
Atomistic Study on Defect–Grain Boundary Interactions in TiVTa Concentrated Solid–Solution Alloys
 , , and
, , and
Abstract
:1. Introduction
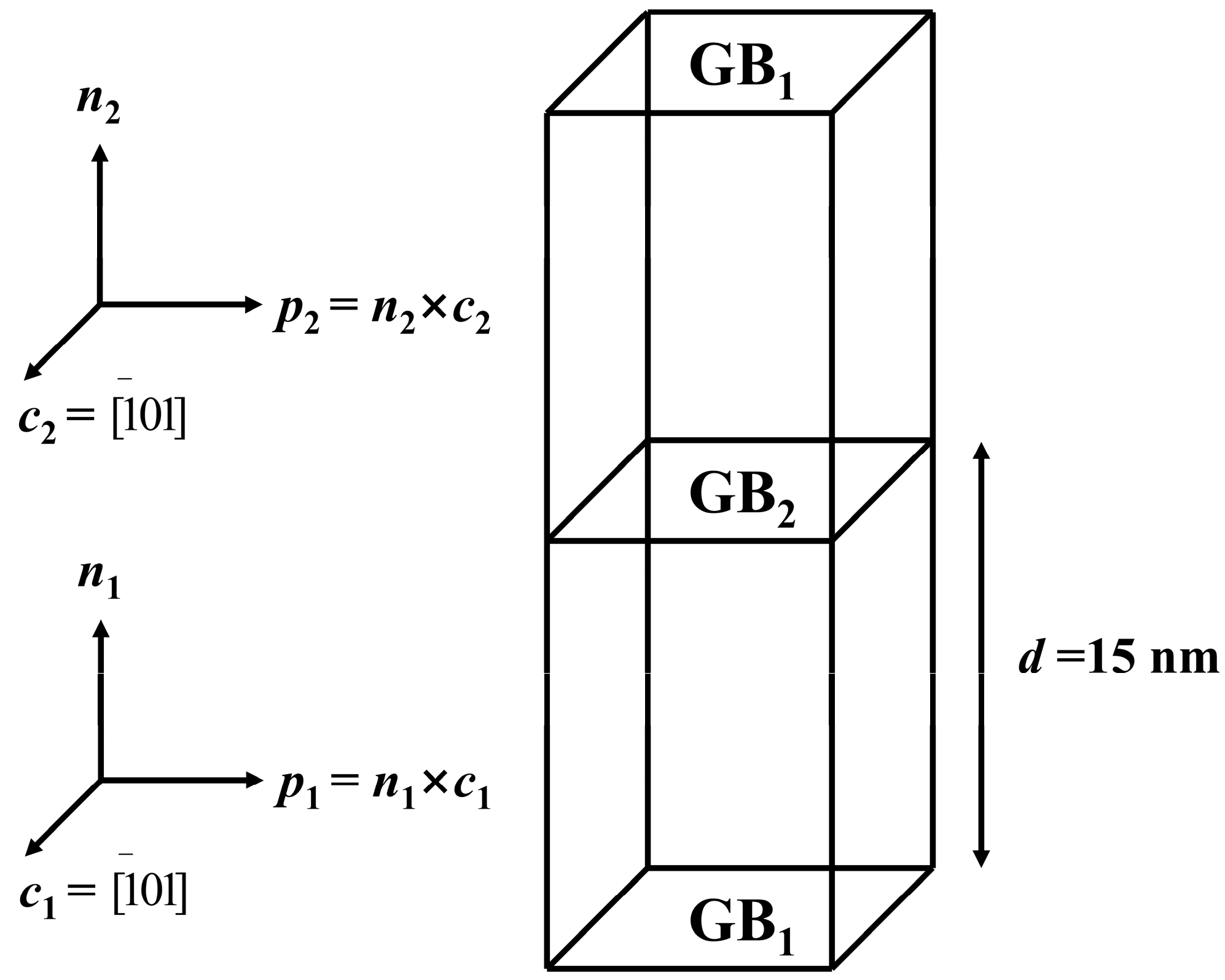
2. Materials and Methods
3. Results
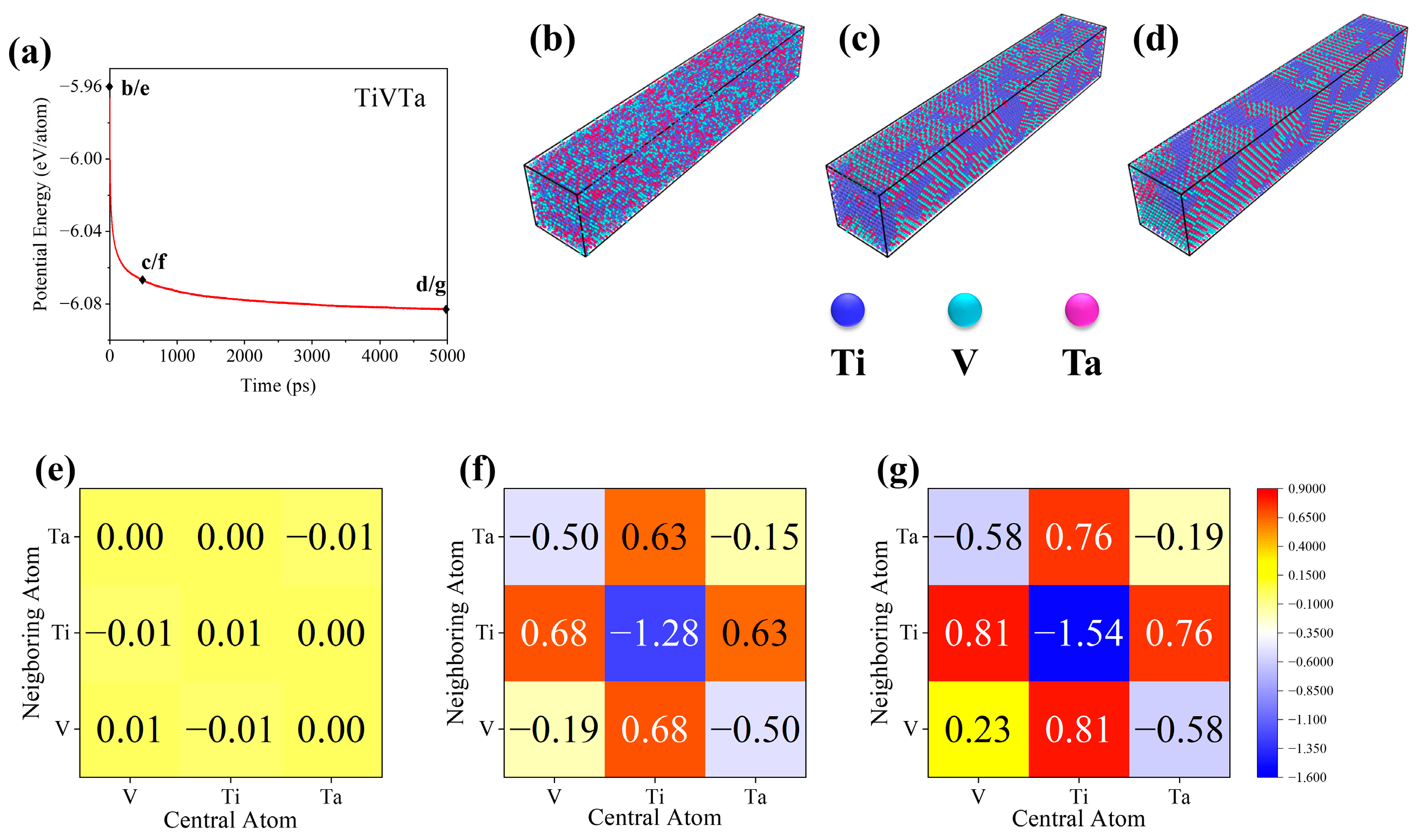
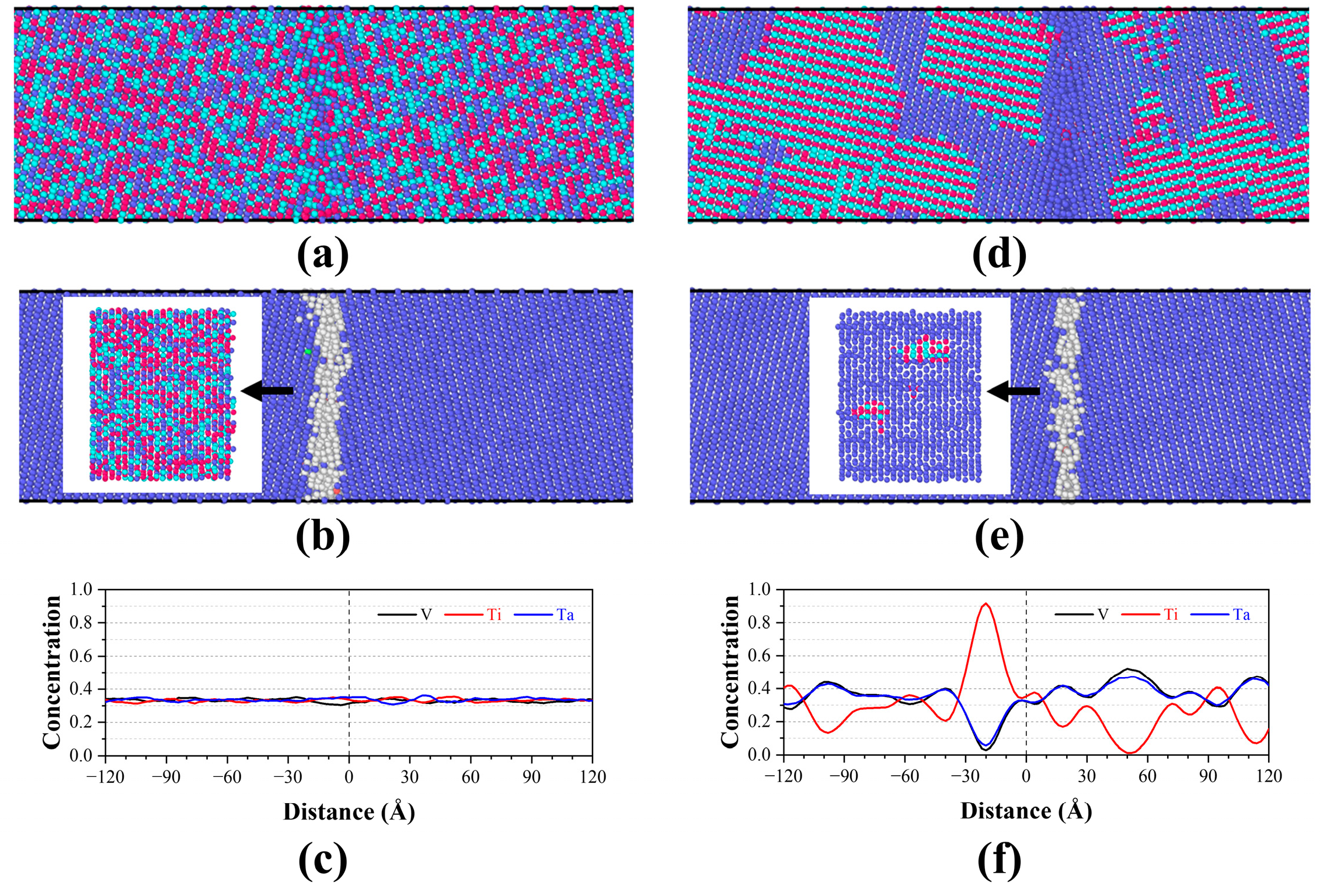
3.1. Grain Boundary Structure
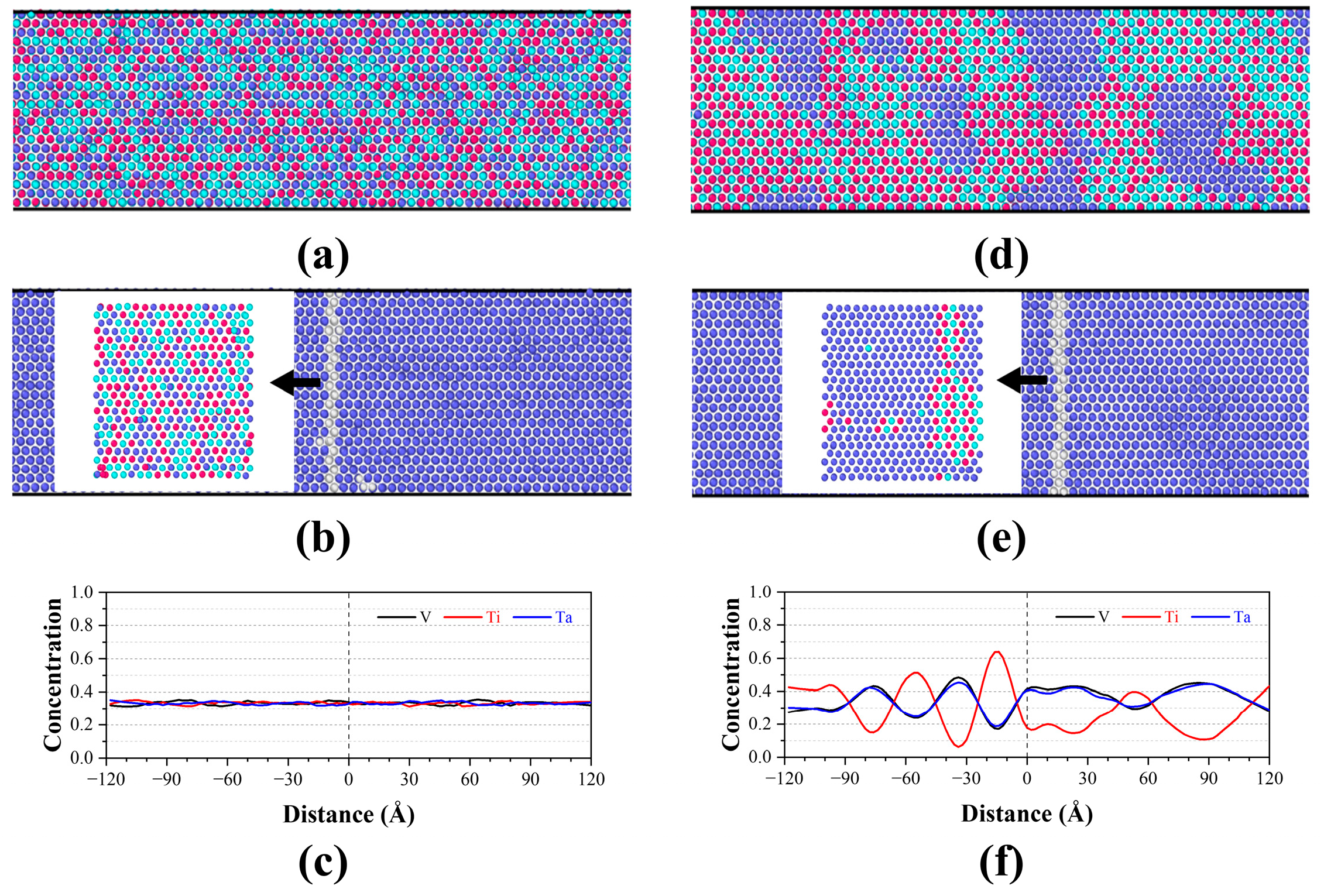
3.2. Grain Boundary Segregation
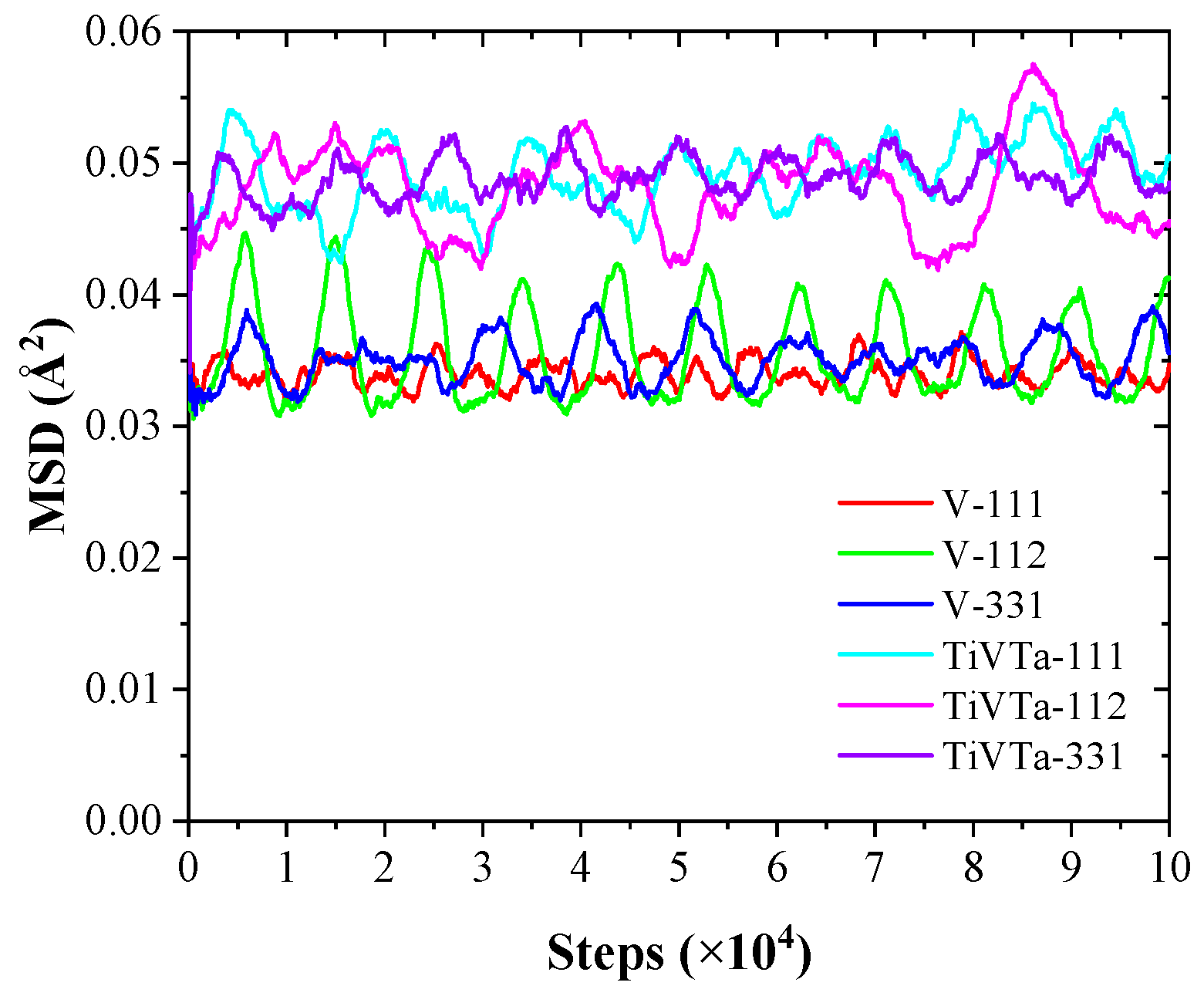
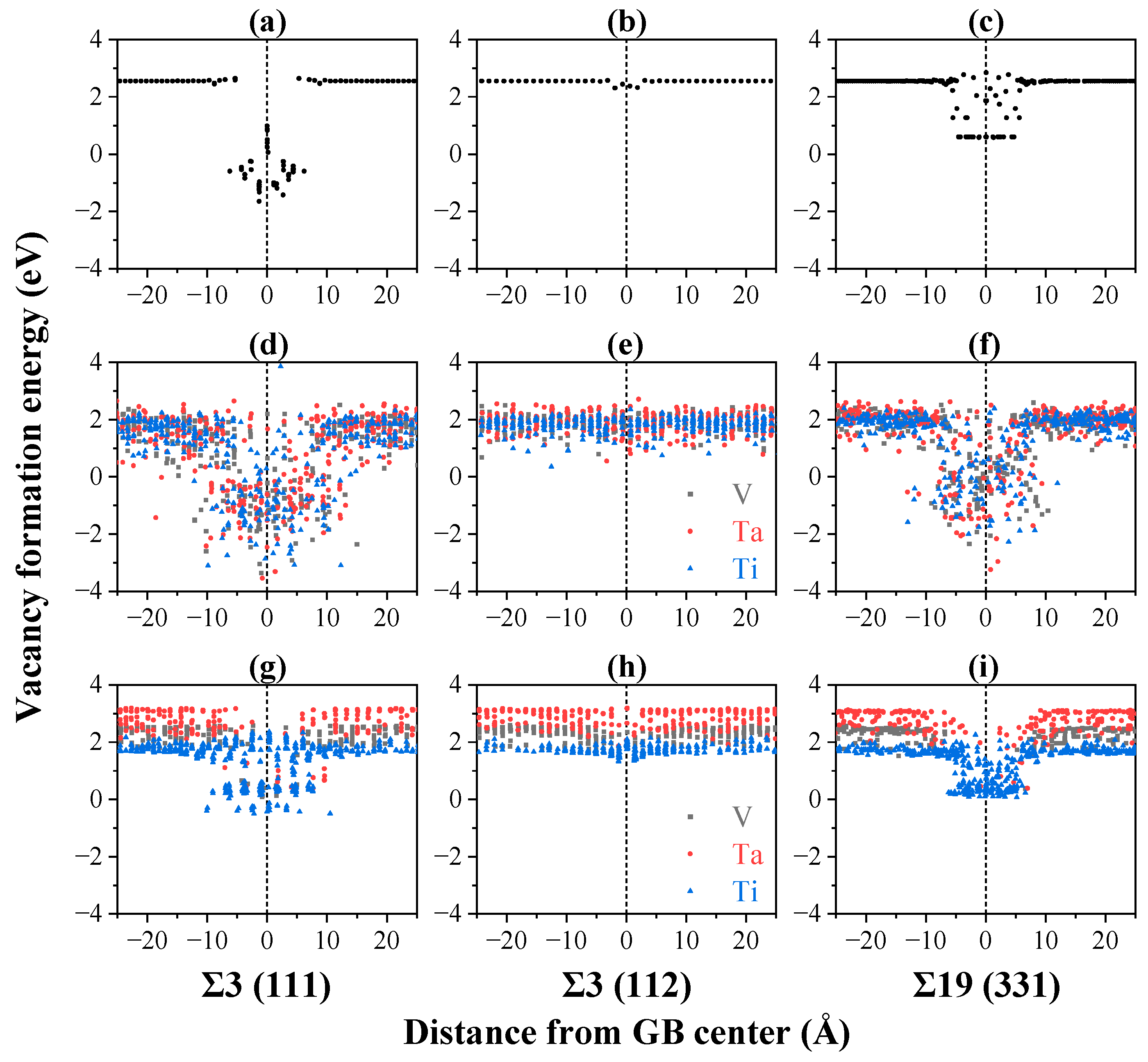
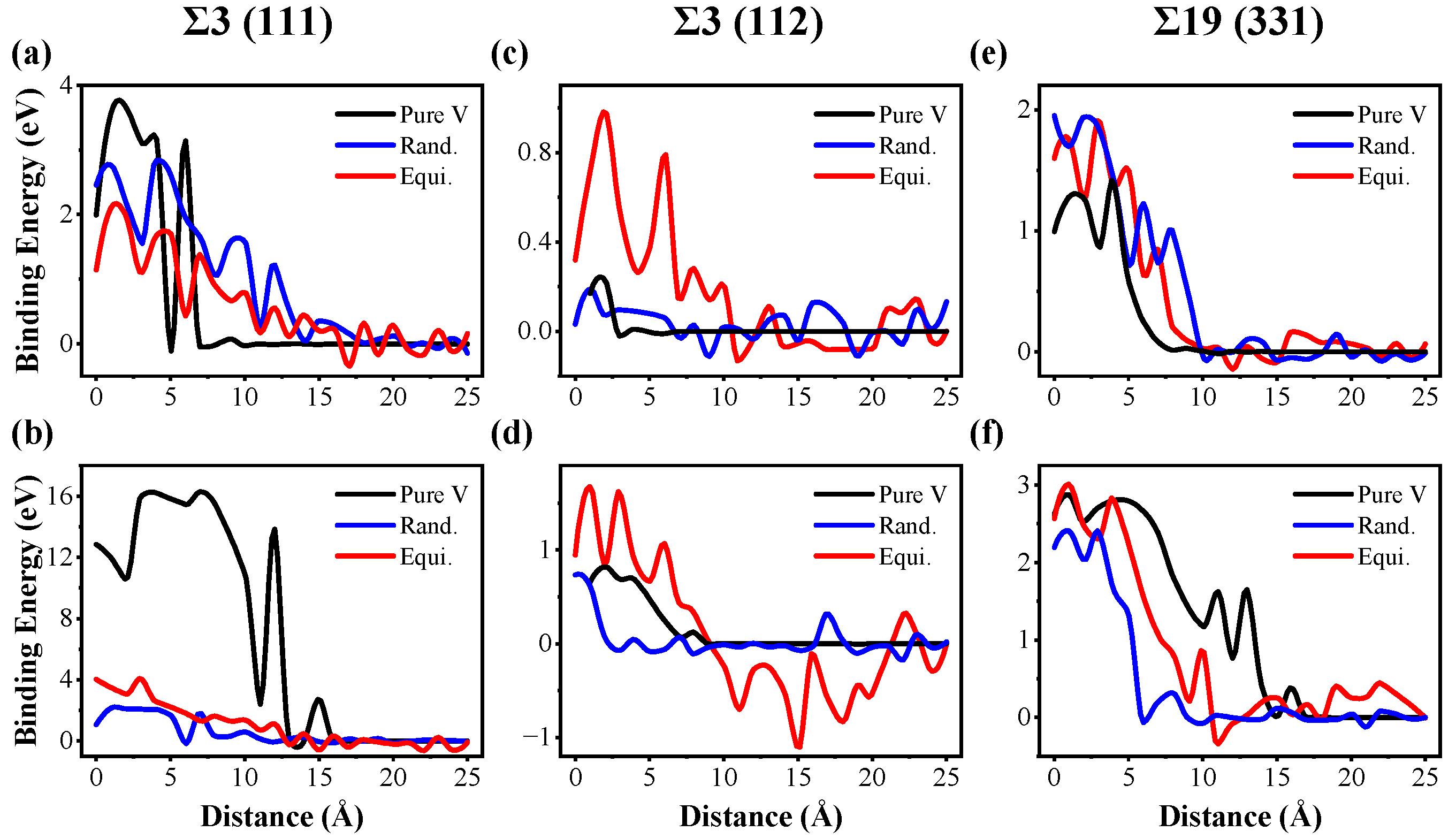
3.3. Defect–Grain Boundary Interactions
4. Discussion
5. Conclusions
- Ti segregation around the GB was observed in TiVTa CSAs, while V and Ta depleted at the GB and uniformly distributed in the matrix with nearly same concentration. Meanwhile, the formation mechanism of SRO (Ti segregation) is elucidated by means of cohesive energy and chemical affinity analyses. In the equilibrated TiVTa CSA, Ti-rich HECs exhibit a preference for localization at GBs, while Ta-/V-rich LECs/MECs tend to be distributed within the matrix. This implies that the chemical-element exclusivity and chemical-affinity disparity serve as plausible driving forces for SRO in refractory CSAs, and this mechanism can be further employed to analyze SRO in future investigations.
- Compared with pure V, the vacancy absorption length scale by GBs is greater, whereas the interstitial absorption length scale by GBs is shorter in TiVTa CSAs, implying an enhanced sink strength towards vacancies and a weakened sink strength towards interstitials due to the lattice distortion and the chemical fluctuation in CSAs. This indicates an enhanced recombination efficiency of point defects due to the reduced disparity in sink strengths between vacancies and interstitials in TiVTa CSAs. This result provides an explanation for the superior irradiation resistance observed in refractory CSAs.
- For the equilibrated CSA, the sink strengths of GBs towards interstitials are enhanced through local ordering with elemental segregation. Conversely, the elemental segregation tends to suppress the sink strength towards vacancies. The bias of GBs towards interstitials and vacancies is thus enhanced, thereby diminishing the efficient annihilation of point defects in the vicinity of GBs. This finding may serve as a valuable reference and theoretical foundation for future investigations on refractory CSAs.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Locatelli, G.; Mancini, M.; Todeschini, N. Generation IV nuclear reactors: Current status and future prospects. Energy Policy 2013, 61, 1503–1520. [Google Scholar] [CrossRef]
