Evaluation of Exfoliated Graphite to Graphene in Polyamide 66 Using Novel High Shear Elongational Flow

 , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
3. Results and Discussion
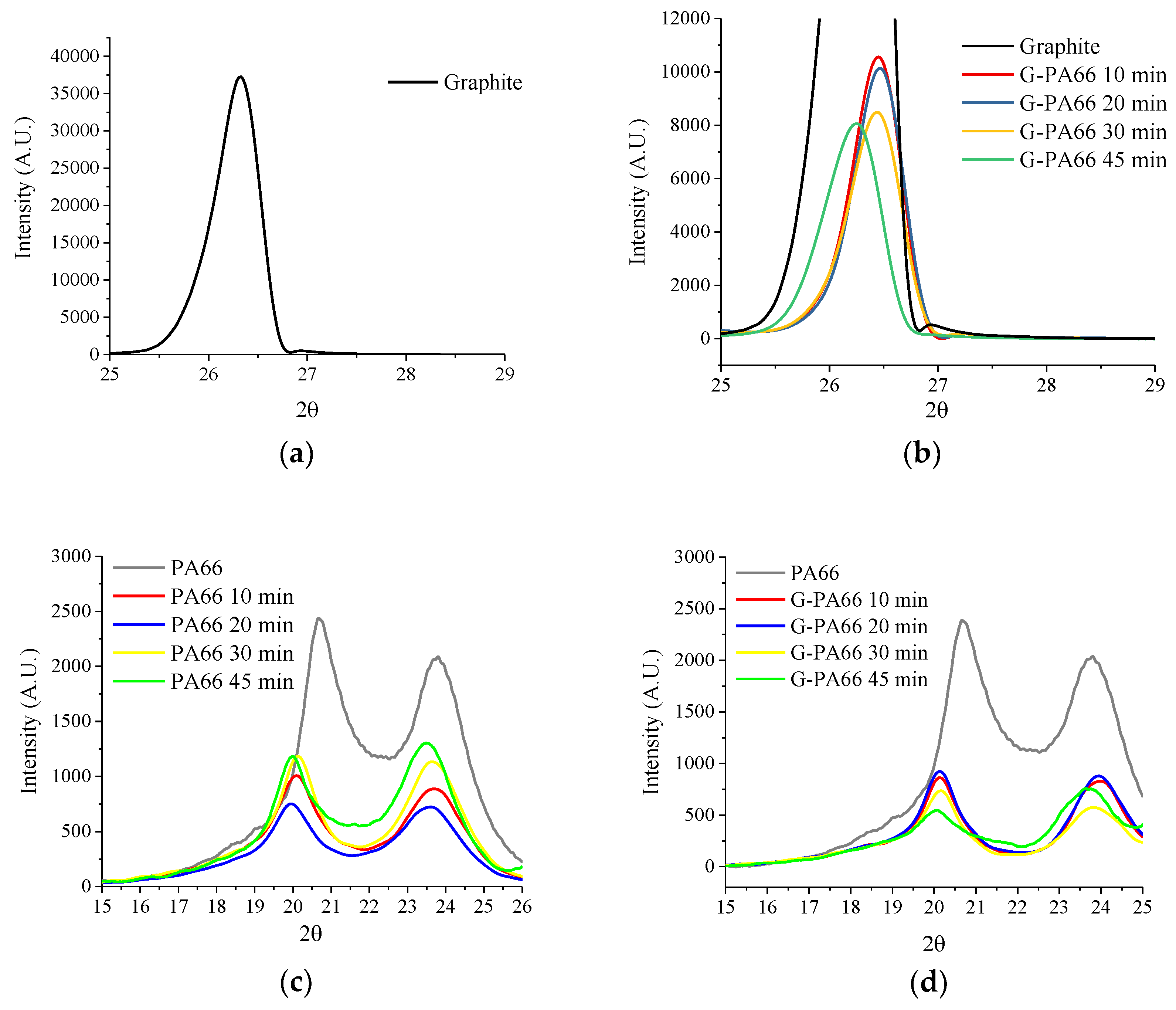
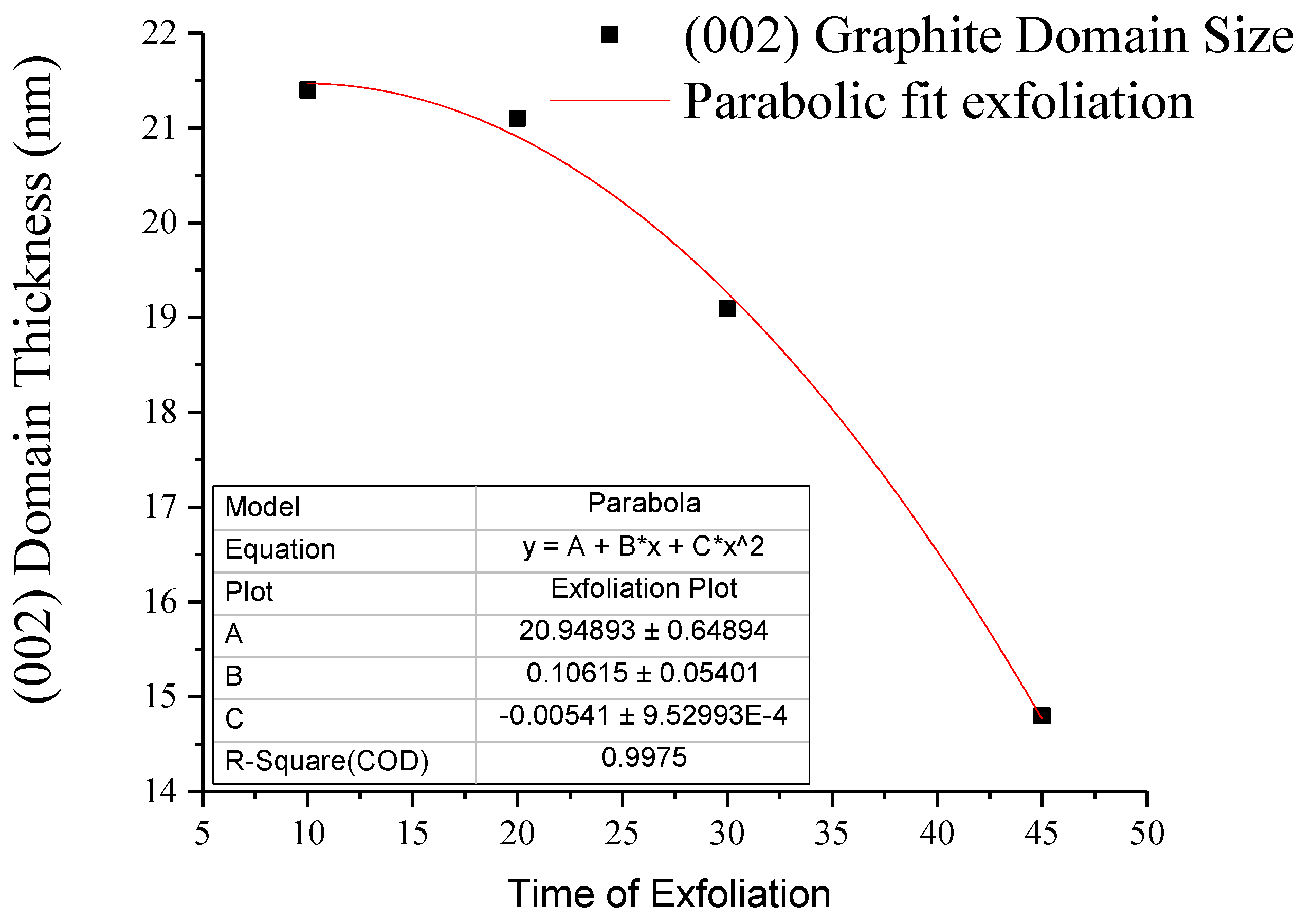
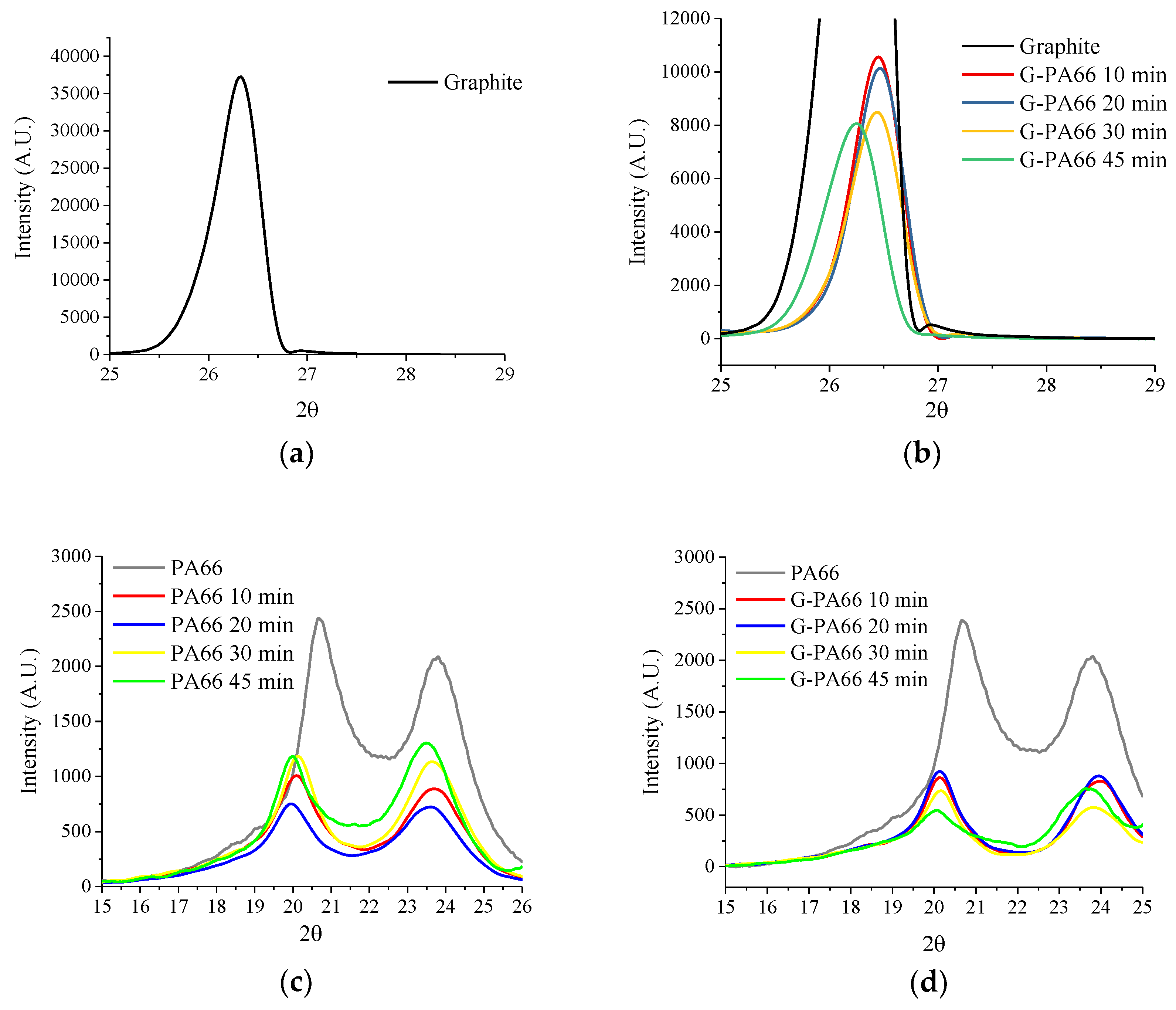
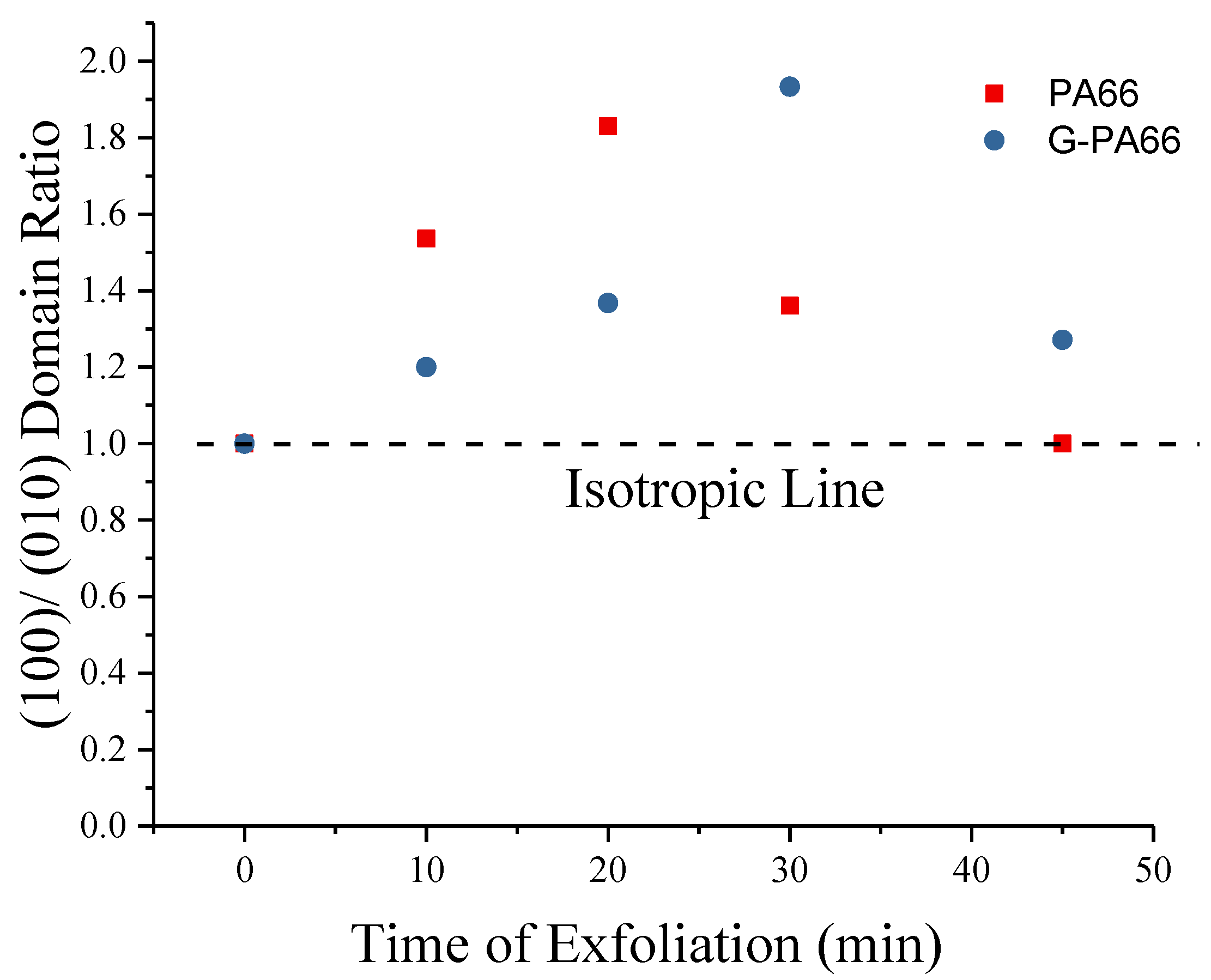
3.1. Wide Angle X-ray Diffraction
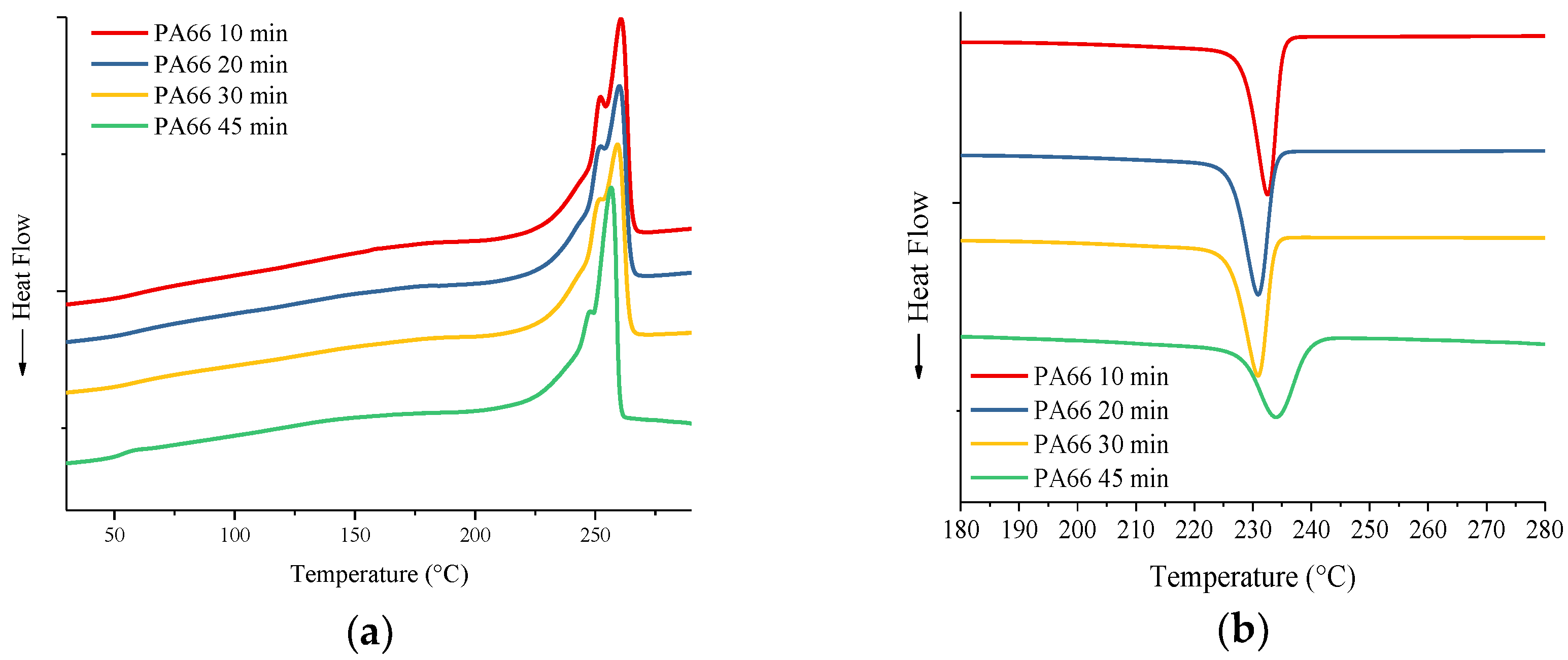
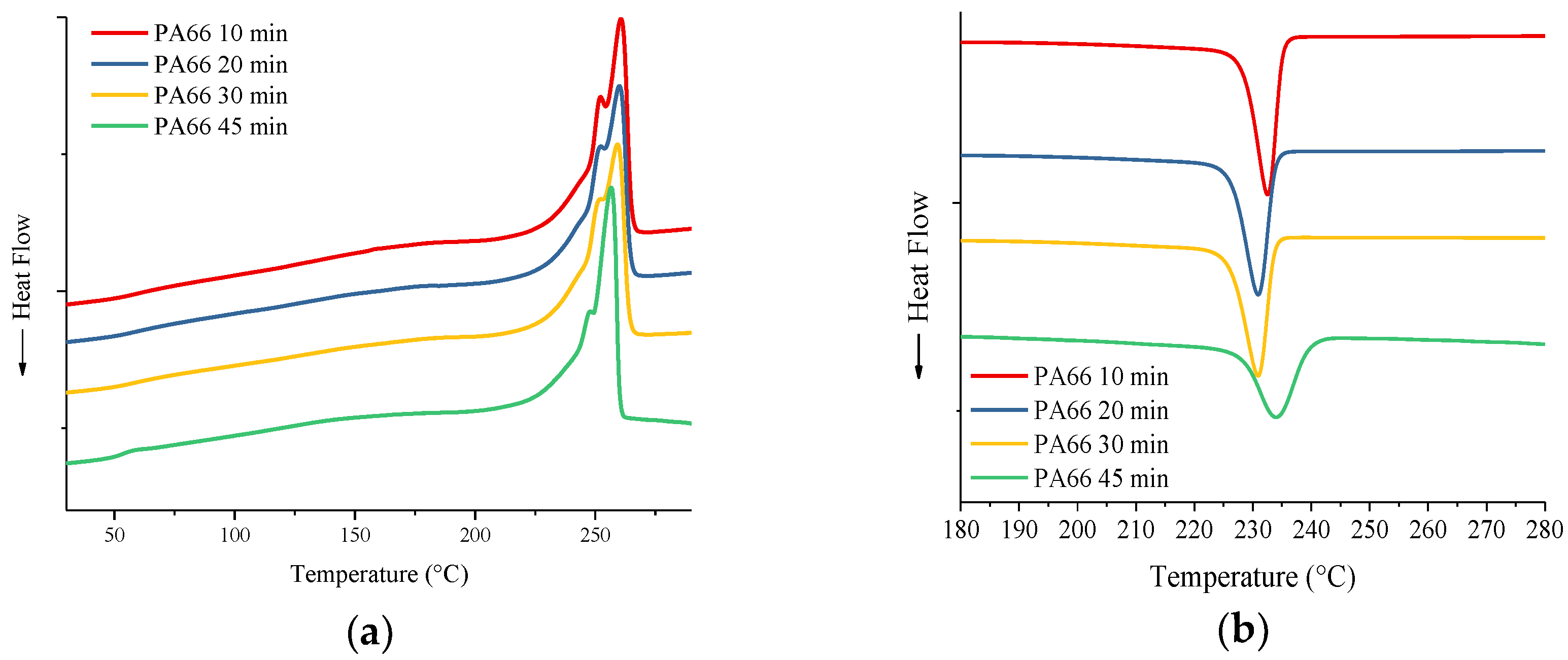
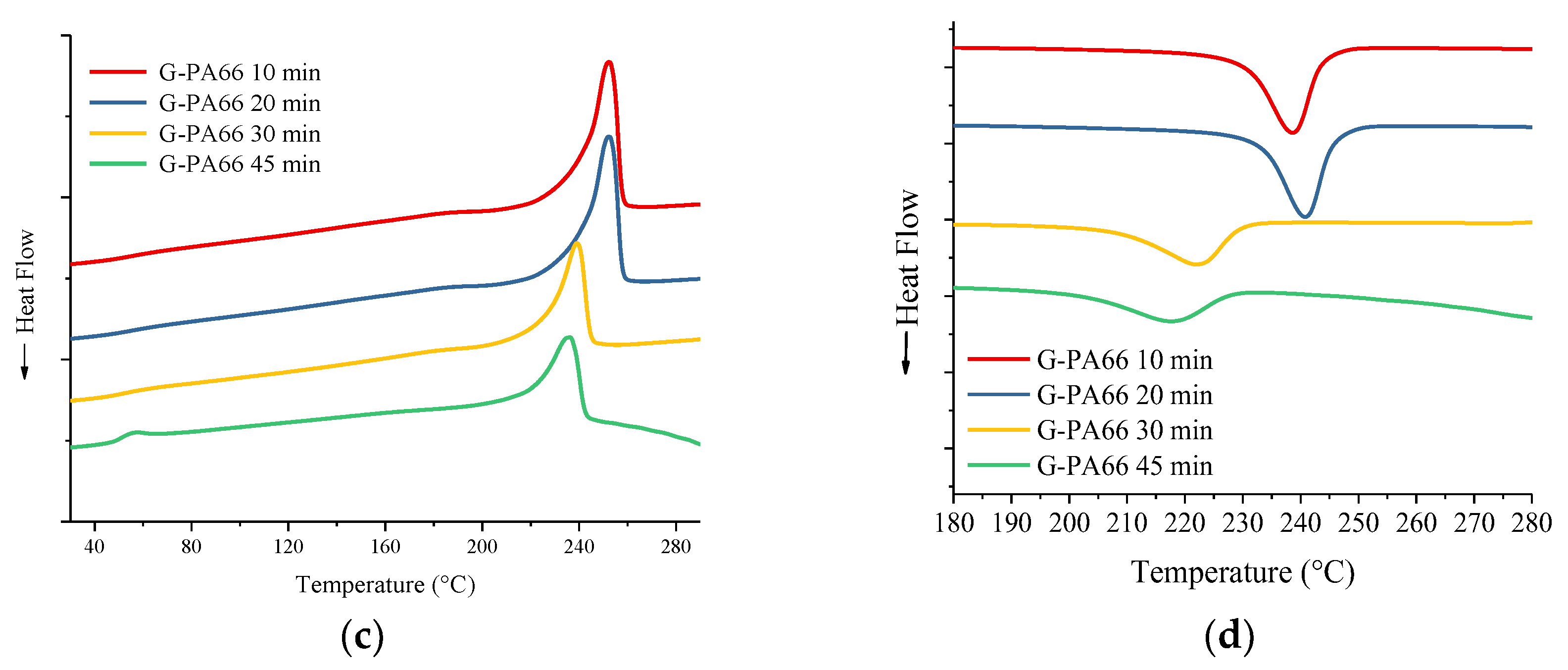
3.2. Differential Scanning Calorimetry
3.3. Scanning Electron Microscopy
3.4. Transmission Electron Microscopy
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yan, Z.; Nika, D.L.; Balandin, A.A. Thermal properties of graphene and few-layer graphene: Applications in electronics. IET Circuits Devices Syst. 2015, 9, 4–12. [Google Scholar] [CrossRef]
- Miculescu, M.; Thakur, V.K.; Miculescu, F.; Voicu, S.I. Graphene-based polymer nanocomposite membranes: A review. Polym. Adv. Technol. 2016, 27, 844–859. [Google Scholar] [CrossRef]
- Kim, H.; Abdala, A.A.; Macosko, C.W. Graphene/Polymer Nanocomposites. Macromolecules 2010, 43, 6515–6530. [Google Scholar] [CrossRef]
- Perreault, F.; de Faria, A.F.; Elimelech, M. Environmental applications of graphene-based nanomaterials. Chem. Soc. Rev. 2015, 44, 5861–5896. [Google Scholar] [CrossRef] [PubMed]
- Talat, M.; Srivastava, O. Deployment of New Carbon Nanostructure: Graphene for Drug Delivery and Biomedical Applications. In Advances in Nanomaterials; Springer: New Delhi, India, 2016; pp. 383–395. [Google Scholar]
