
New Insight into the Electronic and Magnetic Properties of Sub-Stoichiometric WO3: A Theoretical Perspective


Abstract
1. Introduction
2. Method
3. Results
3.1. Stoichiometric WO Bulk
3.1.1. Structural Properties
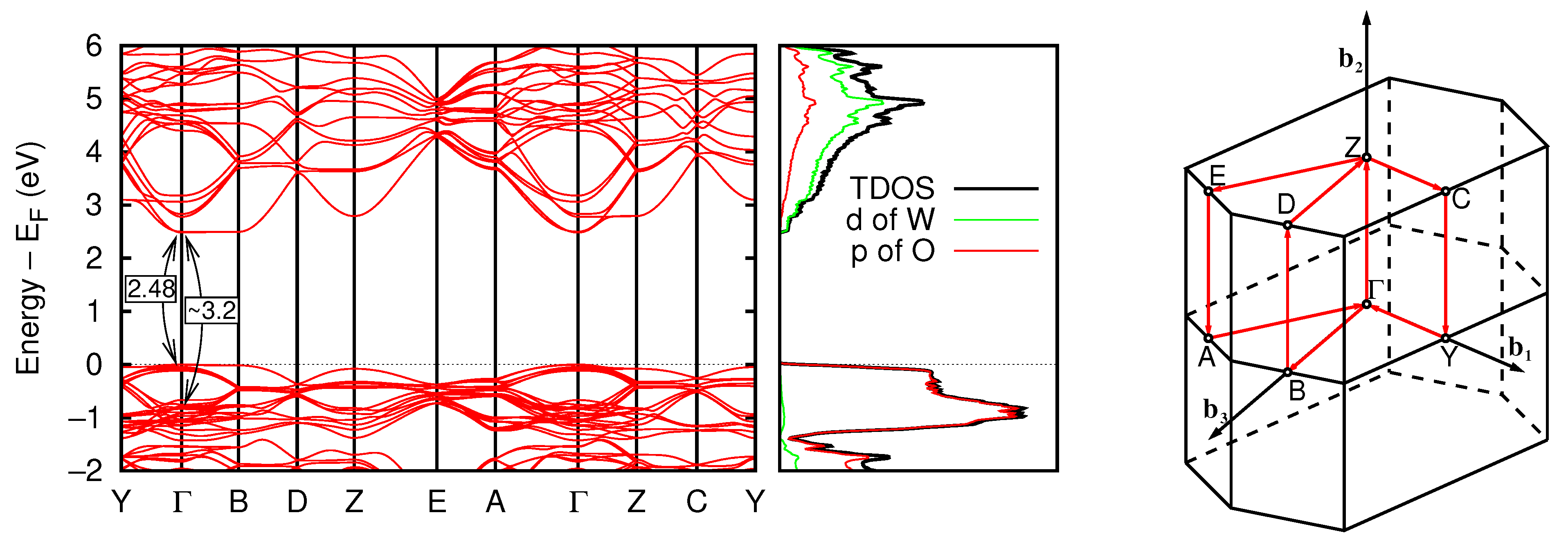
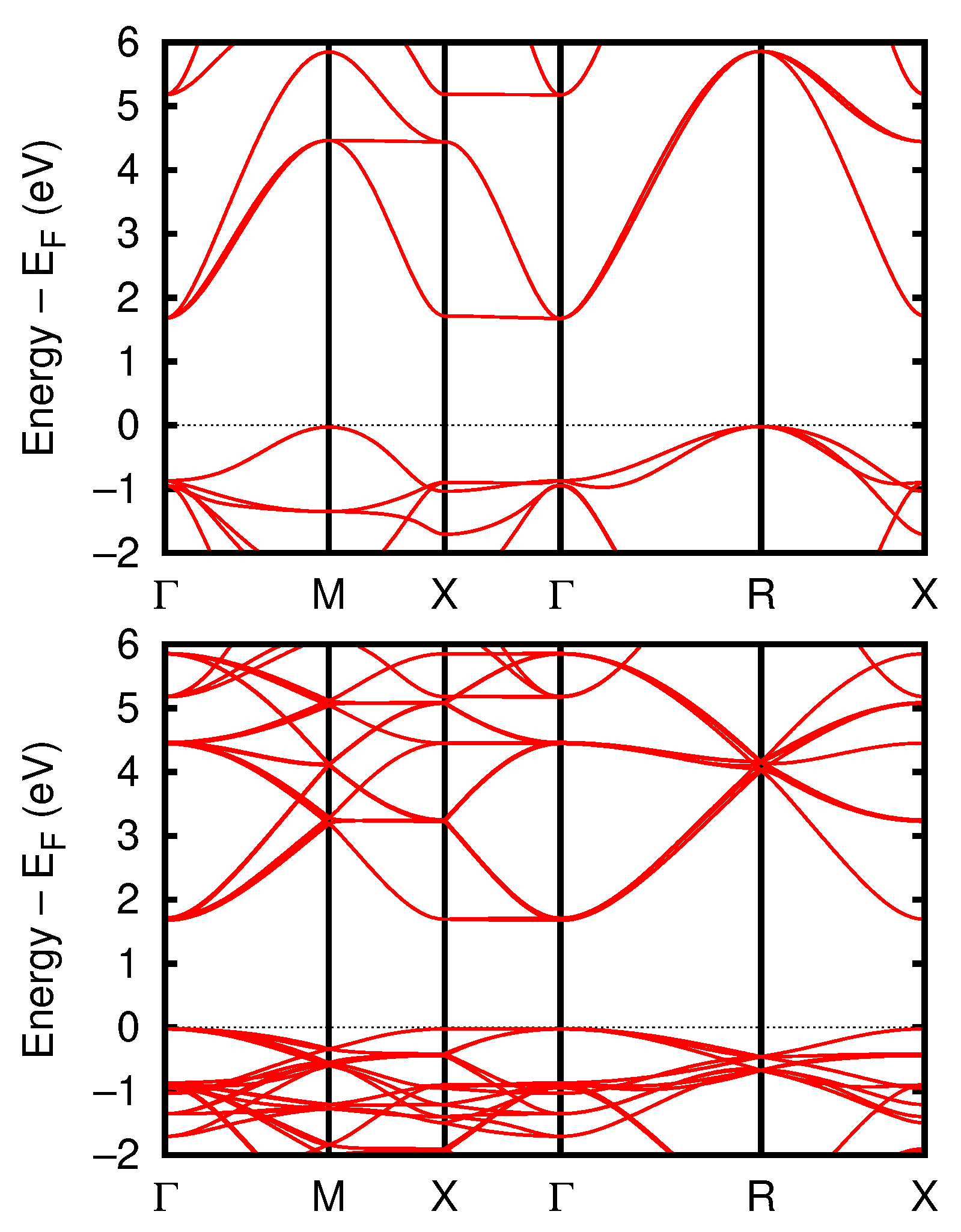
3.1.2. Electronic Properties
3.2. Defective Bulk: WO
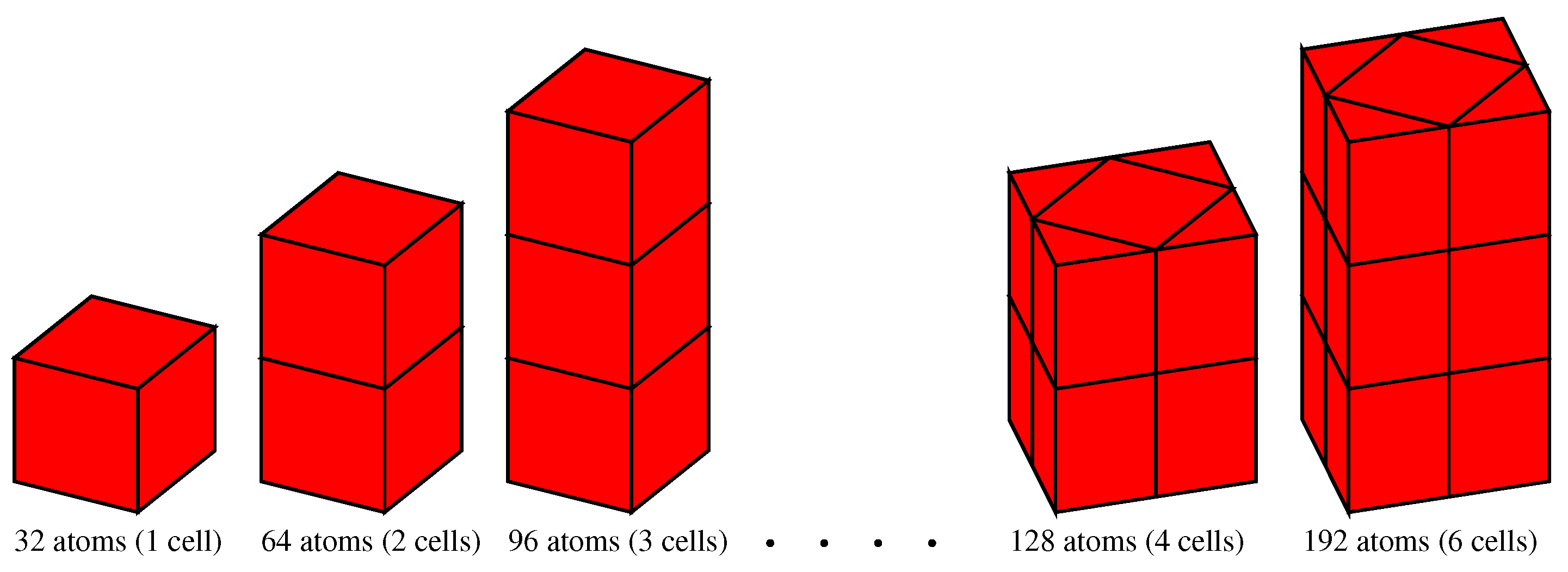
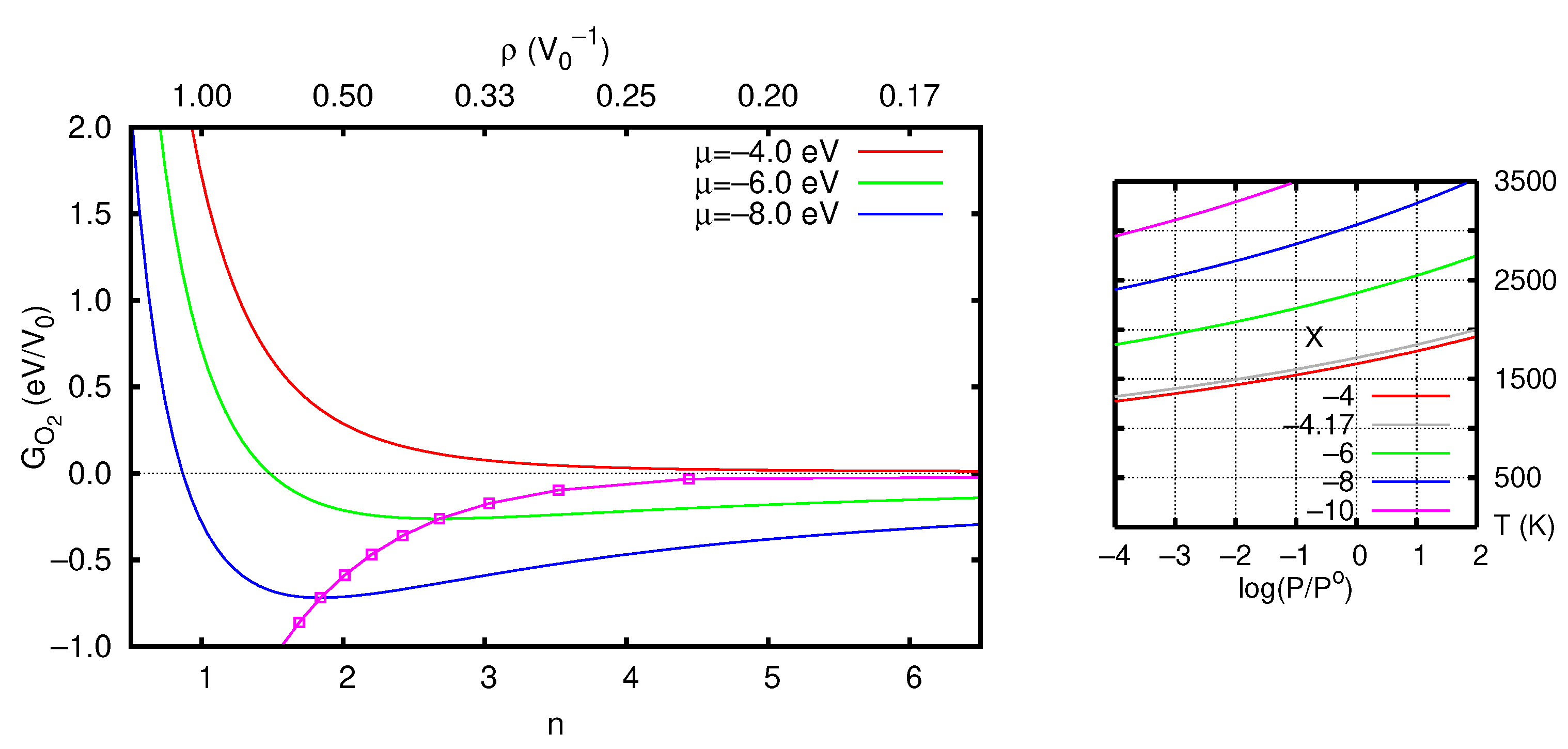
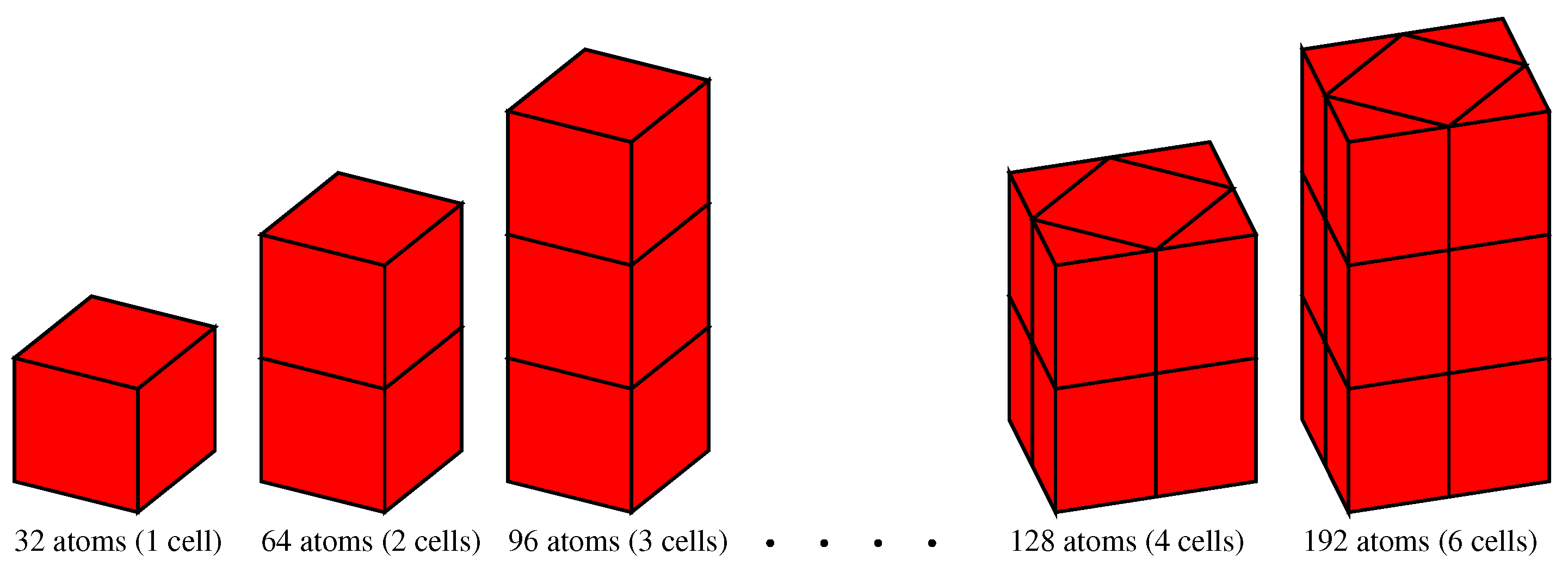
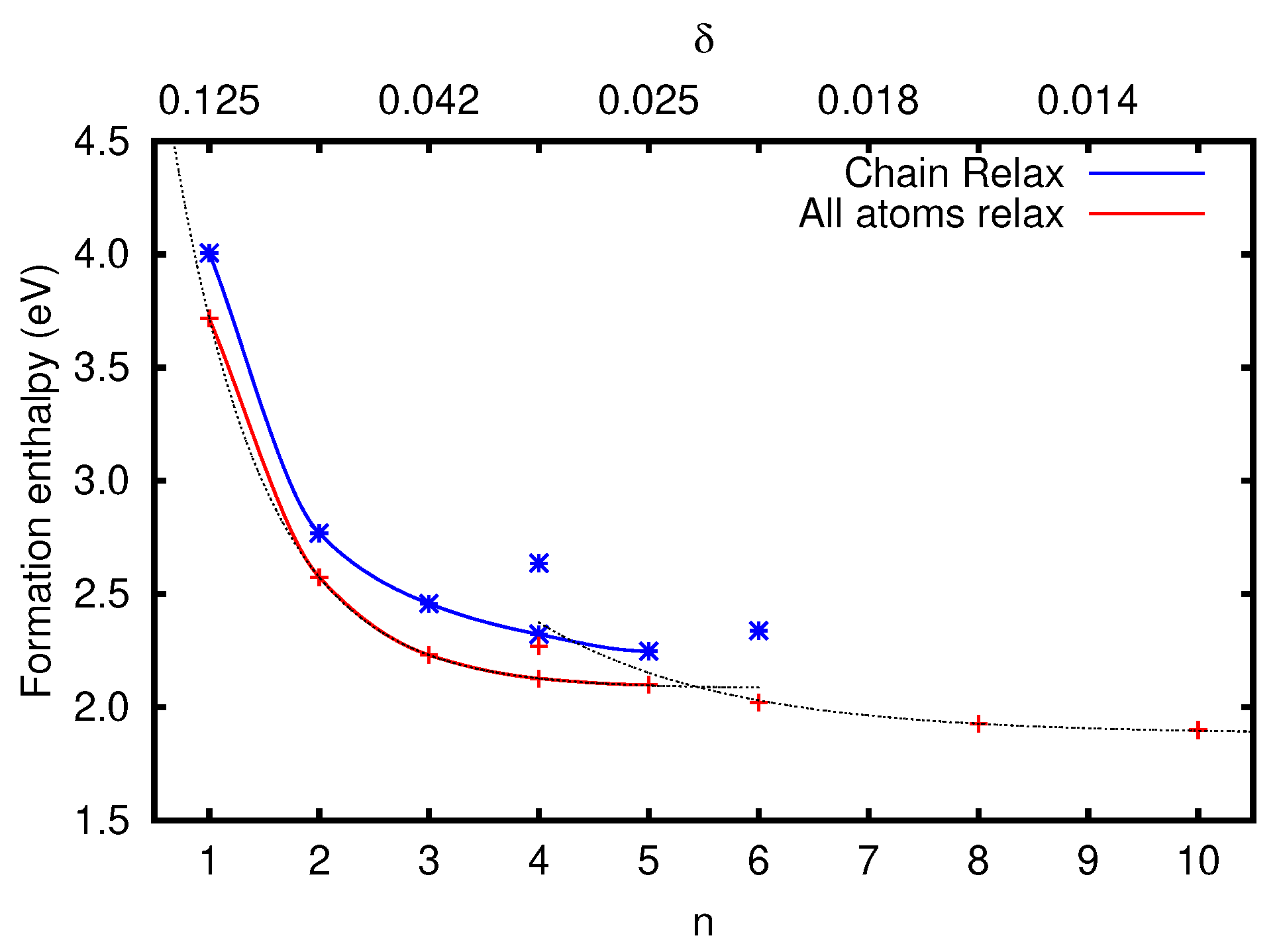
3.2.1. Vacancy Formation Energies
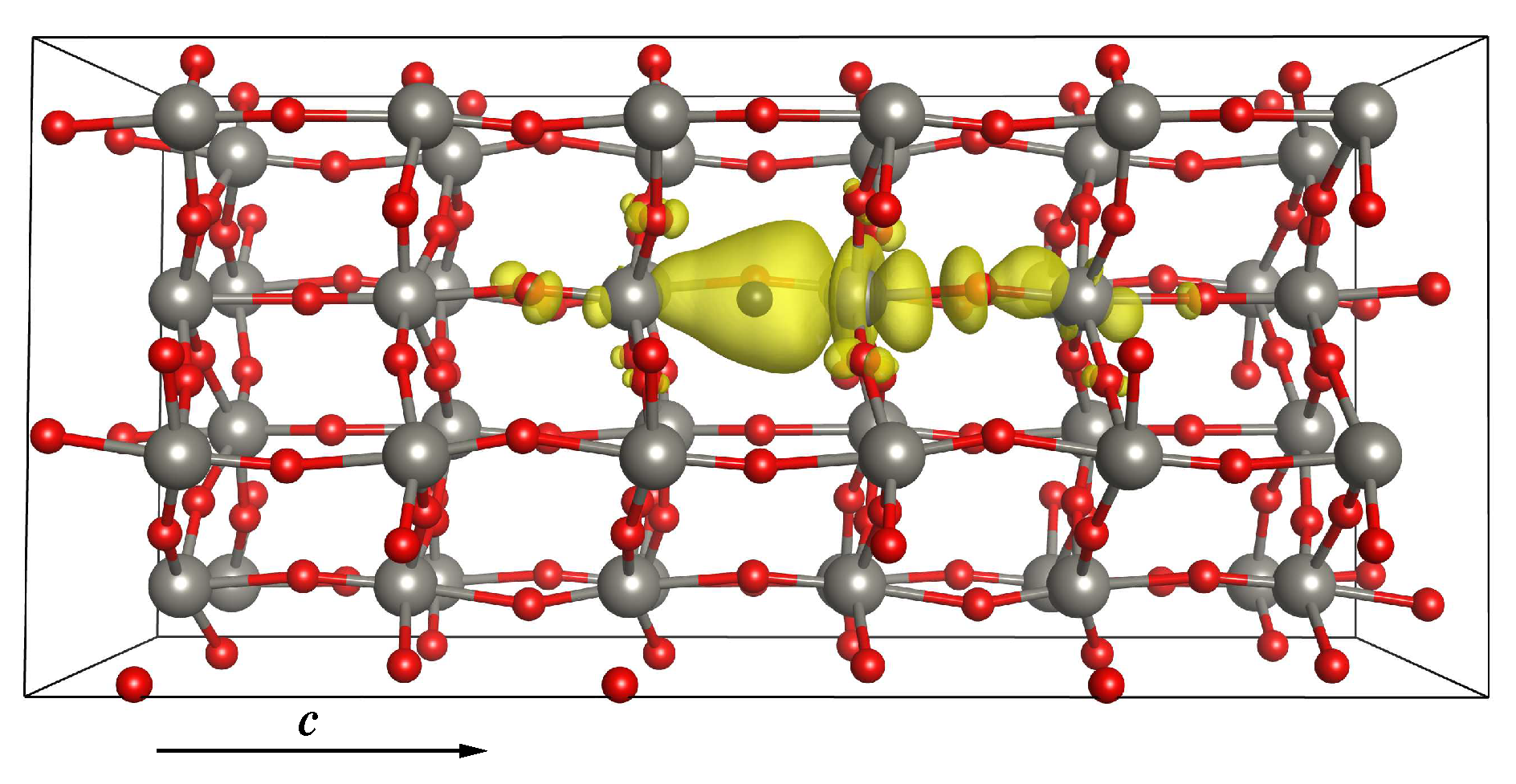
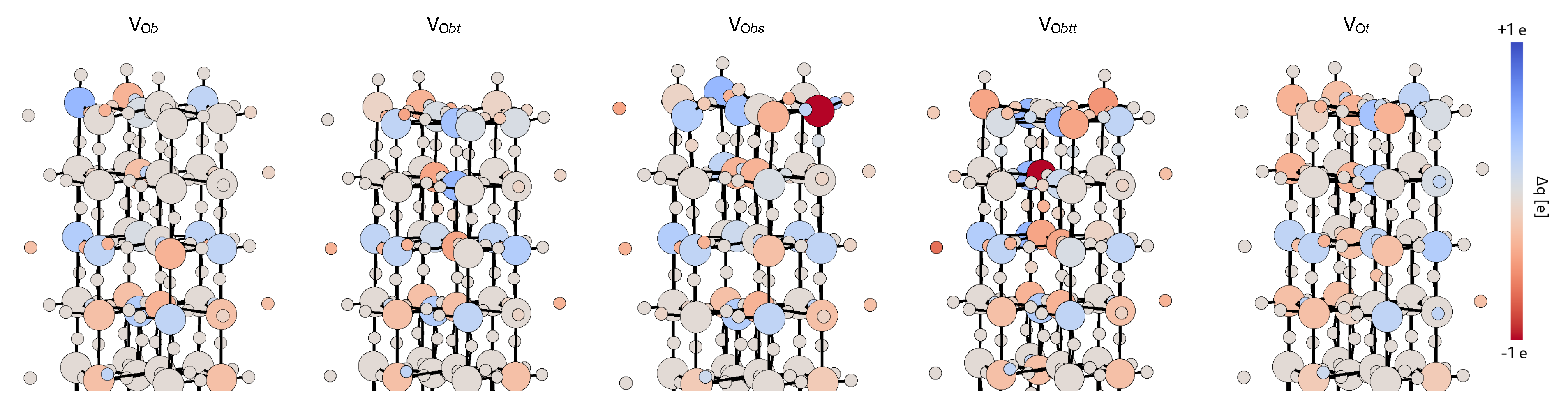
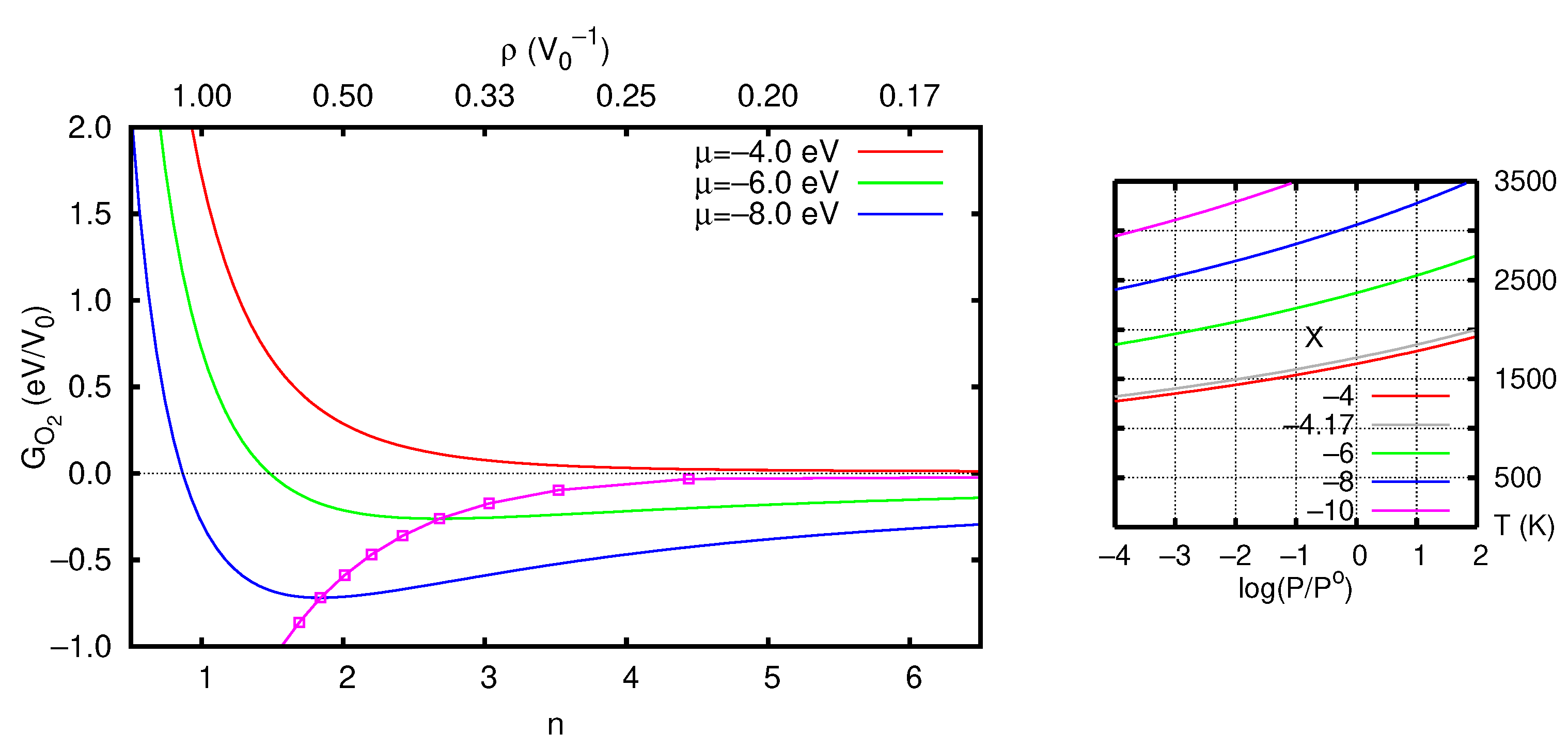
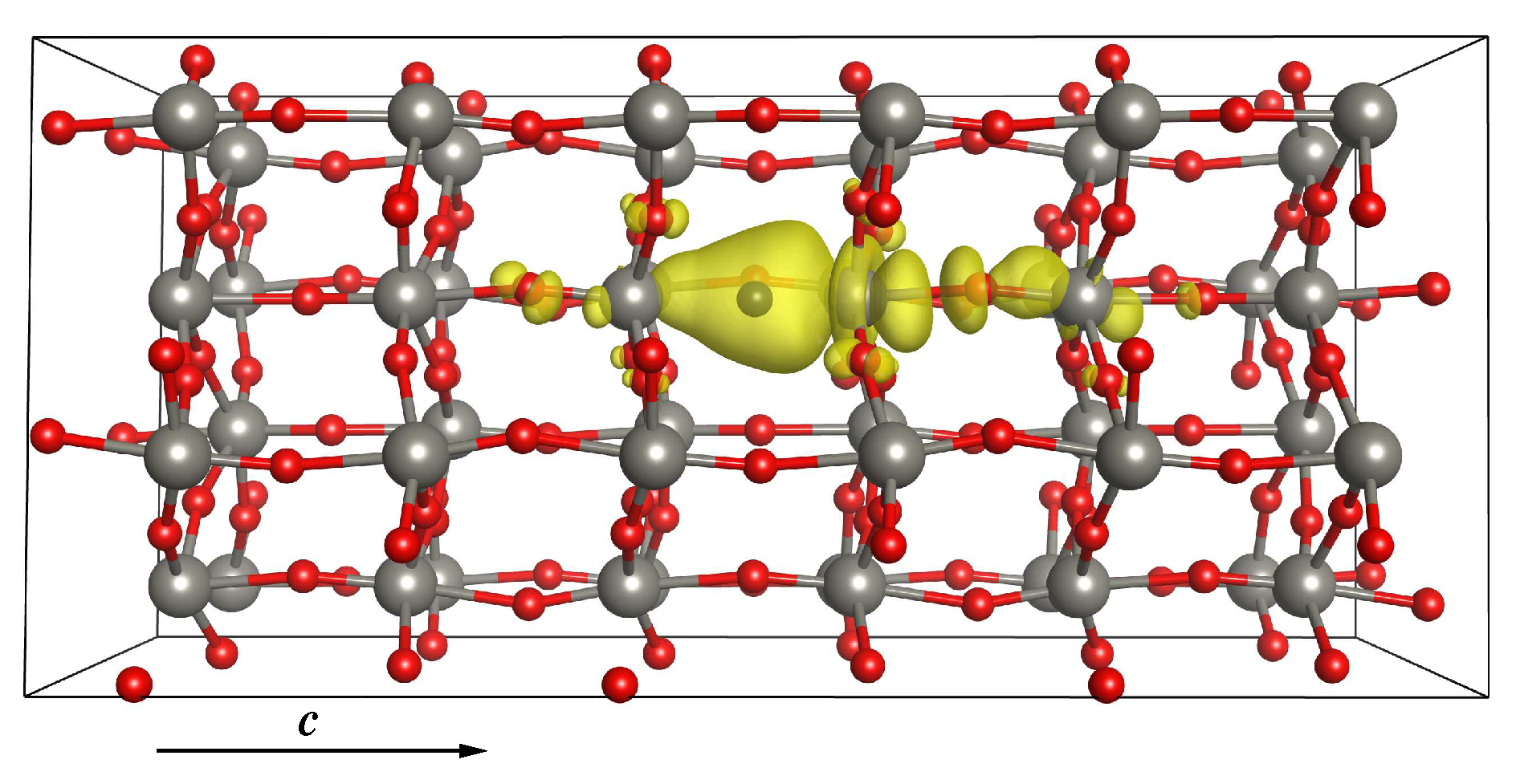
3.2.2. Stability of the O Vacancy
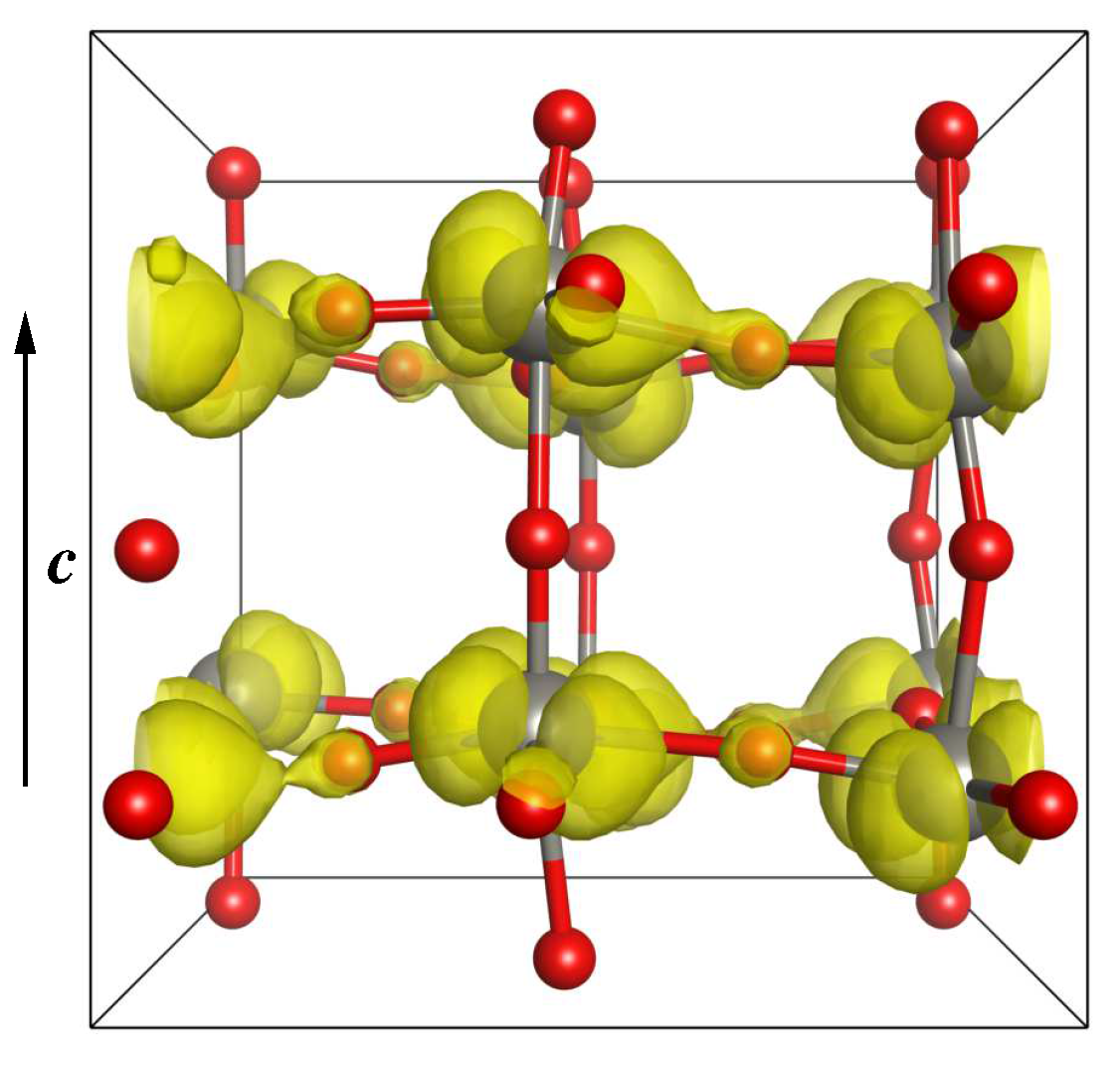
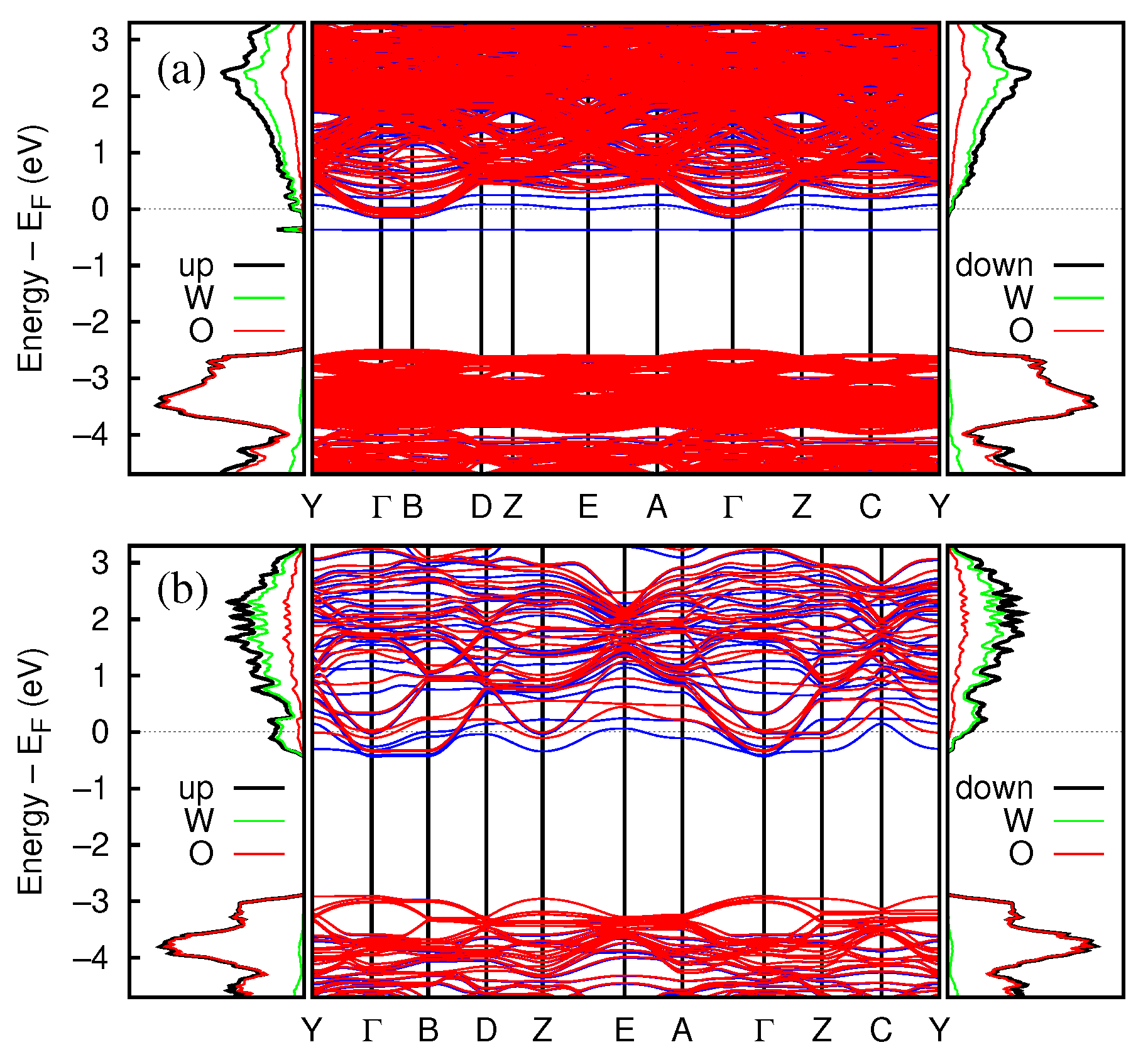
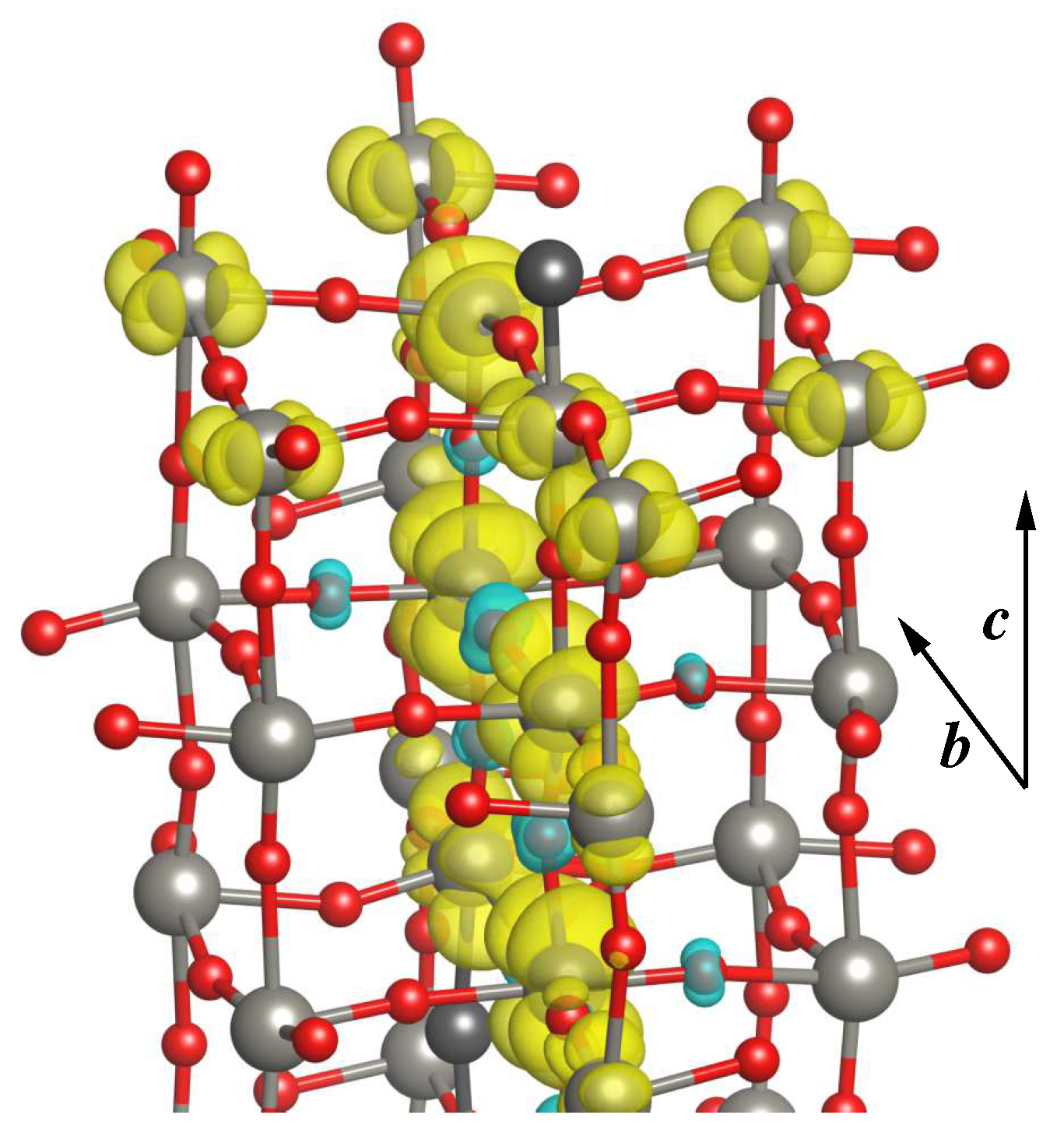
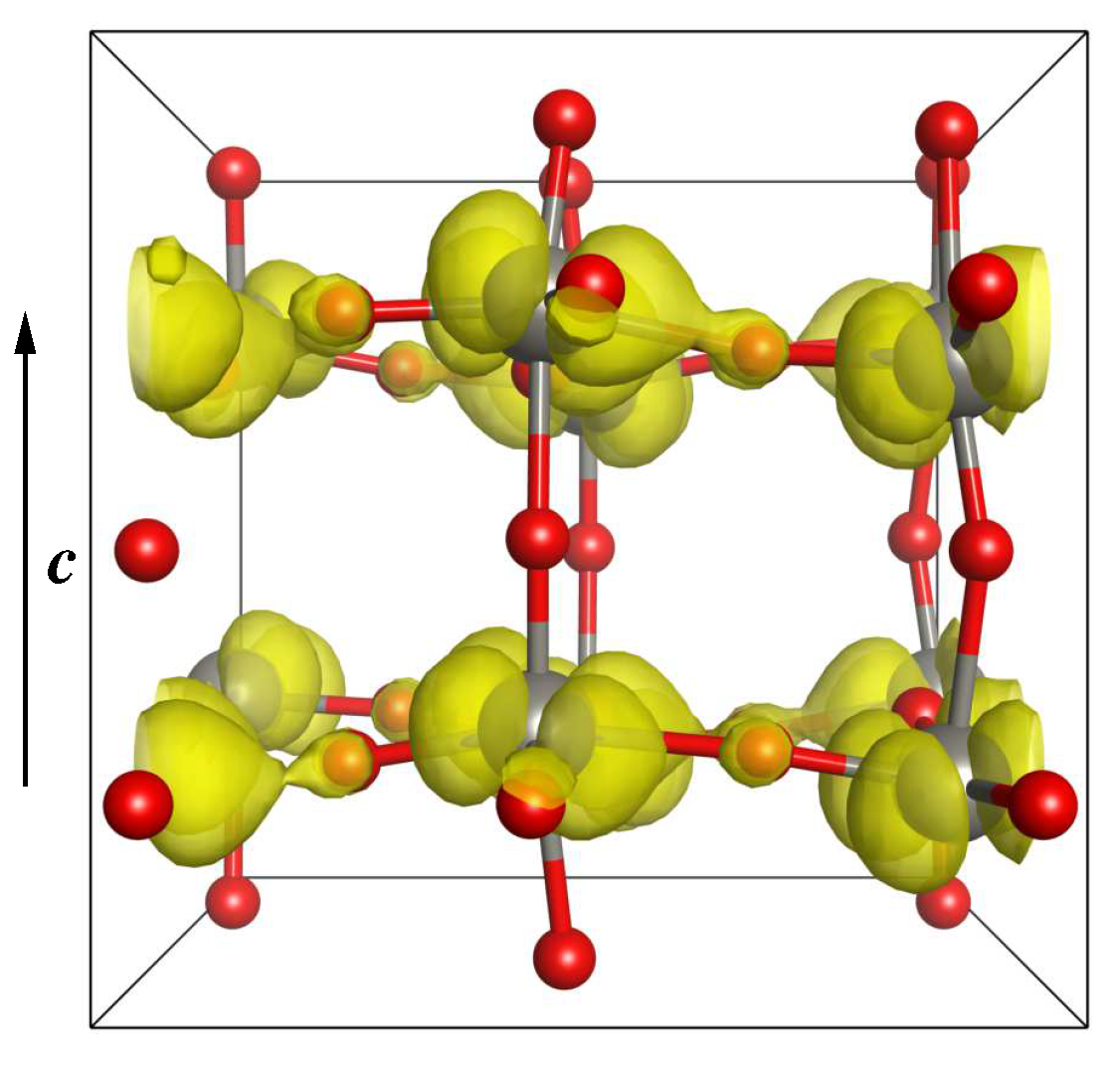
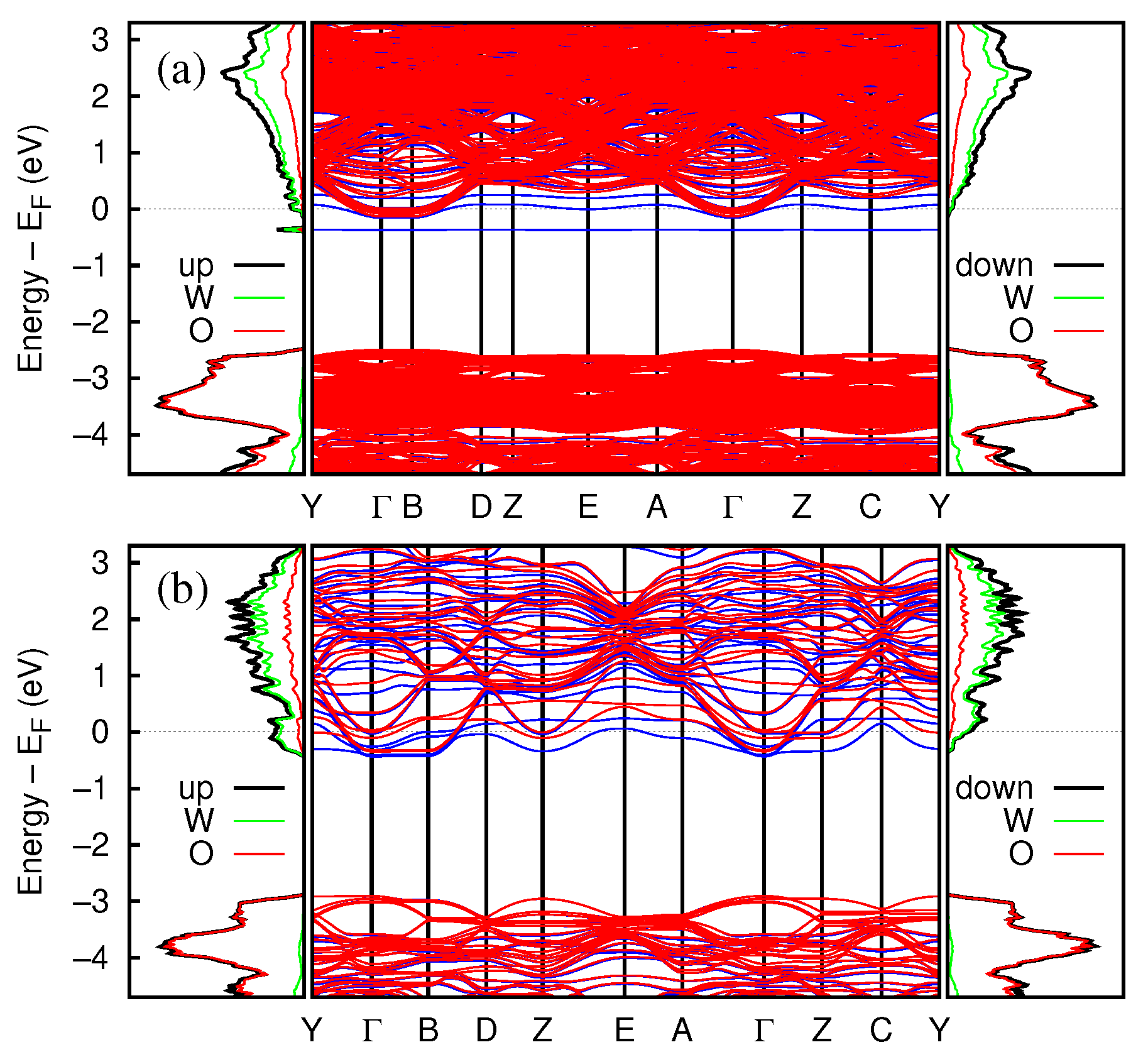
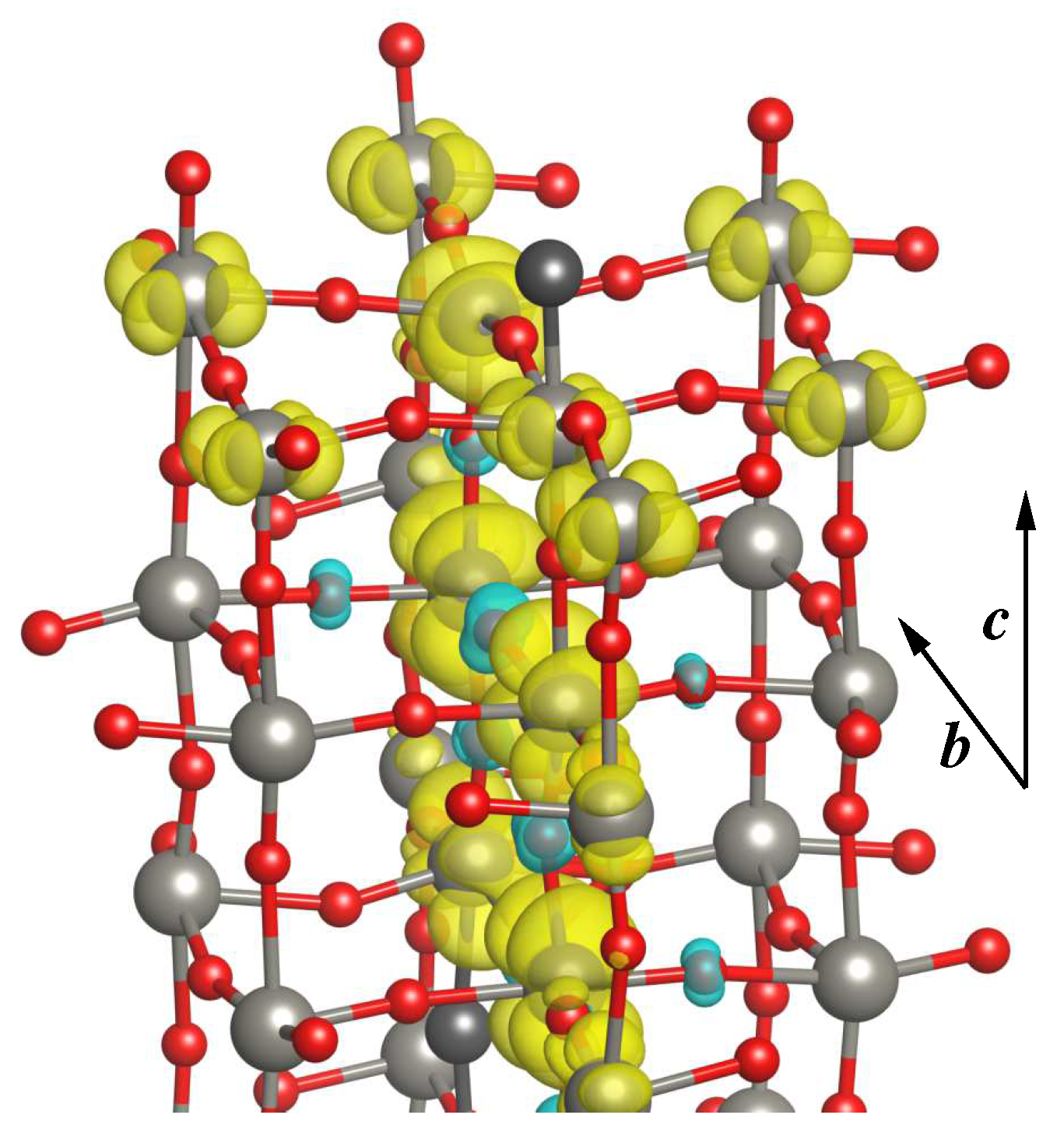
3.2.3. Electronic and Magnetic Properties
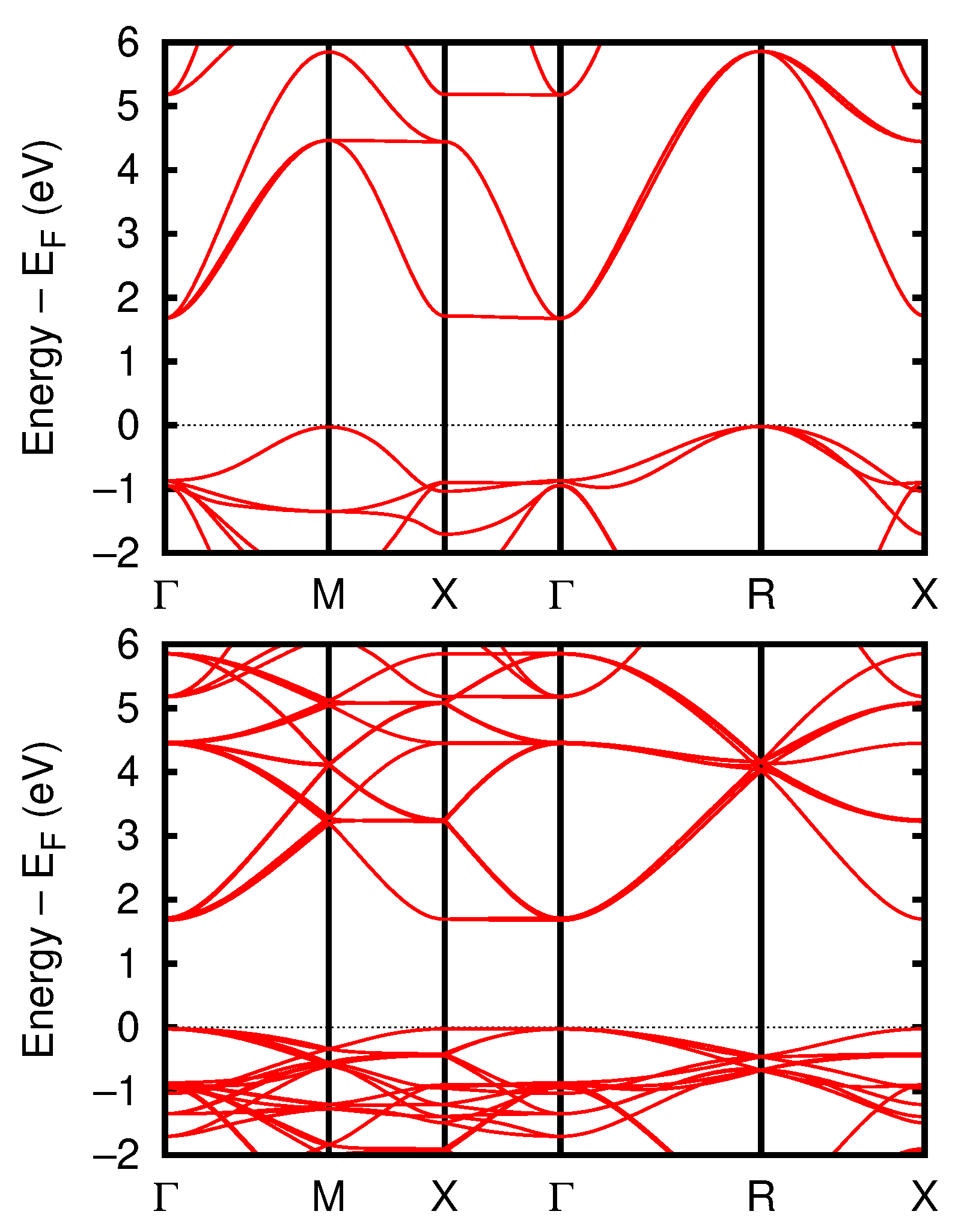
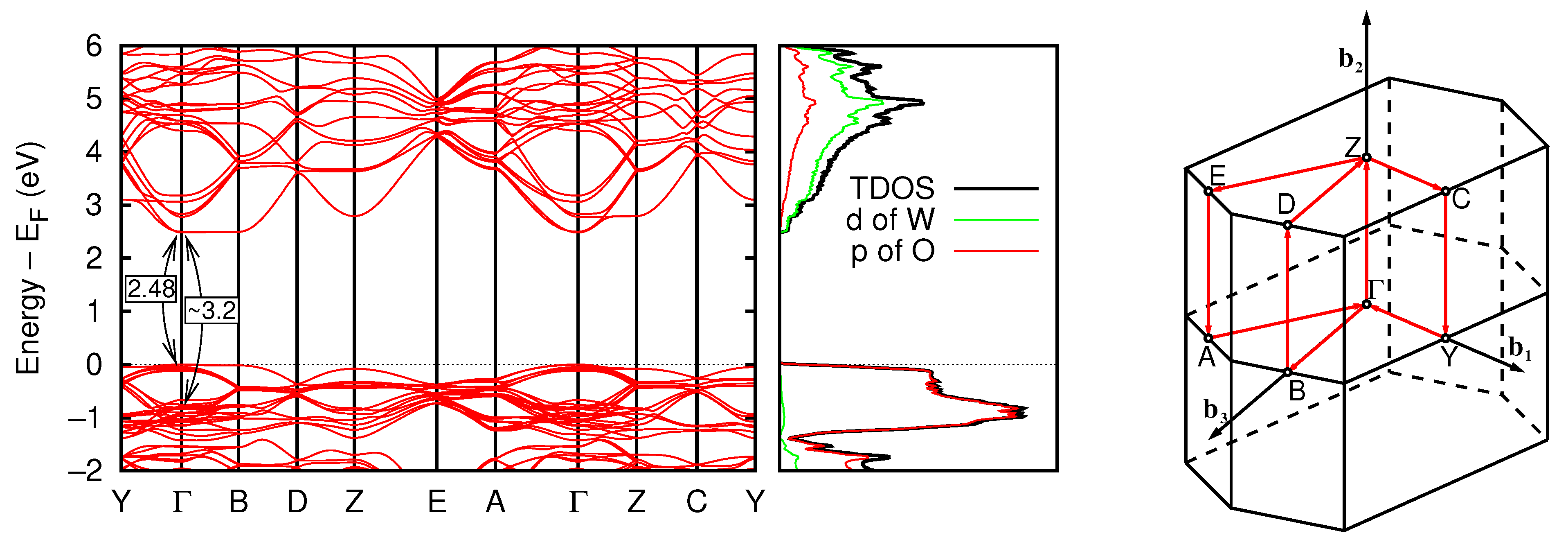
3.2.4. Band Structure
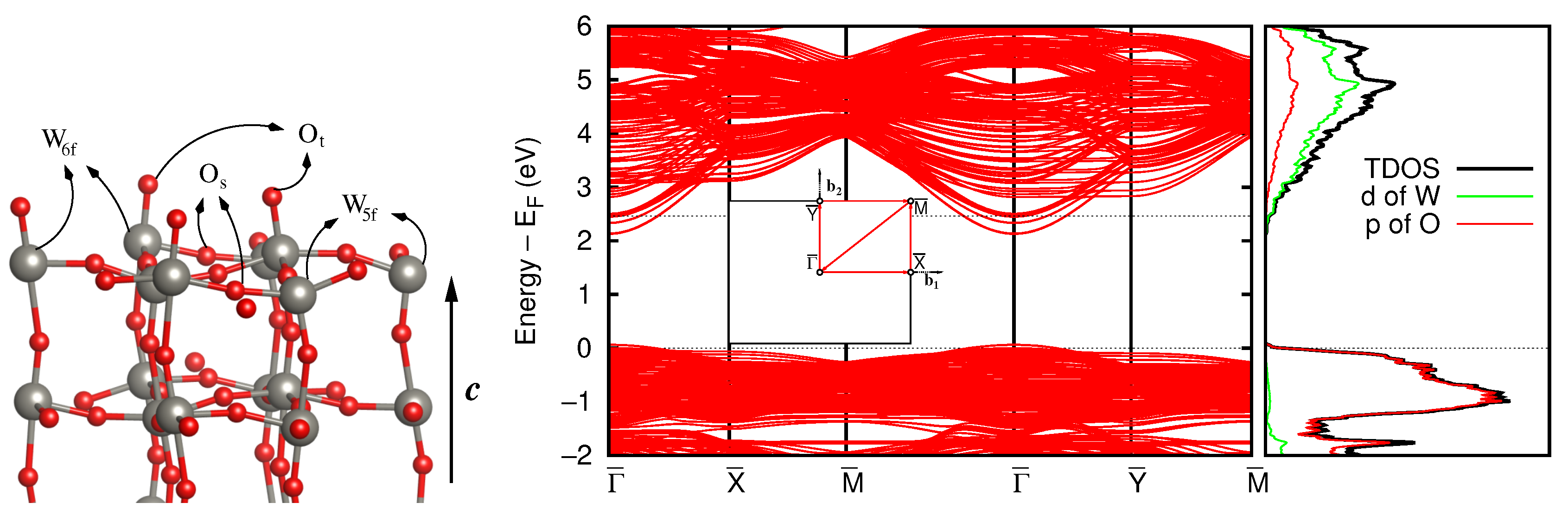
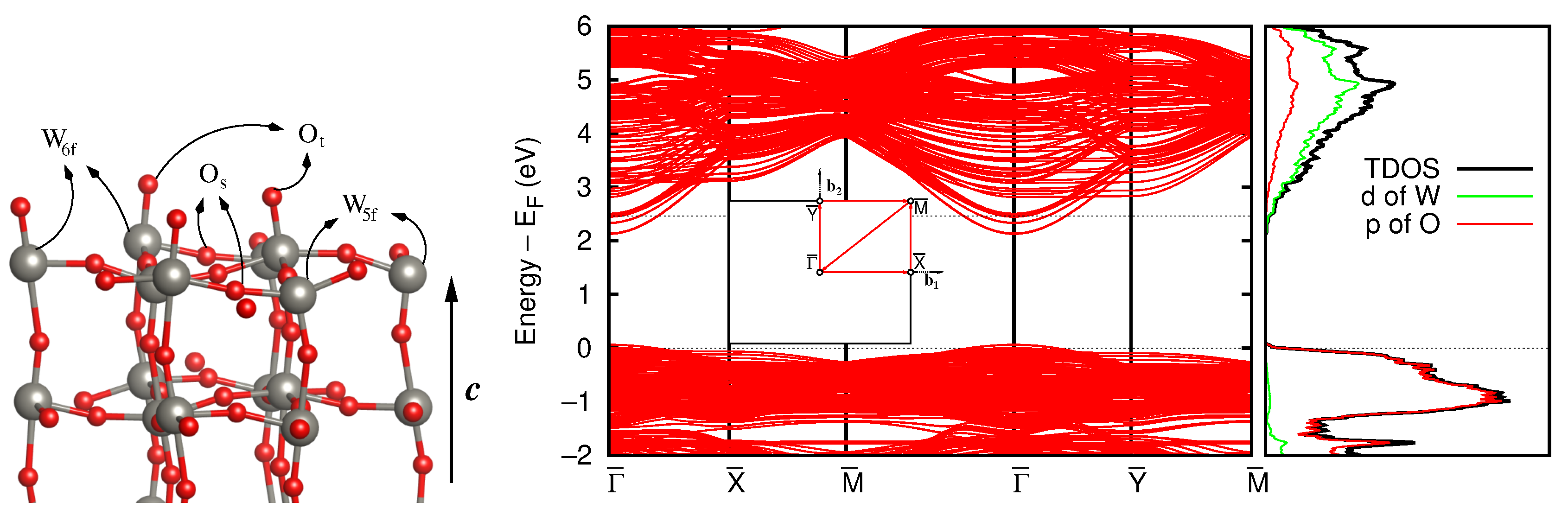
3.3. Stoichiometric WO(001) Surface
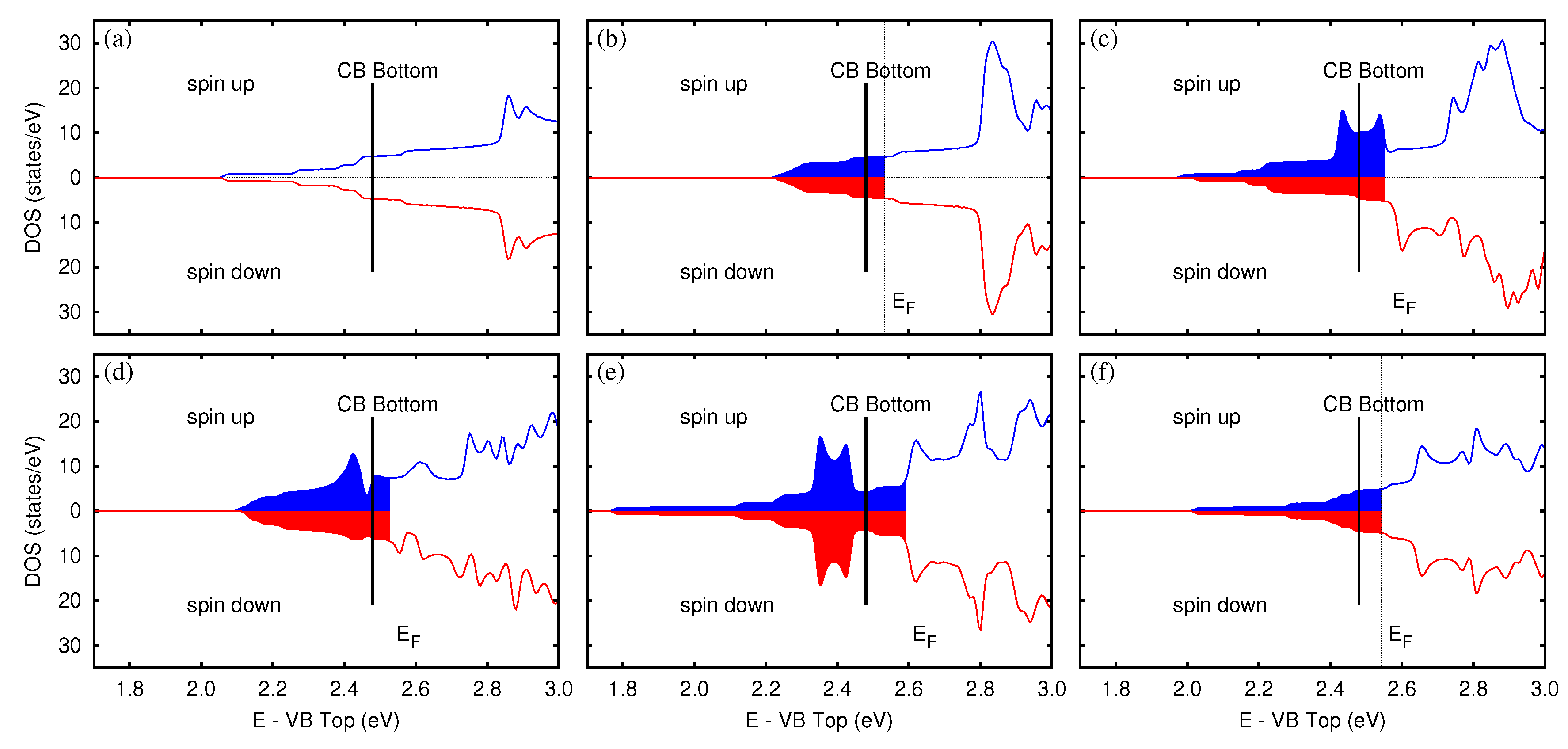
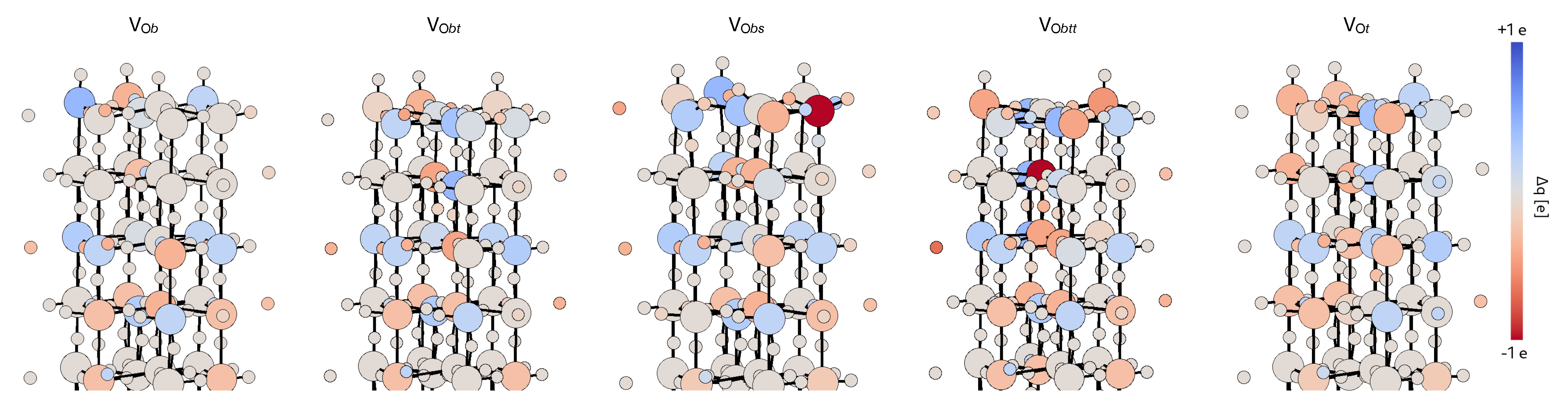
3.4. Defective Surface: WO(001)
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salje, E.K.H.; Rehmann, S.; Pobell, F.; Morris, D.; Knight, K.S.; Herrmannsdörfer, T.; Dove, M.T. Crystal structure and paramagnetic behaviour of ε-WO3−x. J. Phys. Condens. Matter 1997, 9, 6563–6577. [Google Scholar] [CrossRef]
- Matthias, B.T.; Wood, E.A. Low Temperature Polymorphic Transformation in WO3. Phys. Rev. 1951, 84, 1255. [Google Scholar] [CrossRef]
- Woodward, P.M.; Sleight, A.W.; Vogt, T. Structure refinement of triclinic tungsten trioxide. J. Phys. Chem. Solids 1995, 56, 1305–1315. [Google Scholar] [CrossRef]
- Tanisaki, S. On the Phase Transition of Tungsten Trioxide below Room Temperature. J. Phys. Soc. Jpn. 1960, 15, 566–573. [Google Scholar] [CrossRef]
- Diehl, R.; Brandt, G.; Salje, E. The Crystal Structure of Triclinc WO3. Acta Crystallogr. B 1978, 34, 1105–1111. [Google Scholar] [CrossRef]
- Loopstra, B.O.; Rietveld, H.M. Further refinement of the structure of WO3. Acta Crystallogr. B 1969, 25, 1420–1421. [Google Scholar] [CrossRef]
- Loopstra, B.O.; Boldrini, P. Neutron diffraction investigation of WO3. Acta Cryst. 1966, 21, 158–162. [Google Scholar] [CrossRef]
- Salje, E. The orthorhombic phase of WO3. Acta Crystallogr. B 1977, 33, 574–577. [Google Scholar] [CrossRef]
- Tomaszewski, P.E. Structural phase transitions in crystals. I. Database. Phase Transit. 1992, 38, 127–220. [Google Scholar] [CrossRef]
