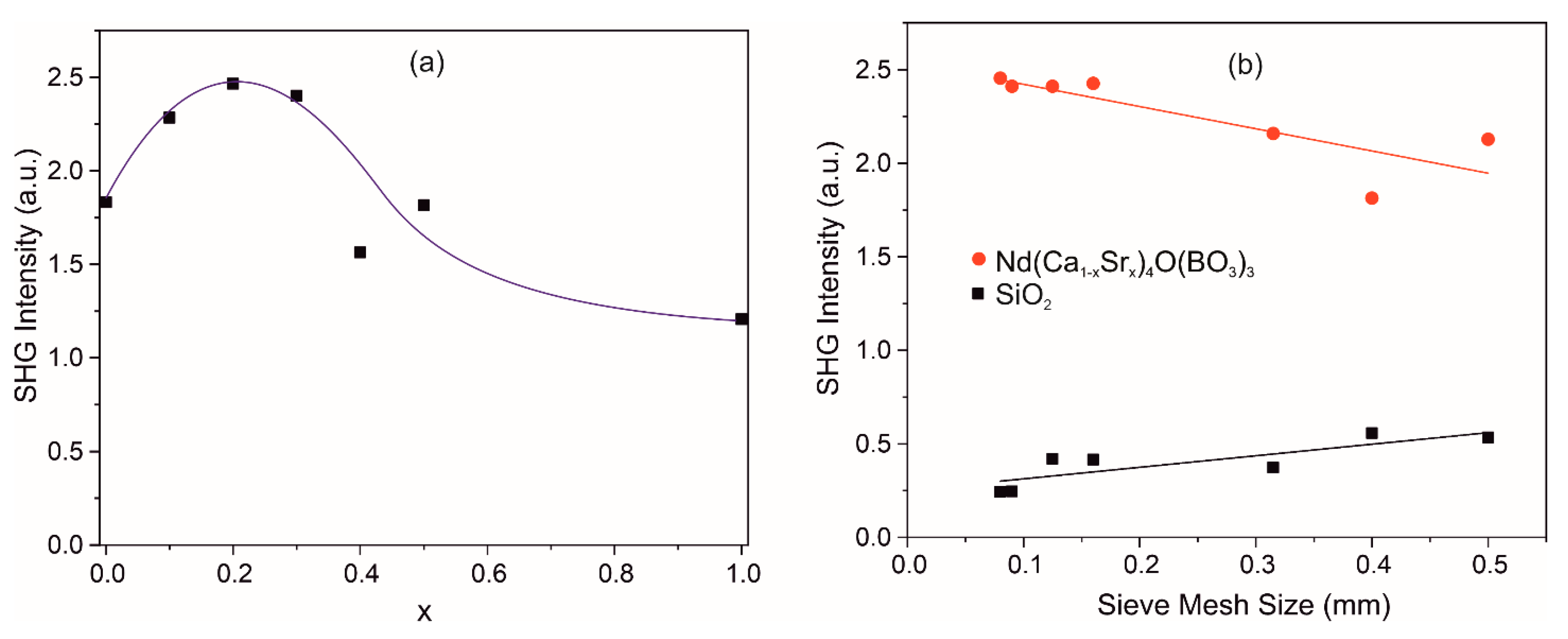
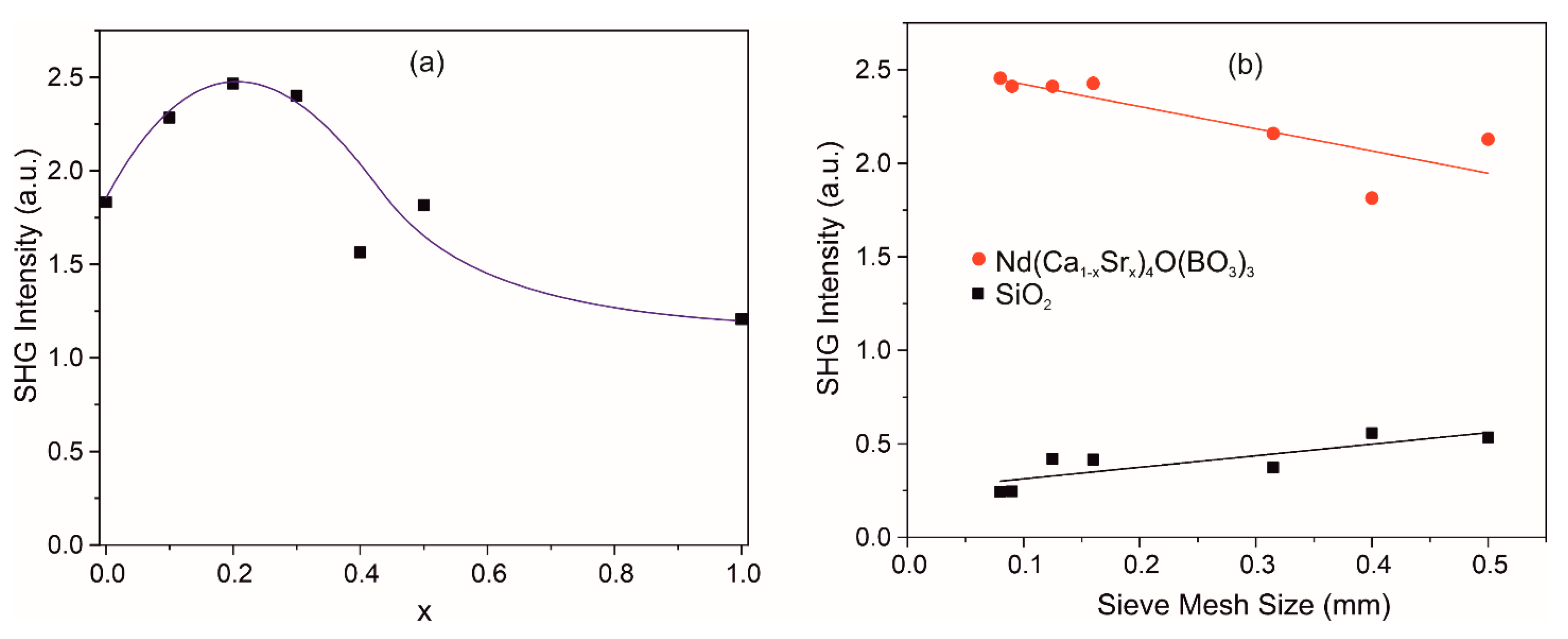
3.7. Second Harmonic Generation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khamaganova, T.N.; Trunov, V.K.; Dzuhrinskii, B.F. The crystal structure of calcium samarium oxide borate Ca8Sm2O2(BO3)6. Russ. J. Inorg. Chem. 1991, 36, 484–485. [Google Scholar]
- Norrestam, R.; Nygren, M.; Bovin, J.-O. Structural investigations of new calcium-rare earth (R) oxyborates with the composition Ca4RO(BO3)3. Chem. Mater. 1992, 4, 737–743. [Google Scholar] [CrossRef]
- Ilyukhin, A.B.; Dzhurinskii, B.F. Crystal structures of binary oxoborates LnCa4O(BO3)3 (Ln = Gd, Tb, and Lu) and Eu2CaO(BO3)2. Russ. J. Inorg. Chem. 1993, 38, 847–850. [Google Scholar]
- Sasaki, T.; Mori, Y.; Yoshimura, M.; Yap, Y.K.; Kamimura, T. Recent development of nonlinear optical borate crystals: Key materials for generation of visible and UV light. Mater. Sci. Eng. R 2000, 30, 1–54. [Google Scholar] [CrossRef]
- Aka, G.; Kahn-Harari, A.; Vivien, D.; Benitez, J.M.; Salin, F.; Godard, J. A new non-linear and neodymium laser self-frequency doubling crystal with congruent melting: Ca4GdO(BO3)3 (GdCOB). Eur. J. Solid State Inorg. Chem. 1996, 33, 727–736. [Google Scholar] [CrossRef]
- Iwai, M.I.M.; Kobayashi, T.K.T.; Furuya, H.F.H.; Mori, Y.M.Y.; Sasaki, T.S.T. Crystal growth and optical characterization of rare-earth (Re) calcium oxyborate ReCa4O(BO3)3 (Re = Y or Gd) as new nonlinear optical material. Jpn. J. Appl. Phys. 1997, 36, L276. [Google Scholar] [CrossRef]
- Aka, G.; Kahn-Harari, A.; Mougel, F.; Vivien, D.; Salin, F.; Coquelin, P.; Damelet, J.P. Linear-and nonlinear-optical properties of a new gadolinium calcium oxoborate crystal, Ca4GdO(BO3)3. J. Opt. Soc. Am. B 1997, 14, 2238–2247. [Google Scholar] [CrossRef]
- Adams, J.J.; Ebbers, C.A.; Schaffers, K.I.; Payne, S.A. Nonlinear optical properties of LaCa4O(BO3)3. Opt. Lett. 2001, 26, 217–219. [Google Scholar] [CrossRef]
- Druon, F.; Balembois, F.; Georges, P.; Brun, A.; Courjaud, A.; Hönninger, C.; Vivien, D. Generation of 90-fs pulses from a mode-locked diode-pumped Yb3+: Ca4GdO(BO3)3 laser. Opt. Lett. 2000, 25, 423–425. [Google Scholar] [CrossRef]