- Hao, F.; Fang, D.; Xu, Z. Mechanical and thermal transport properties of graphene with defects. Appl. Phys. Lett. 2011, 99, 041901. [Google Scholar] [CrossRef]
- Chen, Y.; Tan, C.; Zhang, H.; Wang, L. Two-dimensional graphene analogues for biomedical applications. Chem. Soc. Rev. 2015, 44, 2681–2701. [Google Scholar] [CrossRef] [PubMed]
- Frank, I.W.; Tanenbaum, D.M.; van der Zande, A.M.; McEuen, P.L. Mechanical properties of suspended graphene sheets. J. Vacuum Sci. Technol. B Microelectron. Nanometer Struct. 2007, 25, 2558–2561. [Google Scholar] [CrossRef]
- Yang, S.; Brüller, S.; Wu, Z.-S.; Liu, Z.; Parvez, K.; Dong, R.; Richard, F.; Samorì, P.; Feng, X.; Müllen, K. Organic radical-assisted electrochemical exfoliation for the scalable production of high-quality graphene. J. Am. Chem. Soc. 2015, 137, 13927–13932. [Google Scholar] [CrossRef]
- Bélanger, D. One-Pot Electrochemical Exfoliation and Functionalization of Graphene Sheets. In Proceedings of the 229th ECS Meeting, San Diego, CA, USA, 29 May–2 June 2016. [Google Scholar]
- Wassei, J.K.; Kaner, R.B. Oh, the Places You’ll Go with Graphene. Acc. Chem. Res. 2013, 46, 2244–2253. [Google Scholar] [CrossRef]
- Chua, C.K.; Pumera, M. Chemical reduction of graphene oxide: A synthetic chemistry viewpoint. Chem. Soc. Rev. 2014, 43, 291–312. [Google Scholar] [CrossRef]