- Ueda, R.; Ichinokawa, T. Domain Structure of Tungsten Trioxide. Phys. Rev. 1950, 80, 1106. [Google Scholar] [CrossRef]
- Salje, E.; Viswanathan, K. Physical properties and phase transitions in WO3. Acta Crystallogr. A 1975, A31, 356–359. [Google Scholar] [CrossRef]
- Cazzanelli, E.; Vinegoni, C.; Mariotto, G.; Kuzmin, A.; Purans, J. Raman study of the phase transitions sequence in pure WO3 at high temperature and in HxWO3 with variable hydrogen content. Solid State Ionics 1999, 123, 67–74. [Google Scholar] [CrossRef]
- Kehl, W.L.; Hay, R.G.; Wahl, D. The Structure of Tetragonal Tungsten Trioxide. J. Appl. Phys. 1952, 23, 212–215. [Google Scholar] [CrossRef]
- Wang, B.; Wang, T.; Ma, S.; Bai, J.; Ma, H. Boosted light absorption by WO3−x/Ag/PbS heterostructure for high-efficiency interfacial solar steam generation. J. Colloid Interface Sci. 2024, 660, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Deb, S.K. Opportunities and Challenges in Science and Technology of WO3 for Electrochromic and Related Applications. Sol. Energy Mater. Sol. Cells 2008, 92, 245–258. [Google Scholar] [CrossRef]
- Yoon, S.; Jo, C.; Noh, S.Y.; Lee, C.W.; Song, J.H.; Lee, J. Development of a high-performance anode for lithium ion batteries using novel ordered mesoporous tungsten oxide materials with high electrical conductivity. Phys. Chem. Chem. Phys. 2011, 13, 11060–11066. [Google Scholar] [CrossRef]
- Jin, H.; Zhou, H.; Zhang, Y. Insight into the Mechanism of CO Oxidation on WO3(001) Surfaces for Gas Sensing: A DFT Study. Sensors 2017, 17, 1898. [Google Scholar] [CrossRef]
- Righettoni, M.; Tricoli, A.; Gass, S.; Schmid, A.; Amann, A.; Pratsinis, S.E. Breath acetone monitoring by portable Si:WO3 gas sensors. Anal. Chim. Acta 2012, 738, 69–75. [Google Scholar] [CrossRef]
- Choi, S.J.; Ku, K.H.; Kim, B.J.; Kim, I.D. Novel Templating Route Using Pt Infiltrated Block Copolymer Microparticles for Catalytic Pt Functionalized Macroporous WO3 Nanofibers and Its Application in Breath Pattern Recognition. ACS Sens. 2016, 1, 1124–1131. [Google Scholar] [CrossRef]
- Kim, S.; Park, S.; Park, S.; Lee, C. Acetone sensing of Au and Pd-decorated WO3 nanorod sensors. Sens. Actuators B Chem. 2015, 209, 180–185. [Google Scholar] [CrossRef]
- Americo, S.; Pargoletti, E.; Soave, R.; Cargnoni, F.; Trioni, M.I.; Chiarello, G.L.; Cerrato, G.; Cappelletti, G. Unveiling the acetone sensing mechanism by WO3 chemiresistors through a joint theory-experiment approach. Electrochim. Acta 2021, 371, 137611. [Google Scholar] [CrossRef]
- Niklasson, G.A.; Berggren, L.; Larsson, A.L. Electrochromic tungsten oxide: The role of defects. Sol. Energy Mater. Sol. Cells 2004, 84, 315–328. [Google Scholar] [CrossRef]
