Structural, Vibrational Spectroscopic and Theoretical (DFT) Studies of 4-Chloro- and 5-Chloro-7-azaindole-3-carbaldehydes




Abstract

1. Introduction
2. Materials and Methods
2.1. Preparation of 5Cl7AICA Crystals
2.2. X-ray Structure Determination
2.3. Spectroscopic Measurements
2.4. Theoretical Methods
3. Results and Discussion
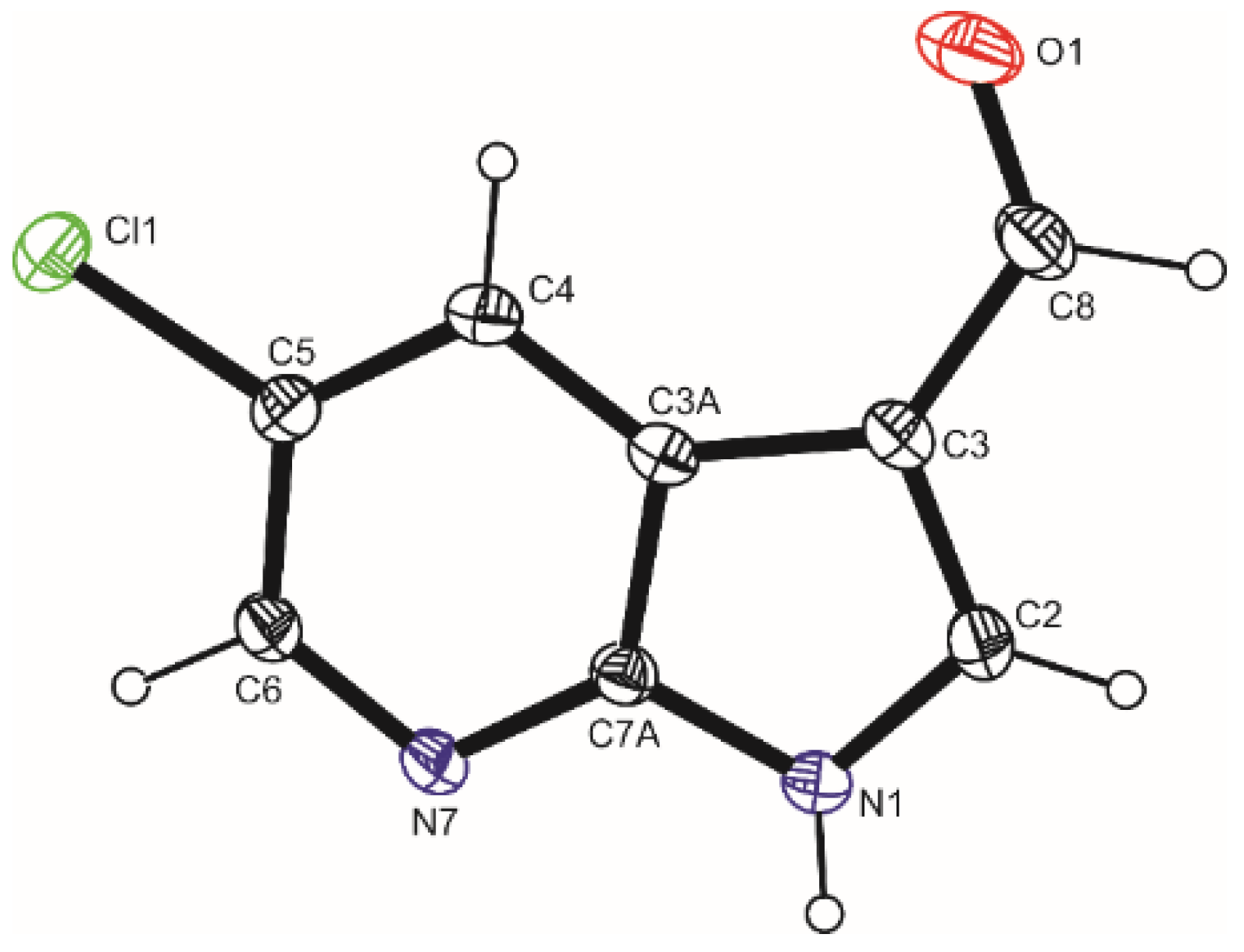
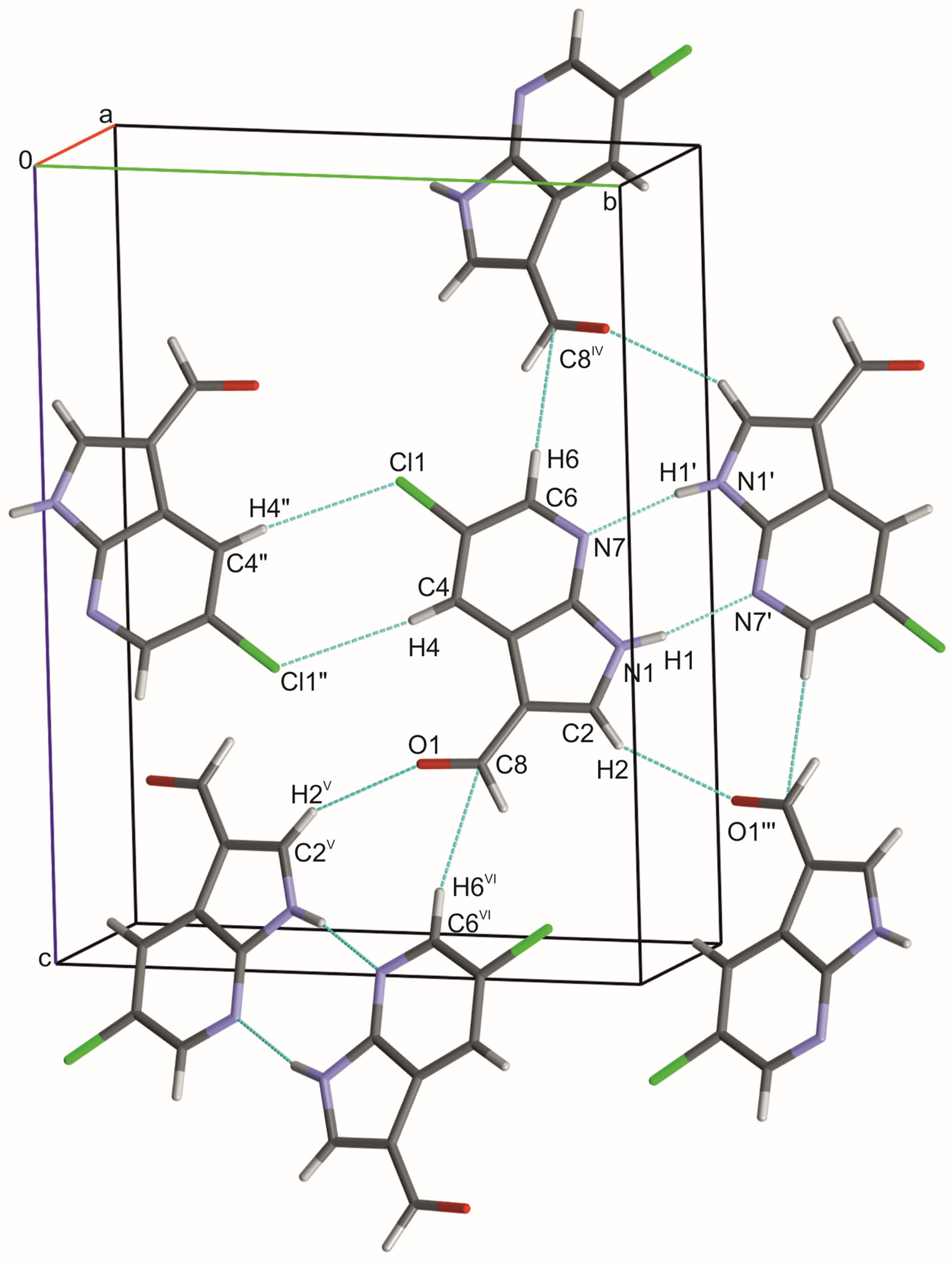
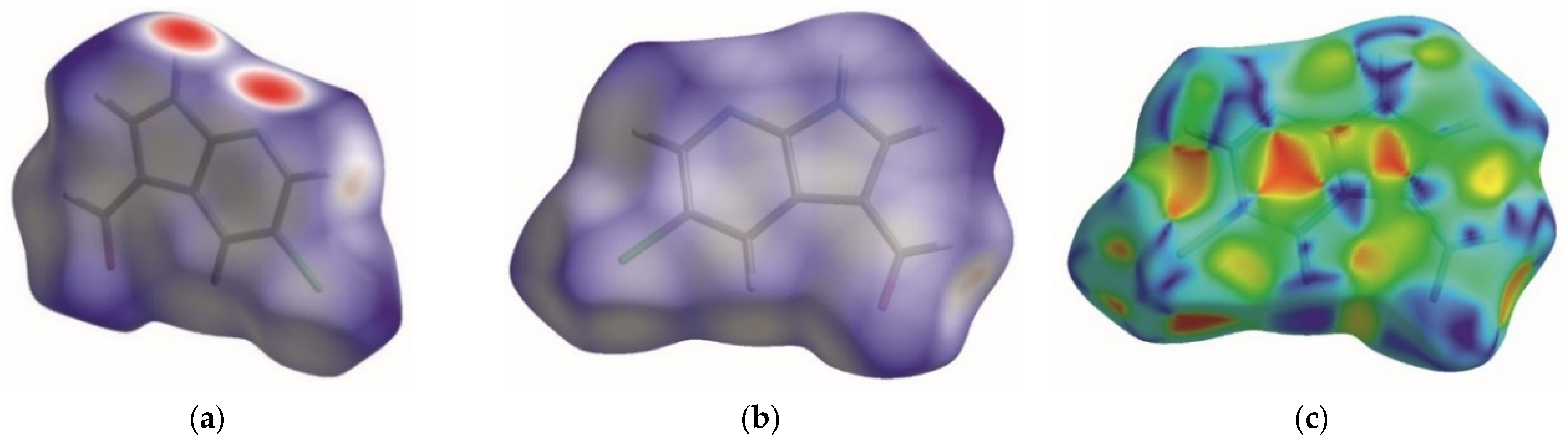
3.1. Crystal Structure of 5Cl7AICA
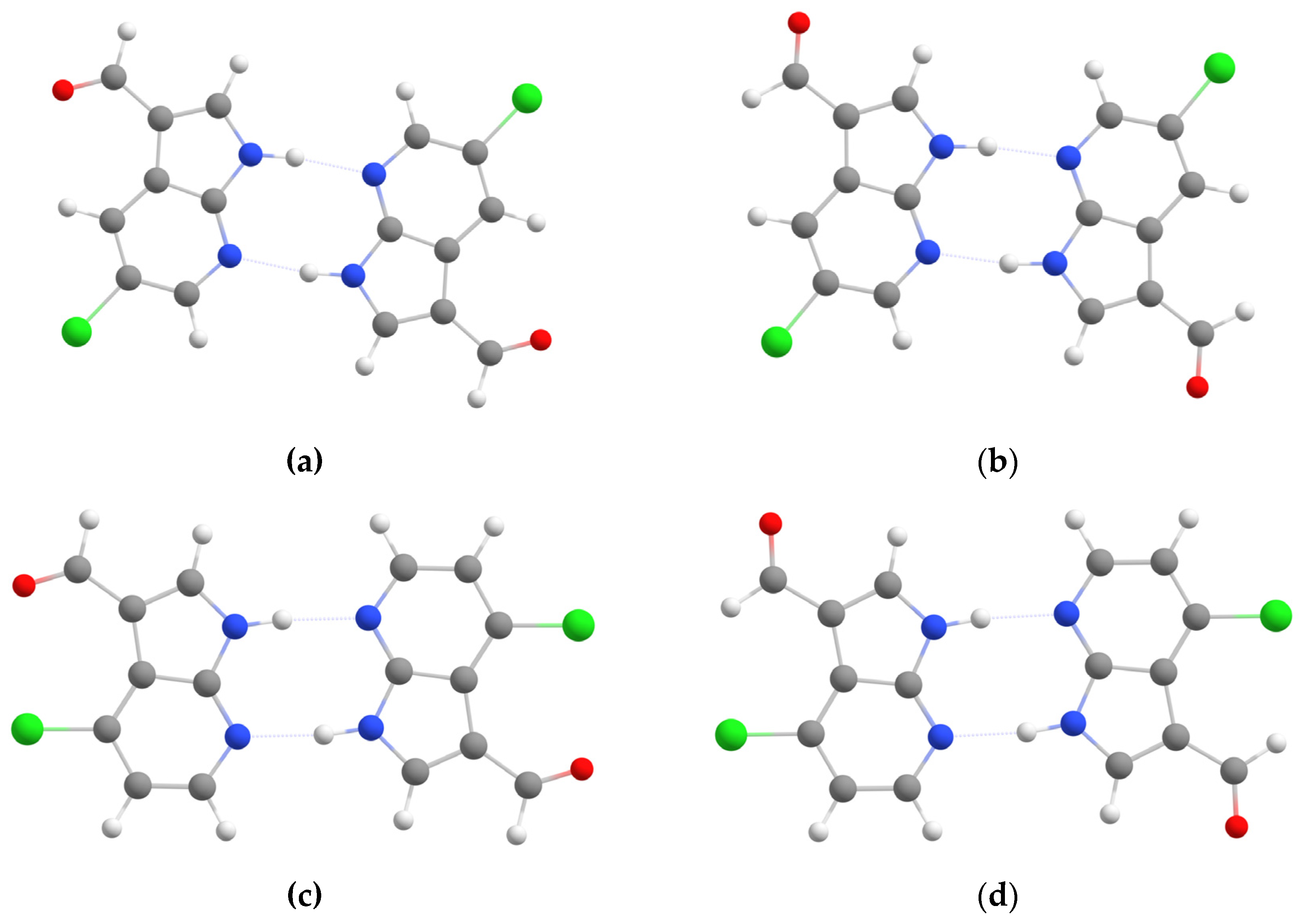
3.2. Structural Analysis of 5Cl7AICA and 4Cl7AICA Dimers Using DFT Methods
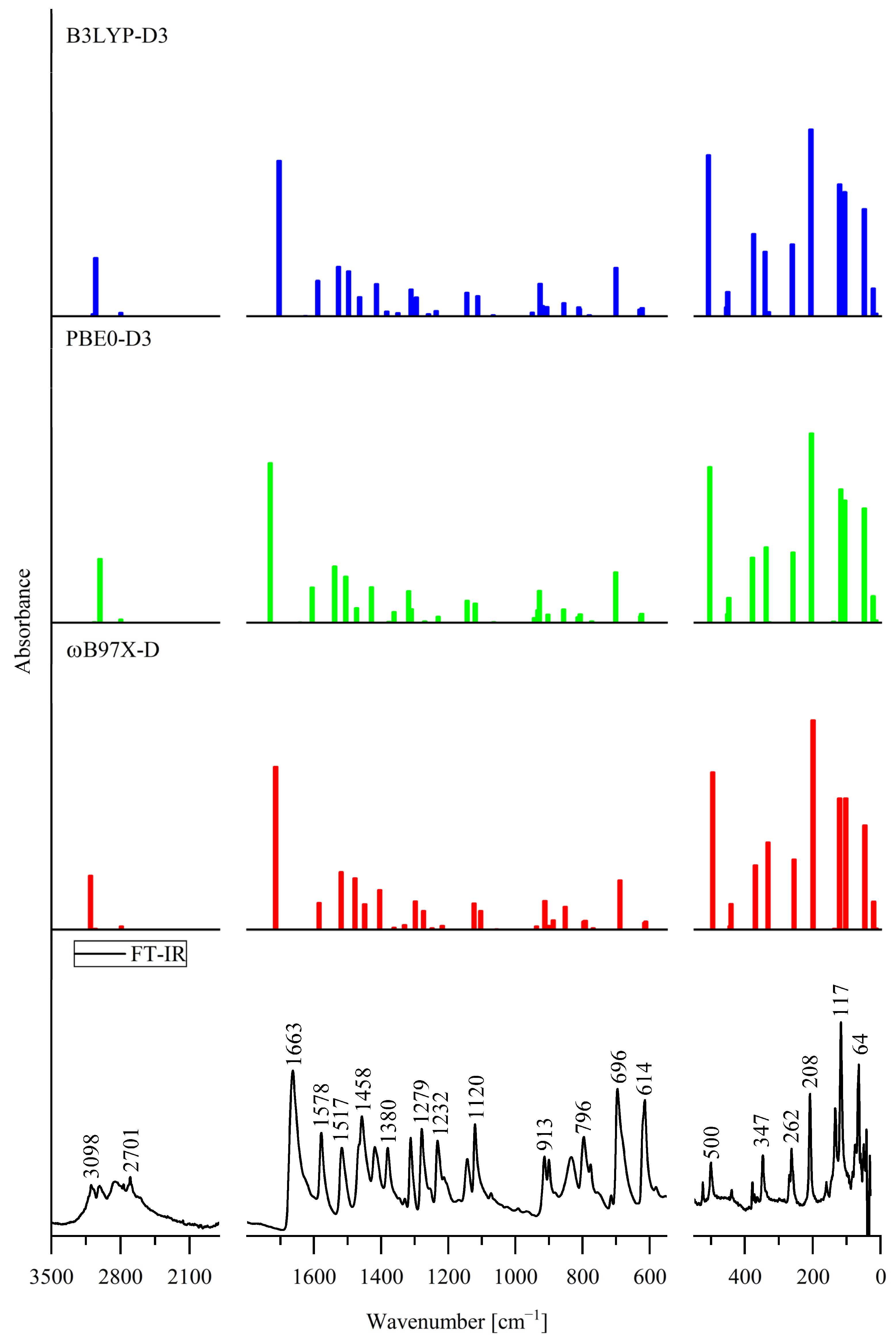
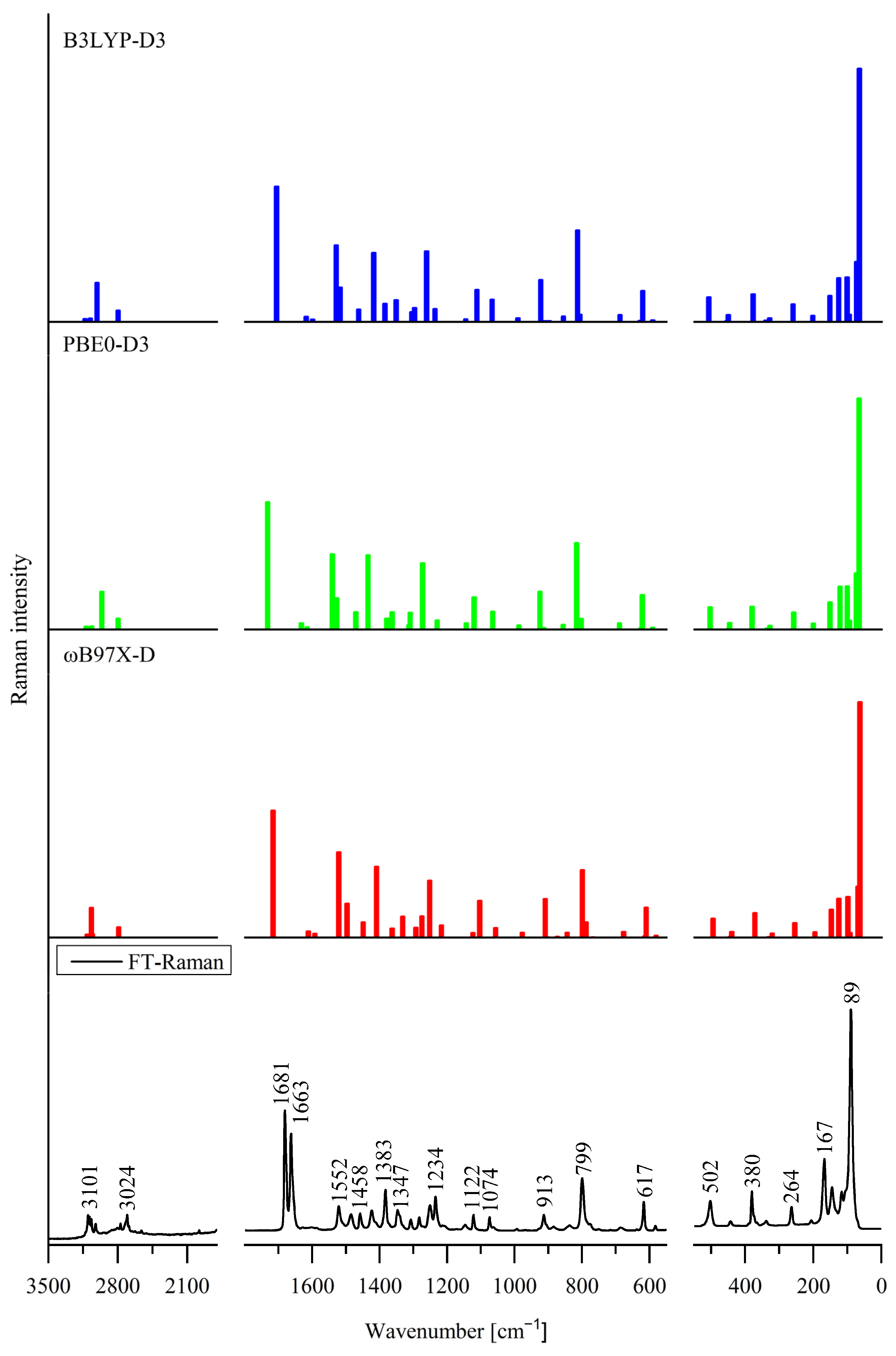
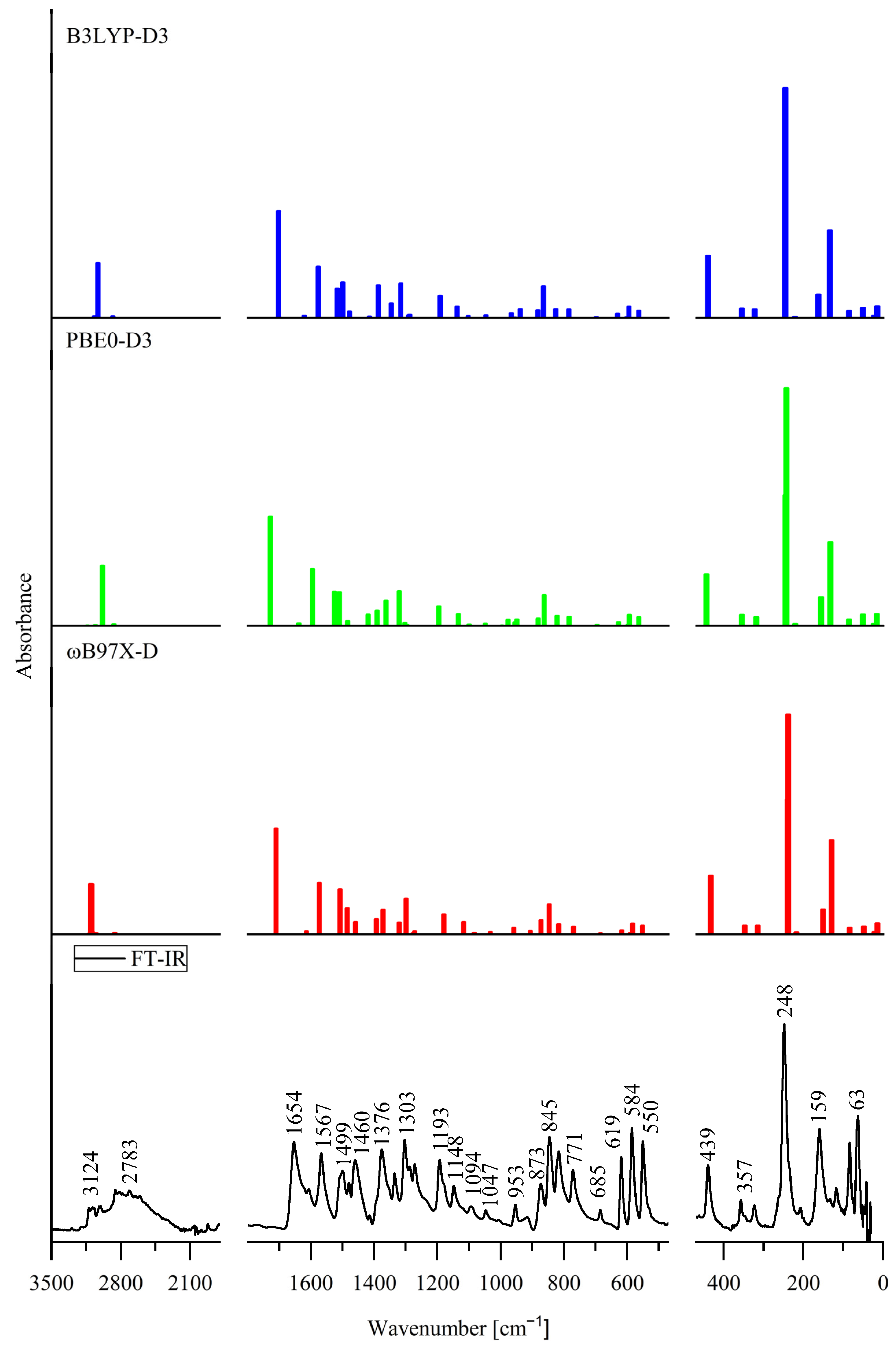
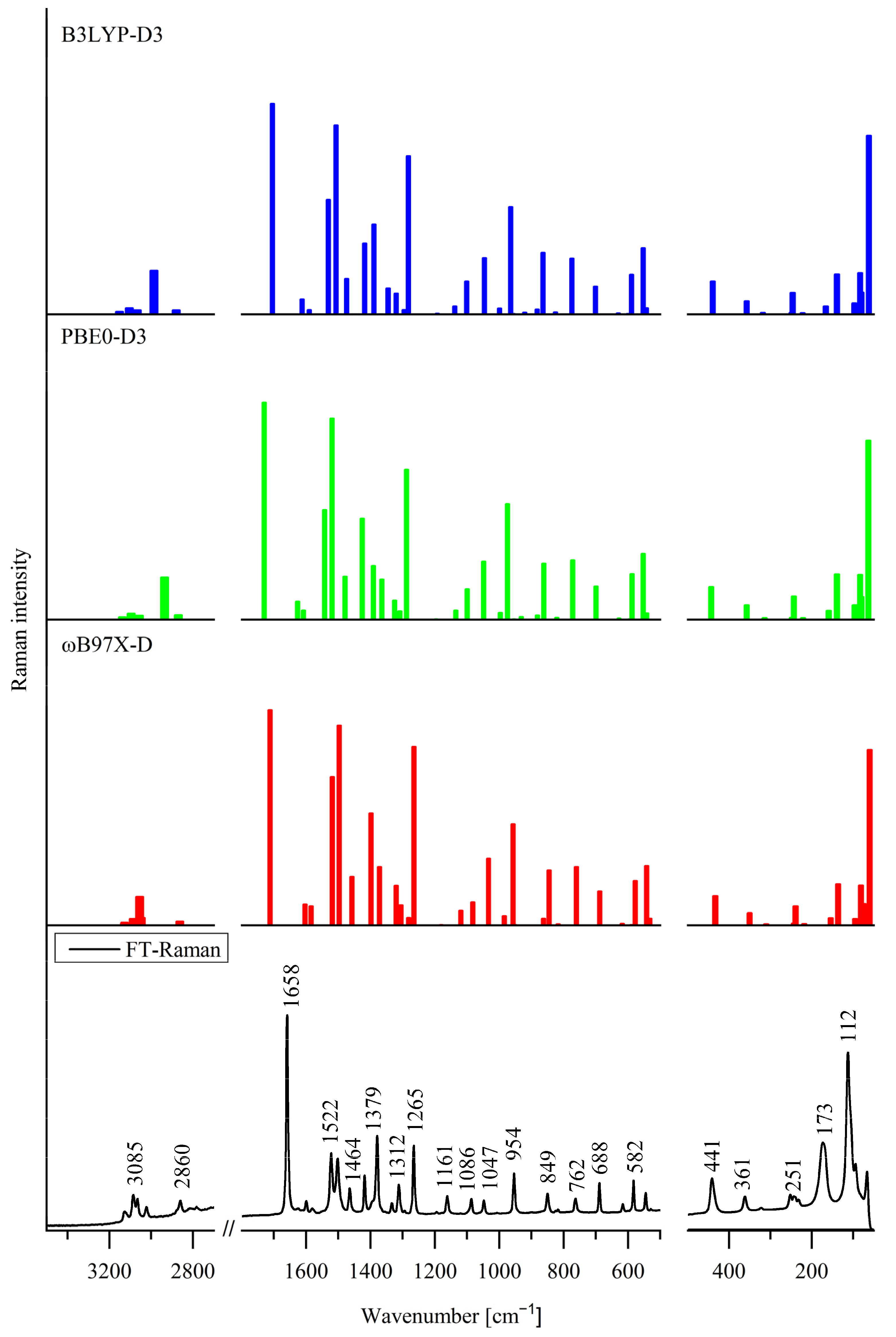
3.3. Effect of Internal Rotation of the Aldehyde Group on Vibrational Spectra of 5Cl7AICA and 4Cl7AICA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Motati, D.R.; Amaradhi, R.; Ganesh, T. Azaindole therapeutic agents. Bioorg. Med. Chem. 2020, 28, 115830. [Google Scholar] [CrossRef]