- Manna, K.; Hsieh, C.-Y.; Lo, S.-C.; Li, Y.-S.; Huang, H.-N.; Chiang, W.-H. Graphene and graphene-analogue nanosheets produced by efficient water-assisted liquid exfoliation of layered materials. Carbon 2016, 105, 551–555. [Google Scholar] [CrossRef]
- Haar, S.; Ciesielski, A.; Clough, J.; Yang, H.; Mazzaro, R.; Richard, F.; Conti, S.; Merstorf, N.; Cecchini, M.; Morandi, V. Graphene: A Supramolecular Strategy to Leverage the Liquid-Phase Exfoliation of Graphene in the Presence of Surfactants: Unraveling the Role of the Length of Fatty Acids (Small 14/2015). Small 2015, 11, 1736. [Google Scholar] [CrossRef]
- Vicarelli, L.; Heerema, S.J.; Dekker, C.; Zandbergen, H.W. Controlling Defects in Graphene for Optimizing the Electrical Properties of Graphene Nanodevices. ACS Nano 2015, 9, 3428–3435. [Google Scholar] [CrossRef] [PubMed]
- Oxfall, H.; Rondin, J.; Bouquey, M.; Muller, R.; Rigdahl, M.; Rychwalski, R.W. Elongational flow mixing for manufacturing of graphite nanoplatelet/polystyrene composites. J. Appl. Polym. Sci. 2013, 128, 2679–2686. [Google Scholar] [CrossRef]
- Ellingham, T.; Duddleston, L.; Turng, L.-S. Sub-critical gas-assisted processing using CO2 foaming to enhance the exfoliation of graphene in polypropylene + graphene nanocomposites. Polymer 2017, 117, 132–139. [Google Scholar] [CrossRef]
- Ibarra-Gómez, R.; Muller, R.; Bouquey, M.; Rondin, J.; Serra, C.A.; Hassouna, F.; Mouedden, Y.E.; Toniazzo, V.; Ruch, D. Processing of nanocomposites PLA/graphite using a novel elongational mixing device. Polym. Eng. Sci. 2015, 55, 214–222. [Google Scholar] [CrossRef]