- Senkov, O.N.; Miracle, D.B.; Chaput, K.J.; Couzinie, J.-P. Development and exploration of refractory high entropy alloys—A review. J. Mater. Res. 2018, 33, 3092–3128. [Google Scholar] [CrossRef]
- Senkov, O.; Gorsse, S.; Miracle, D. High temperature strength of refractory complex concentrated alloys. Acta Mater. 2019, 175, 394–405. [Google Scholar] [CrossRef]
- Gorsse, S.; Miracle, D.B.; Senkov, O.N. Mapping the world of complex concentrated alloys. Acta Mater. 2017, 135, 177–187. [Google Scholar] [CrossRef]
- Gorsse, S.; Couzinié, J.-P.; Miracle, D.B. From high-entropy alloys to complex concentrated alloys. Comptes Rendus Phys. 2018, 19, 721–736. [Google Scholar] [CrossRef]
- Dewangan, S.K.; Mangish, A.; Kumar, S.; Sharma, A.; Ahn, B.; Kumar, V. A review on High-Temperature Applicability: A milestone for high entropy alloys. Eng. Sci. Technol. Int. J. 2022, 35, 101211. [Google Scholar] [CrossRef]
- Zong, Y.; Hashimoto, N.; Oka, H. Study on irradiation effects of refractory bcc high-entropy alloy. Nucl. Mater. Energy 2022, 31, 101158. [Google Scholar] [CrossRef]
- Pan, Q.; Zhang, L.; Feng, R.; Lu, Q.; An, K.; Chuang, A.C.; Poplawsky, J.D.; Liaw, P.K.; Lu, L. Gradient cell–structured high-entropy alloy with exceptional strength and ductility. Science 2021, 374, 984–989. [Google Scholar] [CrossRef]
- Pickering, E.J.; Carruthers, A.W.; Barron, P.J.; Middleburgh, S.C.; Armstrong, D.E.; Gandy, A.S. High-Entropy Alloys for Advanced Nuclear Applications. Entropy 2021, 23, 98. [Google Scholar] [CrossRef]
- El-Atwani, O.; Li, N.; Li, M.; Devaraj, A.; Baldwin, J.K.S.; Schneider, M.M.; Sobieraj, D.; Wróbel, J.S.; Nguyen-Manh, D.; Maloy, S.A.; et al. Outstanding radiation resistance of tungsten-based high-entropy alloys. Sci. Adv. 2019, 5, eaav2002. [Google Scholar] [CrossRef]
- Miracle, D.B.; Senkov, O.N.; Wilks, J. A critical review of high entropy alloys and related concepts. Acta Mater. 2017, 122, 448–511. [Google Scholar] [CrossRef]
- Hu, B.; Yao, B.; Wang, J.; Liu, Y.; Wang, C.; Du, Y.; Yin, H. The Phase Equilibria of the Ti–V–M (M = Si, Nb, Ta) Ternary Systems. Intermetallics 2020, 118, 106701. [Google Scholar] [CrossRef]