- Urvashi; Senthil Kumar, J.B.; Das, P.; Tandon, V. Development of Azaindole-Based Frameworks as Potential Antiviral Agents and Their Future Perspectives. J. Med. Chem. 2022, 65, 6454–6495. [Google Scholar] [CrossRef] [PubMed]
- Abimbola Salubi, C.; Abbo, H.S.; Jahed, N.; Titinchi, S. Medicinal chemistry perspectives on the development of piperazine-containing HIV-1 inhibitors. Bioorg. Med. Chem. 2024, 99, 117605. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ying, Z.; Huang, Y.; Li, Y.; Hu, M.; Kang, K.; Wang, H.; Shao, J.; Wu, G.; Yu, Y.; et al. Synthesis and structure-activity optimization of 7-azaindoles containing aza-β-amino acids targeting the influenza PB2 subunit. Eur. J. Med. Chem. 2023, 250, 115185. [Google Scholar] [CrossRef] [PubMed]
- Reddi, B.; Kishor, C.; Jangam, A.; Bala, S.; Rajeswari, B.; Batchu, U.R.; Gundla, R.; Addlagatta, A. Regioselectivity in inhibition of peptide deformylase from Haemophilus influenzae by 4- vs 5-azaindole hydroxamic acid derivatives: Biochemical, struc-tural and antimicrobial studies. Bioorg. Chem. 2022, 128, 106095. [Google Scholar] [CrossRef] [PubMed]