- Niklasson, G.A.; Granqvist, C.G. Electrochromics for smart windows: Thin films of tungsten oxide and nickel oxide, and devices based on these. J. Mater. Chem. 2007, 17, 127–156. [Google Scholar] [CrossRef]
- Granqvist, C. Electrochromic tungsten oxide films: Review of progress 1993–1998. Sol. Energy Mater. Sol. Cells 2000, 60, 201–262. [Google Scholar] [CrossRef]
- Zhao, Y.; Feng, Z.C.; Liang, Y.; Sheng, H.W. Laser-induced coloration of WO3. Appl. Phys. Lett. 1997, 71, 2227–2229. [Google Scholar] [CrossRef]
- Greiner, M.T.; Chai, L.; Helander, M.G.; Tang, W.M.; Lu, Z.H. Transition Metal Oxide Work Functions: The Influence of Cation Oxidation State and Oxygen Vacancies. Adv. Funct. Mater. 2012, 22, 4557–4568. [Google Scholar] [CrossRef]
- Sachs, M.; Park, J.S.; Pastor, E.; Kafizas, A.; Wilson, A.A.; Francàs, L.; Gul, S.; Ling, M.; Blackman, C.; Yano, J.; et al. Effect of oxygen deficiency on the excited state kinetics of WO3 and implications for photocatalysis. Chem. Sci. 2019, 10, 5667–5677. [Google Scholar] [CrossRef]
- Cheng, C.; Fang, Q.; Fernandez-Alberti, S.; Long, R. Depleted Oxygen Defect State Enhancing Tungsten Trioxide Photocatalysis: A Quantum Dynamics Perspective. J. Phys. Chem. Lett. 2022, 13, 5571–5580. [Google Scholar] [CrossRef]
- Corby, S.; Francàs, L.; Kafizas, A.; Durrant, J.R. Determining the role of oxygen vacancies in the photoelectrocatalytic performance of WO3 for water oxidation. Chem. Sci. 2020, 11, 2907–2914. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Guo, J.; Wang, C.; Zhang, J.; Liu, J.; Dong, G.; Zhong, X.; Diao, X. Essential role of oxygen vacancy in electrochromic performance and stability for WO3−y films induced by atmosphere annealing. Electrochim. Acta 2020, 332, 135504. [Google Scholar] [CrossRef]
- Wang, G.; Ling, Y.; Wang, H.; Yang, X.; Wang, C.; Zhang, J.Z.; Li, Y. Hydrogen-treated WO3 nanoflakes show enhanced photostability. Energy Environ. Sci. 2012, 5, 6180–6187. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Marques, M.; Teles, L.K. Approximation to density functional theory for the calculation of band gaps of semiconductors. Phys. Rev. B 2008, 78, 125116. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Marques, M.; Teles, L.K. Slater half-occupation technique revisited: The LDA-1/2 and GGA-1/2 approaches for atomic ionization energies and band gaps in semiconductors. AIP Adv. 2011, 1, 032119. [Google Scholar] [CrossRef]
- Janak, J.F. Proof that ∂E/∂ni = ϵi in density-functional theory. Phys. Rev. B 1978, 18, 7165–7168. [Google Scholar] [CrossRef]
- Valadares, F.; Guilhon, I.; Teles, L.K.; Marques, M. Electronic Structure Panorama of Halide Perovskites: Approximated DFT-1/2 Quasiparticle and Relativistic Corrections. J. Phys. Chem. C 2020, 124, 18390–18400. [Google Scholar] [CrossRef]