- Jia, N.; Li, Y.; Liu, X.; Zheng, Y.; Wang, B.; Wang, J.; Xue, Y.; Jin, K. Thermal Stability and Mechanical Properties of Low-Activation Single-Phase Ti-V-Ta Medium Entropy Alloys. JOM 2019, 71, 3490–3498. [Google Scholar] [CrossRef]
- Scales, R.J.; Armstrong, D.; Wilkinson, A.J.; Li, B.-S. On the brittle-to-ductile transition of the as-cast TiVNbTa refractory high-entropy alloy. Materialia 2020, 14, 100940. [Google Scholar] [CrossRef]
- Yao, H.W.; Qiao, J.W.; Gao, M.C.; Hawk, J.A.; Ma, S.G.; Zhou, H.F.; Zhang, Y. NbTaV-(Ti,W) refractory high-entropy alloys: Experiments and modeling. Mater. Sci. Eng. A 2016, 674, 203–211. [Google Scholar] [CrossRef]
- Lee, C.; Song, G.; Gao, M.C.; Feng, R.; Chen, P.; Brechtl, J.; Chen, Y.; An, K.; Guo, W.; Poplawsky, J.D.; et al. Lattice distortion in a strong and ductile refractory high-entropy alloy. Acta Mater. 2018, 160, 158–172. [Google Scholar] [CrossRef]
- Kareer, A.; Waite, J.C.; Li, B.; Couet, A.; Armstrong, D.E.J.; Wilkinson, A.J. Short Communication: ‘Low Activation, Refractory, High Entropy Alloys for Nuclear Applications’. J. Nucl. Mater. 2019, 526, 151744. [Google Scholar] [CrossRef]
- An, Z.; Mao, S.; Liu, Y.; Wang, L.; Zhou, H.; Gan, B.; Zhang, Z.; Han, X. A novel HfNbTaTiV high-entropy alloy of superior mechanical properties designed on the principle of maximum lattice distortion. J. Mater. Sci. Technol. 2021, 79, 109–117. [Google Scholar] [CrossRef]
- Sadeghilaridjani, M.; Ayyagari, A.; Muskeri, S.; Hasannaeimi, V.; Salloom, R.; Chen, W.-Y.; Mukherjee, S. Ion irradiation response and mechanical behavior of reduced activity high entropy alloy. J. Nucl. Mater. 2020, 529, 151955. [Google Scholar] [CrossRef]
- Mei, L.; Zhang, Q.; Dou, Y.; Fu, E.; Li, L.; Chen, S.; Dong, Y.; Guo, X.; He, X.; Yang, W.; et al. Distinct ion irradiation response in VTaTi medium entropy alloy from elemental metal V. Scr. Mater. 2023, 223, 115070. [Google Scholar] [CrossRef]
- Jia, N.; Li, Y.; Huang, H.; Chen, S.; Li, D.; Dou, Y.; He, X.; Yang, W.; Xue, Y.; Jin, K. Helium bubble formation in refractory single-phase concentrated solid solution alloys under MeV He ion irradiation. J. Nucl. Mater. 2021, 550, 152937. [Google Scholar] [CrossRef]
- Dou, Y.-K.; Zhao, Y.-P.; He, X.-F.; Gao, J.; Cao, J.-L.; Yang, W. First-principles study of vacancy defects in TiVTa and TiVTaNb concentrated solid-solution alloys. J. Nucl. Mater. 2023, 573, 154096. [Google Scholar] [CrossRef]