- Vincent, B.; Boudrioua, A.; Loulergue, J.C.; Moretti, P.; Tascu, S.; Jacquier, B.; Vivien, D. Channel waveguides in Ca4GdO(BO3)3 fabricated by He+ implantation for blue-light generation. Opt. Lett. 2003, 28, 1025–1027. [Google Scholar] [CrossRef]
- Chai, B.H. Advances in bulk inorganic nonlinear optical materials. Opt. Phot. News 1999, 10, 31. [Google Scholar] [CrossRef]
- Kuz’micheva, G.M.; Kaurova, I.A.; Rybakov, V.B.; Podbel’skiy, V.V. Crystallochemical design of huntite-family compounds. Crystals 2019, 9, 100. [Google Scholar] [CrossRef]
- Kuz’micheva, G.M.; Ageev, A.Y.; Rybakov, V.B.; Panyutin, V.L.; Yu, Y.M.; Chizhikov, V.I. Growth and X-ray diffraction study of YCa4O(BO3)3: Ce, Er, Yb crystals. Inorg. Mater. 2001, 37, 1051–1060. [Google Scholar] [CrossRef]
- Yang, H.C.; Li, C.Y.; He, H.; Tao, Y.; Xu, J.H.; Su, Q. VUV–UV excited luminescent properties of LnCa4O(BO3)3: RE3+ (Ln = Y, La, Gd; Re = Eu, Tb, Dy, Ce). J. Lumin. 2006, 118, 61–69. [Google Scholar] [CrossRef]
- Furuya, H.; Nakao, H.; Yamada, I.; Ruan, Y.F.; Yap, Y.K.; Yoshimura, M.; Mori, Y.; Sasaki, T. Alleviation of photoinduced damage in GdxY1–xCa4O(BO3)3 at elevated crystal temperature for noncritically phase-matched 355-nm generation. Opt. Lett. 2000, 25, 1588–1590. [Google Scholar] [CrossRef] [PubMed]
- Umemura, N.; Nakao, H.; Furuya, H.; Yoshimura, M.; Mori, Y.; Sasaki, T.; Yoshida, K.; Kato, K. 90° phase-matching properties of YCa4O(BO3)3 and GdxY1−xCa4O(BO3)3. Jpn. J. Appl. Phys. 2001, 40, 596. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, X.; Fu, K.; Song, R.; Wang, J.; Wei, J.; Liu, Y.; Shao, Z. Non-critical phase matching of GdxY1−xCa4O(BO3)3 (GdxY1−xCOB) crystal. Solid State Commun. 2001, 120, 397–400. [Google Scholar] [CrossRef]
- Nakao, H.; Kawamura, K.; Furuya, H.; Yamada, I.; Yoshimura, M.; Mori, Y.; Sasaki, T. Investigation of the Gd variation of GdxY1−xCa4O(BO3)3 crystal and its application. Jpn. J. Appl. Phys. 2002, 41, L723. [Google Scholar] [CrossRef]
- Jang, W.; Ye, Q.; Hammons, D.; Eichenholz, J.; Lim, J.; Richardson, M.; Chai, B.H.T.; Van Stryland, E. Improved Second-Harmonic Generation by Selective Yb Ion Doping in a New Nonlinear Optical Crystal YCOB. IEEE J. Quant. Electr. 1999, 35, 1826–1833. [Google Scholar]