- Lahmer, M. Effect of oxygen vacancies on the electronic structure and dielectric properties of SrAl2O4: A first-principles study. J. Phys. Chem. Solids 2022, 160, 110297. [Google Scholar] [CrossRef]
- Matusalem, F.; Bechstedt, F.; Guilhon, I.; Marques, M.; Teles, L.K. DFT-1/2 method applied to 2D topological insulators: Fluorinated and hydrogenated group-IV honeycomb systems. J. Phys. Condens. Matter 2021, 33, 435501. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Long, R. Density Functional Theory Half-Electron Self-Energy Correction for Fast and Accurate Nonadiabatic Molecular Dynamics. J. Phys. Chem. Lett. 2021, 12, 10886–10892. [Google Scholar] [CrossRef]
- Traore, B.; Even, J.; Pedesseau, L.; Képénekian, M.; Katan, C. Band gap, effective masses, and energy level alignment of 2D and 3D halide perovskites and heterostructures using DFT-1/2. Phys. Rev. Mater. 2022, 6, 014604. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Monographs on Chemistry; Oxford University Press: Oxford, UK, 1990; Volume 22. [Google Scholar]
- Kong, H.; Yang, H.; Park, J.S.; Chae, W.S.; Kim, H.Y.; Park, J.; Lee, J.H.; Choi, S.Y.; Park, M.; Kim, H.; et al. Spatial Control of Oxygen Vacancy Concentration in Monoclinic WO3 Photoanodes for Enhanced Solar Water Splitting. Adv. Funct. Mater. 2022, 32, 2204106. [Google Scholar] [CrossRef]
- Li, W.; Yuan, X.; Qiu, S.; Liu, X. First-principles study of the effect of non-stoichiometry on sensing mechanism of NOx on WO3(001) surfaces. Mater. Today Commun. 2022, 33, 104926. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter 2002, 14, 2745–2779. [Google Scholar] [CrossRef]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993–2006. [Google Scholar] [CrossRef] [PubMed]
- Rivero, P.; García-Suárez, V.M.; Pereñiguez, D.; Utt, K.; Yang, Y.; Bellaiche, L.; Park, K.; Ferrer, J.; Barraza-Lopez, S. Systematic pseudopotentials from reference eigenvalue sets for DFT calculations. Comput. Mater. Sci. 2015, 98, 372–389. [Google Scholar] [CrossRef]
- Lambert-Mauriat, C.; Oison, V. Density-functional study of oxygen vacancies in monoclinic tungsten oxide. J. Phys. Condens. Matter 2006, 18, 7361–7371. [Google Scholar] [CrossRef]
- Otero-de-la Roza, A.; Johnson, E.R.; Luaña, V. Critic2: A program for real-space analysis of quantum chemical interactions in solids. Comput. Phys. Commun. 2014, 185, 1007–1018. [Google Scholar] [CrossRef]
- Vogt, T.; Woodward, P.M.; Hunter, B.A. The High-Temperature Phases of WO3. J. Solid State Chem. 1999, 144, 209–215. [Google Scholar] [CrossRef]
- Howard, C.J.; Luca, V.; Knight, K.S. High-temperature phase transitions in tungsten trioxide—The last word? J. Phys. Condens. Matter 2002, 14, 377–387. [Google Scholar] [CrossRef]