- Zhao, Y.-P.; Dou, Y.-K.; He, X.-F.; Deng, H.-Q.; Wang, L.-F.; Yang, W. An atomic scale study on self-interstitial formation and diffusion behaviors in TiVTa and TiVTaNb concentrated solid-solution alloys. Comput. Mater. Sci. 2023, 218, 111943. [Google Scholar] [CrossRef]
- Maiti, S.; Steurer, W. Structural-disorder and its effect on mechanical properties in single-phase TaNbHfZr high-entropy alloy. Acta Mater. 2016, 106, 87–97. [Google Scholar] [CrossRef]
- Zhang, F.X.; Zhao, S.; Jin, K.; Xue, H.; Velisa, G.; Bei, H.; Huang, R.; Ko, J.Y.P.; Pagan, D.C.; Neuefeind, J.C.; et al. Local Structure and Short-Range Order in a NiCoCr Solid Solution Alloy. Phys. Rev. Lett. 2017, 118, 205501. [Google Scholar] [CrossRef] [PubMed]
- Cowley, J.M. X-Ray Measurement of Order in Single Crystals of Cu3Au. J. Appl. Phys. 2004, 21, 24–30. [Google Scholar] [CrossRef]
- Chen, S.; Aitken, Z.H.; Pattamatta, S.; Wu, Z.; Yu, Z.G.; Srolovitz, D.J.; Liaw, P.K.; Zhang, Y.-W. Simultaneously enhancing the ultimate strength and ductility of high-entropy alloys via short-range ordering. Nat. Commun. 2021, 12, 4953. [Google Scholar] [CrossRef]
- Ji, W.; Wu, M.S. Inhibiting the inverse Hall-Petch behavior in CoCuFeNiPd high-entropy alloys with short-range ordering and grain boundary segregation. Scr. Mater. 2022, 221, 114950. [Google Scholar] [CrossRef]
- Li, Q.-J.; Sheng, H.; Ma, E. Strengthening in multi-principal element alloys with local-chemical-order roughened dislocation pathways. Nat. Commun. 2019, 10, 3563. [Google Scholar] [CrossRef]
- Zhao, S.; Osetsky, Y.N.; Zhang, Y. Atomic-scale dynamics of edge dislocations in Ni and concentrated solid solution NiFe alloys. J. Alloys Compd. 2017, 701, 1003–1008. [Google Scholar] [CrossRef]
- Zhao, S. Effects of local elemental ordering on defect-grain boundary interactions in high-entropy alloys. J. Alloys Compd. 2021, 887, 161314. [Google Scholar] [CrossRef]
- Niu, C.; Zaddach, A.J.; Oni, A.A.; Sang, X.; Hurt, J.W.; LeBeau, J.M.; Koch, C.C.; Irving, D.L. Spin-driven ordering of Cr in the equiatomic high entropy alloy NiFeCrCo. Appl. Phys. Lett. 2015, 106, 161906. [Google Scholar] [CrossRef]