- Kelly, N.D.; Savvin, S.; Dutton, S.E. Crystal structure and specific heat of calcium lanthanide oxyborates Ca4LnO(BO3)3. Z. Kristallogr. 2022, 237, 317–327. [Google Scholar] [CrossRef]
- Möckel, R.; Reuther, C.; Götze, J. REECOB: 20 years of rare earth element calcium oxoborates crystal growth research. Cryst. Growth Des. 2013, 371, 70–76. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Z.; Zhang, S.; Wang, J.; Chen, H.; Shao, Z.; Jiang, M. Second-harmonic generation of 1.06 μm in Sr doped GdCa4O(BO3)3 crystal. Opt. Commun. 2001, 195, 267–271. [Google Scholar] [CrossRef]
- Achim, A.; Gheorghe, L.; Voicu, F.; Stanciu, G. Blue light production by type-I non-critical phase matching second-harmonic generation in La(Ca1−xSrx)4O(BO3)3 single crystals. Cryst. Eng. Comm. 2015, 17, 4098–4101. [Google Scholar] [CrossRef]
- Weigel, T.; Mehner, E.; Stöcker, H.; Meyer, D.C.; Götze, J.; Hanzig, J. Crystal Structure of Gd(Ca3.319Sr0.681)O[BO3]3 and Gd(Ca2.592Sr1.408)O[BO3]3. Cryst. Res. Technol. 2023, 58, 2200255. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A Found. Crystallogr. 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Münchhalfen, M.; Schreuer, J.; Reuther, C.; Mehner, E.; Stöcker, H. Elastic, piezoelectric, and dielectric properties of rare-earth calcium oxoborates RCa4O(BO3)3 (R = Er, Y, Dy, Gd, Sm, Nd, La). J. Appl. Phys. 2021, 130, 095102. [Google Scholar] [CrossRef]
- Mougel, F.; Kahn-Harari, A.; Aka, G.; Pelenc, D. Structural and thermal stability of Czochralski grown GdCOB oxoborate single crystals. J. Mater. Chem. 1998, 8, 1619–1623. [Google Scholar] [CrossRef]
- Dirksen, G.E.; Blasse, G. Tetracalcium gadolinium oxoborate (Ca4GdO(BO3)3) as a new host lattice for luminescent materials. J. Alloys Compd. 1993, 191, 121–126. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y. Photo-luminescent properties of Eu3+ ion-doped Ca4REO(BO3)3 (RE La, Y, Gd). J. Lumin. 2005, 113, 45–49. [Google Scholar] [CrossRef]
- Kuo, T.W.; Chen, T.M. Synthesis and luminescence of Ca4YO(BO3)3: Eu3+ for fluorescent lamp application. Opt. Mater. 2010, 32, 882–885. [Google Scholar] [CrossRef]
- Ju, G.; Hu, Y.; Chen, L.; Wang, X.; Mu, Z.; Wu, H.; Kang, F. The luminescence of bismuth and europium in Ca4YO(BO3)3. J. Lumin. 2012, 132, 717–721. [Google Scholar] [CrossRef]