- Butler, M.A. Photoelectrolysis and physical properties of the semiconducting electrode WO3. J. Appl. Phys. 1977, 48, 1914–1920. [Google Scholar] [CrossRef]
- Koffyberg, F.P.; Dwight, K.; Wold, A. Interband transitions of semiconducting oxides determined from photoelectrolysis spectra. Solid State Commun. 1979, 30, 433–437. [Google Scholar] [CrossRef]
- Miyake, K.; Kaneko, H.; Sano, M.; Suedomi, N. Physical and electrochromic properties of the amorphous and crystalline tungsten oxide thick films prepared under reducing atmosphere. J. Appl. Phys. 1984, 55, 2747–2753. [Google Scholar] [CrossRef]
- González-Borrero, P.P.; Sato, F.; Medina, A.N.; Baesso, M.L.; Bento, A.C.; Baldissera, G.; Persson, C.; Niklasson, G.A.; Granqvist, C.G.; Ferreira da Silva, A. Optical band-gap determination of nanostructured WO3 film. Appl. Phys. Lett. 2010, 96, 061909. [Google Scholar] [CrossRef]
- Valyukh, I.; Green, S.; Arwin, H.; Niklasson, G.; Wäckelgård, E.; Granqvist, C. Spectroscopic ellipsometry characterization of electrochromic tungsten oxide and nickel oxide thin films made by sputter deposition. Sol. Energy Mater. Sol. Cells 2010, 94, 724–732. [Google Scholar] [CrossRef]
- Shen, Z.; Zhao, Z.; Wen, J.; Qian, J.; Peng, Z.; Fu, X. Role of Oxygen Vacancies in the Electrical Properties of WO3−x Nano/Microrods with Identical Morphology. J. Nanomater. 2018, 2018, 7802589. [Google Scholar] [CrossRef]
- Rinaldi, F.G.; Arutanti, O.; Arif, A.F.; Hirano, T.; Ogi, T.; Okuyama, K. Correlations between Reduction Degree and Catalytic Properties of WOx Nanoparticles. ACS Omega 2018, 3, 8963–8970. [Google Scholar] [CrossRef] [PubMed]
- De Wijs, G.A.; de Boer, P.K.; de Groot, R.A.; Kresse, G. Anomalous behavior of the semiconducting gap in WO3 from first-principles calculations. Phys. Rev. B 1999, 59, 2684–2693. [Google Scholar] [CrossRef]
- Huda, M.N.; Yan, Y.; Moon, C.Y.; Wei, S.H.; Al-Jassim, M.M. Density-functional theory study of the effects of atomic impurity on the band edges of monoclinic WO3. Phys. Rev. B 2008, 77, 195102. [Google Scholar] [CrossRef]
- Migas, D.B.; Shaposhnikov, V.L.; Rodin, V.N.; Borisenko, V.E. Tungsten oxides. I. Effects of oxygen vacancies and doping on electronic and optical properties of different phases of WO3. J. Appl. Phys. 2010, 108, 093713. [Google Scholar] [CrossRef]
- Wang, F.; Di Valentin, C.; Pacchioni, G. Electronic and Structural Properties of WO3: A Systematic Hybrid DFT Study. J. Phys. Chem. C 2011, 115, 8345–8353. [Google Scholar] [CrossRef]
- Alvarez-Quiceno, J.C.; Dalpian, G.M.; Osorio-Guillén, J.M. A systematic first-principles study of the tungsten trioxide polymorphs. Phys. Status Solidi B 2015, 252, 2290–2295. [Google Scholar] [CrossRef]
