
Machine Learning-Based Predictions of Power Factor for Half-Heusler Phases

Abstract

1. Introduction
2. Computational Details
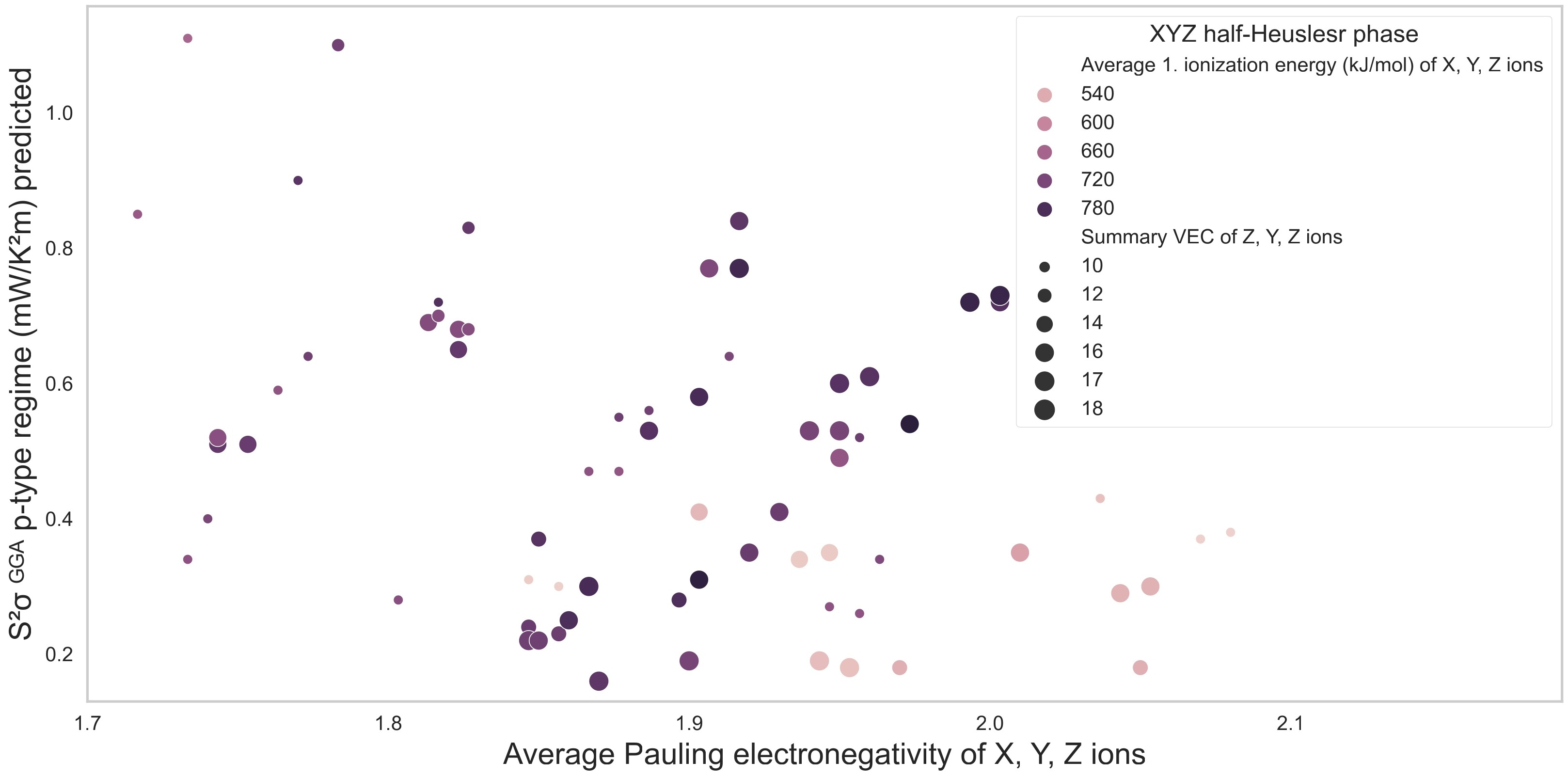
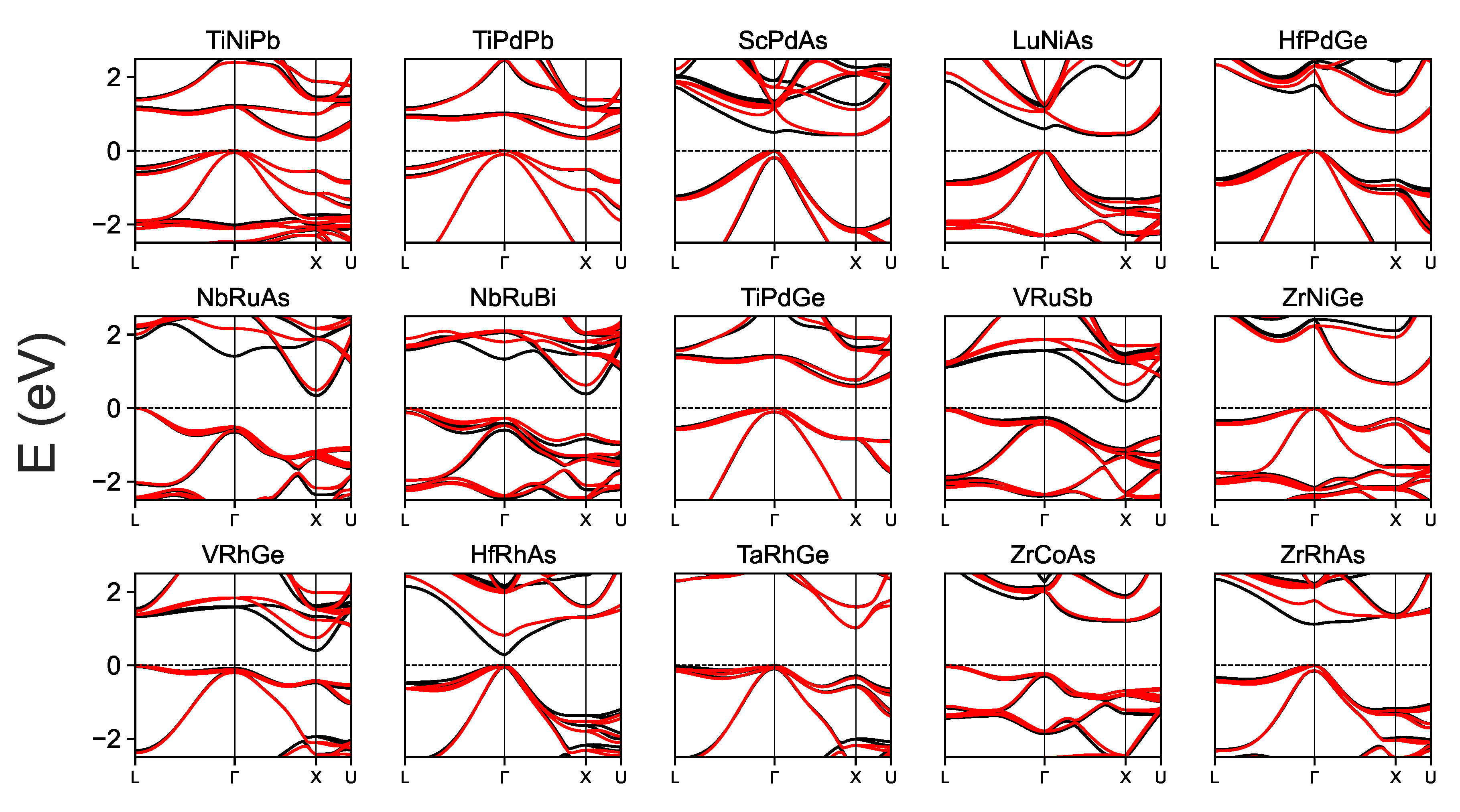
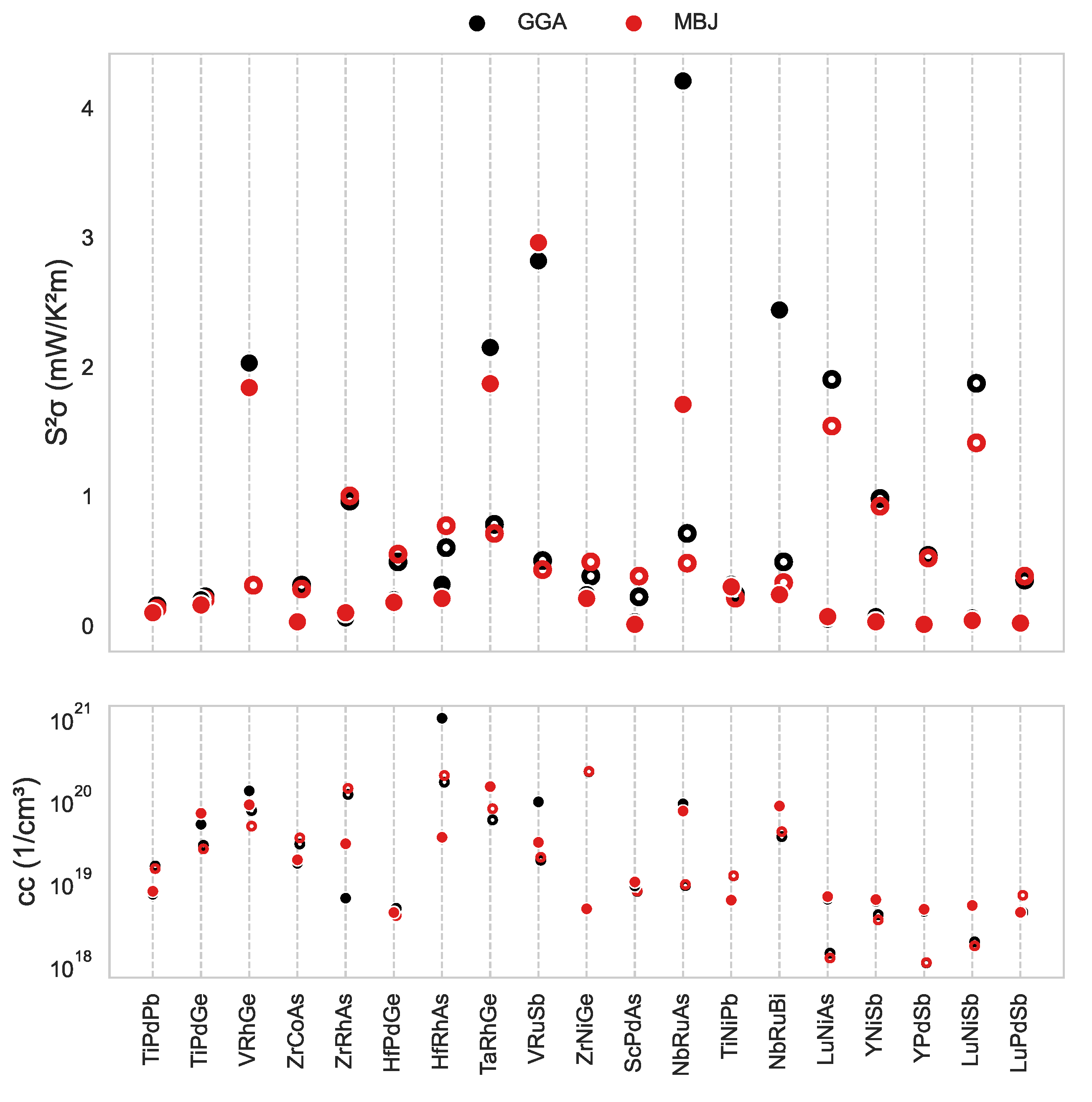
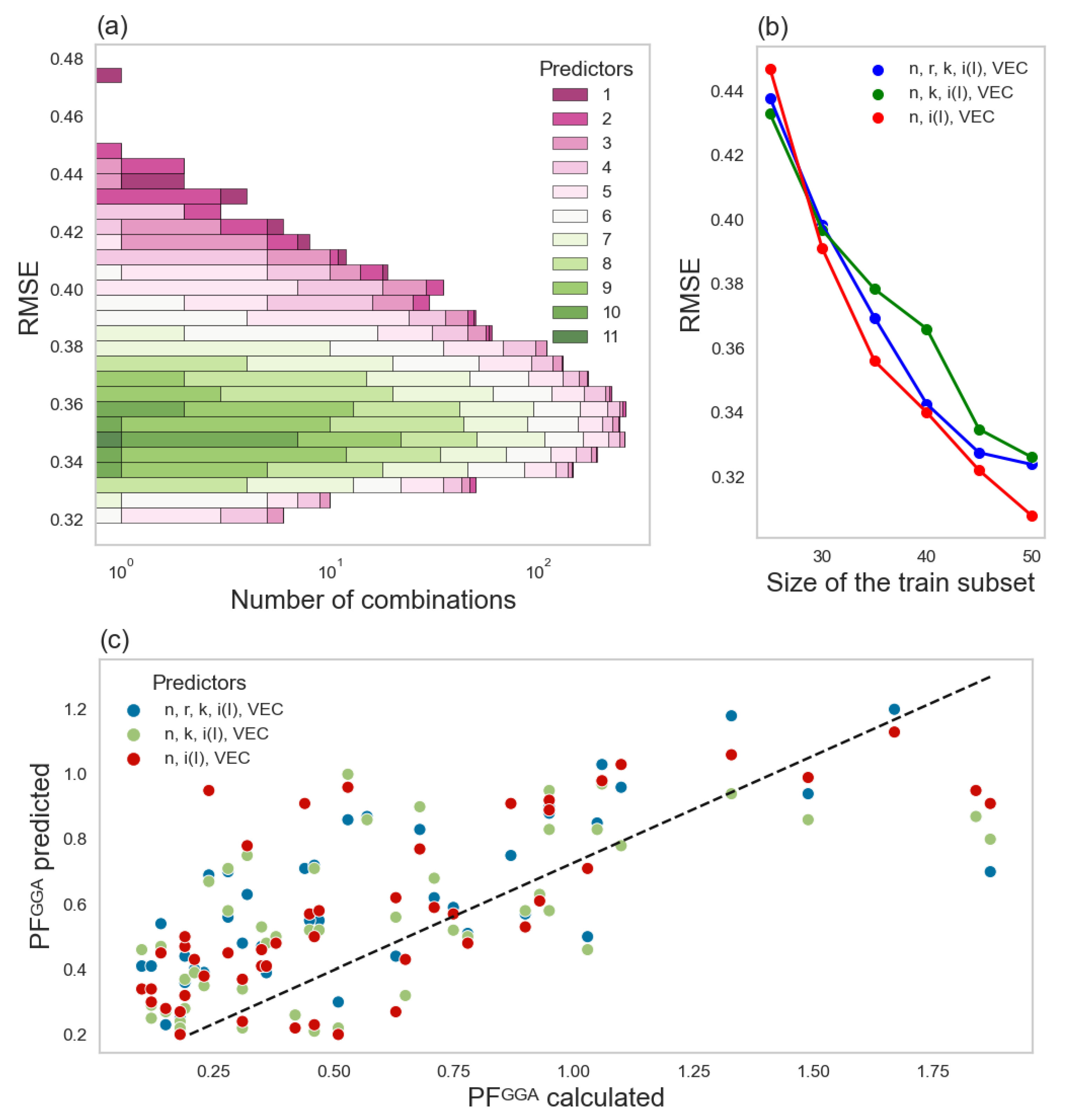
3. Results
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Carrete, J.; Li, W.; Mingo, N.; Wang, S.; Curtarolo, S. Finding unprecedentedly low-thermal-conductivity half-Heusler semiconductors via high-throughput materials modeling. Phys. Rev. X 2014, 4, 011019. [Google Scholar] [CrossRef]
- Miyazaki, H.; Tamura, T.; Mikami, M.; Watanabe, K.; Ide, N.; Ozkendir, O.M.; Nishino, Y. Machine learning based prediction of lattice thermal conductivity for half-Heusler compounds using atomic information. Sci. Rep. 2021, 11, 13410. [Google Scholar] [CrossRef] [PubMed]
- Bilińska, K.; Winiarski, M.J. Machine Learning-Based Predictions for Half-Heusler Phases. Inorganics 2024, 12, 5. [Google Scholar] [CrossRef]