- Nosker, T.; Lynch, J.; Hendrix, J.; Kear, B.; Chiu, G.; Tse, S.; Rutgers State University of New Jersey. In situ exfoliation method to fabricate a graphene-reinforced polymer matrix composite (G-PMC). U.S. Patent 9,896,565, 20 February 2018. [Google Scholar]
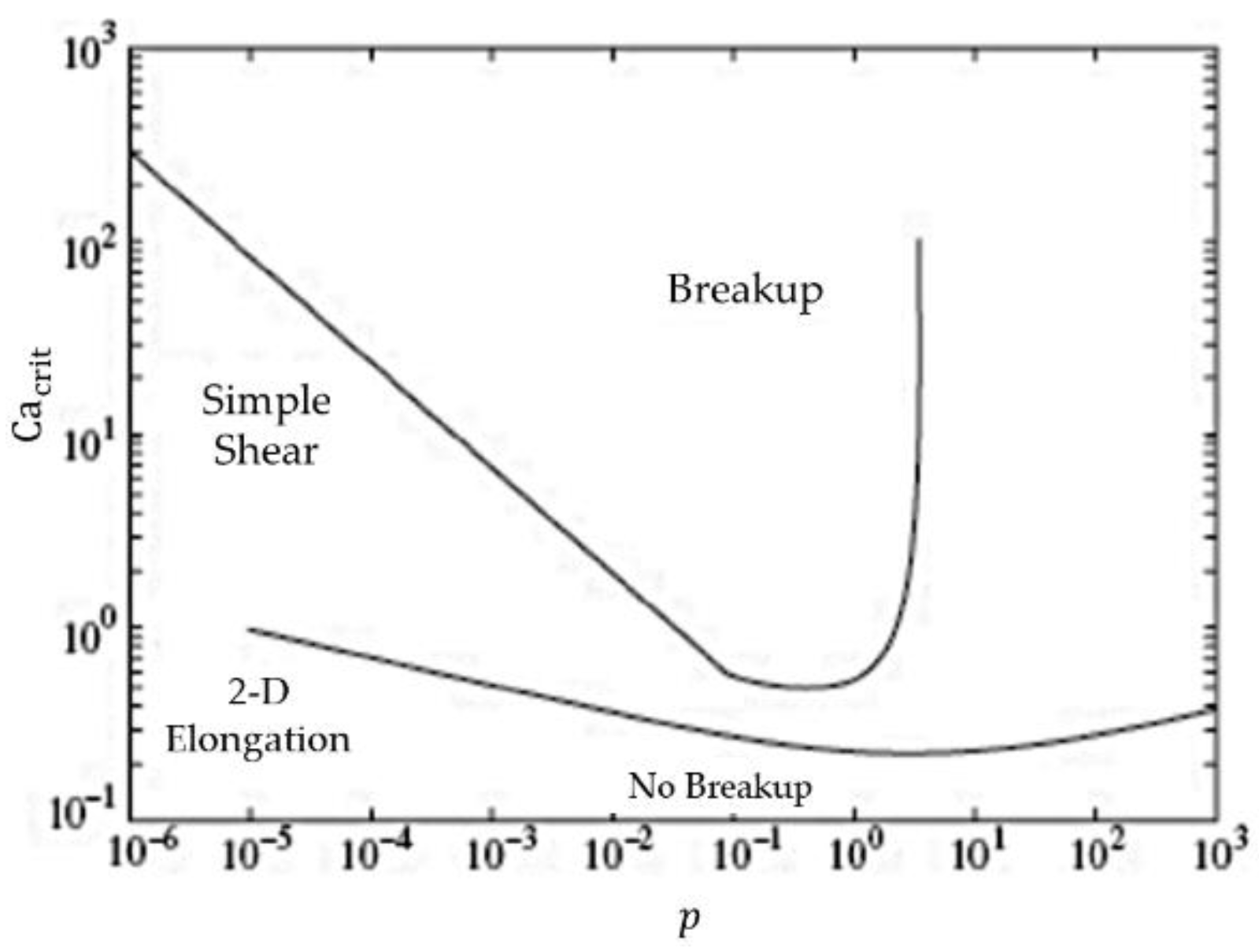
- Taylor, G.I. The Formation of Emulsions in Definable Fields of Flow. Proc. R. Soc. Lond. 1934, 146, 501–523. [Google Scholar] [CrossRef] [Green Version]
- Rumscheidt, F.D.; Mason, S.G. Particle motions in sheared suspensions XII. Deformation and burst of fluid drops in shear and hyperbolic flow. J. Colloid Sci. 1961, 16, 238–261. [Google Scholar] [CrossRef]
- Aguilera, J.M.; Simpson, R.; Welti-Chanes, J.; Aguirre, D.B.; Barbosa-Cánovas, G.V. Food Engineering Interfaces; Springer: New York, NY, USA, 2010. [Google Scholar]
- Grace, H.P. Dispersion phenomena in high viscosity immiscible fluid systems and application of static mixers as dispersion devices in such systems. Chem. Eng. Commun. 1982, 14, 225–277. [Google Scholar] [CrossRef]
- Delhaes, P. Graphite and Precursors; Taylor & Francis: London, UK, 2000. [Google Scholar]
- Varin, R.A.; Czujko, T.; Wronski, Z.S. Nanomaterials for Solid State Hydrogen Storage; Springer: New York, NY, USA, 2009. [Google Scholar]
- Goto, M.; Smith, J.M.; McCoy, B.J. Parabolic profile approximation (linear driving-force model) for chemical reactions. Chem. Eng. Sci. 1990, 45, 443–448. [Google Scholar] [CrossRef]