- Fang, G.; Chen, H.; Cheng, Z.; Tang, Z.; Wan, Y. Azaindole derivatives as potential kinase inhibitors and their SARs elucidation. Eur. J. Med. Chem. 2023, 258, 115621. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Gao, X.; Duan, X.; Liang, H.; Gao, M.; Dong, D.; Guo, C.; Huang, L. Design, synthesis and SAR of novel 7-azaindole derivatives as potential Erk5 kinase inhibitor with anticancer activity. Bioorg. Med. Chem. 2023, 95, 117503. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Chaudhary, A.; Sachdeva, M. An insight into the structure–activity relationship studies of anticancer medicinal attributes of 7-azaindole derivatives: A review. Future Med. Chem. 2023, 15, 24. [Google Scholar] [CrossRef] [PubMed]
- Gorecki, L.; Muthna, D.; Merdita, S.; Andrs, M.; Kucera, T.; Havelek, R.; Muckova, L.; Kobrlova, T.; Soukup, J.; Krupa, P.; et al. 7-Azaindole, 2,7-diazaindole, and 1H-pyrazole as core structures for novel anticancer agents with potential chemosensitizing properties. Eur. J. Med. Chem. 2022, 240, 114580. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.H.; Jang, D.J. Proton Transfer of Excited 7-Azaindole in Reverse-Micellar Methanol Nanopools: Even Faster than in Bulk Methanol. J. Phys. Chem. B 2005, 109, 8049–8052. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S. Concerted-asynchronous reaction path of the excited-state double proton transfer in the 7-azaindole homodimer and 6H-indolo[2,3-b]quinoline/7-azaindole heterodimer. Chem. Phys. 2018, 515, 768–778. [Google Scholar] [CrossRef]
- Wang, C.; He, F.; Sun, K.; Guo, K.; Lu, S.; Wu, T.; Gao, X.; Fang, M. Identification and characterization of 7-azaindole derivatives as inhibitors of the SARS-CoV-2 spike-hACE2 protein interaction. Int. J. Biol. Macromol. 2023, 244, 125182. [Google Scholar] [CrossRef] [PubMed]