- Yu, F.; Zhang, S.; Zhao, X.; Guo, S.; Duan, X.; Yuan, D.; Shrout, T.R. Investigation of the dielectric and piezoelectric properties of ReCa4O(BO3)3 crystals. J. Phys. D Appl. Phys. 2011, 44, 135405. [Google Scholar] [CrossRef]
- Münchhalfen, M.; Schreuer, J.; Reuther, C.; Möckel, R.; Götze, J.; Mehner, E.; Stöcker, H.; Meyer, D. Order/disorder processes and electromechanical properties of monoclinic GdCa4O(BO3)3. Z. Kristallogr. 2019, 234, 707–723. [Google Scholar] [CrossRef]
- Kelly, N.D.; Dutton, S.E. Magnetic properties of quasi-one-dimensional lanthanide calcium oxyborates Ca4LnO(BO3)3. Inorg. Chem. 2020, 59, 9188–9195. [Google Scholar] [CrossRef] [PubMed]
- Kalakkodu, S. ICDD, PDF-2 2011, Database; ICDD: Newtown Square, PA, USA, 2011. [Google Scholar]
- Bubnova, R.S.; Firsova, V.A.; Volkov, S.N.; Filatov, S.K. RietveldToTensor: Program for processing powder X-ray diffraction data under variable conditions. Glass Phys. Chem. 2018, 44, 33–40. [Google Scholar] [CrossRef]
- Palatinus, L.; Chapuis, G. SUPERFLIP—A computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Crystallogr. 2007, 40, 786–790. [Google Scholar] [CrossRef]
- Petříček, V.; Dušek, M.; Palatinus, L. Crystallographic Computing System JANA2006: General features. Z. Kristallogr. Cryst. Mater. 2014, 229, 345–352. [Google Scholar] [CrossRef]
- VESTA: Visualization for Electronic and Structural Analysis. Available online: https://jp-minerals.org/vesta/en/ (accessed on 17 November 2021).
- Zhang, Y.; Chen, X.L.; Liang, J.K.; Cao, Y.G.; Xu, T. Phase relations in the system La2O3–CaO–B2O3. J. Alloys Compd. 2001, 315, 198–202. [Google Scholar] [CrossRef]
- Reuther, C.; Moeckel, R.; Hengst, M.; Goetze, J.; Schwarzer, A.; Schmidt, H. Growth and structure of Ca4La[O(BO)3)3]. J. Cryst. Growth 2011, 320, 90–94. [Google Scholar] [CrossRef]
- Crossno, S.K. Tetracalcium Lanthanide Borate Oxide: Structures and Optical Properties. Master’s Thesis, Oregon State University, Corvallis, ON, USA, 1997. [Google Scholar]
- Filatov, S.K. Nekotoryye strukturno-geometricheskiye izmereniya deformatsiy kristallov pri ispol’zovanii temperatury, davleniya i khimizm. Kristallogr. Kristallokh. 1973, 2, 5. [Google Scholar]