- Christofides, P.D.; Daoutidis, P. Nonlinear control of diffusion-convection-reaction processes. Comput. Chem. Eng. 1996, 20, S1071–S1076. [Google Scholar] [CrossRef]
- Buzanowski, M.A.; Yang, R.T. Approximations for intraparticle diffusion rates in cyclic adsorption and desorption. Chem. Eng. Sci. 1991, 46, 2589–2598. [Google Scholar] [CrossRef]
- Jones, N.A.; Atkins, E.D.T.; Hill, M.J. Investigation of solution-grown, chain-folded lamellar crystals of the even-even nylons: 6 6, 8 6, 8 8, 10 6, 10 8, 10 10, 12 6, 12 8, 12 10, and 12 12. J. Polym. Sci. Part B Polym. Phys. 2000, 38, 1209–1221. [Google Scholar] [CrossRef]
- Starr, F.W.; Schrøder, T.B.; Glotzer, S.C. Molecular Dynamics Simulation of a Polymer Melt with a Nanoscopic Particle. Macromolecules 2002, 35, 4481–4492. [Google Scholar] [CrossRef] [Green Version]
- Lopes dos Santos, J.M.B.; Peres, N.M.R.; Castro Neto, A.H. Graphene Bilayer with a Twist: Electronic Structure. Phys. Rev. Lett. 2007, 99, 256802. [Google Scholar] [CrossRef]
- Haberkorn, H.; Illers, K.H.; Simak, P. Molekülordnung und Kristallinität in Polyhexamethylenadipamid. Colloid Polym. Sci. 1979, 257, 820–840. [Google Scholar] [CrossRef]
- Khanna, Y.P. Overview of transition phenomenon in nylon 6. Macromolecules 1992, 25, 3298–3300. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, B.; Man, R.; Peng, Z. Isothermal crystallization kinetics of in situ Nylon 6/graphene composites by differential scanning calorimetry. Polym. Eng. Sci. 2014, 54, 2610–2616. [Google Scholar] [CrossRef]