- Gautier, R.; Zhang, X.; Hu, L.; Yu, L.; Lin, Y.; Sunde, T.O.; Chon, D.; Paeppelmeirer, K.; Zunger, A. Prediction and accelerated laboratory discovery of previously unknown 18-electron ABX compounds. Nat. Chem. 2015, 7, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Gzyl, A.S.; Oliynyk, A.O.; Adutwum, L.A.; Mar, A. Solving the Coloring Problem in Half-Heusler Structures: Machine-Learning Predictions and Experimental Validation. Inorg. Chem. 2019, 58, 9280–9289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, X. Machine learning modeling of lattice constants for half-Heusler alloys. AIP Adv. 2020, 10, 045121. [Google Scholar] [CrossRef]
- Tranås, R.; Løvvik, O.M.; Tomic, O.; Berland, K. Lattice thermal conductivity of half-Heuslers with density functional theory and machine learning: Enhancing predictivity by active sampling with principal component analysis. Comput. Mater. Sci. 2022, 202, 110938. [Google Scholar] [CrossRef]
- Tranås, R.; Løvvik, O.M.; Berland, K. Lattice Thermal Conductivity from First Principles and Active Learning with Gaussian Process Regression. arXiv 2023, arXiv:2309.06786. [Google Scholar]
- Dylla, M.T.; Dunn, A.; An, S.; Jain, A.; Snyder, G.J. Machine learning chemical guidelines for engineering electronic structures in half-heusler thermoelectric materials. Research 2020, 2020, 6375171. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, M.K.; Raj, V.A.; Ravindran, P. Composition and Structure Based GGA Bandgap Prediction Using Machine Learning Approach. arXiv 2023, arXiv:2309.07424. [Google Scholar]
- Kurniawan, I.; Miura, Y.; Hono, K. Machine learning study of highly spin-polarized Heusler alloys at finite temperature. Phys. Rev. Mater. 2022, 6, L091402. [Google Scholar] [CrossRef]
- Fu, C.; Zhu, T.; Liu, Y.; Xie, H.; Zhao, X. Band engineering of high performance p-type FeNbSb based half-Heusler thermoelectric materials for figure of merit zT > 1. Energy Environ. Sci. 2015, 8, 216–220. [Google Scholar] [CrossRef]
- Fang, T.; Zheng, S.; Chen, H.; Cheng, H.; Wang, L.; Zhang, P. Electronic structure and thermoelectric properties of p-type half-Heusler compound NbFeSb: A first-principles study. RSC Adv. 2016, 6, 10507–10512. [Google Scholar] [CrossRef]
- Bilińska, K.; Winiarski, M.J. High-Throughput Exploration of Half-Heusler Phases for Thermoelectric Applications. Crystals 2023, 13, 1378. [Google Scholar] [CrossRef]
- Saal, J.E.; Kirklin, S.; Aykol, M.; Meredig, B.; Wolverton, C. Materials Design and Discovery with High-Throughput Density Functional Theory: The Open Quantum Materials Database (OQMD). JOM 2013, 2013 65, 1501–1509. [Google Scholar] [CrossRef]