- Hazen, R.M. Temperature, pressure, and composition: Structurally analogous variables. Phys. Chem. Miner. 1977, 1, 83–94. [Google Scholar] [CrossRef]
- Hazen, R.M.; Finger, L.W. Comparative Crystal Chemistry; John Wiley & Sons: New York, NY, USA, 1982; Volume 231. [Google Scholar]
- Filatov, S.K. Vysokotemperaturnaya kristallokhimiya; Nedra: Leningrad, Russia, 1990; Volume 288. [Google Scholar]
- Hazen, R.M.; Downs, R.T.; Prewitt, C.T. Principles of comparative crystal chemistry. Rev. Mineral. Geochem. 2000, 41, 1–33. [Google Scholar] [CrossRef]
- Bubnova, R.S.; Filatov, S.K. High-temperature borate crystal chemistry. Z. Kristallogr. 2013, 228, 395–428. [Google Scholar] [CrossRef]
- Hazen, R.M.; Prewitt, C.T. Effects of temperature and pressure on interatomic distances in oxygen-based mineral. Amer. Miner. 1977, 62, 309–315. [Google Scholar]
- Cruickshank, D.W.J. Errors in bond lengths due to rotational oscillations of molecules. Acta Cryst. 1956, 9, 757–758. [Google Scholar] [CrossRef]
- Busing, W.R.; Levy, H.A. The effect of thermal motion on the estimation of bond lengths from diffraction measurements. Acta Cryst. 1964, 17, 142–146. [Google Scholar] [CrossRef]
- Biryukov, Y.P.; Bubnova, R.S.; Krzhizhanovskaya, M.G.; Filatov, S.K.; Povolotskiy, A.V.; Ugolkov, V.L. Thermal behavior of polymorphic modifications of LuBO3. Solid State Sci. 2020, 99, 106061. [Google Scholar] [CrossRef]
- Yasseri, M.; Schüpfer, D.; Chen, L.; Kamila, H.; Müller, E.; de Boor, J.; Klar, P.J. Raman Spectroscopic Study of the Optical Phonons of Mg2Si1−xSnx Solid Solutions. Phys. Status Solidi. 2020, 14, 1900574. [Google Scholar] [CrossRef]
- Wang, X.; Qi, H.; Li, Y.; Yu, F.; Wang, H.; Chen, F.; Zhao, X. Synthesis and characterization of new Sr3(BO3)2 crystal for stimulated Raman scattering applications. Crystals 2017, 7, 125. [Google Scholar] [CrossRef]
- Chen, C.; Sasaki, T.; Li, R.; Wu, Y.; Lin, Z.; Mori, Y.; Hu, Z.; Wang, J.; Uda, S.; Yoshimura, M.; et al. Nonlinear Optical Borate Crystals: Principals and Applications; John Wiley & Sons: New York, NY, USA, 2012; Volume 406. [Google Scholar]
- Becker, P. Borate materials in nonlinear optics. Adv. Mater. 1998, 10, 979–992. [Google Scholar] [CrossRef]