- Aoyama, S.; Park, Y.T.; Ougizawa, T.; Macosko, C.W. Melt crystallization of poly(ethylene terephthalate): Comparing addition of graphene vs. carbon nanotubes. Polymer 2014, 55, 2077–2085. [Google Scholar] [CrossRef]
- Kim, S.; Poostforush, M.; Kim, J.; Lee, S. Thermal diffusivity of in-situ exfoliated graphite intercalated compound/polyamide and graphite/polyamide composites. Express Polym. Lett. 2012, 6, 476–484. [Google Scholar] [CrossRef]
- Sawyer, L.C. Polymer Microscopy; Springer: Dordrecht, The Netherlands, 2012. [Google Scholar]
- Stankovich, S.; Dikin, D.A.; Piner, R.D.; Kohlhaas, K.A.; Kleinhammes, A.; Jia, Y.; Wu, Y.; Nguyen, S.T.; Ruoff, R.S. Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide. Carbon 2007, 45, 1558–1565. [Google Scholar] [CrossRef]
- Eda, G.; Fanchini, G.; Chhowalla, M. Large-area ultrathin films of reduced graphene oxide as a transparent and flexible electronic material. Nat. Nanotechnol. 2008, 3, 270. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.; Cheng, H.-M.; Enoki, T.; Gogotsi, Y.; Hurt, R.H.; Koratkar, N.; Kyotani, T.; Monthioux, M.; Park, C.R.; Tascon, J.M.D.; et al. All in the graphene family—A recommended nomenclature for two-dimensional carbon materials. Carbon 2013, 65, 1–6. [Google Scholar] [CrossRef]