- Ariza, M.; Kolb, H.C.; Moechars, D.; Rombouts, F.; Andrés, J.I. Tau Positron Emission Tomography (PET) Imaging: Past, Pre-sent, and Future. J. Med. Chem. 2015, 58, 4365–4382. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Wang, Z.; Yang, G.; Yang, H. A Novel Electrochemical Sensor for the Determination of Aflatoxin B1 Based on Polymerized 4-Amino-7-azaindole-Nitrogen-Doped Graphene. Int. J. Electrochem. Sci. 2020, 15, 11092–11101. [Google Scholar] [CrossRef]
- Cardoza, S.; Das, P.; Tandon, V. Pd-Catalyzed Sequential Arylation of 7-Azaindoles: Aggregate-Induced Emission of Tetra-Aryl 7-Azaindoles. J. Org. Chem. 2019, 84, 14015–14029. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yue, Z.Z.; Feng, J.M.; He, Q.; Miao, Z.H.; Yang, C.H. Design and synthesis of pyrido[3,2-α]carbazole derivatives and their analogues as potent antitumour agents. Eur. J. Med. Chem. 2013, 66, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Morzyk-Ociepa, B.; Szmigiel, K.; Turowska-Tyrk, I.; Michalska, D. X-ray crystal structure, infrared, Raman and density functional studies of 7-azaindole-3-carboxaldehyde. J. Mol. Struct. 2017, 1128, 186–194. [Google Scholar] [CrossRef]