- Wang, W.; Janotti, A.; Van de Walle, C.G. Role of oxygen vacancies in crystalline WO3. J. Mater. Chem. C 2016, 4, 6641–6648. [Google Scholar] [CrossRef]
- Hamdi, H.; Salje, E.K.H.; Ghosez, P.; Bousquet, E. First-principles reinvestigation of bulk WO3. Phys. Rev. B 2016, 94, 245124. [Google Scholar] [CrossRef]
- Chatten, R.; Chadwick, A.V.; Rougier, A.; Lindan, P.J.D. The Oxygen Vacancy in Crystal Phases of WO3. J. Phys. Chem. B 2005, 109, 3146–3156. [Google Scholar] [CrossRef] [PubMed]
- Bondarenko, N.; Eriksson, O.; Skorodumova, N.V. Polaron mobility in oxygen-deficient and lithium-doped tungsten trioxide. Phys. Rev. B 2015, 92, 165119. [Google Scholar] [CrossRef]
- Wang, F.; Di Valentin, C.; Pacchioni, G. Semiconductor-to-metal transition in WO3−x: Nature of the oxygen vacancy. Phys. Rev. B 2011, 84, 073103. [Google Scholar] [CrossRef]
- Gerosa, M.; Di Valentin, C.; Onida, G.; Bottani, C.E.; Pacchioni, G. Anisotropic Effects of Oxygen Vacancies on Electrochromic Properties and Conductivity of γ-Monoclinic WO3. J. Phys. Chem. C 2016, 120, 11716–11726. [Google Scholar] [CrossRef]
- Aroyo, M.I.; Orobengoa, D.; de la Flor, G.; Tasci, E.S.; Perez-Mato, J.M.; Wondratschek, H. Brillouin-zone database on the Bilbao Crystallographic Server. Acta Crystallogr. A 2014, 70, 126–137. [Google Scholar] [CrossRef]
- Allpress, J.; Tilley, R.; Sienko, M. Examination of substoichiometric WO3−x crystals by electron microscopy. J. Solid State Chem. 1971, 3, 440–451. [Google Scholar] [CrossRef]
- Bousquet, E.; Hamdi, H.; Aguado-Puente, P.; Salje, E.K.H.; Artacho, E.; Ghosez, P. First-principles characterization of single-electron polaron in WO3. Phys. Rev. Res. 2020, 2, 012052. [Google Scholar] [CrossRef]
- Le, H.M.; Vu, N.H.; Phan, B.T. Migrations of oxygen vacancy in tungsten oxide (WO3): A density functional theory study. Comput. Mater. Sci. 2014, 90, 171–176. [Google Scholar] [CrossRef]
- Wassmann, T.; Seitsonen, A.P.; Saitta, A.M.; Lazzeri, M.; Mauri, F. Structure, Stability, Edge States, and Aromaticity of Graphene Ribbons. Phys. Rev. Lett. 2008, 101, 096402. [Google Scholar] [CrossRef] [PubMed]
- Chase, M.W. NIST-JANAF Thermochemical Tables; American Chemical Society: New York, NY, USA, 1998. [Google Scholar] [CrossRef]
- Wang, L.; Maxisch, T.; Ceder, G. Oxidation energies of transition metal oxides within the GGA+U framework. Phys. Rev. B 2006, 73, 195107. [Google Scholar] [CrossRef]
- Fang, Z.; Jiao, S.; Wang, B.; Yin, W.; Liu, S.; Gao, R.; Liu, Z.; Pang, G.; Feng, S. Synthesis of reduced cubic phase WO3−x nanosheet by direct reduction of H2WO4·H2O. Mater. Today Energy 2017, 6, 146–153. [Google Scholar] [CrossRef]