- Shenoy, V.B.; Reddy, C.D.; Zhang, Y.-W. Spontaneous Curling of Graphene Sheets with Reconstructed Edges. ACS Nano 2010, 4, 4840–4844. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mixing time (min) | Control materials | Composite materials |
|---|---|---|
| 10 | PA66 | G-PA66 |
| 20 | PA66 | G-PA66 |
| 30 | PA66 | G-PA66 |
| 45 | PA66 | G-PA66 |
| Materials | Mixing time (min) | PA66 d (100) (nm) | PA66 (100) Domain (nm) | PA66 d (010)/(110) (nm) | PA66 (010)/(110) Domain (nm) | Graphite d (002) (nm) | Graphite (002) Domain (nm) |
|---|---|---|---|---|---|---|---|
| Graphite | - | - | - | - | - | 0.3344 | 62.7 (4.1) |
| PA66 | - | 0.4407 | 4.4 (0.2) | 0.3878 | 4.4 (0.7) | - | - |
| PA66 | 10 | 0.4427 | 8.6 (0.4) | 0.3753 | 5.6 (0.2) | - | - |
| PA66 | 20 | 0.4453 | 9.7 (0.6) | 0.3768 | 5.3 (0.2) | - | - |
| PA66 | 30 | 0.4414 | 8.3 (0.4) | 0.3758 | 6.1 (0.2) | - | - |
| PA66 | 45 | 0.4435 | 2.9 (0.9) | 0.3796 | 2.9 (0.5) | - | - |
| G-PA66 | 10 | 0.4404 | 8.4 (0.4) | 0.3719 | 7.0 (0.2) | 0.3360 | 21.4 (0.3) |
| G-PA66 | 20 | 0.4405 | 9.3 (0.5) | 0.3720 | 6.8 (0.1) | 0.3358 | 21.1 (0.2) |
| G-PA66 | 30 | 0.4393 | 11.6 (0.8) | 0.3735 | 6.0 (0.1) | 0.3363 | 19.1 (0.2) |
| G-PA66 | 45 | 0.4423 | 8.9 (1.7) | 0.3772 | 7.0 (0.3) | 0.3386 | 14.8 (0.1) |
| Material | Mixing time (min) | Tg (°C) | Onset melting (°C) | Tm (°C) | ΔHm (J/g) | Onset crystallization (°C) | Tc (°C) | ΔHc (J/g) | Crystallinity % |
|---|---|---|---|---|---|---|---|---|---|
| PA66 | 10 | 60 | 246 | 261 | 72 | 235 | 233 | 70 | ~36 |