- Kirklin, S.S.; Saal, J.E.; Meredig, B.; Thompson, A.; Doak, J.W.; Aykol, M.; Rühl, S.; Wolverton, C. The Open Quantum Materials Database (OQMD): Assessing the accuracy of DFT formation energies. npj Comput. Mater. 2015, 1, 15010. [Google Scholar] [CrossRef]
- Aykol, M.; Dwaraknath, S.S.; Sun, W.; Persson, K.A. Thermodynamic limit for synthesis of metastable inorganic materials. Sci. Adv. 2018, 4, eaaq0148. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B 1993, 48, 13115. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Tran, F.; Blaha, P. Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential. Phys. Rev. Lett. 2009, 102, 226401. [Google Scholar] [CrossRef] [PubMed]
- Bardeen, J.; Shockley, W.J.P.R. Deformation potentials and mobilities in non-polar crystals. Phys. Rev. 1950, 1950 80, 72. [Google Scholar] [CrossRef]
- Slack, G.A. Nonmetallic crystals with high thermal conductivity. J. Phys. Chem. Solids 1973, 34, 321–335. [Google Scholar] [CrossRef]
- Madsen, G.K.H.; Carrete, J.; Verstraete, M.J. BoltzTraP2, a program for interpolating band structures and calculating semi-classical transport coefficients. Comput. Phys. Commun. 2018, 231, 140–145. [Google Scholar] [CrossRef]
- Smola, A.J.; Schölkopf, B. A tutorial on support vector regression. Stat. Comput. 2004, 14, 199–222. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Rohmah, M.F.; Putra, I.K.G.D.; Hartati, R.S.; Ardiantoro, L. Comparison four kernels of svr to predict consumer price index. J. Phys. Conf. Ser. 2021, 1737, 012018. [Google Scholar] [CrossRef]
- Fornberg, B.; Larsson, E.; Flyer, N. Stable computations with Gaussian radial basis functions. SIAM J. Sci. Comput. 2011, 33, 869–892. [Google Scholar] [CrossRef]
- Awad, M.; Khanna, R.; Awad, M.; Khanna, R. Support vector regression. In Efficient Learning Machines: Theories, Concepts, and Applications for Engineers and System Designers; Springer: Berlin/Heidelberg, Germany, 2015; pp. 67–80. [Google Scholar]
- Elisseeff, A.; Pontil, M. Leave-one-out error and stability of learning algorithms with applications. NATO Sci. Ser. III Comput. Syst. Sci. 2003, 190, 111–130. [Google Scholar]
- WebElements. Available online: https://www.webelements.com (accessed on 13 November 2023).