- Chen, S.; Aitken, Z.H.; Pattamatta, S.; Wu, Z.; Yu, Z.G.; Srolovitz, D.J.; Liaw, P.K.; Zhang, Y.-W. Short-range ordering alters the dislocation nucleation and propagation in refractory high-entropy alloys. Mater. Today 2023, 65, 14–25. [Google Scholar] [CrossRef]
- Moon, J.; Jang, M.J.; Bae, J.W.; Yim, D.; Park, J.M.; Lee, J.; Kim, H.S. Mechanical behavior and solid solution strengthening model for face-centered cubic single crystalline and polycrystalline high-entropy alloys. Intermetallics 2018, 98, 89–94. [Google Scholar] [CrossRef]
- Zhang, Y.; A Tunes, M.; Crespillo, M.L.; Zhang, F.; Boldman, W.L.; Rack, P.D.; Jiang, L.; Xu, C.; Greaves, G.; E Donnelly, S.; et al. Thermal stability and irradiation response of nanocrystalline CoCrCuFeNi high-entropy alloy. Nanotechnology 2019, 30, 294004. [Google Scholar] [CrossRef] [PubMed]
- Wynblatt, P.; Chatain, D. Modeling grain boundary and surface segregation in multicomponent high-entropy alloys. Phys. Rev. Mater. 2019, 3, 054004. [Google Scholar] [CrossRef]
- Li, L.; Kamachali, R.D.; Li, Z.; Zhang, Z. Grain boundary energy effect on grain boundary segregation in an equiatomic high-entropy alloy. Phys. Rev. Mater. 2020, 4, 053603. [Google Scholar] [CrossRef]
- Li, X.-G.; Chen, C.; Zheng, H.; Zuo, Y.; Ong, S.P. Complex strengthening mechanisms in the NbMoTaW multi-principal element alloy. npj Comput. Mater. 2020, 6, 70. [Google Scholar] [CrossRef]
- Ackland, G. Controlling Radiation Damage. Science 2010, 327, 1587–1588. [Google Scholar] [CrossRef]
- Han, W.; Demkowicz, M.; Fu, E.; Wang, Y.; Misra, A. Effect of grain boundary character on sink efficiency. Acta Mater. 2012, 60, 6341–6351. [Google Scholar] [CrossRef]
- Lejcek, P. Grain Boundary Segregation in Metals; Springer Series in Materials Science; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Tschopp, M.A.; Solanki, K.N.; Gao, F.; Sun, X.; Khaleel, M.A.; Horstemeyer, M.F. Probing Grain Boundary Sink Strength at the Nanoscale: Energetics and Length Scales of Vacancy and Interstitial Absorption by Grain Boundaries in A-Fe. Phys. Rev. B 2012, 85, 064108. [Google Scholar] [CrossRef]
- Bai, X.-M.; Voter, A.F.; Hoagland, R.G.; Nastasi, M.; Uberuaga, B.P. Efficient Annealing of Radiation Damage Near Grain Boundaries via Interstitial Emission. Science 2010, 327, 1631–1634. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Wang, J.; Chen, T.; Shao, L. Defect annihilation at grain boundaries in alpha-Fe. Sci. Rep. 2013, 3, srep01450. [Google Scholar]