| PA66 | 20 | 56 | 244 | 260 | 69 | 233 | 231 | 68 | ~35 |
| PA66 | 30 | 57 | 243 | 259 | 71 | 233 | 231 | 66 | ~36 |
| PA66 | 45 | 53 | 245 | 256 | 73 | 238 | 234 | 59 | ~37 |
| G-PA66 | 10 | 56 | 241 | 252 | 97 | 243 | 239 | 85 | ~49 |
| G-PA66 | 20 | 57 | 243 | 254 | 103 | 245 | 241 | 94 | ~52 |
| G-PA66 | 30 | 51 | 226 | 239 | 69 | 229 | 222 | 74 | ~35 |
| G-PA66 | 45 | 50 | 218 | 236 | 48 | 227 | 217 | 38 | ~24 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hendrix, J.W.; Szeto, R.; Nosker, T.; Lynch-Branzoi, J.; Emge, T.J. Evaluation of Exfoliated Graphite to Graphene in Polyamide 66 Using Novel High Shear Elongational Flow. Polymers 2018, 10, 1399. https://doi.org/10.3390/polym10121399
Hendrix JW, Szeto R, Nosker T, Lynch-Branzoi J, Emge TJ. Evaluation of Exfoliated Graphite to Graphene in Polyamide 66 Using Novel High Shear Elongational Flow. Polymers. 2018; 10(12):1399. https://doi.org/10.3390/polym10121399
Chicago/Turabian StyleHendrix, Justin W., Ryan Szeto, Thomas Nosker, Jennifer Lynch-Branzoi, and Thomas J. Emge. 2018. "Evaluation of Exfoliated Graphite to Graphene in Polyamide 66 Using Novel High Shear Elongational Flow" Polymers 10, no. 12: 1399. https://doi.org/10.3390/polym10121399
APA StyleHendrix, J. W., Szeto, R., Nosker, T., Lynch-Branzoi, J., & Emge, T. J. (2018). Evaluation of Exfoliated Graphite to Graphene in Polyamide 66 Using Novel High Shear Elongational Flow. Polymers, 10(12), 1399. https://doi.org/10.3390/polym10121399