- Shen, Y.; Zhao, S.; Luo, J. The role of cations in second-order nonlinear optical materials based on π-conjugated [BO3]3− groups. Coord. Chem. Rev. 2018, 366, 1–28. [Google Scholar] [CrossRef]
- Gong, P.F.; Liu, X.M.; Kang, L.; Lin, Z.S. Inorganic planar π-conjugated groups in nonlinear optical crystals: Review and outlook. Inorg. Chem. Front. 2020, 7, 839–852. [Google Scholar] [CrossRef]
- Mutailipu, M.; Poeppelmeier, K.R.; Pan, S. Borates: A Rich Source for Optical Materials. Chem. Rev. 2021, 121, 1130–1202. [Google Scholar] [CrossRef]
- Bubnova, R.; Volkov, S.; Albert, B.; Filatov, S.K. Borates—Crystal Structures of Prospective Nonlinear Optical Materials: High Anisotropy of the Thermal Expansion Caused by Anharmonic Atomic Vibrations. Crystals 2017, 7, 93. [Google Scholar] [CrossRef]
- Bubnova, R.S.; Shablinskii, A.P.; Stefanovich, S.Y.; Arsent’ev, M.Y.; Krzhizhanovskaya, M.G.; Lazoryak, B.I.; Ugolkov, V.L.; Filatov, S.K. Expanding gaudefroyite family to Sr2MBi(REEO)3(BO3)4 (M = Ca, Sr, Ba; REE = Y, Eu) borates with large second harmonic generation responses. Ceram. Intern. 2023, 49, 15082–15090. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| xSr | 0.0 [21] | 0.2 * | 0.5 * | 1.0 * |
|---|---|---|---|---|
| Crystal data | ||||
| Mr | 535 | 592.1 | 687.1 | |
| Crystal system, space group, Z | Monoclinic, Cm, 2 | |||
| Temperature (K) | 293 | |||
| a (Å) | 8.12367 (5) | 8.1882 (2) | 8.2832 (3) | 8.3311 (2) |
| b (Å) | 16.04587 (9) | 16.1822 (4) | 16.3631 (6) | 16.4552 (4) |
| c (Å) | 3.595667 (19) | 3.6238 (1) | 3.6797 (2) | 3.7085 (1) |
| β (°) | 101.3937 (4) | 101.444 (2) | 101.671 (4) | 101.767 (2) |
| V (Å3) | 459.457 (4) | 470.62 (2) | 488.43 (4) | 497.71 (2) |
| Radiation type | Mo Ka | |||
| µ (mm−1) | 17.21 | 11.79 | 26.46 | |
| Crystal size (mm) | 0.2 × 0.4 × 0.3 | 0.2 × 0.4 × 0.2 | 0.3 × 0.2 × 0.1 | |
| Data collection | ||||
| Diffractometer | XtaLAB Synergy, single source at home/near, HyPix | |||
| Absorption correction | Multi-scan | |||
| No. of measured, independent and observed [I > 3σ(I)] reflections | 1683, 1682, 1655 | 1361, 1360, 1285 | 22454, 2447, 2416 | |
| Rint | 0.03 | 0.04 | 0.045 | |
| (sin θ/λ)max (Å−1) | 0.778 | 0.703 | 0.848 | |
| Refinement | ||||