- Zhang, J.; He, H.; Liu, W.; Kang, L.; Yun, D.; Chen, P. Effects of grain boundaries on the radiation-induced defects evolution in BCC Fe–Cr alloy: A molecular dynamics study. Nucl. Mater. Energy 2020, 22, 100726. [Google Scholar] [CrossRef]
- Kumar, N.A.P.K.; Li, C.; Leonard, K.J.; Bei, H.; Zinkle, S.J. Microstructural stability and mechanical behavior of FeNiMnCr high entropy alloy under ion irradiation. Acta Mater. 2016, 113, 230–244. [Google Scholar] [CrossRef]
- Barr, C.M.; Nathaniel, J.E.; Unocic, K.A.; Liu, J.; Zhang, Y.; Wang, Y.; Taheri, M.L. Exploring Radiation Induced Segregation Mechanisms at Grain Boundaries in Equiatomic Cocrfenimn High Entropy Alloy under Heavy Ion Irradiation. Scr. Mater. 2018, 156, 80–84. [Google Scholar] [CrossRef]
- Song, H.; Tian, F.; Hu, Q.-M.; Vitos, L.; Wang, Y.; Shen, J.; Chen, N. Local lattice distortion in high-entropy alloys. Phys. Rev. Mater. 2017, 1, 023404. [Google Scholar] [CrossRef]
- He, M.-R.; Wang, S.; Shi, S.; Jin, K.; Bei, H.; Yasuda, K.; Matsumura, S.; Higashida, K.; Robertson, I.M. Mechanisms of Radiation-Induced Segregation in Crfeconi-Based Single-Phase Concentrated Solid Solution Alloys. Acta Mater. 2017, 126, 182–193. [Google Scholar] [CrossRef]
- Shi, T.; Su, Z.; Li, J.; Liu, C.; Yang, J.; He, X.; Yun, D.; Peng, Q.; Lu, C. Distinct point defect behaviours in body-centered cubic medium-entropy alloy NbZrTi induced by severe lattice distortion. Acta Mater. 2022, 229, 117806. [Google Scholar] [CrossRef]
- Zhang, P.; Jiang, L.; Yang, J.; Su, Z.; Wang, J.; Shi, T.; Lu, C. Research Progress in Refractory High Entropy Alloys for Nuclear Applications. Mater. Rep. 2022, 36, 5–26. [Google Scholar]
- Chen, S.; Aitken, Z.H.; Pattamatta, S.; Wu, Z.; Yu, Z.G.; Banerjee, R.; Srolovitz, D.J.; Liaw, P.K.; Zhang, Y.-W. Chemical-Affinity Disparity and Exclusivity Drive Atomic Segregation, Short-Range Ordering, and Cluster Formation in High-Entropy Alloys. Acta Mater. 2021, 206, 116638. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Qiu, R.; Chen, Y.; Liao, X.; He, X.; Yang, W.; Hu, W.; Deng, H. Finnis–Sinclair-type potential for atomistic simulation of defects behaviour in V-Ti-Ta ternary system. J. Nucl. Mater. 2021, 557, 153231. [Google Scholar] [CrossRef]
- Tschopp, M.A.; Mcdowell, D.L. Asymmetric tilt grain boundary structure and energy in copper and aluminium. Philos. Mag. 2007, 87, 3871–3892. [Google Scholar] [CrossRef]