- Morzyk-Ociepa, B.; Szmigiel, K.; Turowska-Tyrk, I.; Malik-Gajewska, M.; Banach, J.; Wietrzyk, J. New mono- and dinuclear complexes of 7-azaindole-3-carboxaldehyde with palladium(II): Crystal structure, IR and Raman spectra, DFT calculations and in vitro antiproliferative activity. Polyhedron 2018, 153, 88–98. [Google Scholar] [CrossRef]
- Morzyk-Ociepa, B.; Szmigiel-Bakalarz, K.; Nentwig, M.; Oeckler, O.; Malik-Gajewska, M.; Turlej, E.; Wietrzyk, J.; Michalska, D. Platinum(II) and copper(II) complexes of 7-azaindole-3-carboxaldehyde: Crystal structures, IR and Raman spectra, DFT calcu-lations and in vitro antiproliferative activity of the platinum(II) complex. Inorg. Chim. Acta 2019, 490, 68–77. [Google Scholar] [CrossRef]
- Smith, B.R.; Eastman, C.M.; Njardarson, J.T. Beyond C, H, O, and N! Analysis of the Elemental Composition of U.S. FDA Approved Drug Architectures. J. Med. Chem. 2014, 57, 9764–9773. [Google Scholar] [CrossRef] [PubMed]
- Rigaku Oxford Diffraction. CrysAlis PRO; Rigaku Oxford Diffraction Ltd.: Oxfordshire, UK, 2015. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Kleemiss, F.; Dolomanov, O.V.; Bodensteiner, M.; Peyerimhoff, N.; Midgley, L.; Bourhis, L.J.; Genoni, A.; Malaspina, L.A.; Jayatilaka, D.; Spencer, J.L.; et al. Accurate crystal structures and chemical properties from NoSpherA2. Chem. Sci. 2021, 12, 1675–1692. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment—Olex2 dissected. Acta Cryst. 2015, A71, 59–75. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system—Version 5.0. WIREs Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2000, 53, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 ele-ments H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wavefunctions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Chemcraft. Graphical Software for Visualization of Quantum Chemistry Computations. Version 1.8, Build 682. Available online: https://www.chemcraftprog.com (accessed on 27 February 2024).