- Xie, F.Y.; Gong, L.; Liu, X.; Tao, Y.T.; Zhang, W.H.; Chen, S.H.; Meng, H.; Chen, J. XPS studies on surface reduction of tungsten oxide nanowire film by Ar+ bombardment. J. Electron Spectrosc. Relat. Phenom. 2012, 185, 112–118. [Google Scholar] [CrossRef]
- Qian, J.; Peng, Z.; Shen, Z.; Zhao, Z.; Zhang, G.; Fu, X. Positive impedance humidity sensors via single-component materials. Sci. Rep. 2016, 6, 25574. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Shen, Z.; Peng, F.; Lu, Y.; Ge, M.; Fu, X.; Sun, Y.; Chen, X.; Dai, N. Improving gas sensing performance by oxygen vacancies in sub-stoichiometric WO3−x. RSC Adv. 2019, 9, 7723–7728. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Dong, X.; Zhang, S.; Yang, J. Defect-induced ferromagnetism in W18O49 nanowires. J. Alloys Compd. 2020, 818, 152894. [Google Scholar] [CrossRef]
- Oliver, P.M.; Parker, S.C.; Egdell, R.G.; Jones, F.H. Computer simulation of the surface structures of WO3. J. Chem. Soc. Faraday Trans. 1996, 92, 2049–2056. [Google Scholar] [CrossRef]
- Wang, F.; Di Valentin, C.; Pacchioni, G. DFT Study of Hydrogen Adsorption On the Monoclinic WO3 (001) Surface. J. Phys. Chem. C 2012, 116, 10672–10679. [Google Scholar] [CrossRef]
- Migas, D.B.; Shaposhnikov, V.L.; Borisenko, V.E.; Skorodumova, N.V. The Surface Energy and Band Structure of γ-WO3 Thin Films. Sci. Adv. Mater. 2017, 9, 469–474. [Google Scholar] [CrossRef]
- Lambert-Mauriat, C.; Oison, V.; Saadi, L.; Aguir, K. Ab initio study of oxygen point defects on tungsten trioxide surface. Surf. Sci. 2012, 606, 40–45. [Google Scholar] [CrossRef]
- Halek, G.; Baikie, I.; Teterycz, H.; Halek, P.; Suchorska-Woźniak, P.; Wiśniewski, K. Work function analysis of gas sensitive WO3 layers with Pt doping. Sens. Actuators B Chem. 2013, 187, 379–385. [Google Scholar] [CrossRef]
- Albanese, E.; Di Valentin, C.; Pacchioni, G. H2O Adsorption on WO3 and WO3−x (001) Surfaces. ACS Appl. Mater. Interfaces 2017, 9, 23212–23221. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a | b | c | (Degree) | Vol * | Gap (eV), (D) Direct (I) Indirect | |
|---|---|---|---|---|---|---|
| Ref. [6] [Exp.] | 7.306 (1) | 7.540 (1) | 7.692 (1) | 90.881 (5) | 52.96 (7) | – |
| Ref. [3] [Exp.] | 7.3008 (5) | 7.5388 (1) | 7.6896 (3) | 90.892 (1) | 52.898 | – |
| Ref. [49] [Exp.] | 7.3271 (2) | 7.5644 (2) | 7.7274 (3) | 90.488 (3) | 53.535 (3) | – |
| Ref. [50] [Exp.] | 7.3033 (2) | 7.5375 (2) | 7.6920 (2) | 90.855 (1) | 52.923 | – |
| Ref. [51] [Exp.] | 2.65, 2.70, 2.77 (I) | |||||
| Ref. [52] [Exp.] | 2.62 ± 0.04 (I), 3.53 ± 0.02 (D) | |||||
| Ref. [53] [Exp.] | 2.57, 2.70, 2.75 (I) | |||||
| Ref. [54] [Exp.] | 2.70–2.75 ± 0.06 | |||||
| Ref. [55] [Exp.] | 3.150 ± 0.003 (D) | |||||
| Ref. [56] [Exp.] | 2.75 | |||||
| Ref. [57] [Exp.] | 2.61 | |||||
| Ref. [21] [Exp.] | 2.6 | |||||