| R[F2 > 2σ(F2)], wR(F2), S | 0.028, 0.035, 2.05 | 0.034, 0.043, 2.25 | 0.028, 0.038, 3.12 | |
| No. of reflections | 1682 | 1360 | 2447 | |
| No. of parameters | 88 | 85 | 84 | |
| xSr | 0 [21] | 0.2 | 0.5 | 1.0 |
|---|---|---|---|---|
| <M1—O>6/BVS | 2.393 | 2.404/2.665 | 2.414/2.561 | 2.422/2.729 |
| <M2—O>6/BVS | 2.354 | 2.375/2.185 | 2.413/2.228 | 2.435/2.385 |
| <M3—O>8/BVS | 2.556 | 2.586/1.970 | 2.628/1.992 | 2.649/1.981 |
| <B1—O>3/BVS | 1.381 | 1.376/2.957 | 1.379/2.933 | 1.366/2.968 |
| <B2—O>3/BVS | 1.381 | 1.378/2.944 | 1.381/2.921 | 1.379/2.923 |
| M1: Occ Nd | 0.942 (8) | 0.8977 | 0.8728 | 1.0 |
| Occ Ca | 0.058 | 0.1023 | 0.1272 | 0.0 |
| Occ Sr | 0.0 | 0.0 | 0.0 | 0.0 |
| M2: Occ Nd | 0.013 | 0 | 0 | 0 |
| Occ Ca | 0.987 | 0.8096 | 0.5303 | 0 |
| Occ Sr | 0 | 0.1904 | 0.4697 | 1.0 |
| M3: Occ Nd | 0.017 | 0.0511 | 0.0636 | 0 |
| Occ Ca | 0.983 | 0.7393 | 0.4061 | 0 |
| Occ Sr | 0 | 0.2096 | 0.5303 | 1.0 |
| x | α11 | α22 | α33 | μ1a | μ3c | αa | αc | αβ | αV |
|---|---|---|---|---|---|---|---|---|---|
| T = 25 °C | |||||||||
| 0 | 9.02 | 6.94 | 11.38 | 21.1 | 9.7 | 9.33 | 11.31 | 0.668 | 27.35 |
| 0.2 | 10.22 | 7.46 | 13.17 | 26.09 | 15.5 | 10.83 | 12.96 | 1.10 | 30.85 |
| 0.5 | 8.41 | 6.59 | 11.51 | 31.1 | 19.5 | 9.24 | 11.17 | 1.32 | 26.52 |
| T = 400 °C | |||||||||
| 0 | 9.78 | 8.38 | 14.04 | 26.9 | 15.5 | 10.65 | 13.74 | 1.59 | 32.2 |
| 0.2 | 9.22 | 7.83 | 14.19 | 25.5 | 14.0 | 10.15 | 13.90 | 1.75 | 31.24 |
| 0.5 | 9.36 | 7.41 | 14.12 | 22.5 | 10.8 | 10.06 | 13.95 | 1.44 | 30.89 |
| T = 600 °C | |||||||||
| 0 | 10.14 | 9.10 | 15.38 | 28.2 | 16.8 | 11.30 | 14.94 | 2.05 | 34.61 |
| 0.2 | 8.69 | 8.02 | 14.73 | 25.2 | 13.6 | 9.77 | 14.40 | 2.09 | 31.45 |
| 0.5 | 9.82 | 7.84 | 15.54 | 20.1 | 8.4 | 10.49 | 15.41 | 1.51 | 33.19 |
| T = 800 °C | |||||||||
| 0 | 10.48 | 9.81 | 16.70 | 29.2 | 17.7 | 11.96 | 16.13 | 2.51 | 36.99 |
| 0.2 | 9.56 | 8.29 | 15.48 | 24.9 | 13.1 | 9.33 | 14.94 | 2.77 | 31.65 |
| 0.5 | 10.28 | 8.20 | 16.94 | 18.1 | 6.0 | 11.03 | 16.89 | 1.67 | 35.87 |
| Formula | Coefficient Tensor | Region | |||||
|---|---|---|---|---|---|---|---|
| α11 | α22 | α33 | µ1a | αaver | αV | T (°C) | |
| Main parameters of the thermal expansion tensor α (×106 °C−1)) | |||||||