- Marian, J.; Wirth, B.; Odette, G.; Perlado, J. Cu diffusion in α-Fe: Determination of solute diffusivities using atomic-scale simulations. Comput. Mater. Sci. 2004, 31, 347–367. [Google Scholar] [CrossRef]
- Zhao, Y.-P.; Dou, Y.-K.; He, X.-F.; Cao, H.; Wang, L.-F.; Deng, H.-Q.; Yang, W. Molecular Dynamics Study of Primary Radiation Damage in Tivta Concentrated Solid-Solution Alloy. Chin. Phys. B 2023. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO—The Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2009, 18, 015012. [Google Scholar] [CrossRef]
- Larsen, P.M.; Schmidt, S.; Schiøtz, J. Robust structural identification via polyhedral template matching. Model. Simul. Mater. Sci. Eng. 2016, 24, 05500. [Google Scholar] [CrossRef]
- Stukowski, A. Computational Analysis Methods in Atomistic Modeling of Crystals. JOM 2014, 66, 399–407. [Google Scholar] [CrossRef]
- Li, C.; Lu, S.; Divinski, S.; Vitos, L. Theoretical and experimental grain boundary energies in body-centered cubic metals. Acta Mater. 2023, 255, 119074. [Google Scholar] [CrossRef]
- Utt, D.; Stukowski, A.; Albe, K. Grain boundary structure and mobility in high-entropy alloys: A comparative molecular dynamics study on a Σ11 symmetrical tilt grain boundary in face-centered cubic CuNiCoFe. Acta Mater. 2020, 186, 11–19. [Google Scholar] [CrossRef]
- Bristowe, P.D.; Crocker, A.G.; Norgett, M.J. The structure of twin boundaries in body centred cubic metals. J. Phys. F: Met. Phys. 1974, 4, 1859–1864. [Google Scholar] [CrossRef]
- Bristowe, P.D.; Crocker, A.G. A computer simulation study of the structures of twin boundaries in body-centred cubic crystals. Philos. Mag. 1975, 31, 503–517. [Google Scholar] [CrossRef]
- Nilles, J.L.; Olson, D.L. Energy of a bcc Iron Deformation Twin Boundary. J. Appl. Phys. 2003, 41, 531–532. [Google Scholar] [CrossRef]
- Vystavél, T.; Pénisson, J.M.; Gemperle, A. High-Resolution and Conventional Electron Microscopy Study of a Σ = 3, [101]{121} Twin Grain Boundary in Molybdenum. Philos. Mag. A 2001, 81, 417–429. [Google Scholar] [CrossRef]
- Kittel, C. Introduction to Solid State Physics; John Wiley & Sons: New York, NY, USA, 1956. [Google Scholar]
- Aray, Y.; Rodriguez, J.; Vega, D. Topology of the Electron Density and Cohesive Energy of the Face-Centered Cubic Transition Metals. J. Phys. Chem. B 2000, 104, 4608–4612. [Google Scholar] [CrossRef]