| This work [PBE] | 7.46 | 7.68 | 7.88 | 90.4 | 56.5 | 2.48 (I), 3.2 (D) |
| Ref. [58] [PW91] | 7.55 | 7.62 | 7.83 | 90.2 | 56.3 | 0.90 |
| Ref. [58] [LDA] | 7.37 | 7.46 | 7.64 | 90.6 | 52.3 | 1.10 |
| Ref. [47] [PBE] | 7.48 | 7.66 | 7.85 | 90.4 | 56.2 | ∼1.0 |
| Ref. [59] [LDA] | 7.381 | 7.472 | 7.633 | – | ∼52.6 | 1.31 |
| Ref. [60] [PBE] | 7.498 | 7.649 | 7.789 | 90.61 | 55.8 | 1.40 |
| Ref. [61] [HSE06] | 7.39 | 7.64 | 7.75 | 90.3 | 54.7 | 2.80 |
| Ref. [61] [B3LYP] | 7.44 | 7.73 | 7.91 | 90.2 | 56.9 | 3.13, 2.80 |
| Ref. [62] [PBEsol] | 7.492 | 7.546 | 7.757 | 90.06 | 54.8 | 1.31, 2.76 |
| Ref. [63] [HSE06] | 7.41 | 7.63 | 7.79 | 90.15 | 55.1 | 2.56 |
| Ref. [64] [B1-WC] | 7.359 | 7.486 | 7.544 | 91.31 | 51.9 | 2.85 |
| Along a (Å) | Along b (Å) | Along c (Å) | |
|---|---|---|---|
| Ref. [6] [Exp.] | 1.861 (15)–1.940 (16) | 1.741 (15)–2.119 (15) | 1.736 (10)–2.176 (10) |
| Ref. [49] [Exp.] | 1.860 (11)–1.926 (11) | 1.813 (17)–2.051 (17) | 1.713 (11)–2.197 (11) |
| This work [PBE] | 1.919–1.934 | 1.820–2.093 | 1.793–2.189 |
| Ref. [47] [PBE] | 1.916–1.940 | 1.819–2.102 | 1.791–2.178 |
| Ref. [65] [PBE] | 1.865–1.937 | 1.785–2.087 | 1.795–2.166 |
| Ref. [61] [B3LYP] | 1.90–1.91 | 1.77–2.15 | 1.75–2.22 |
| Ref. [66] [PBE+U] | 1.89–1.90 | 1.84–2.04 | 1.80–2.10 |
| n | |||
|---|---|---|---|
| 1 | 0.1250 | 4.01 | 3.72 |
| 2 | 0.0625 | 2.77 | 2.57 |
| 3 | 0.0417 | 2.46 | 2.23 |
| 4 | 0.0313 | 2.32 | 2.13 |
| 5 | 0.0250 | 2.25 | 2.10 |
| 2 × 2 | 0.0313 | 2.64 | 2.27 |
| 3 × 2 | 0.0208 | 2.34 | 2.02 |
| N. of V | (eV) (per vac.) | ||
|---|---|---|---|
| V | 1 | 2.27 | 0.00 |
| V | 2 | 4.20 (2.10) | 1.04 |
| V | 2 | 4.48 (2.24) | 0.41 |
| V | 3 | 6.16 (2.05) | 0.00 |
| V | 1 | 1.40 | 0.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trioni, M.I.; Cargnoni, F.; Americo, S.; Soave, R. New Insight into the Electronic and Magnetic Properties of Sub-Stoichiometric WO3: A Theoretical Perspective. Crystals 2024, 14, 372. https://doi.org/10.3390/cryst14040372
Trioni MI, Cargnoni F, Americo S, Soave R. New Insight into the Electronic and Magnetic Properties of Sub-Stoichiometric WO3: A Theoretical Perspective. Crystals. 2024; 14(4):372. https://doi.org/10.3390/cryst14040372
Chicago/Turabian StyleTrioni, Mario Italo, Fausto Cargnoni, Stefano Americo, and Raffaella Soave. 2024. "New Insight into the Electronic and Magnetic Properties of Sub-Stoichiometric WO3: A Theoretical Perspective" Crystals 14, no. 4: 372. https://doi.org/10.3390/cryst14040372
APA StyleTrioni, M. I., Cargnoni, F., Americo, S., & Soave, R. (2024). New Insight into the Electronic and Magnetic Properties of Sub-Stoichiometric WO3: A Theoretical Perspective. Crystals, 14(4), 372. https://doi.org/10.3390/cryst14040372