| NdCa4O(BO3)3 Nd(Ca0.8Sr0.2)4O(BO3)3 Nd(Ca0.5Sr0.5)4O(BO3)3 | 9.78 (7) 9.2 (3) 9.10 (6) | 8.45 (6) 7.97 (26) 7.35 (5) | 14.06 (10) 14.20 (46) 13.98 (9) | 27.2 25.9 23.1 | 10.38 10.32 10.14 | 32.3 (2) 31.4 (10) 30.4 (2) | 20–800 20–800 20–800 |
| Coefficient tensor | Region | ||||||
| γ11 | γ22 | γ33 | µ1a | γaver | γV | x, at% Sr | |
| Main parameters of the compositional deformation tensor γ (×0.01/at% Sr) | |||||||
| Nd(Ca0.5Sr0.5)4O(BO3)3 | 32.01(1) | 35.75(1) | 47.44(2) | 27.0 | 38.69 | 115.20(5) | 0–0.5 |
| Coefficient tensor | Region | ||||||
| (α/γ)11 | (α/γ)22 | (α/γ)33 | (α/γ)aver | (α/γ)V | |||
| Compositional equivalents of the thermal expansion: α/γ (×10−6 °C−1/0.01 at% Sr) | |||||||
| Nd(Ca0.5Sr0.5)4O(BO3)3 | 0.28 | 0.21 | 0.295 | 0.262 | 0.264 | 20–800 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bubnova, R.; Yukhno, V.; Yurev, A.; Povolotskiy, A.; Krzhizhanovskaya, M.; Volkov, S.; Ugolkov, V.; Filatov, S. Novel Non-Centrosymmetric NdSr4O(BO3)3 Borate and Nd(Ca1−xSrx)4O(BO3)3 Solid Solutions: Preparation, Crystal Structures, Thermal Expansion and Optical Properties. Crystals 2023, 13, 1395. https://doi.org/10.3390/cryst13091395
Bubnova R, Yukhno V, Yurev A, Povolotskiy A, Krzhizhanovskaya M, Volkov S, Ugolkov V, Filatov S. Novel Non-Centrosymmetric NdSr4O(BO3)3 Borate and Nd(Ca1−xSrx)4O(BO3)3 Solid Solutions: Preparation, Crystal Structures, Thermal Expansion and Optical Properties. Crystals. 2023; 13(9):1395. https://doi.org/10.3390/cryst13091395
Chicago/Turabian StyleBubnova, Rimma, Valentina Yukhno, Artem Yurev, Alexey Povolotskiy, Maria Krzhizhanovskaya, Sergey Volkov, Valery Ugolkov, and Stanislav Filatov. 2023. "Novel Non-Centrosymmetric NdSr4O(BO3)3 Borate and Nd(Ca1−xSrx)4O(BO3)3 Solid Solutions: Preparation, Crystal Structures, Thermal Expansion and Optical Properties" Crystals 13, no. 9: 1395. https://doi.org/10.3390/cryst13091395
APA StyleBubnova, R., Yukhno, V., Yurev, A., Povolotskiy, A., Krzhizhanovskaya, M., Volkov, S., Ugolkov, V., & Filatov, S. (2023). Novel Non-Centrosymmetric NdSr4O(BO3)3 Borate and Nd(Ca1−xSrx)4O(BO3)3 Solid Solutions: Preparation, Crystal Structures, Thermal Expansion and Optical Properties. Crystals, 13(9), 1395. https://doi.org/10.3390/cryst13091395