- Winiarski, M.J.; Bilińska, K. High termoelectric power factors of p-type half-Heusler alloys YNiSb, LuNiSb, YPdSb, and LuPdSb. Intermetallics 2019, 108, 55–60. [Google Scholar] [CrossRef]
- Winiarski, M.J.; Bilińska, K.; Kaczorowski, D.; Ciesielski, K. Thermoelectric performance of p-type half-Heusler alloys ScMSb (M = Ni, Pd, Pt) by ab initio calculation. J. Alloys Compd. 2018, 762, 901–905. [Google Scholar] [CrossRef]
- Winiarskia, M.J.; Bilinska, K. Power Factors of p-type Half-Heusler Alloys ScNiBi, YNiBi, and LuNiBi by ab initio Calculations. Acta Phys. Pol. A. 2020, 138, 533–538. [Google Scholar] [CrossRef]
- Pei, Y.; LaLonde, A.D.; Wang, H.; Snyder, G.J. Low effective mass leading to high thermoelectric performance. Energy Environ. Sci. 2012, 5, 7963–7969. [Google Scholar] [CrossRef]
- Bilińska, K.; Winiarski, M.J. Search for semiconducting materials among 18-electron half-Heusler alloys. Solid State Commun. 2023, 365, 115133. [Google Scholar] [CrossRef]
- Kalita, D.; Limbu, N.; Ram, M.; Saxena, A. DFT study of structural, mechanical, thermodynamic, electronic, and thermoelectric properties of new PdTi Z (Z= Ge and Pb) half Heusler compounds. Int. J. Quantum Chem. 2022, 122, e26951. [Google Scholar] [CrossRef]
- Solola, G.T.; Bamgbose, M.K.; Adebambo, P.O.; Ayedun, F.; Adebayo, G.A. First-principles investigations of structural, electronic, vibrational, and thermoelectric properties of half-Heusler VYGe (Y= Rh, Co, Ir) compounds. Comput. Condens. Matter 2023, 36, e00827. [Google Scholar] [CrossRef]
- Bendahma, F.; Mana, M.; Terkhi, S.; Cherid, S.; Bestani, B.; Bentata, S. Investigation of high figure of merit in semiconductor XHfGe (X= Ni and Pd) half-Heusler alloys: Ab-initio study. Comput. Condens. Matter 2019, 21, e00407. [Google Scholar] [CrossRef]
- Kaur, K.; Kumar, R. On the possibility of thermoelectricity in half Heusler XRuSb (X= V, Nb, Ta) materials: A first principles prospective. J. Phys. Chem. Solids 2017, 110, 108–115. [Google Scholar] [CrossRef]
- Cherifi, F.; Mostefa, Z.; Boukra, A.; Meghoufel, Z.F.; Bouattou, M.; Kadi Allah, F.; Terki, F. Thermoelectric Transport Parameters of p-Type RuVAs and RuNbAs Heusler Alloys. Phys. Status Solidi B Basic Res. 2020, 257, 2000271. [Google Scholar] [CrossRef]
- Hong, D.; Zeng, W.; Xin, Z.; Liu, F.S.; Tang, B.; Liu, Q.J. First-principles calculations of structural, mechanical and electronic properties of TiNi-X (X= C, Si, Ge, Sn, Pb) alloys. Int. J. Mod. Phys. B 2019, 33, 1950167. [Google Scholar] [CrossRef]
- Narducci, D. Do we really need high thermoelectric figures of merit? A critical appraisal to the power conversion efficiency of thermoelectric materials. Appl. Phys. Lett. 2011, 99, 102104. [Google Scholar] [CrossRef]
- Touia, A.; Benyahia, K.; Tekin, A. First-principles calculations of structural, electronic, optical, and thermoelectric properties of LuNiBi and LuNiSb half-heusler. J. Supercond. Nov. Magn. 2021, 34, 2689–2698. [Google Scholar] [CrossRef]