- Zapata Trujillo, J.C.; McKemmish, L.K. Model chemistry recommendations for scaled harmonic frequency calculations: A benchmark study. J. Phys. Chem. 2023, A127, 1715–1735. [Google Scholar] [CrossRef] [PubMed]
- Legler, C.R.; Brown, N.R.; Dunbar, R.A.; Harness, M.D.; Nguyen, K.; Oyewole, O.; Collier, W.B. Scaled Quantum Mechanical scale factors for vibrational calculations using alternate polarized and augmented basis sets with the B3LYP density functional calculation model. Spectrochim. Acta A 2015, 145, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Morzyk-Ociepa, B.; Dysz, K.; Turowska-Tyrk, I.; Michalska, D. X-ray crystal structure, vibrational spectra and DFT calculations of 3-chloro-7-azaindole: A case of dual N–H⋯N hydrogen bonds in dimers. Spectrochim. Acta 2015, 136A, 405–415. [Google Scholar] [CrossRef]
- Hazra, D.K.; Mukherjee, A.K.; Helliwell, M.; Mukherjee, M. Topological features and electronic structure of 4-chloro-1H-pyrrolo[2{,}3-b]pyridine: Experimental charge density analysis and DFT studies. CrystEngComm 2012, 14, 993–1000. [Google Scholar] [CrossRef]
- Morzyk-Ociepa, B.; Dysz, K.; Turowska-Tyrk, I.; Michalska, D. Crystal structures, vibrational spectra and DFT calculations of five halogeno-derivatives of 7-azaindole (3Br7AI, 4Br7AI, 4Cl7AI, 3Br4Cl7AI and 5Br3Cl7AI): A comparative study. J. Mol. Struct. 2018, 1152, 386–398. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond in Structural Chemistry and Biology; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Cryst. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Janiak, C. A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands. J. Chem. Soc. Dalton Trans. 2000, 3885–3896. [Google Scholar] [CrossRef]
- Tureski, R.E.; Tanski, J.M. 2-bromo-5-fluorobenzaldehyd. Acta Cryst. 2013, E69, o1246. [Google Scholar] [CrossRef]
- Dreyer, J. Unraveling the structure of hydrogen bond stretching mode infrared absorption bands: An anharmonic density functional theory study on 7-azaindole dimers. J. Chem. Phys. 2007, 127, 054309. [Google Scholar] [CrossRef]
- Morzyk-Ociepa, B.; Dysz, K.; Turowska-Tyrk, I.; Michalska, D. Reinvestigation of the crystal structure, vibrational spectroscopic studies and DFT calculations of 5-bromo-7-azaindole with dual N–H···N hydrogen bonds in dimers. J. Mol. Struct. 2015, 1101, 91–100. [Google Scholar] [CrossRef]
- Hiremath, C.S.; Tonannavar, J. Vibrational assignments and effect of aldehyde rotation on substituents in some trisubstituted benzaldehydes. Spectrochim. Acta 2009, 73A, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Hiremath, C.S.; Yenagi, J.; Tonannavar, J. FT-Raman and infrared spectra and vibrational assignments for 3-chloro-4-methoxybenzaldehyde, as supported by ab initio, hybrid density func-tional theory and normal coordinate calculations. Spectrochim. Acta 2007, 68A, 710–717. [Google Scholar] [CrossRef]
- Binoy, J.; Joe, I.H.; Jayakumar, V.S. Changes in the vibrational spectral modes by the nonbonded interactions in the NLO crystal vanillin. J. Raman Spectrosc. 2005, 36, 1091–1100. [Google Scholar] [CrossRef]
- Rogojerov, M.; Keresztury, G.; Jordanov, B. Vibrational spectra of partially oriented molecules having two conformers in nematic and isotropic solutions: Furfural and 2-chlorobenzaldehyde. Spectrochim. Acta 2005, 61A, 1661–1670. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D–H | H···A | D···A | D–H···A | |
|---|---|---|---|---|
| N1–H1∙∙∙N7′, exp. | 1.024(17) | 1.880(17) | 2.8834(14) | 165.8(13) |
| N1–H1∙∙∙N7, ωB97X-D | 1.03 | 1.88 | 2.908 | 172 |
| N1–H1∙∙∙N7, PBE0-D3 | 1.04 | 1.84 | 2.870 | 172 |
| N1–H1∙∙∙N7, B3LYP-D3 | 1.04 | 1.87 | 2.898 | 172 |
| C2–H2∙∙∙O1′′′ exp. | 1.087(12) | 2.554(12) | 3.5285(17) | 148.8(9) |
| C6–H6∙∙∙C8iv exp. | 1.081(13) | 2.752(14) | 3.8051(19) | 164.7(10) |
| C4–H4∙∙∙Cl1″ exp. | 1.080(11) | 2.789(12) | 3.8317(13) | 162.2(10) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mucha, W.; Bąkowicz, J.; Malik, M.; Morzyk-Ociepa, B. Structural, Vibrational Spectroscopic and Theoretical (DFT) Studies of 4-Chloro- and 5-Chloro-7-azaindole-3-carbaldehydes. Crystals 2024, 14, 631. https://doi.org/10.3390/cryst14070631
Mucha W, Bąkowicz J, Malik M, Morzyk-Ociepa B. Structural, Vibrational Spectroscopic and Theoretical (DFT) Studies of 4-Chloro- and 5-Chloro-7-azaindole-3-carbaldehydes. Crystals. 2024; 14(7):631. https://doi.org/10.3390/cryst14070631
Chicago/Turabian StyleMucha, Wiktor, Julia Bąkowicz, Magdalena Malik, and Barbara Morzyk-Ociepa. 2024. "Structural, Vibrational Spectroscopic and Theoretical (DFT) Studies of 4-Chloro- and 5-Chloro-7-azaindole-3-carbaldehydes" Crystals 14, no. 7: 631. https://doi.org/10.3390/cryst14070631
APA StyleMucha, W., Bąkowicz, J., Malik, M., & Morzyk-Ociepa, B. (2024). Structural, Vibrational Spectroscopic and Theoretical (DFT) Studies of 4-Chloro- and 5-Chloro-7-azaindole-3-carbaldehydes. Crystals, 14(7), 631. https://doi.org/10.3390/cryst14070631