- Starace, A.K.; Neal, C.M.; Cao, B.; Jarrold, M.F.; Aguado, A.; López, J.M. Correlation between the Latent Heats and Cohesive Energies of Metal Clusters. J. Chem. Phys. 2008, 129, 144702. [Google Scholar] [CrossRef] [PubMed]
- Yan, S. Fundamentals of Solid State Physics; Peking University Press: Beijing, China, 2011. [Google Scholar]
- Sorkin, V.; Chen, S.; Tan, T.L.; Yu, Z.; Man, M.; Zhang, Y. First-principles-based high-throughput computation for high entropy alloys with short range order. J. Alloys Compd. 2021, 882, 160776. [Google Scholar] [CrossRef]
- Rajagopalan, M.; Tschopp, M.A.; Solanki, K.N. Grain Boundary Segregation of Interstitial and Substitutional Impurity Atoms in Alpha-Iron. JOM 2014, 66, 129–138. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atomic Pairs | Ecohe | Chemical Affinity [33] | ||
|---|---|---|---|---|
| This Work | MD [33] | DFT [33] | ||
| Ti–Ti | −4.8 | −4.9 | −5.3 | 0.0 |
| Ti–V | −5.0 | \ | \ | \ |
| Ti–Ta | −6.4 | −6.3 | −6.6 | 0.4 |
| V–V | −5.3 | \ | −5.4 | 0.0 |
| V–Ta | −6.7 | \ | −6.9 | 0.4 |
| Ta–Ta | −8.1 | −8.1 | −8.1 | 0.8 |
| Vacancies | Interstitials | ||||||
|---|---|---|---|---|---|---|---|
| Random CSA | (111) | 3.48 | 1.75 | −1.73 | 2.22 | 2.20 | −0.02 |
| (112) | 0.19 | 1.88 | 1.69 | 0.73 | 2.21 | 1.48 | |
| (331) | 2.29 | 1.92 | −0.37 | 2.40 | 2.19 | −0.21 | |
| Equilibrated CSA | (111) | 2.11 | 2.58 | 0.47 | 4.10 | 4.15 | 0.05 |
| (112) | 0.99 | 2.48 | 1.49 | 1.67 | 3.52 | 1.85 | |
| (331) | 1.92 | 2.27 | 0.35 | 3.01 | 3.39 | 0.38 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Zhao, Y.; Dou, Y.; He, X.; Zhang, Z.; Chen, M.; Deng, H.; Yang, W. Atomistic Study on Defect–Grain Boundary Interactions in TiVTa Concentrated Solid–Solution Alloys. Crystals 2024, 14, 166. https://doi.org/10.3390/cryst14020166
Wang L, Zhao Y, Dou Y, He X, Zhang Z, Chen M, Deng H, Yang W. Atomistic Study on Defect–Grain Boundary Interactions in TiVTa Concentrated Solid–Solution Alloys. Crystals. 2024; 14(2):166. https://doi.org/10.3390/cryst14020166
Chicago/Turabian StyleWang, Linfeng, Yongpeng Zhao, Yankun Dou, Xinfu He, Zhongao Zhang, Mengyao Chen, Huiqiu Deng, and Wen Yang. 2024. "Atomistic Study on Defect–Grain Boundary Interactions in TiVTa Concentrated Solid–Solution Alloys" Crystals 14, no. 2: 166. https://doi.org/10.3390/cryst14020166