- Singh, S.; Zeeshan, M.; Brink, J.V.D.; Kandpal, H.C. Ab initio study of Bi-based half Heusler alloys as potential thermoelectric prospects. arXiv 2019, arXiv:1904.02488. [Google Scholar]
- Yazdani-Kachoei, M.; Li, S.; Sun, W.; Allaei, S.M.V.; Di Marco, I. Role of volume change on the physics of thermoelectric half-Heusler compounds. Phys. Rev. Mater. 2023, 7, 104602. [Google Scholar] [CrossRef]
- Li, S.; Zhao, H.; Li, D.; Jin, S.; Gu, L. Synthesis and thermoelectric properties of half-Heusler alloy YNiBi. J. Appl. Phys. 2015, 117, 205101. [Google Scholar] [CrossRef]
- Fang, T.; Zheng, S.; Zhou, T.; Yan, L.; Zhang, P. Computational prediction of high thermoelectric performance in p-type half-Heusler compounds with low band effective mass. PCCP 2017, 19, 4411–4417. [Google Scholar] [CrossRef]
- Akinlami, J.O.; Odeyemi, O.O.; Omeike, M.O.; Adebayo, G.A. First principle calculations of the structural, elastic, electronic and transport properties of XRuAs (X = Ta and V). Mater. Sci. Semicond. 2022, 148, 106837. [Google Scholar] [CrossRef]
- Jaishi, D.R.; Sharma, N.; Karki, B.; Belbase, B.P.; Adhikari, R.P.; Ghimire, M.P. Electronic structure and thermoelectric properties of half-Heusler alloys NiTZ. AIP Adv. 2021, 11, 025304. [Google Scholar] [CrossRef]
- Mafe, A.S.; Shogo, O.E.; Bello, B.W.; Musari, A.A. Systematic study of stable palladium and nickel based half-Heusler compounds for thermoelectric generators. Solid State Sci. 2024, 149, 107451. [Google Scholar] [CrossRef]
- Kaur, K.; Kumar, R. Ti based half Heusler compounds: A new on the screen with robustic thermoelectric performance. J. Alloys Compd. 2017, 727, 1171–1177. [Google Scholar] [CrossRef]
- Benallou, Y.; Amara, K.; Doumi, B.; Arbouche, O.; Zemouli, M.; Bekki, B.; Mokaddem, A. Structural stability, electronic structure, and novel transport properties with high thermoelectric performances of ZrIrX (X = As, Bi, and Sb). J. Comput. Electron. 2017, 16, 1–11. [Google Scholar] [CrossRef]
- Rani, B.; Khandy, S.A.; Singh, J.; Verma, A.S.; Ali, A.M.; Dhiman, S.; Kaur, K. Electronic structure, elastic and transport properties of new Palladium-based Half-Heusler Materials for Thermoelectric Applications. Mater. Today Commun. 2023, 2023, 106461. [Google Scholar] [CrossRef]
- Lazab, M.; Djebour, B.; Bouafia, H.; Bousmaha, M.; Sahli, B.; Boudia, K. Mechanical and dynamical stability, electronic and bonding properties of a new narrow-gap semiconductor YPdAs Half-Heusler: DFT and QTAIM study. Mater. Sci. Semicond. 2024, 173, 108160. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | a | B | ||||||
|---|---|---|---|---|---|---|---|---|
| TiPdPb | 6.328 | 103.88 | 0.352 | 0.324 | 0.65 | 0.70 | 0.66 | 0.64 |
| TiPdGe | 5.964 | 131.38 | 0.619 | 0.584 | 0.61 | 0.57 | 0.61 | 0.57 |
| VRhGe | 5.796 | 172.54 | 0.433 | 0.748 | 1.20 | 0.21 | 1.10 | 0.24 |
| ZrCoAs | 5.831 | 147.58 | 1.203 | 1.229 | 1.02 | 3.49 | 1.05 | 3.31 |
| ZrRhAs | 6.110 | 143.97 | 1.117 | 1.292 | 0.36 | 0.98 | 0.30 | 1.70 |
| HfPdGe | 6.142 | 133.71 | 0.552 | 0.506 | 0.40 | 0.61 | 0.31 | 0.61 |
| HfRhAs | 6.063 | 160.02 | 0.282 | 0.816 | 0.43 | 0.29 | 0.28 | 0.32 |
| TaRhGe | 5.973 | 185.13 | 1.044 | 1.026 | 0.79 | 0.14 | 0.83 | 0.15 |
| VRuSb | 6.044 | 165.64 | 0.189 | 0.631 | 0.53 | 0.19 | 0.52 | 0.23 |
| ZrNiGe | 5.893 | 141.57 | 0.679 | 0.654 | 0.83 | 0.71 | 0.60 | 0.74 |
| ScPdAs | 6.099 | 111.31 | 0.432 | 0.451 | 0.35 | 3.17 | 0.20 | 4.23 |
| NbRuAs | 5.961 | 183.13 | 0.337 | 0.506 | 0.36 | 0.11 | 0.37 | 0.12 |
| TiNiPb | 6.038 | 115.66 | 0.341 | 0.292 | 0.66 | 0.42 | 0.67 | 0.41 |
| NbRuBi | 6.307 | 151.77 | 0.383 | 0.557 | 0.42 | 0.13 | 0.52 | 0.16 |
| LuNiAs | 5.989 | 106.94 | 0.409 | 0.475 | 0.20 | 4.03 | 0.18 | 3.06 |
| YNiSb | 6.350 | 93.02 | 0.270 | 0.283 | 0.26 | 2.63 | 0.22 | 4.16 |
| YPdSb | 6.608 | 88.15 | 0.147 | 0.160 | 0.17 | 5.06 | 0.14 | 4.94 |
| LuNiSb | 6.269 | 97.16 | 0.207 | 0.193 | 0.21 | 2.70 | 0.18 | 3.56 |
| LuPdSb | 6.544 | 89.30 | 0.106 | 0.103 | 0.25 | 6.17 | 0.16 | 4.56 |
| Compd | Compd | Compd | Compd | Compd | |||||
|---|---|---|---|---|---|---|---|---|---|
| HfNiGe | 0.40 | NbIrPb | 0.29 | TaIrSn | 0.53 | VCoSn | 0.68 | YPdAs | 0.30 |
| HfRhBi | 0.47 | NbCoSn | 0.69 | TaIrPb | 0.35 | VRhSn | 0.26 | YPtSb | 0.37 |
| HfNiPb | 0.31 | NbCoPb | 0.34 | TaRuSb | 0.84 | VIrSn | 0.41 | ZrNiSn | 0.34 |
| HfRhSb | 0.55 | NbOsSb | 0.60 | TaRuBi | 0.77 | VRuAs | 0.72 | ZrNiPb | 0.30 |
| HfPdPb | 0.19 | NbFeSb | 0.83 | TaOsSb | 0.77 | VOsSb | 0.61 | ZrPdGe | 0.22 |
| HfPtSn | 0.24 | NbFeBi | 0.70 | TaCoPb | 0.41 | VRuBi | 0.49 | ZrPdPb | 0.18 |
| HfPtPb | 0.18 | ScNiAs | 0.72 | TaFeBi | 1.10 | VOsAs | 0.73 | ZrPtSn | 0.23 |
| HfCoSb | 0.51 | ScNiSb | 0.64 | TiNiSn | 0.28 | VOsBi | 0.53 | ZrCoSb | 0.51 |
| LuNiBi | 1.11 | ScNiBi | 0.59 | TiPdSn | 0.19 | VCoPb | 0.35 | ZrCoBi | 0.52 |
| NbRhGe | 0.34 | ScPdSb | 0.16 | TiPtPb | 0.18 | VRhPb | 0.38 | ZrRhSb | 0.56 |
| NbOsAs | 0.72 | ScPtSb | 0.28 | TiCoSb | 0.65 | VIrPb | 0.30 | ZrRhBi | 0.47 |
| NbOsBi | 0.53 | TaRhSn | 0.64 | TiRhSb | 0.52 | VFeBi | 0.68 | ZrIrAs | 0.31 |
| NbRhSn | 0.27 | TaRhPb | 0.43 | TiIrAs | 0.54 | YNiAs | 0.90 | ZrIrSb | 0.25 |
| NbRhPb | 0.37 | TaIrGe | 0.58 | TiIrBi | 0.35 | YNiBi | 0.85 | ZrIrBi | 0.22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bilińska, K.; Winiarski, M.J. Machine Learning-Based Predictions of Power Factor for Half-Heusler Phases. Crystals 2024, 14, 354. https://doi.org/10.3390/cryst14040354
Bilińska K, Winiarski MJ. Machine Learning-Based Predictions of Power Factor for Half-Heusler Phases. Crystals. 2024; 14(4):354. https://doi.org/10.3390/cryst14040354
Chicago/Turabian StyleBilińska, Kaja, and Maciej J. Winiarski. 2024. "Machine Learning-Based Predictions of Power Factor for Half-Heusler Phases" Crystals 14, no. 4: 354. https://doi.org/10.3390/cryst14040354
APA StyleBilińska, K., & Winiarski, M. J. (2024). Machine Learning-Based Predictions of Power Factor for Half-Heusler Phases. Crystals, 14(4), 354. https://doi.org/10.3390/cryst14040354